
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrbital analysis of bonding in diarylhalonium salts and relevance to periodic trends in structure and reactivity†
Shubhendu S.
Karandikar
a,
Avik
Bhattacharjee
a,
Bryan E.
Metze
a,
Nicole
Javaly
 a,
Edward J.
Valente
*b,
Theresa M.
McCormick
*a and
David R.
Stuart
*a
a,
Edward J.
Valente
*b,
Theresa M.
McCormick
*a and
David R.
Stuart
*a
aDepartment of Chemistry, Portland State University, Portland, OR 97201, USA. E-mail: dstuart@pdx.edu
bDepartment of Chemistry, University of Portland, Portland, OR 97203, USA
First published on 19th May 2022
Abstract
Diarylhalonium compounds provide new opportunities as reagents and catalysts in the field of organic synthesis. The three center, four electron (3c–4e) bond is a center piece of their reactivity, but structural variation among the diarylhaloniums, and in comparison with other λ3-iodanes, indicates that the model needs refinement for broader applicability. We use a combination of Density Functional Theory (DFT), Natural Bond Orbital (NBO) Theory, and X-ray structure data to correlate bonding and structure for a λ3-iodane and a series of diarylchloronium, bromonium, and iodonium salts, and their isoelectronic diarylchalcogen counterparts. This analysis reveals that the s-orbital on the central halogen atom plays a greater role in the 3c–4e bond than previously considered. Finally, we show that our revised bonding model and associated structures account for both kinetic and thermodynamic reactivity for both acyclic phenyl(mesityl)halonium and cyclic dibenzohalolium salts.
Introduction
Diarylhalonium salts continue to emerge as molecular scaffolds of importance in organic synthesis.1 Diaryliodonium salts have historically dominated research in this area and the hypervalent bond is a central design element for new reactions with these reagents/catalysts (Fig. 1a).2 Although known for some time,3 advances with the lighter halonium salts have only appeared in recent years,1c–g and notable differences in reactivity between diaryliodonium and lighter diarylbromonium and diarylchloronium compounds have been reported (Fig. 1b). For instance, Uchiyama and co-workers reported mesityl transfer to pyridine at room temperature via a chlorobenzene leaving group (Fig. 1b).1f Analogous aryl transfer from diaryliodonium salts to pyridine has not been reported under metal-free conditions, but rather requires higher temperature and a copper catalyst.4 Additionally, Wencel-Delord and co-workers recently described the generation and trapping of arynes with mild base from cyclic dibenzobromolium salts (Fig. 1b);1c and it was reported that the corresponding dibenziodolium salt was unreactive under identical conditions. Finally, Yoshida and co-workers recently demonstrated that dibenzobromolium triflate is an effective Lewis acid catalyst for the conjugate addition of indole to enones, whereas the corresponding iodolium salt does not act as a catalyst (Fig. 1b).1b In each case, substantial differences in reactivity are observed, and here we bring together empirical observations and theoretical insight to reconcile these differences.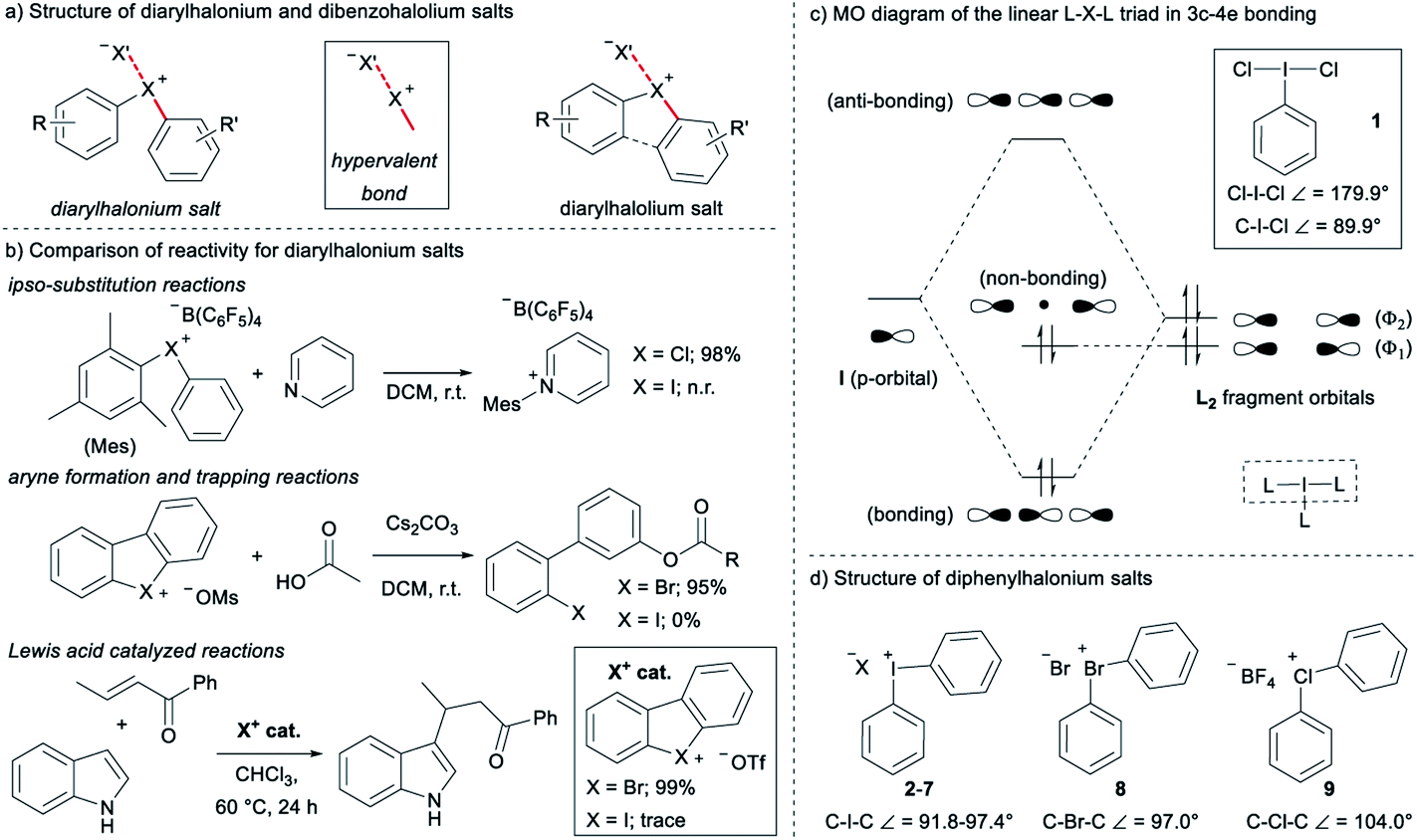 | ||
| Fig. 1 Structure of diarylhalonium salts and bonding models. | ||
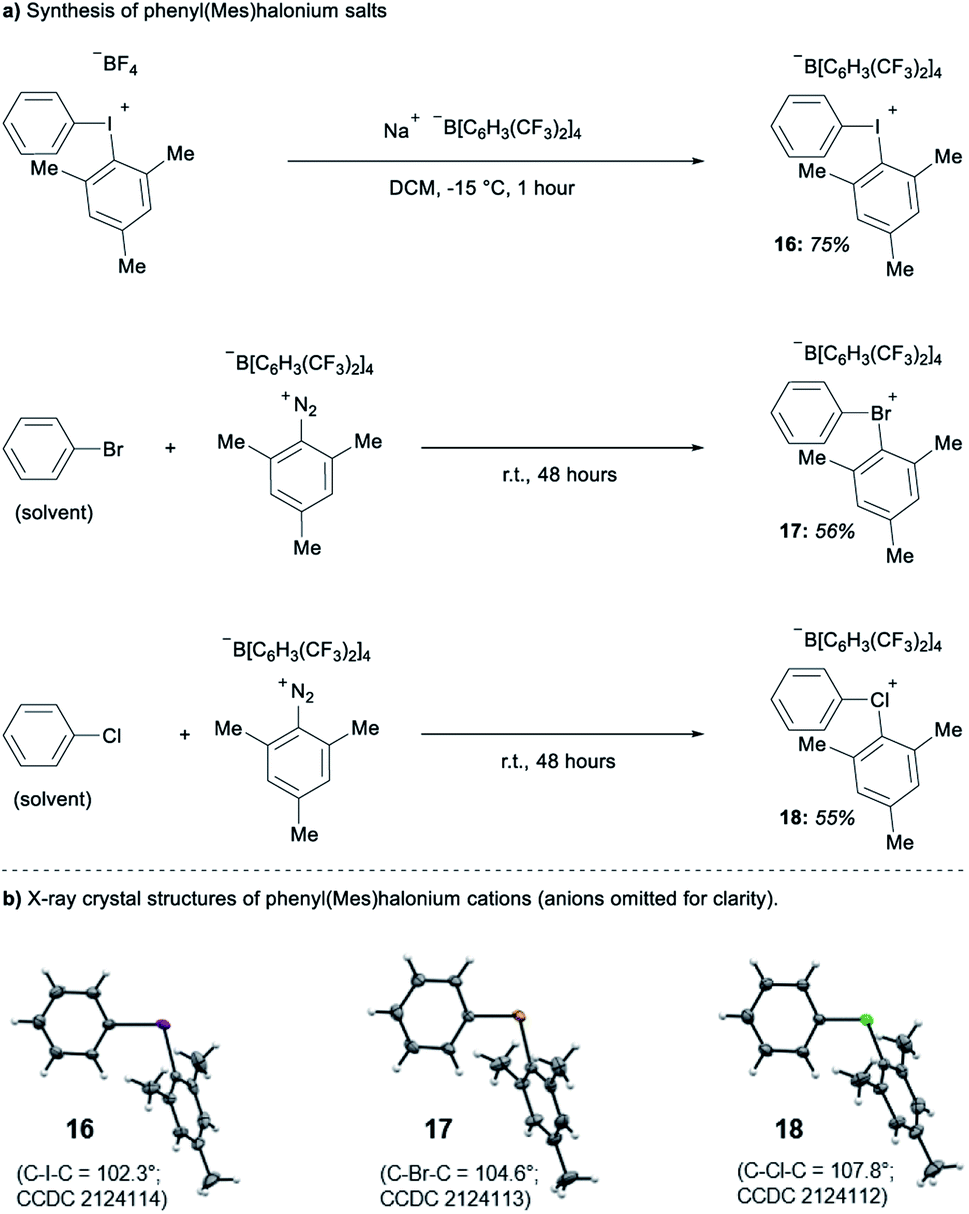 | ||
| Scheme 1 Synthesis and structures of 16–18. | ||
