
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the lifetime of cucurbit[8]uril based self-abolishing nanozymes†
Saurav
Das
 ,
Tanushree
Das
,
Priyam
Das
and
Debapratim
Das
*
,
Tanushree
Das
,
Priyam
Das
and
Debapratim
Das
*
Department of Chemistry, Indian Institute of Technology Guwahati, Assam 781039, India. E-mail: ddas@iitg.ac.in
First published on 14th February 2022
Abstract
Nature has evolved a unique mechanism of self-regulatory feedback loops that help in maintaining an internal cellular environment conducive to growth, healing and metabolism. In biology, enzymes display feedback controlled switchable behaviour to upregulate/downregulate the generation of metabolites as per the need of the cells. To mimic the self-inhibitory nature of certain biological enzymes under laboratory settings, herein, we present a cucurbit[8]uril based pH responsive supramolecular peptide amphiphile (SPA) that assembles into hydrolase mimetic vesicular nanozymes upon addition of alkaline TRIS buffer (activator) but disintegrates gradually owing to the catalytic generation of acidic byproducts (deactivator). The lifetime of these nanozymes could be manipulated in multiple ways, either by varying the amount of catalytic groups on the surface of the vesicles, by changing the acid generating substrate, or by changing the ratio between the activator and the substrate. The self-inhibitory nanozymes displayed highly tunable lifetimes ranging from minutes to hours, controlled and in situ generation of deactivating agents and efficient reproducibility across multiple pH cycles.
Introduction
Self-regulatory feedback and feedforward mechanisms are ubiquitous in living systems. These feedback loops help in maintaining homeostasis by controlling various chemical signalling pathways.1–3 For example, signal amplification in positive feedback loops leads to enhanced secretion of oxytocin during child birth4 and rapid activation of platelets for clotting5 in the event of an injury. On the other hand, negative feedback loops exert a decelerating effect on certain metabolic processes that help in maintaining a stable physiological environment through regulation of body temperature, blood pressure, and blood sugar level, among other processes.6 Likewise, in the self-inhibitory feedback mechanism, the activity of certain enzymes is inhibited by their own metabolic end product. Feedback inhibition helps in modulating the activity of enzymes according to the need of the cell and prevents accumulation of harmful metabolites in the cellular micro-compartments.Akin to the biochemical reaction networks, a few instances of chemically triggered feedback loops have been exploited in synthetic organic systems as well.7–10 Chemical reaction relays in synthetic systems have profusely been used for designing out-of-equilibrium self-assemblies wherein rationally designed feedback mechanisms regulate the lifetime of the assemblies.11–18 These non-equilibrium systems, however, invariably employ a slow deactivating agent as a key component of the dissipating reaction and thus, the concentration of the deactivator and/or the rate of the backward dissipative reaction dictates the lifetime of the self-assembly. Biomimetic transient self-assembly processes driven by negative feedback reaction relays wherein the self-assembled structures catalyse their own decomposition have rarely been explored and thus, development of autocatalytic self-destructive molecular assemblies that follow non-equilibrium kinetic pathways still remains highly elusive. Moreover, with proper adjustment of the self-inhibitory activity, control over the lifetime of the assemblies can be achieved without the need of any external deactivating agent.
Environmental factors such as pH, temperature, ionic and redox strength play key roles in driving the self-assembly of supramolecular ensembles with emergent functional attributes and have been exploited on many occasions to generate transient self-assembled nanostructures.19–23 Chemically triggered cyclic pH changes have been exploited as an efficient technique to instigate self-assemblies in a temporally controlled fashion.24–29 Following our previous findings,27 herein, we report a cucurbit[8]uril (CB[8])30,31 based supramolecular peptide amphiphile (SPA)32,33 with an intermediate pH responsive imine linkage that assembles into transient vesicles with hydrolase like catalytic properties (nanozyme) under alkaline conditions. The nanozyme catalyses its own degradation owing to the generation of deactivating acidic products (and thereby changing the pH) in the system (Scheme 1). Moreover, by simple changes in the headgroup composition or by changing the substrate for the catalysis, control over the lifetime of the nanozyme can be achieved for a wide time range.