The hypervalent bond is a focal point of diarylhalonium chemistry. The three centre, four electron (3c–4e) bonding model is historically used to describe the orbital interactions in hypervalent compounds,5 including generally hypervalent iodine2a,c,6 and specifically diaryliodonium salts.2b In this model, when applied to hypervalent iodine, an unhybridized p-orbital on iodine interacts with two ligands in a linear L–I–L triad (Fig. 1c, dashed box). The part of the molecular orbital (MO) diagram for the L–I–L triad shows that, because of symmetry considerations, the iodine p-orbital interacts with the Φ2 ligand fragment, but not the Φ1 ligand fragment (Fig. 1c). Consequently, there are two electrons in a bonding MO and two electrons in a non-bonding MO that is ligand based (Fig. 1c). In 1, in which L = Cl and the Cl–I–Cl triad is symmetrical, similar Cl–I bond lengths (∼2.5 Å) are observed and the ligand-centred non-bonding electrons are shared equally between the two Cl atoms.7 Valence bond resonance is an alternative model used to describe the distribution of electrons in these systems.8 Moreover, the bond angles observed in the solid state for 1 reflect the p-orbital contribution in the 3c–4e bond. The Cl–I–Cl bond angle in 1 is 179.9° and the C–I–Cl bond angle is 89.9°,7 consistent with the iodine p-orbital dominating the orbital interaction with the chlorine ligands. Diaryliodonium salts broadly emulate these features and therefore are generally considered to be well described with the 3c–4e bond model, though some structural deviations have been observed.
The recent work of Uchiyama brings to light key structural differences between diaryliodonium and diarylchloronium salts, and a breakdown of the hypervalent bond was suggested for the latter.1f In diphenyliodonium salts 2–7 the solid-phase C–I–C bond angles, determined by single crystal X-ray diffraction, range from 91.8° (X = Br), which is similar to the C–I–Cl bond angle of 1, to 97.4° (X = PF6) depending on the counter anion (X− in Fig. 1d).9 Additionally, typical X⋯I and I–Cphenyl bond distances for the almost linear X⋯I–C fragment are ∼3 Å and 2 Å, respectively. Therefore, diaryliodonium salts 2–7 are believed to have highly unsymmetrical 3c–4e bonds in which the non-bonding electrons are expected to be located on the counter anion X.10 Very few of the lighter diarylhalonium salts have been characterized by X-ray diffraction, but two representative members provide evidence for periodic deviations from the 3c–4e bond. Diphenylbromonium bromide 8 has a C–Br–C bond angle of 97.0° and diphenylchloronium tetrafluoroborate 9 has a C–Cl–C bond angle of 104.0° (Fig. 1d).3a Collectively, there is considerable variation in C–X–C bond angles (12.2°) of the diphenylhalonium salt series,3a,9 which we suspect is the result of differences in bonding among this series and may also describe the aforementioned differences in reactivity.
Given the variation observed in C–X–C bond angles for diarylhalonium cations, we have investigated the halonium orbitals used for bonding and show a departure from the traditional 3c–4e bonding model that is typically suggested for these compounds. We used Density Functional Theory (DFT) and Natural Bond Orbital (NBO) Theory approaches to correlate bonding and structure in diarylhalonium salts and diarylchalcogens, which are isoelectronic to the diarylhalonium cations. In addition, we synthesized a series of analogous phenyl(mesityl)chloronium, bromonium, and iodonium salts, and cyclic dibenzochlorolium, bromolium, and iodolium salts, and assessed their kinetic and thermodynamic reactivity as a function of structure and bonding.
Results and discussion
Computational studies
The link between orbitals used in bonding and molecular geometry is a pillar of organic chemistry, which is largely based on Pauling's theory of orbital hybridization.11 A more nuanced view on the role of atomic orbitals in bonding was developed by Bent,12 and accurately describes deviations from ideal geometry (i.e., tetrahedral = 109.5°). The IUPAC Gold Book defines Bent's rule as: “In a molecule, smaller bond angles are formed between electronegative ligands since the central atom, to which the ligands are attached, tends to direct bonding hybrid orbitals of greater p-character towards its more electronegative substituents”.13 In other words, bond angles become larger as more s-orbital (spherical, non-directional) character is involved in bonding. Bent's rule has been used effectively to analyse bonding in other main group compounds,14 and here we explore its use to describe structure/reactivity periodic trends in the diarylhalonium series.We selected a range of compounds to test our hypothesis that the composition of orbitals can explain trends in the structure of diarylhalonium compounds (i.e., C–X–C bond angle). In addition to diarylhalonium salts, compound 1 was included in this study as the prototypical hypervalent iodine compound engaging in a traditional 3c–4e bond proposed to involve an unhybridized p-orbital (Fig. 1). A variety of diphenyliodonium salts are known with different counter anions and we included both halide and non-halide counter anions to assess their potential impact on structure (2–7, Fig. 1). Diphenylbromonium and diphenylchloronium 8 and 9 were selected to assess how the identity of the central atom impacts structure (Fig. 1). Finally, Bent previously discussed the relationship between isoelectronic molecules and structure,12 and accordingly we included several diphenyl and diaryl chalcogens, 10–15, which are isoelectronic with the diphenylhalonium cations. By design, compounds 1–15 used in our computational study have been previously characterized by X-ray diffraction and the C–E–C bond angle is known.3a,7,9,15
Energy minimized structures for compounds 1–15 were obtained in Gaussian 09,16 by DFT using B3LYP/cc-PVTZ, for the lighter atoms, and Def2QZVPP for Te and I.17 The computed C–E–C bond angles correlate well with the known bond angles from X-ray crystal structures.18 NBO analysis quantifies the composition of orbitals used in bonding.19 Using the NBO 3.1 module in Gaussian 09,16 we specifically assessed the orbitals on the central halogen (or chalcogen) directed toward the ligands involved in the hypervalent bond.20 We observed a negligible contribution from d-orbitals in our NBO analysis, which is consistent with previous studies on bonding in main group elements.14 Notably, the contribution from s- and p-orbitals to bonding with the ligand varies depending on the position of the central halogen, or chalcogen, in the periodic table (Table 1). The bonding in compound 1, which is a prototypical hypervalent λ3-iodane, is consistent with the traditional 3c–4e bond as the iodine orbital directed at the Cl ligands is essentially an unhybridized p-orbital (99.95% p-character; Table 1). This is also consistent with the 89.8° bond angle formed by the C–I–Cl linkage (Table 1). Our calculations reveal that the diaryliodonium salts 2–7 incorporate 5.5–9.2% s-character in the orbital on iodine that is directed at the phenyl group trans to the anion (X−), and consequently the C–I–C bond angle becomes more obtuse, consistent with Bent's rule (90.5–96.6°, Table 1). Notably, the identity of the counter anion (X−) does effect the C–I–C bond angle, which will be discussed in more detail below. The bromine atom of diphenylbromonium cation uses more s-character in bonding than the iodine in the analogous iodonium (cf.8 and 3, Table 1), and consequently a wider C–Br–C bond angle is observed. Likewise comparison of analogous diphenyliodonium and diphenylchloronium compounds with tetrafluoroborate counter anions reveals differences in s-orbital character and bond angles. The chlorine atom of 9 uses 19.1% s-character in bonding with the phenyl groups compared to 8.6% in 6, which results in C–Cl–C bond angle of 106.4° (Table 1). The periodic trend observed for the diphenylhalonium salts is that the lighter halogens use more s-orbital character in bonding with the phenyl groups which results in wider C–E–C bond angles. This trend is also observed for the chalcogens (10–15, Table 1). Of the chalcogen series, 10 has the most acute C–E–C bond angle, which is also only slightly larger than the iodoniums (97.7°, Table 1). The extent of s-character involved in bonding increases moving up to lighter chalcogens and the C–E–C bond angle increase accordingly. Our calculations show that the oxygen atom of diphenyl ether 15 use more than 30% s-character in the bond to the phenyl groups and the C–O–C bond angle is 121.2° (Table 1). Collectively, we observed a linear correlation between the extent of s-character used by the central atom (E) and the C–E–C bond angle (Fig. 2).
|
|
|||||
|---|---|---|---|---|---|
| Compound | E | A | C–E–C bond angle (DFT, °) | % s-orbital on E (NBO) | % p-orbital on E (NBO) |
| a C–I–Cl bond angle. b R = 2-NO2. c R = 4-NO2. d R = 4-CO2H. | |||||
| 1 | I | — | 89.8a | 0.05 | 99.95 |
| 2 | I | Cl | 91.7 | 5.5 | 94.5 |
| 3 | I | Br | 91.0 | 5.6 | 94.4 |
| 4 | I | I | 90.5 | 5.6 | 94.4 |
| 5 | I | ClO4 | 94.6 | 7.7 | 92.3 |
| 6 | I | BF4 | 95.9 | 8.6 | 91.5 |
| 7 | I | PF6 | 96.6 | 9.2 | 90.8 |
| 8 | Br | Br | 94.4 | 7.4 | 92.6 |
| 9 | Cl | BF4 | 106.4 | 19.1 | 80.9 |
| 10 | Te | — | 97.7 | 11.6 | 88.4 |
| 11 | Se | — | 101.2 | 14.8 | 85.2 |
| 12 | S | — | 101.2 | 16.5 | 83.5 |
| 13 | O | — | 122.2 | 32.6 | 67.4 |
| 14 | O | — | 122.0 | 32.6 | 67.4 |
| 15 | O | — | 121.2 | 32.2 | 67.8 |
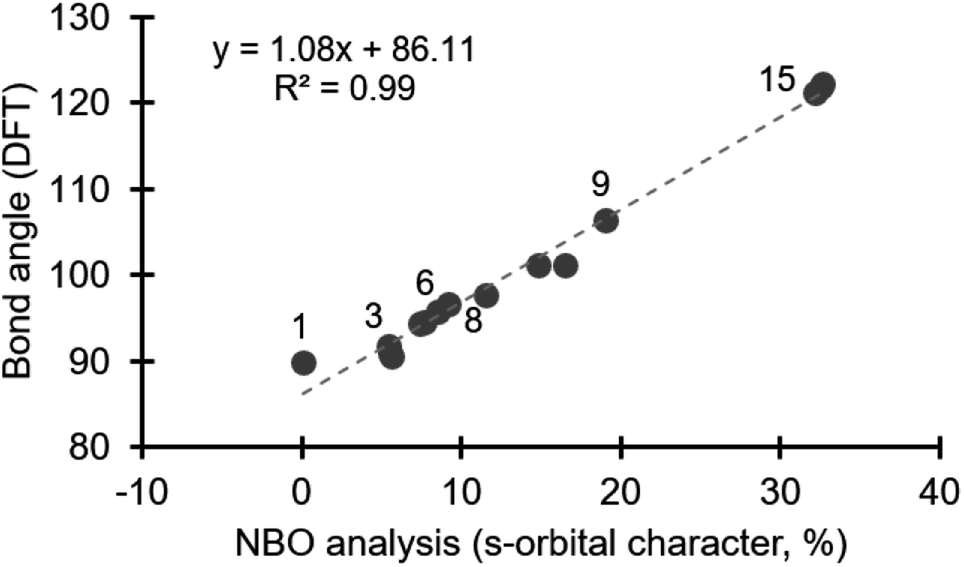 | ||
| Fig. 2 Correlation of s-orbital character in bonding and bond angle. | ||
The impact of counter anion on structure and bonding warrants closer inspection (Fig. 3). First, analysis of the orbitals on iodine used to form the bonds a and b in compound 3 show that there is less s-orbital character and therefore greater p-orbital character in bond a, which participates in a 3c–4e bond with the counter ion, Br (Fig. 3). This trend is also observed in compounds 6 and 8 (bonds c/d and e/f, Fig. 3). However, NBO analysis on the DFT-computed structure of 9 indicates that the s-character of the orbitals on the central chlorine atom are equal (Fig. 3). In solution, the interaction of the counter anion with different regions of the cation are expected to be much more dynamic and influenced by the identity of the solvent. Second, comparison of 3 and 6 suggest that less coordinating ligands (cf. Br vs. BF4) participating in the 3c–4e bond result in more s-character and less p-character on the iodine orbital involved in the 3c–4e bond (cf. c = 8.6% s and a = 5.6% s; Fig. 3). Extrapolating this trend to diphenyl telluride 10, which does not have a counter anion, shows even more s-character and larger C–Te–C bond angle (Table 1). Therefore, the orbitals used by the central halogen in bonding depend on the relative ability of the counter anion (or other Lewis base) to donate electrons; more p-character on the halogen is used to accommodate stronger coordination by the anion. Third, the positive charge of the diarylhalonium cation is primarily, but not exclusively, concentrated on the central halogen atom. We calculated Hirshfeld charges to determine the distribution of positive charge on the central halogen atoms. This analysis revealed that there is greater positive charge concentrated on the iodine relative to bromine or chlorine (cf.3 with 8 and 6 with 9, Fig. 3). The magnitude of the Hirshfeld charge is also impacted by the identity of the counter anion, and less coordinating counter anions result in more positive charge on the central halogen (cf.3 with 6, Fig. 3).