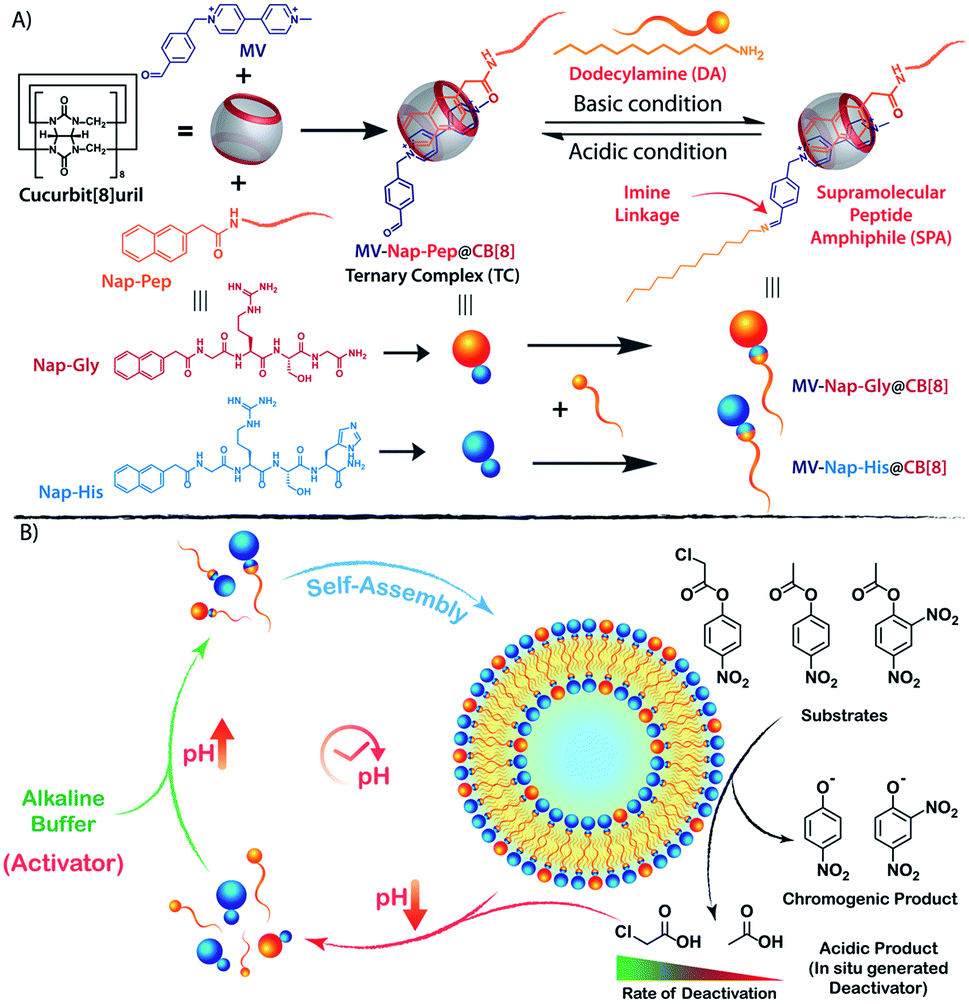 | ||
| Scheme 1 Chemical structures of different components and graphical presentation of (A) the formation of CB[8] assisted SPA under alkaline condition and (B) the self-inhibitory feedback driven temporal formation of nanozymes showing hydrolase like catalytic activity. | ||
Results and discussion
Design of the SPA and the corresponding vesicle formation
The vesicle formation by CB[8] assisted ternary complexation of the asymmetric viologen amphiphile and an electron rich second guest is very well documented in the literature.34–38 We took the help of this phenomenon and designed our transient nanozyme accordingly. As depicted in Scheme 1A, the head group of the SPA was constructed with the help of well-established hetero-ternary complex formation by CB[8].39–41 An electron rich guest for CB[8] was designed wherein naphthalene is connected to a hydrophilic catalytic peptide sequence bearing Histidine (His) at the C-terminal (Nap-His). The naphthalene unit of the peptide forms a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 heteroternary complex (termed TC, hereafter) with electron deficient viologen (MV) units inside the hydrophobic cavity of CB[8]. The complexation is stabilized through charge transfer interaction between the naphthalene and viologen units.42 It is worth mentioning here that entrance of both the guests from the same side of CB[8] is not energetically favourable mainly because of the steric crowding. Moreover, the ternary complexation where guests enter the CB[8] cavity from opposite directions is very well documented in the literature.30,43,44 The peptide was designed in a way to incorporate catalytic amino acid residues (His and Serine (Ser)) commonly found in the catalytic pockets of hydrolases to induce hydrolase like catalytic activity.45,46 The presence of the Arginine (Arg) unit not only provides hydrophilicity but also enhances substrate binding because of its strong H-bond donating propensity.46 On the other hand, the viologen units (MV) carry an aldehyde group at one end. The incorporation of this aldehyde unit was judicially done to form imine linkages with alkyl amine (dodecylamine, DA) and form the SPA as discussed later.
:1:1 heteroternary complex (termed TC, hereafter) with electron deficient viologen (MV) units inside the hydrophobic cavity of CB[8]. The complexation is stabilized through charge transfer interaction between the naphthalene and viologen units.42 It is worth mentioning here that entrance of both the guests from the same side of CB[8] is not energetically favourable mainly because of the steric crowding. Moreover, the ternary complexation where guests enter the CB[8] cavity from opposite directions is very well documented in the literature.30,43,44 The peptide was designed in a way to incorporate catalytic amino acid residues (His and Serine (Ser)) commonly found in the catalytic pockets of hydrolases to induce hydrolase like catalytic activity.45,46 The presence of the Arginine (Arg) unit not only provides hydrophilicity but also enhances substrate binding because of its strong H-bond donating propensity.46 On the other hand, the viologen units (MV) carry an aldehyde group at one end. The incorporation of this aldehyde unit was judicially done to form imine linkages with alkyl amine (dodecylamine, DA) and form the SPA as discussed later.
Formation of the TC was monitored utilizing various standard techniques. UV-Visible absorption spectra of 1:1:1 CB[8], Nap-His and MV showed the appearance of the characteristic charge transfer band at 394 nm which was not present for any other combinations as can be seen in Fig. 1A.42 A prominent colour change was also observed when all these three components were mixed and confirms the formation of the CT complex (inset of Fig. 1A). Further, the emission of the naphthalene moiety of Nap-His at 333 nm decreased drastically upon formation of TC with MV and CB[8] which indicates the incorporation of MV and Nap units of the peptide inside the CB[8] cavity (Fig. 1B).47,48 Moreover, isothermal calorimetric titration (ITC) of the 1:1 Nap-His@CB[8] solution with the MV solution shows 1:1 binding between these two components with a binding constant of 3.44 × 105 M−1 and thereby confirms the formation of TC (Fig. 1C).