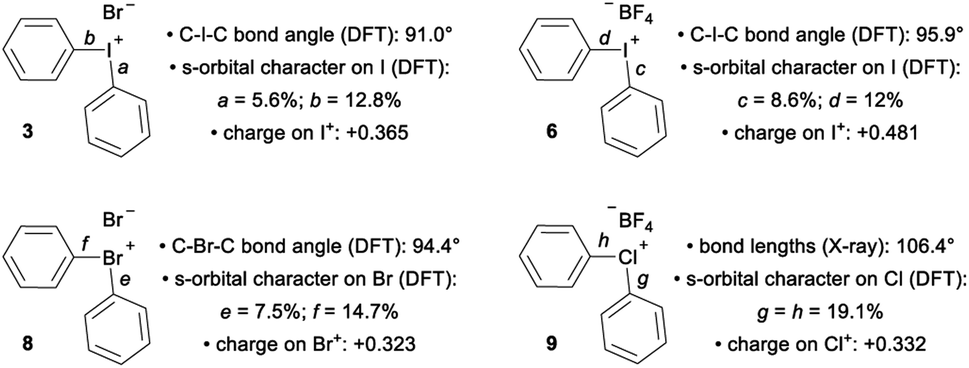 | ||
| Fig. 3 Impact of counter anion on structure and bonding in diphenylhalonium salts. | ||
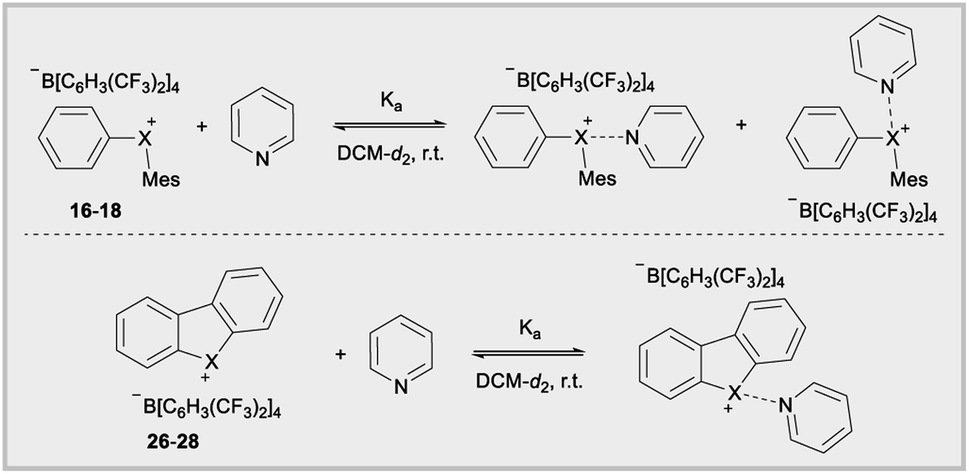
Synthetic and reactivity studies
Parallel to synthetic applications of diarylhalonium salts,1 systematic studies that unify theoretical bonding models and reactivity provide key insight into the most salient features of these systems for future advances.21 Toward this end, we synthesized a series of phenyl(mesityl)halonium BArF salts (BArF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) 16–18 (Scheme 1). Although salts containing phenyl(mesityl)halonium cations have been recently synthesized,1f the structural characterization of an analogous series via X-ray diffraction and study of the relative reactivity has not been reported. We obtained X-ray quality crystals of 16–18 and the structure of the cations are shown in Scheme 1 (see the ESI† for full structures). The C–X–C bond angles observed for this series are consistent with the periodic trends described above; C–X–C increases moving up toward the lighter the halogens (X = I < Br < Cl; Scheme 1). The C–Cl–C bond angle observed for 18 is almost identical to that obtained previously for phenyl(mesityl)chloronium salts containing a different tetraaryl borate counter anion (107°).1f However, the C–I–C bond angle obtained for 16 is larger than that obtained previously for phenyl(mesityl)iodonium tetrachloroaurate salt,22 and may be the largest C–I–C bond angle recorded to date for a diaryliodonium salt (cf., 102.3° vs. 98.3°). The discrepancy in C–I–C bond angle between 16 and phenyl(mesityl)iodonium tetrachloroaurate may be a result of the phenyl torsion angle and the phenyl C–H bond pointing into the mesityl ring in 16 (Scheme 1). Incidentally, a similar torsion angle is also observed in the X-ray structures of 17 and 18 (Scheme 1). The orbital contribution on X (I, Br, Cl) directed at the aryl ligands was determined by NBO analysis for BF4 analogues of 16–18. The calculated s-character on I, Br, and Cl is ∼10%, 14%, and 20% for 16, 17, and 18, respectively; values that correlate well with both the calculated DFT and experimental X-ray C–X–C bond angles (see Table S1† in the ESI). With compounds 16–18 in hand, we investigated both the kinetic and thermodynamic reactivity of these species in order to identify possible correlations with structure and bonding.Diaryliodonium salts are extensively used as aryl transfer reagents,2b,d and there are a few reports of aryl transfer from a diarylchloronium and bromonium salts.1f,3b,21 We considered both ipso-substitution and aryne formation as aryl transfer reactions to investigate periodic trends and the connection between structure, bonding, and kinetic reactivity. Notably, in both cases dramatic differences in reactivity have been observed between diaryliodonium and the lighter halonium salts (Fig. 1b). First, pyridine was selected as a representative nucleophile for this study because Uchiyama established precedent for mesitylation of pyridine with 18.1f We initiated a systematic study of kinetic reactivity by repeating Uchiyama's protocol and observed a very clean, albeit slow, mesityl transfer from 18 to pyridine to yield 19 (Scheme 2a).1f The mechanism of arylation with diaryliodonium salts generally proceeds through a ligand coupling pathway mediated by iodine(III),2 although direct ipso substitution has also been proposed (Scheme 3).23 In either case a bimolecular reaction would be expected, in which the rate depends on both halonium electrophile and pyridine nucleophile concentrations. We observed that the rate of formation of 19 was indeed dependent on the concentration of pyridine consistent with a bimolecular reaction and further rules out the very unlikely possibility of an SN1Ar mechanism, in which aryl cation formation would be rate determining (Scheme 2b, cf. orange triangles and blue squares). Therefore, we favour a ligand coupling pathway or direct ipso-substitution. We observed a significantly slower rate of formation of 19 when we subjected bromonium salt 17 to identical reaction conditions (Scheme 2, grey circles), and use of iodonium 16 did not result in observable formation of 19 under these metal-free conditions.4 The relative kinetic arylation reactivity follows that 18 reacts faster than 17 and 16 is unreactive. This trend aligns with the s-orbital contribution to bonding observed in 16–18. Specifically, the X-orbitals directed toward the mesityl group have 9.3%, 13.7%, and 19.7% s-character for 16, 17, and 18, respectively (Scheme 3). Therefore, when cleavage of the X–Cmesityl bond occurs in a heterolytic manner the electron pair migrating onto the halogen of the aryl halide leaving group is better stabilized by an orbital with higher s-character (Scheme 3).
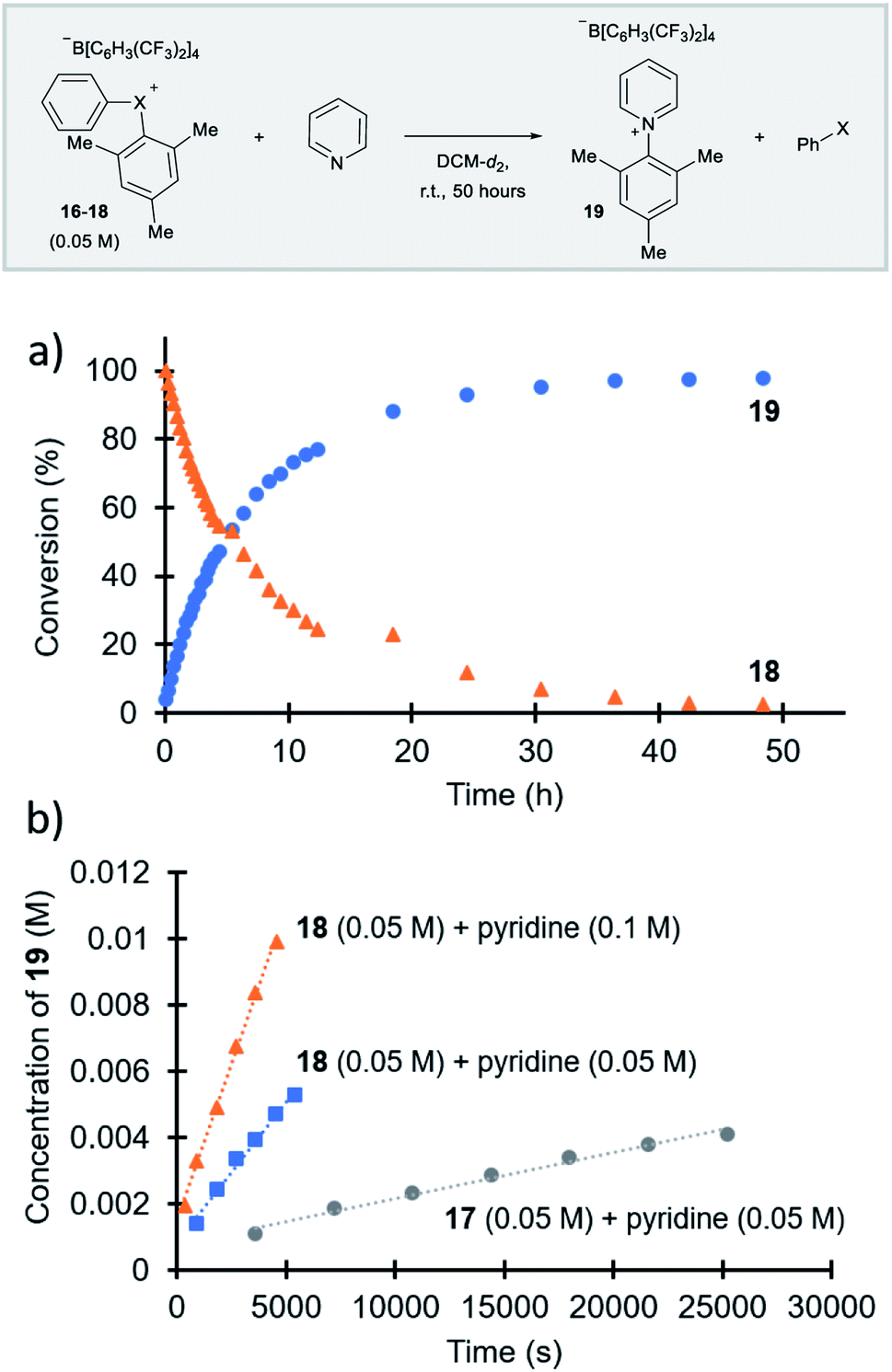 | ||
| Scheme 2 Mesitylation of pyridine. (a) Full reaction profile with for the reaction of 18 with pyridine. (b) Initial rates of reaction for 17 and 18 with pyridine. | ||
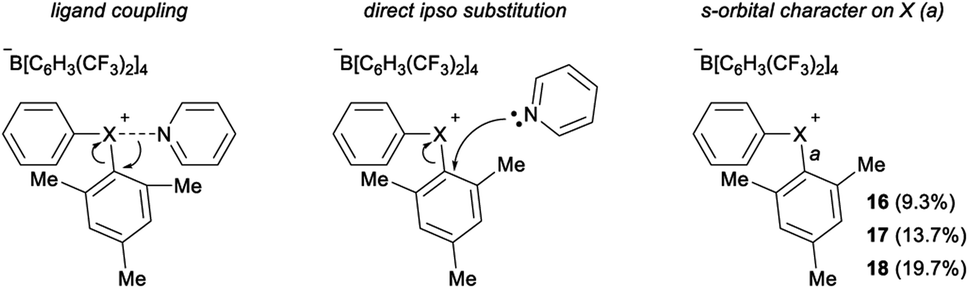 | ||
| Scheme 3 Relationship between s-orbital character and leaving group ability of PhX. | ||
Diaryliodonium salts are now established aryne precursors and operate via deprotonation with relatively strong base (NaOt-Bu) at the ortho-position to an aryl iodide leaving group.24 More recently, Wencel-Delord and co-works reported that cyclic dibenzobromolium salts extrude arynes when treated with milder base (Cs2CO3),1c,e and the corresponding cyclic dibenzoiodolium salts were unreactive under these milder conditions (Fig. 1b). Although the C–X–C bond angles are constrained by the cyclic nature of dibenzohalolium salts, we were intrigued to see if there was a periodic trend in the composition of orbitals used by the central halogen and if this trend is reflected in the kinetic reactivity of aryne formation. We synthesized an analogous series of dibenzohalolium mesylate salts 20–22 and performed DFT and NBO calculations to determine the percentage of orbitals used by the central halogen in bonding to the aryl carbon ligands (Table 2). Indeed, the C–X–C bond angles of 20–22 are smaller than 16–18 due to the cyclic structures of the former, but the trend in bond angles is preserved in this series (Table 2). Additionally, the periodic trend observed for the acyclic diarylhalonium salts is also observed here: the lighter halogens use more s-character in bonding to the carbon ligands (Table 2). Under conditions previously developed by Wencel-Delord, we used furan as an aryne trap so that the mechanism of the reaction was unambiguous (Table 2).1c,e Consistent with previous observations, no product 23 was observed when iodolium 20 was used in the reaction. The bromolium 21 is reactive under these conditions and biaryl 24 is obtained in 60%, 78%, and 80% after 1, 2, and 8 hours, respectively (Table 2). In line with the kinetic reactivity observed for ipso-substitution, chlorolium 22 reacts faster than 21 under these conditions and the yield of 25 is 86%, 87%, and 91% after 1, 2, and 8 hours time points. Therefore, this is another example where heterolytic cleavage of the C–X bond is accelerated when the bonding orbitals from the X-atom have greater s-orbital character. Conceptually, the leaving group ability of aryl halides, in both ipso-substitution and aryne formation, is analogous to the increasing kinetic acidity of hydrocarbons with increasing s-orbital character in hybrid orbitals: sp3 (25% s) < sp2 (33% s) < sp (50% s).