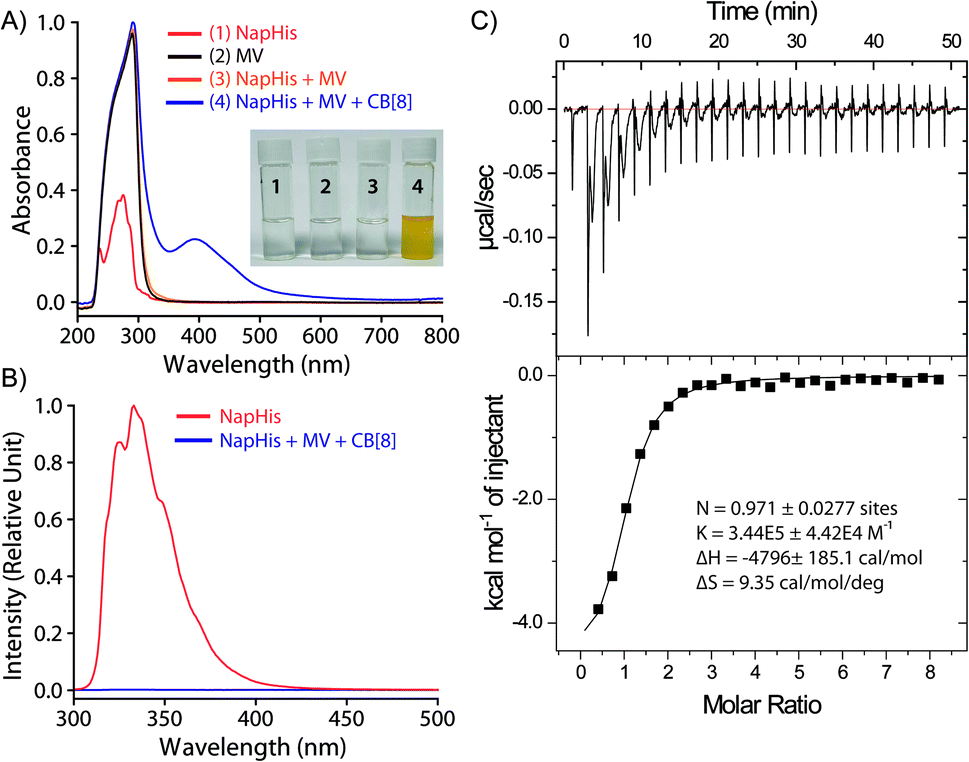 | ||
| Fig. 1 Formation of the CB[8] assisted charge transfer complex as observed from (A) UV-Visible spectra, and (B) fluorescence spectra of different components of TC (0.2 mM), (C) ITC thermograms (top) and binding isotherms (bottom) of Nap-His@CB[8] with MV at 298 K. | ||
Next, the formation of the SPA and the corresponding vesicles was confirmed. The imine bond formation under aqueous basic pH and hydrolysis of the same at lower pH are well documented in the literature.49 We have previously shown that such head groups result in SPAs upon linking with dodecylamine (DA) at basic pH and assemble into higher order aggregates.27,50 The 1H NMR study shows the disappearance of the aldehyde signal (δ = 9.96 ppm) and appearance of the imine peak (δ = 9.38 ppm) as we move from neutral to basic condition and confirms the formation of the imine bond between the aldehyde group of MV and the amine of DA (Fig. 2A). Upon acidification of the system, the disappearance of the imine signal and reappearance of the aldehyde proton peak demonstrate the instability of the imine linkage under acidic conditions. Dynamic light scattering (DLS) studies showed an increase in the hydrodynamic size of the assemblies generated by TC and DA from ∼20 nm at pH 6 to ∼250 nm at pH 7–9, indicating the formation of ordered assemblies under alkaline conditions (Fig. 2B). The vesicular nature of the assemblies was ascertained by microscopic techniques likes field emission scanning electron microscopy (FESEM) and field emission transmission electron microscopy (FETEM) (Fig. 2C and D). A thin dark boundary around the spherical aggregates of ∼200 nm size in the FETEM images confirmed the formation of vesicles.
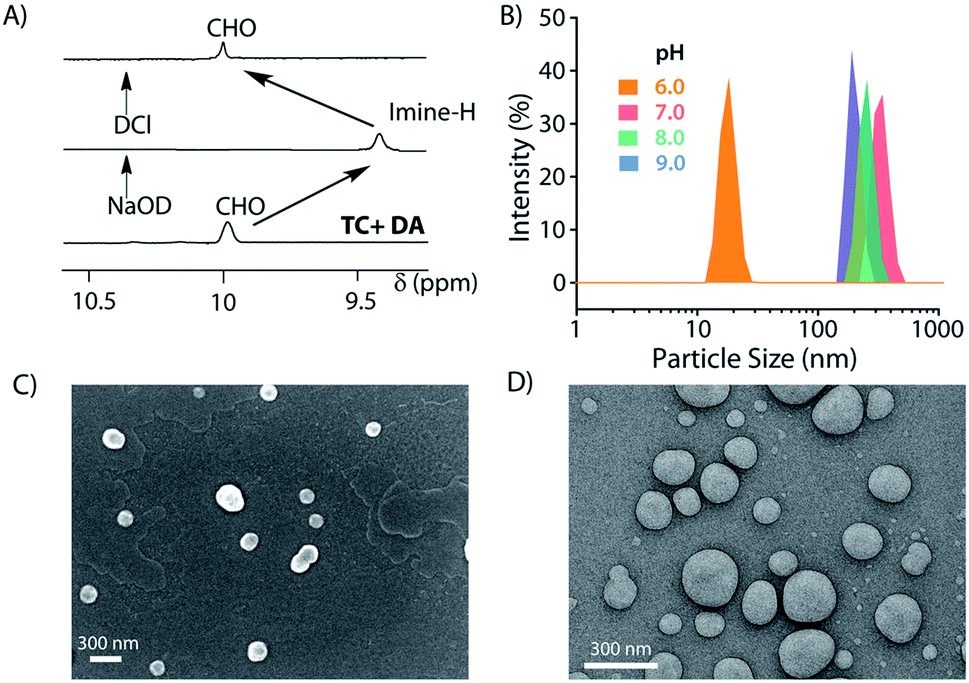 | ||
| Fig. 2 Formation of the SPA vesicles. (A) Partial 1H-NMR showing formation/disruption of the imine linked SPAs, (B) DLS profile of assemblies formed at different pH values, (C) FESEM, and (D) FETEM images of the vesicles formed at pH 8. | ||
Catalytic activity of the vesicular nanozymes
It is well known that enzyme mimicking peptides show significant improvement in the catalytic efficiency when they are in the aggregated state compared to that in the monomeric state.51 In the present case, the proximate localization of the His groups on the surface of the vesicles was expected to show cooperative interactions amongst them wherein a pair of neighbouring His units simultaneously can act as a nucleophile and proton donor and thereby enhance the rate of hydrolysis. To establish the catalytic ability of the SPA vesicles, a pH irresponsive control viologen derivative, hexadecyl-methylviologen (HDMV), was used to form the control SPA using CB[8] and Nap-His. The resultant pH insensitive SPA vesicles were used to assess the catalytic proficiency of the proximal histidine units. A bell-shaped curve for the rate of hydrolysis of p-nitrophenylacetate (pNPA) at different pH values was obtained with a maximum at around pH 7 (close to the pKa of His) (Fig. S1, ESI†). These observations pointed towards the cooperative catalysis mechanism.52–54However, when the catalytic activity was monitored in the pH responsive system (TC + DA), it displayed negligible catalytic activity at pH 6 (Vmax = 0.378 μM min−1, Fig. 3A). At this pH, the formation of the SPA and the consequent vesicles is not possible as the imine bonds are cleaved under acidic conditions.27 Thus the molecules (TC and DA) remain in non-associated form and thereby the cooperative interactions are essentially absent to promote effective catalysis. Formation of the vesicles at pH 8, however, led to a 38-fold jump in activity (Vmax = 14.388 μM min−1) signifying the cooperative effect in the vesicular aggregates. Moreover, switching the pH of the solution between acidic and alkaline range allowed efficient switching of the activity of the nanozyme over multiple cycles (Fig. 3B). To confirm that the values obtained are not arising from the auto-hydrolysis due to the basic conditions, two control experiments were performed. The hydrolysis rates in pH 8 buffer (Vmax = 1.562 μM min−1) and the activity of the catalytically inert control SPA (Vmax = 5.01 μM min−1) containing glycine (Gly) instead of His (Nap-Gly, Scheme 1) in the head group were found to be significantly lower than those of the vesicles with Nap-His at the surface.