|
|
|||||
|---|---|---|---|---|---|
| Reactant/product | C–X–C bond angle (DFT, °) | % s-orbital on X (NBO)b | Yield at 1 h | Yield at 2 h | Yield at 8 h |
| a Conditions: 20–22 (0.1 mmol, 1 equiv.), furan (0.5 mmol, 5 equiv.), Cs2CO3 (0.3 mmol, 3 equiv.), DCM (1 mL), r.t., 1–8 h. b Average % s-orbital character on X over both X–C bonds. | |||||
| 20/23 (X = I) | 80.7 | 10.1 | n.r. | ||
| 21/24 (X = Br) | 85.8 | 12.2 | 60% | 78% | 80% |
| 22/25 (X = Cl) | 90.5 | 16.5 | 86% | 87% | 91% |
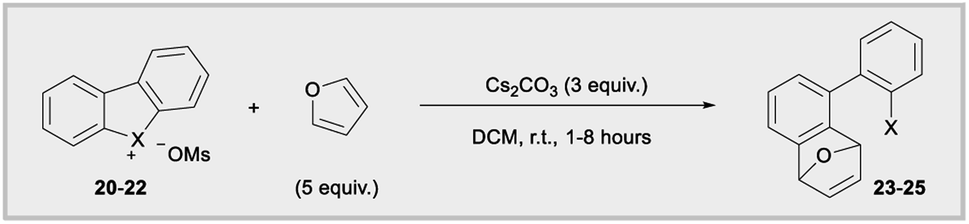
The Lewis acidity of diarylhalonium salts is an emerging property of particular relevance to catalysis.1b,d,25 Pioneering studies by Ochiai and more recent developments by Legault have established an empirical relative Lewis acidity scale.26 Additionally, Legault and Huber have conducted theoretical studies to glean insight into the fundamental basis for the strength of halogen bond donors.27 Common bonding models for secondary bonding, which includes both hypervalent and halogen bonding have been summarized by Crabtree.28 Given the similarities in orbital interaction between hypervalent and halogen bonding, especially with regard to charge transfer,29 we assessed the relevance of orbital contribution on the central halogen to periodic trends in Lewis acidity of diarylhaloniums. To the best of our knowledge no direct experimental comparison of Lewis acidities for an analogous series of diarylhalonium or cyclic diarylhalolium salts has been previously reported based on association constants with Lewis bases. We started with the acyclic series 16–18 and measured their thermodynamic reactivity by quantifying association constants with pyridine via1H NMR titration. We are cognizant that pyridine could bind opposite the phenyl and mesityl groups and that both are likely present in solution due to the dynamic nature of these interactions (Table 3). However, the 1H NMR titration experiment does not distinguish these two possible binding interactions, and the trends in the composition of orbitals on X that are directed toward the phenyl and mesityl groups are similar.18 We present the average % s- and p-orbital contribution on X directed toward the aryl groups in Table 3. Notably, in 16–18 the s-orbital contribution increases and p-orbital contribution decreases moving from heavier to lighter halogens (Table 3, 16–18). There is also an observed decrease in association constant (Ka) that trends with decreasing p-orbital character on X for compounds 16–18 (Table 3). This aligns with greater charge transfer (n → σ*) in secondary bonding, halogen bonding in this case, occurring for orbitals with greater p-character.
|
|
|||
|---|---|---|---|
| Compound | % s-orbital on X (NBO)b | % p-orbital on X (NBO)b | K a/M−1 |
| a Conditions: 16–18 and 26–28 (10 mM), pyridine (0–40 equiv.), DCM-d2 (for 16–18) or CDCl3 (for 26–28), r.t. b Average orbital contribution used by central atom X for the corresponding BF4 salt. | |||
| 16 (X = I) | 10.6 | 89.4 | 22.8 |
| 17 (X = Br) | 13.8 | 86.2 | 1.4 |
| 18 (X = Cl) | 18.9 | 81.1 | 0.4 |
| 26 (X = I) | 9.8 | 90.2 | 129.5 |
| 27 (X = Br) | 14.3 | 85.7 | 25.7 |
| 28 (X = Cl) | 17.5 | 82.5 | 7.0 |
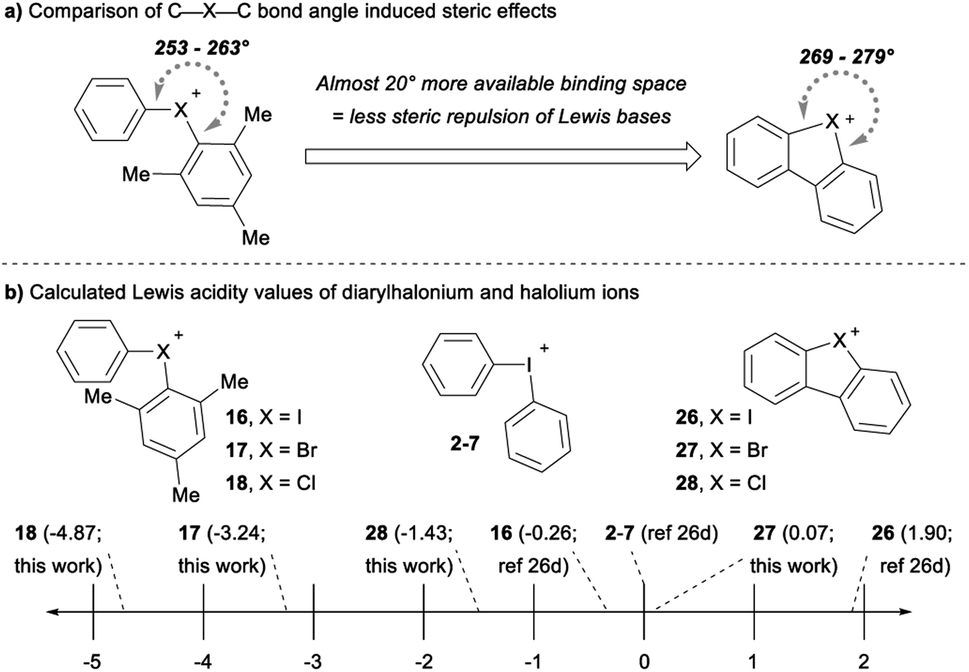
The cyclic dibenzohalolium salts display a similar periodic trend in orbital composition as the acyclic compounds (cf.26–28vs.16–18, Table 3). The s-orbital character increases and p-orbital character decreases on moving from iodolium 26 to chlorolium 28 (Table 3). Similar to the acyclic series a decrease in the Ka value measured with pyridine is observed in the order 26 > 27 > 28, which aligns with the extent of p-orbital character and charge transfer (Table 3). However, there are marked differences in measured Ka values between the acyclic haloniums and their cyclic halolium counterparts despite having similar p-character in orbitals engaged in secondary bonding (Table 3). While the C–X–C bond angle correlates with orbital composition, it may also play a role in steric repulsion of Lewis bases (Fig. 4a). The DFT calculated C–X–C bond angle in the acyclic series 16–18 is between 97–107°, therefore there is ∼253–263° of available space within the C–X–C plane for a Lewis base to bind (Fig. 4a). On the other hand, the C–X–C bond angle in the cyclic series 26–28 ranges from 81–91°, and therefore these more acute bond angles allow a larger portion of the arc on the C–X–C plane (269–279°) exposed for binding with Lewis bases. In summary greater steric repulsion by the aryl groups in 16–18 may partially account for the reduced Lewis acidity observed relative to cyclic analogues 26–28. Bolotin has also suggested that cooperative hydrogen bonding by ortho-Hs with Lewis bases in 26 contributes to greater Lewis acidity relative to diphenyliodonium 2–7.25g Collectively these results point toward the likely-hood that this phenomenon will be best described by a multi-variate model.
 | ||
| Fig. 4 Comparison of Lewis acidity of acyclic and cyclic halonium cations. | ||
The Lewis acidity (LA) scale previously determined by Legault and co-workers provides Lewis acidity parameters for the diphenyliodonium cation 2–7, 16, and 26 (Fig. 4b).26d Using these values, we estimated the Lewis basicity (LB) and sensitivity (sI) of pyridine in DCM, which agreed well with related species.26d The LB values for pyridine, together with the Ka values measured with pyridine, allowed us to estimate the relative Lewis acidity of 17, 18, 27 and 28 (Fig. 4b). Notably, the cyclic bromolium 27 has a similar LA value to the diphenyliodonium cation, but the cyclic chlorolium 28 and the acyclic bromonium 17 and chloronium 18 cations have negative LA values (Fig. 4b).
Revised bonding model and implications for reactivity
The 3c–4e bond has remained a useful model for hypervalent main group compounds because it accurately predicts both structure and reactivity. Indeed, our NBO analysis of compound 1 reveals 99.95% p-orbital contribution to the orbital on iodine directed at the chlorine ligands consistent with the 3c–4e bond model (Fig. 1). However, the large deviations in structure for the diarylhalonium series indicates that this model, as originally proposed, involving an unhybridized p-orbital, is not adequate for this class of formally hypervalent compounds. Zhdankin aptly described the geometry of the diaryliodonium cation as “pseudo-tetrahedral”,2c and indeed there is significant distortion from the ideal tetrahedral geometry of 109.5° among the diarylhalonium salts investigated here. Incidentally, there are similar levels of tetrahedral distortion in the diaryl chalcogens. It is intriguing to note that the C–Cl–C bond angle of 9 (106°) is similar to the H–O–H bond angle of water (105°) and both central atoms (Cl and O) use 19% s-orbital and 81% p-orbital in the orbitals used for bonding with the C- or H-groups.30 Therefore, it is appropriate to consider the symmetry and bonding in diarylhalonium cations to be similar to that of water and the related chalcogens in which the periodic trend of decreasing contribution of the s-orbital in bonding is observed moving down the halogens and chalcogens.A bonding model involving both s- and p-orbital contribution to the orbitals on the central halogen atom directed at the carbon ligand impacts both kinetic and thermodynamic reactivity. Although this model may not account for minute differences in reactivity within the individual halogens (i.e., aryl transfer selectivity),31 it clearly describes periodic trends in reactivity over the synthetically accessible halogen series.32 Greater s-orbital character on diarylchloroniums than on the heavier halogens results in significantly higher leaving group ability of aryl chloride, and faster reactions in both ipso-substitution and aryne formation. Therefore, synthetic chemists with an eye to arylation of very weak nucleophiles or generation of arynes under exceedingly mild (and rapid) conditions may look to use diarylchloronium salts, and improved synthetic access to these compounds will make such transformations a reality. Additionally, consistent with non-hypervalent halogen bond donors,33 we provide the first experimental evidence that diaryliodoniums are stronger halogen bond donors than the corresponding bromoniums and chloroniums based on association constants with pyridine. This trend correlates with the extent of p-character in the orbital on the central halogen X directed at the aryl ligands and aligns with the n → σ* charge transfer component of halogen bonding.29 Although the orbital composition does not account for the significant difference in Lewis acidity between cyclic and acyclic diarylhalonium compounds, the increased steric repulsion between the aryl ligands and approaching Lewis bases that is a result of a more obtuse C–X–C bond angle in acyclic compounds 16–18 may be an important factor.34 Considering the design or selection of Lewis acid catalysts, one might anticipate that a stronger Lewis acid is more advantageous, and therefore the iodoniums would be favoured. However, there is at least one reported case in which a bromonium catalyst resulted in a higher yield of product than the corresponding iodonium catalyst highlighting the complexity of catalytic cycles.1b Therefore, knowledge of the relative Lewis acidities of the diarylhalolium series, as provided here, empowers synthetic chemists to dial-in the appropriate Lewis acidity for a given reaction.
Conclusions
On the basis of this study, we find that the central halogen atom of diarylhalonium salts uses, not only p-orbitals, but rather mixed s/p orbitals in bonding with both aryl ligands. Moreover, a periodic trend is observed such that the extent of s-character decreases down the halogen group, paralleling the chalcogens, so that diaryliodonium bonding approaches the traditional 3c–4e bond. This revised bonding model accurately predicts trends in structures determined by X-ray crystallography and DFT. The kinetic reactivity, assessed by rate of pyridine mesitylation and aryne formation, also correlates with the extent of s-character in the mixed s/p orbital of the aryl halide leaving group. The thermodynamic reactivity of phenyl(mesityl)halonium salts 16–18 and cyclic dibenzohalolium salts 26–28, manifested as the strength of a Lewis acid–base interaction, correlates with the extent of p-character in the mixed s/p orbital on the central halogen, which is consistent with Bent's rule. This work illuminates p-orbital character as a parameter to describe halogen bonding, and may be applied to other types of secondary bonding (i.e., chalcogen, pnictogen, and tetrel) in main group compounds.Data availability
All of the relevant experimental data is in the ESI.† Crystal structures have been deposited with CCDC.Author contributions
SSK and DRS conceived of the project. SSK, BEM, and NJ conducted synthetic studies. AB and NJ conducted the computational studies with guidance from TMM. EJV solved the X-ray structures of 16–18 and provided critical analysis on their structural features. DRS wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Science Foundation (NSF, CHE #1856705, CHE #1800599) for support of this work. SSK is grateful to the El-Mansy family for a summer fellowship at PSU. The NSF provided instrument funding for the BioAnalytical Mass Spectrometry Facility (MRI #1828753) and the high-performance computing cluster (DMS #1624776) at PSU, and the X-ray diffraction centre at the University of Portland (MRI #0604188). We thank Souradeep Basu for assistance with conductance measurements and non-linear curve fitting.Notes and references
- (a) M. Kuriyama, N. Hanazawa, Y. Abe, K. Katagiri, S. Ono, K. Yamamoto and O. Onomura, Chem. Sci., 2020, 11, 8295 RSC; (b) Y. Yoshida, S. Ishikawa, T. Mino and M. Sakamoto, Chem. Commun., 2021, 57, 2519 RSC; (c) M. Lanzi, Q. Dherbassy and J. Wencel-Delord, Angew. Chem., Int. Ed., 2021, 60, 14852 CrossRef CAS PubMed; (d) Y. Yoshida, T. Mino and M. Sakamoto, ACS Catal., 2021, 11, 13028 CrossRef CAS; (e) M. Lanzi, R. A. A. Abdine, M. De Abreu and J. Wencel-Delord, Org. Lett., 2021, 23, 9047 CrossRef CAS PubMed; (f) M. Nakajima, K. Miyamoto, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2019, 141, 6499 CrossRef CAS PubMed; (g) K. Miyamoto and M. Uchiyama, Chem. Lett., 2021, 50, 832 CrossRef CAS.
- (a) M. Ochiai, in Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, ed. T. Wirth, Springer, New York, 2003, pp. 5–68 Search PubMed; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (c) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, United Kingdom, 2014 Search PubMed; (d) B. Olofsson, Hypervalent Iodine Chemistry, in Topics in Current Chemistry, ed. T. Wirth, Springer, Cham, 2015, vol. 373, pp. 135–166 Search PubMed.
- (a) A. N. Nesmeyanov, T. L. Khotsyanova, V. V. Saatsazov, T. P. Tolstaya and L. S. Isaeva, Dokl. Akad. Nauk SSSR, 1974, 218, 140 CAS; (b) M. Ochiai, Synlett, 2009, 159 CAS.
- B. Feng, D. Wan, L. Yan, V. D. Kadam, J. You and G. Gao, RSC Adv., 2016, 6, 66407 RSC.
- (a) R. E. Rundle, J. Am. Chem. Soc., 1947, 69, 1327 CrossRef CAS; (b) G. C. Pimentel, J. Chem. Phys., 1951, 19, 446 CrossRef CAS; (c) R. E. Rundle, J. Am. Chem. Soc., 1964, 85, 112 CrossRef.
- A. Bauer and N. Maulide, Chem. Sci., 2021, 12, 853 RSC.
- J. V. Carey, P. A. Chaloner, P. B. Hitchcock and K. R. Sneddon, J. Chem. Res., 1996, 358, 2031 Search PubMed.
- C. A. Coulson, J. Chem. Soc., 1964, 1142 Search PubMed.