 | ||
| Fig. 3 pH dependent catalytic activity of the nanozymes (A) Michaelis–Menten plot for the hydrolysis of pNPA using the NapHis/NapGly (control) head group by the assembled/disassembled nanozyme (100 μM) at pH 8/6, respectively. (B) Switchable catalytic activity of the nanozyme (100 μM) at pH 6 and 8 using 0.5 mM pNPA. | ||
Temporal existence and self-destruction of the nanozyme
To introduce transient and self-destructing features into the hydrolase nanozyme, a self-inhibitory pH feedback response was established by using p-nitrophenol and 2,4-dinitrophenol ester substrates that generate acidic entities upon hydrolysis. These esters remain stable in a solution of the inactive nanozyme components (TC and DA) at pH ∼5 but undergo hydrolysis as soon as the nanozyme formation is triggered by the addition of a specified amount of alkaline TRIS buffer (pH 9) as an activator. The nanozyme catalyses the generation of acid from the esters which in turn perturbs the environment conducive to its own existence. Thus, a pH drop is registered as long as the nanozyme is active and below a certain pH value, the nanozyme is dissipated by its own catalytic by-products. pNPA underwent very slow hydrolysis in the presence of the nanozyme at pH 8 (V0 = 3.327 ± 0.148 μM min−1) as compared to p-nitrophenylchloroacetate (pNPCAA) (V0 = 19.719 ± 0.255 μM min−1) (Fig. 4A). However, both the esters displayed meagre hydrolytic decomposition at acidic pH. The pH clock established by adding 1 mM pH 9 TRIS buffer to 100 μM nanozyme solution at pH 5 containing 5 mM pNPA or pNPCAA led to a rapid jump in pH followed by a slow decay. It is to be noted that the concentrations mentioned (for buffer and substrates) here and hereafter represent the overall concentrations of the components in the system and not of the stock solutions added. Owing to the slow hydrolysis of pNPA to acetic acid, the pH dropped to 6 in 194 min whereas pNPCAA (chloroacetic acid as the hydrolysis product) presented a much faster pH drop to 5.73 in 34 min and thereafter a gradual decline owing to slow background hydrolysis of pNPCAA at low pH (Fig. 4B). The faster drop in pH in the case of choloracetic acid (pKa = 2.68) as compared to acetic acid (pKa = 4.56) may also be attributed to the stronger acidic nature of the former.55 On account of the faster kinetics and more prominent pH change by pNPCAA, it was used as a substrate for subsequent pH kinetic studies.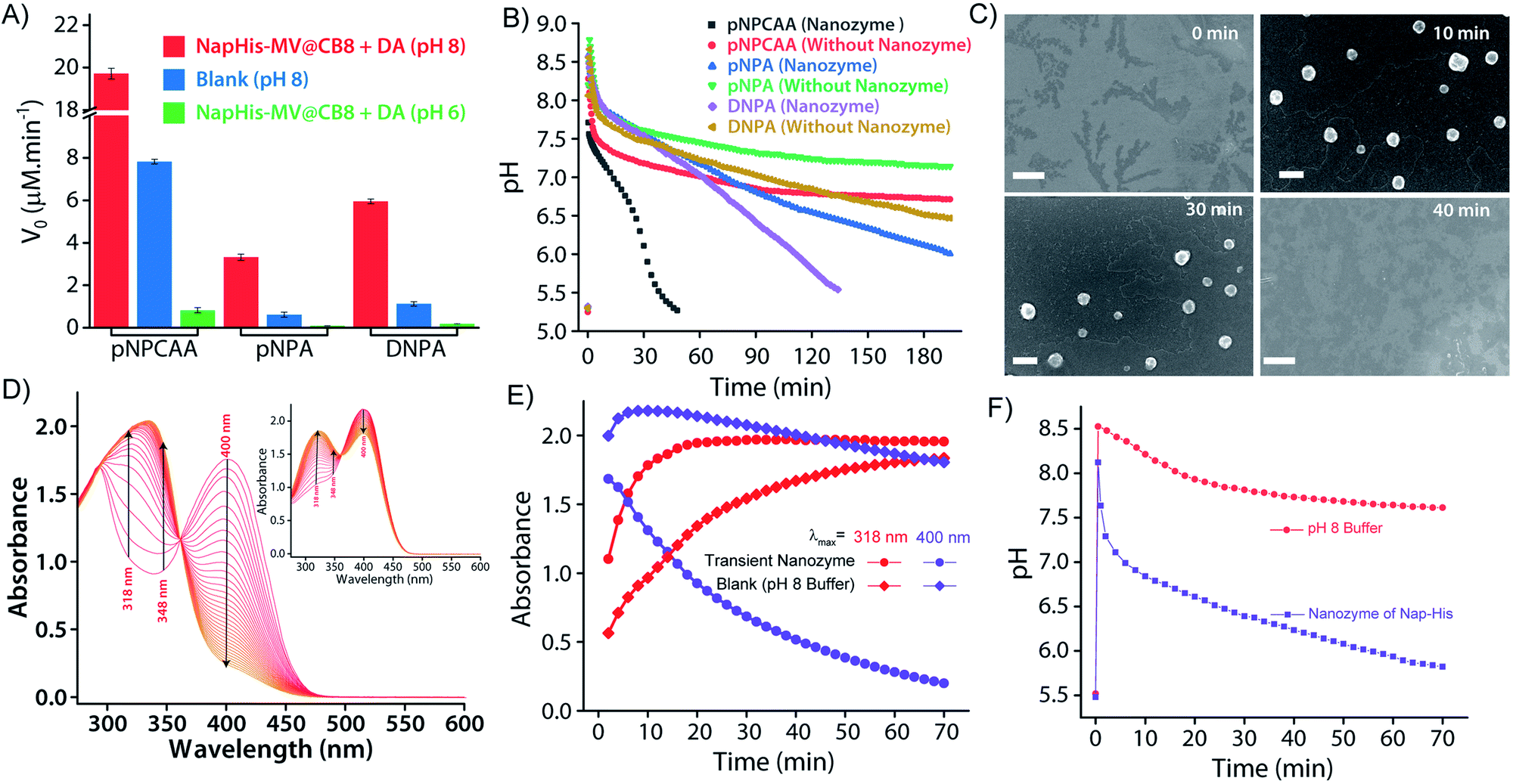 | ||
| Fig. 4 (A) Rate of hydrolysis of pNPA/pNPCAA/DNPA (0.5 mM) by the alkaline buffer (blank, pH 8) and the assembled/disassembled nanozyme (100 μM) at pH 8/6, respectively. (B) pH clock with/without the nanozyme (100 μM) using pNPA/pNPCAA/DNPA (5 mM) as substrates and 1 mM pH 9 TRIS buffer as the activator. (C) FESEM images of the transient vesicles furnished by SPAs accommodating equimolar (0.1 mM) amounts of CB[8], Nap-His and MV–DA formed during the course of the pH cycle (scale bar = 500 nm). [pH 9 TRIS buffer] = 1 mM, [pNPCAA] = 5 mM. (D) Time dependent UV-Visible spectra obtained at different time intervals after the addition of 0.5 mM pH 9 TRIS buffer to a solution of 1 mM pNPCAA in the presence and absence (inset of D) of the nanozyme (100 μM), respectively. (E) Change in absorbance at λmax = 318 nm and 400 nm as observed from D. (F) Time dependent variation in pH for the systems in D. | ||
Time-gated formation of the SPAs and their subsequent hydrolysis were further monitored through FESEM microscopic analysis. In this case, freeze dried samples of the system (Nap-His + CB[8] + MV + DA + pNPCAA) collected at different time points along the course of the pH cycle were analysed and as can be seen from Fig. 4C, no self-assembled structures were visible prior to the initiation of the pH cycle. After initiation, as the pH clock progressed, vesicular assemblies were observed which slowly disintegrated with the course of time, indicating the transient formation of the nanozymes.
The in situ generated p-nitrophenol (pNP) chromogenic product served as a pH indicator and enabled visual monitoring of the pH cycles. pNP is colourless at acidic pH (pH < 6) and shows absorption maxima at 318 nm. p-Nitrophenolate ions, formed in alkaline solution (pH > 7), however, strongly absorb at λmax = 400 nm and appear yellow in colour. The absorption intensity at 318 nm and 400 nm, thus, gives an approximation of the pH of the solution. The isosbestic point of pNP at 348 nm, on the other hand, is pH independent and allows quantification of the amount of pNP present/generated in the solution. UV-visible spectrophotometric analyses of the designed pH clock showed that upon adding 0.5 mM pH 9 TRIS buffer to 1 mM pNPCAA, the nanozyme (100 μM) resulted in a significant decrease in absorbance at 400 nm and an enhancement in absorbance at 318 nm over a period of 70 minutes (Fig. 4D and E). The blank solution (without nanozyme), however, displayed only a slight fall in the absorbance at 400 nm (Fig. 4D (inset) and E). Although, both systems showed increment in absorbance at 348 nm indicating the formation of pNP, as expected, the extent of the increment in the case of the nanozyme was significantly higher. Furthermore, HPLC kinetics studies using the nanozyme indicated gradual consumption of pNPCAA and formation of pNP as the pH cycle progressed (Fig. S2, ESI†). These results show that the nanozyme simultaneously catalyses the hydrolysis of pNPCAA and leads to a fall in the pH of the solution to the point when the nanozyme is no more existent. The uncatalyzed alkali mediated hydrolysis of pNPCAA in the absence of the nanozyme failed to produce similar results. These observations were further corroborated by the pH kinetics experiment where a significant drop in pH (8.29 to 5.81) was observed in the case of the nanozyme as compared to the blank solution (8.51 to 7.23) (Fig. 4F). Also, the transient formation of the nanozyme was confirmed by time dependent fluorescence kinetics studies using diphenylhexatriene (DPH) as a lipophilic probe.56 The emission intensity of DPH enhances when trapped in vesicular aggregates.27,56 In the present case, as expected, DPH showed enhanced emission upon addition of the activator as it gets trapped inside the vesicles (Fig. S3, ESI†). The enhanced emission was followed by a slow decay indicating the disruption of the vesicles. The transient aggregation process was also monitored through confocal laser scanning microscopy (CLSM) using Nile Red as the fluorescent probe which shows enhanced emission upon intercalation in lipid bilayers.27 Appearance of fluorescent assemblies shortly after the addition of the activator that slowly disappeared within 60 minutes indicated the formation of transient vesicular nanozymes (Fig. 5). Particle count analyses of the CLSM images also confirmed the transient emergence of vesicular aggregates (Fig. S4, ESI†).