- (a) N. W. Alcock and R. M. Countryman, J. Chem. Soc., Dalton Trans., 1977, 217 RSC; (b) G. Cavallo, J. S. Murray, P. Politzer, T. Pilati, M. Ursini and G. Resnati, IUCrJ, 2017, 4, 411 CrossRef CAS PubMed; (c) Y. T. Struchkov and T. L. Khotsyanova, Izv. Akad. Nauk SSSR, Ser. Khim., 1960, 821 CAS.
- A. C. Reiersølmoen, S. Battaglia, S. Øien-Ødegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdélyi, Chem. Sci., 2020, 11, 7979 RSC.
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 1367 CrossRef CAS.
- H. Bent, Chem. Rev., 1960, 60, 275 Search PubMed.
- IUPAC, Compendium of Chemical Terminology, compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781 CrossRef CAS PubMed.
- (a) S. Bhandary, A. Sirohiwal, R. Kadu, S. Kumar and D. Chopra, Cryst. Growth Des., 2018, 18, 3734 CrossRef CAS; (b) A. Kucsman, I. Kapovits, L. Parkanyi, G. Argay and A. Kalman, J. Mol. Struct., 1984, 125, 331 CrossRef CAS; (c) A. Dey and G. R. Desiraju, Chem. Commun., 2005, 2486 RSC; (d) A. R. Choudhury, K. Islam, M. T. Kirchner, G. Mehta and T. N. Guru Row, J. Am. Chem. Soc., 2004, 126, 12274 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- See the ESI† for further details.
- (a) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS; (b) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (c) J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41 CrossRef; (d) F. Weinhold and J. E. Carpenter, The Structure of Small Molecules and Ions, ed. R. Naaman and Z. Vager, Springer US, Boston, MA, 1988, pp. 227–236 Search PubMed.
- For the chalcogen series there is no hypervalent bond and therefore the orbital contribution on E is the same for the orbitals directed at both aryl ligands.
- (a) G. A. Olah, T. Sakakibara and G. Asensio, J. Org. Chem., 1978, 43, 463 CrossRef CAS; (b) T. P. Tolstaya, I. I. Demkina, V. V. Grushin and A. N. Vanchikov, J. Org. Chem., 1989, 25, 2305 Search PubMed; (c) V. V. Grushin, Acc. Chem. Res., 1992, 25, 529 CrossRef CAS.
- I. S. Aliyarova, D. M. Ivanov, N. S. Soldatova, A. S. Novikov, P. S. Postnikov, M. S. Yusubov and V. Y. Kukushkin, Cryst. Growth Des., 2021, 21, 1136 CrossRef CAS.
- E. Stridfeldt, E. Lindstedt, M. Reitti, J. Blid, P.-O. Norrby and B. Olofsson, Chem.–Eur. J., 2017, 23, 13249 CrossRef CAS PubMed.
- (a) S. K. Sundalam, A. Nilova, T. L. Seidl and D. R. Stuart, Angew. Chem., Int. Ed., 2016, 55, 8431 CrossRef CAS PubMed; (b) M. Wang and Z. Huang, Org. Biomol. Chem., 2016, 14, 10185 RSC; (c) D. R. Stuart, Synlett, 2017, 28, 275 CrossRef CAS; (d) Z. Zhang, X. Wu, J. Han, W. Wu and L. Wang, Tetrahedron Lett., 2018, 59, 1737 CrossRef CAS; (e) H. Chen, J. Han and L. Wang, Beilstein J. Org. Chem., 2018, 14, 354 CrossRef CAS PubMed; (f) A. Nilova, P. A. Sibbald, E. J. Valente, G. A. Gonzalez-Montiel, H. C. Richardson, K. S. Brown, P. H. Y. Cheong and D. R. Stuart, Chem.–Eur. J., 2021, 27, 7168 CrossRef CAS PubMed; (g) A. Nilova, B. Metze and D. R. Stuart, Org. Lett., 2021, 23, 4813 CrossRef CAS PubMed.
- (a) Y. Zhang, J. Han and Z.-J. Liu, RSC Adv., 2015, 5, 25485 RSC; (b) F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830 CrossRef CAS PubMed; (c) R. Haraguchi, T. Nishikawa, A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 4185 CrossRef CAS; (d) F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069 CrossRef CAS PubMed; (e) Y. Nishida, T. Suzuki, Y. Takagi, E. Amma, R. Tajima, S. Kuwano and T. Arai, ChemPlusChem, 2021, 86, 741 CrossRef CAS PubMed; (f) S. N. Yunusova, A. S. Novikov, N. S. Soldatova, M. A. Vovk and D. S. Bolotin, RSC Adv., 2021, 11, 4574 RSC; (g) M. V. Il'in, A. A. Sysoeva, A. S. Novikov and D. S. Bolotin, J. Org. Chem., 2022, 87, 4569 CrossRef PubMed.
- (a) M. Ochiai, T. Suefuji, K. Miyamoto, N. Tada, S. Goto, M. Shiro, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 769 CrossRef CAS PubMed; (b) T. Suefuji, M. Shiro, K. Yamaguchi and M. Ochiai, Heterocycles, 2006, 67, 391 CrossRef CAS; (c) A. Labattut, P.-L. Tremblay, O. Moutounet and C. Y. Legault, J. Org. Chem., 2017, 82, 11891 CrossRef CAS PubMed; (d) R. J. Mayer, A. R. Ofial, H. Mayr and C. Y. Legault, J. Am. Chem. Soc., 2020, 142, 5221 CrossRef CAS PubMed.
- (a) E. Engelage, D. Reinhard and S. M. Huber, Chem.–Eur. J., 2020, 26, 3843 CrossRef CAS PubMed; (b) R. Robidas, C. Y. Legault and S. M. Huber, Phys. Chem. Chem. Phys., 2021, 23, 3041 RSC; (c) R. Robidas, D. L. Reinhard, C. Y. Legault and S. M. Huber, Chem. Rec., 2021, 21, 1912 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720 RSC.
- J. Rezac and A. de la Lande, Phys. Chem. Chem. Phys., 2017, 19, 791 RSC.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS.
- Hammett σ-parameters are more likely to account for these smaller differences in reactivity as previously demonstrated for unsymmetrical diaryliodoniums, see:, H. Pinto de Magalhaes, H. P. Luthi and A. Togni, Org. Lett., 2012, 14, 3830 CrossRef PubMed.
- For comment on the synthesis and characterization of organofluoroniums, including diarylfluoroniums, see: (a) N. E. Shchepina, V. D. Nefedov, M. A. Toropova, G. A. Badun, V. V. Avrorin and V. M. Fedoseev, Radiochemistry, 2002, 44, 378 CrossRef CAS; (b) M. D. Struble, M. G. Holl, M. T. Scerba, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2015, 137, 11476 CrossRef CAS PubMed; (c) K. O. Christie, R. Haiges, M. Rahm, D. A. Dixon and M. Vasiliu, J. Fluorine Chem., 2017, 204, 6 CrossRef; (d) M. G. Holl, C. R. Pitts and T. Lectka, Acc. Chem. Res., 2020, 53, 265 CrossRef CAS PubMed; (e) K. F. Hoffmann, A. Wiesner, C. Muller, S. Steinhauer, H. Beckers, M. Kazim, C. R. Pitts, T. Lectka and S. Riedel, Nat. Commun., 2021, 12, 5275 CrossRef CAS PubMed.
- M. S. Taylor, Nat. Chem., 2014, 6, 1029 CrossRef CAS PubMed.
- R. Robidas, D. L. Reinhard, S. M. Huber and C. Y. Legault, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-jtt63 ; This content is a preprint and has not been peer-reviewed..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2124112–2124114. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02332f |
| This journal is © The Royal Society of Chemistry 2022 |