 | ||
| Fig. 5 CLSM images showing the transient formation of nile red (NR) (10 μM) encapsulated vesicular nanozymes. [CB8]/[Nap-His]/[MV]/[DA] = 100 μM, [pNPCAA] = 5 mM, [pH 9 TRIS buffer] = 1 mM. Scale bars: 25 μm. | ||
Controlling the lifetime
As discussed in the Introduction, though creation of artificial transient assemblies has been reported on several occasions in the last decade, not much attention has been paid to create systems where the lifetime of these assemblies can be catalytically controlled by the assemblies itself. In the present work, we put an effort to gain control over the lifetime of the formed nanozyme through a self-inhibitory mechanism. The change in the pH of the system is a good indicator of the existence/disappearance of the nanozyme. As no particle distribution corresponding to the vesicles was observed at pH 6 in the DLS measurement (Fig. 2B), we consider reaching pH 6 by the system during the deactivation process as a good consideration of the complete destruction of the nanozymes. Thus, the corresponding timeframe can be considered as the lifetime of the transient nanozymes.As mentioned earlier, Nap-Gly containing vesicles displayed little catalytic behaviour and thus we envisioned that a combination of Nap-His and Nap-Gly peptides to construct the nanozyme may allow us to tune the lifetime of the nanozyme. As can be seen from Fig. 6A, various combinations of Nap-His/Nap-Gly containing head groups were tested for this purpose using 3 mM pNPCAA as the substrate. With the increase in Nap-Gly content, the nanozymes displayed progressively lowering activities and longer lifetimes (Fig. 6A and B). The presence of Nap-Gly along with Nap-His disrupts the cooperative effect that was gained by the assembly process in the case of the Nap-His based nanozyme. Thus, by varying the head group composition, a wide range of lifetimes (52 min to 145 min) could be achieved. Moreover, the purely NapGly containing nanozyme presented an insignificant fall in pH (similar to the blank solution) and retained its vesicular form over an extended period of time.
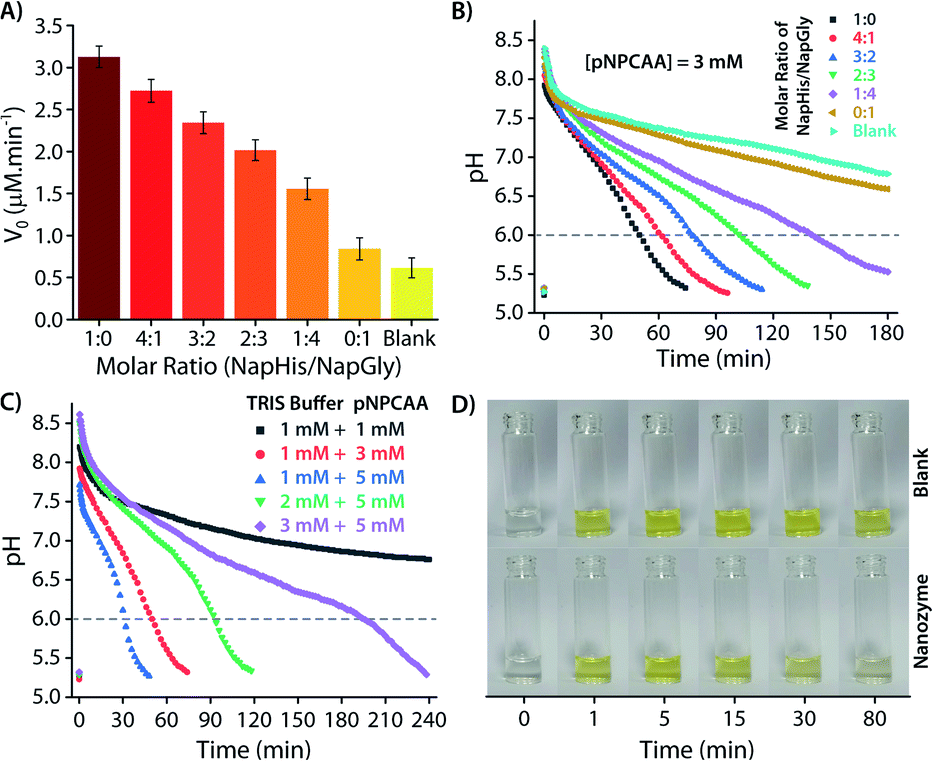 | ||
| Fig. 6 (A) Rate of hydrolysis of pNPA (0.5 mM) using different molar ratios of NapHis/NapGly ([nanozyme] = 100 μM) in pH 8 buffer. (B) pH clock using different molar ratios of NapHis/NapGly nanozyme (100 μM), 1 mM pH 9 TRIS buffer and 3 mM pNPCAA. (C) pH clock using various molar ratios of pH 9 TRIS buffer and pNPCAA in the presence of 100 μM nanozyme. (D) Photographic images of the nanozyme and blank (without nanozyme) solution captured along the course of the pH clock triggered by using 0.5 mM pH 9 TRIS buffer and 1 mM pNPCAA. | ||
Another approach to tune the lifetime of the transient state is to vary the molar ratio of the activator (pH 9 TRIS buffer) and the substrate (pNPCAA). For a fixed concentration of TRIS buffer (1 mM), a higher concentration of pNPCAA resulted in shorter lifetimes of the nanozymes owing to the faster rate of hydrolysis (Fig. 6C). Higher concentrations of TRIS buffer against a fixed concentration of pNPCAA (5 mM), however, resulted in longer lifetimes on account of the decrease in the ability of the generated acid to neutralize higher concentrations of the alkaline buffer. Visual inspection of the nanozyme and blank (without nanozyme) solutions subjected to the pH cycle by adding 0.5 mM pH 9 TRIS buffer and 1 mM pNPCAA showed that the nanozyme as well as the blank solution both turned yellow soon after the addition of the activator owing to the formation of pNP (Fig. 6D, ESI Video SV1†). However, as the pH became acidic in the case of the nanozyme solution, the solution slowly turned clear whereas the blank solution retained its yellow colour over a prolonged duration of time. This clearly indicates the hydrolytic prowess of the nanozyme which is in fact responsible for driving the pH cycle.
As we were able to construct nanozymes with a lifetime range of 34 min to more than 3 h, we focused on preparing systems that can exist for a much shorter period. Working with esters of stronger acids like trifluoroacetic acid (pKa = 0.0), trinitrobenzoic acid (pKa = 0.42) or dichloroacetic acid (pKa = 0.87) etc. is extremely difficult both in terms of synthesis and extremely fast hydrolysis of the corresponding esters.55 In this regard, we realized that the hydrolysis product, p-nitrophenol, though acidic in nature, is not necessarily contributing toward the acidification of the system mainly because of its higher pKa value (7.2).55 However, if both the products can contribute (the acid and the phenol) to the self-destruction of the nanozyme, a much lower lifetime can be achieved. We found that dinitrophenol (DNP) has a much lower pKa (4.09)57 compared to the lowest pH (<6) where the nanozyme can exist. Based on this fact, esters of DNP were prepared and used as substrates for the nanozyme. The chloroacetic ester derivative of DNP, however, was hydrolytically unstable and underwent hydrolysis even in the absence of the vesicular nanozyme. Based on the results obtained from experiments with pNPCAA, a combination of 1 mM TRIS buffer and 5 mM acetate of DNP (DNPA) was used (Fig. 4B). As expected, a fast decrease in the pH was observed compared to the case of pNPA (lifetime, 194 min) and the entire process took ∼110 min to reach a pH value of 6. Although, we were not able to achieve a shorter lifetime than that in the case of pNPCAA, the use of different substrates significantly enhanced the scope of tuning the lifetimes of the nanozymes.
Recyclability
An interesting feature of natural enzymes is their reactivation and recyclability under favourable environmental conditions. Akin to natural counterparts, vesicular nanozymes could be reactivated by fresh addition of the activator after consecutive pH cycles. Briefly, addition of 1 mM TRIS buffer (pH 9) to the nanozyme solution (100 μM) containing 5 mM pNPCAA triggered a pH cycle that ended in 34 min (Fig. 7A). Subsequent addition of 1 mM TRIS buffer enacted two more consecutive pH cycles without any further addition of pNPCAA. The lifetimes of the successive pH cycles, however, increased owing to the consumption of pNPCAA in each cycle. Fresh addition of 5 mM pNPCAA and 1 mM TRIS buffer after the third cycle produced similar changes to those observed initially (first cycle). DLS measurements at different time points along the course of the pH clocks indicated the transient and repeated formation of the nanozymes. The HPLC kinetics experiment also presented progressive formation of pNP towards the beginning of the pH cycle and slower kinetics at the end of the pH clock (Fig. S5, ESI†). Addition of TRIS buffer after the first pH cycle again resulted in a jump in the rate of formation of pNP, indicating the regeneration of the nanozymes. The kinetic fluorescence experiment using DPH also indicated the reproducibility of the system across three pH cycles (Fig. 7B). It is worth mentioning that although the nanozyme displayed appreciable reproducibility owing to the self-catalysed degradation pathway, the accumulation of wastes in the system led to minor dampening in subsequent pH cycles.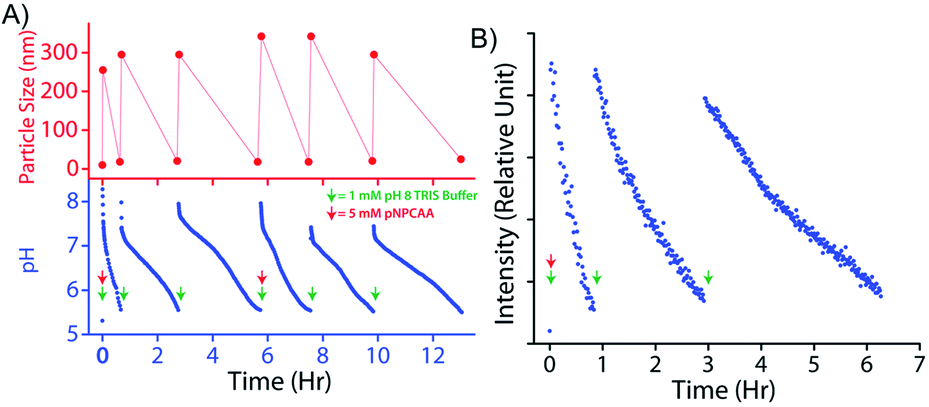 | ||
| Fig. 7 (A) Time dependent variation in pH (bottom) and particle size (top) across six consecutive pH cycles. Note: 1 mM pH 9 TRIS buffer was added to 100 μM nanozyme solution to initiate each cycle and 5 mM pNPCAA was added before initiating the 1st and 4th cycles. (B) Fluorescence kinetics across three consecutive cycles using the DPH lipophilic probe. [DPH] = 10 μM, λex (DPH) = 355 nm, λem (DPH) = 428 nm. | ||
Conclusions
In conclusion, an imine linked CB[8] based pH responsive SPA is reported which displays alkaline buffer (activator) triggered vesicular assembly in aqueous medium. The histidine groups localized on the surface of the vesicles impart catalytic properties to the vesicle through a cooperative mechanism. The catalytic prowess of the assembled nanozyme leads to in situ generation of the deactivating agent (acid) from the substrates and results in disintegration of the nanozyme due to hydrolysis of the imine linkages at acidic pH. The denatured nanozymes could be reactivated by adding fresh pulses of the activator and the transient formation of the nanozymes could be repeated across multiple pH cycles. Importantly, we have shown how to modulate the lifetime of the nanozymes. By varying the composition of catalytic groups on the nanozyme surface, the catalytic efficiency can be controlled and thereby formation of acid and subsequent self-destruction of the nanozyme can be manipulated. Another approach is to vary the substrate for the catalytic reaction and thereby increase or decrease the strength of the generated acid. The rate of production of the acid and its strength eventually determine the lifetime of the nanozyme. In another approach, the ratio of the activator (buffer) and the substrates was varied to achieve a wide range of lifetimes for the nanozyme. The self-inhibitory feedback mechanism employed herein presents a new approach to generation of transient functional assemblies and may serve as a novel addition to the progressively developing field of out-of-equilibrium systems chemistry.Data availability
The datasets supporting this article have been uploaded as part of this manuscript and its corresponding ESI.†Author contributions
SD and DD conceived the idea. SD, TD and PD executed the work and analysed the data along with DD. SD and DD wrote the manuscript with the help of TD and PD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD acknowledges financial support from the SERB (CRG 2020/002030) and FIST program for the Department of Chemistry, IIT Guwahati.Notes and references
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed.
- M. Freeman, Nature, 2000, 408, 313–319 CrossRef CAS PubMed.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- J. A. Russell, G. Leng and A. J. Douglas, Front. Neuroendocrinol., 2003, 24, 27–61 CrossRef CAS.
- J. Jesty and E. Beltrami, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 2463–2469 CrossRef CAS PubMed.
- H. Modell, W. Cliff, J. Michael, J. McFarland, M. P. Wenderoth and A. Wright, Adv. Physiol. Educ., 2015, 39, 259–266 CrossRef PubMed.
- S. P. Afrose, S. Bal, A. Chatterjee, K. Das and D. Das, Angew. Chem., Int. Ed., 2019, 58, 15783–15787 CrossRef CAS PubMed.
- S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed.
- M. N. Z. Yurt, Y. Cakmak, G. Tekin, S. Karakurt and S. Erbas-Cakmak, ACS Omega, 2019, 4, 12293–12299 CrossRef CAS PubMed.
- W. Huang, Y. Zhou, Y. Deng and Y. He, Phys. Chem. Chem. Phys., 2018, 20, 4347–4350 RSC.
- K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- L. S. Kariyawasam and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 11949–11955 CrossRef CAS PubMed.
- L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, Angew. Chem., Int. Ed., 2021, 60, 12648–12658 CrossRef CAS PubMed.
- P. S. Schwarz, M. Tena-Solsona, K. Dai and J. Boekhoven, Chem. Commun., 2022, 58, 1284–1297 RSC.
- M. P. van der Helm, C.-L. Wang, B. Fan, M. Macchione, E. Mendes and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 20604–20611 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem.–Eur. J., 2021, 27, 8928–8939 CrossRef CAS PubMed.
- J. Leira-Iglesias, A. Sorrenti, A. Sato, P. A. Dunne and T. M. Hermans, Chem. Commun., 2016, 52, 9009–9012 RSC.
- K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC.
- J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS PubMed.
- W. A. Ogden and Z. Guan, ChemSystemsChem, 2020, 2, e1900030 CrossRef CAS.
- X. Fan and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 11398–11405 CrossRef CAS PubMed.
- T. Heuser, A.-K. Steppert, C. Molano Lopez, B. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS PubMed.
- S. Panja, K. Boháčová, B. Dietrich and D. J. Adams, Nanoscale, 2020, 12, 12840–12848 RSC.
- P. Dowari, S. Das, B. Pramanik and D. Das, Chem. Commun., 2019, 55, 14119–14122 RSC.
- S. Panja, C. Patterson and D. J. Adams, Macromol. Rapid Commun., 2019, 40, 1900251 CrossRef PubMed.
- X. Hao, K. Yang, H. Wang, F. Peng and H. Yang, Angew. Chem., Int. Ed., 2020, 59, 4314–4319 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- D. Das, K. I. Assaf and W. M. Nau, Front. Chem., 2019, 7, 619 CrossRef CAS PubMed.
- A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704–10724 CrossRef CAS PubMed.
- S. Das and D. Das, Front. Chem., 2021, 9, 770102 CrossRef CAS PubMed.
- J. H. Mondal, S. Ahmed and D. Das, Langmuir, 2014, 30, 8290–8299 CrossRef CAS PubMed.
- J. H. Mondal, S. Ahmed, T. Ghosh and D. Das, Soft Matter, 2015, 11, 4912–4920 RSC.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- Y. J. Jeon, P. K. Bharadwaj, S. Choi, J. W. Lee and K. Kim, Angew. Chem., Int. Ed., 2002, 41, 4474–4476 CrossRef CAS PubMed.
- J. H. Mondal, T. Ghosh, S. Ahmed and D. Das, Langmuir, 2014, 30, 11528–11534 CrossRef CAS PubMed.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS PubMed.
- S. Ahmed, N. Singha, B. Pramanik, J. H. Mondal and D. Das, Polym. Chem., 2016, 7, 4393–4401 RSC.
- Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305–1315 RSC.
- F. Biedermann, U. Rauwald, J. M. Zayed and O. A. Scherman, Chem. Sci., 2011, 2, 279–286 RSC.
- Y. H. Ko, K. Kim, J.-K. Kang, H. Chun, J. W. Lee, S. Sakamoto, K. Yamaguchi, J. C. Fettinger and K. Kim, J. Am. Chem. Soc., 2004, 126, 1932–1933 CrossRef CAS PubMed.
- N. Ma, F. Li, S. Li, S. Chu, L. Han, S. Liu, T. Yan, R. Tian, Q. Luo and J. Liu, Nanoscale, 2019, 11, 3521–3526 RSC.
- M. D. Nothling, Z. Xiao, N. S. Hill, M. T. Blyth, A. Bhaskaran, M.-A. Sani, A. Espinosa-Gomez, K. Ngov, J. White, T. Buscher, F. Separovic, M. L. O'Mara, M. L. Coote and L. A. Connal, Sci. Adv., 2020, 6, eaaz0404 CrossRef CAS PubMed.
- L. Yang, H. Yang, F. Li and X. Zhang, Langmuir, 2013, 29, 12375–12379 CrossRef CAS PubMed.
- M. E. Bush, N. D. Bouley and A. R. Urbach, J. Am. Chem. Soc., 2005, 127, 14511–14517 CrossRef CAS PubMed.
- B. Pramanik and D. Das, J. Phys. Chem. C, 2018, 122, 3655–3661 CrossRef CAS.
- S. Das, P. Das, P. Dowari, B. K. Das and D. Das, J. Colloid Interface Sci., 2022, 614, 172–180 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, A. A. Orr, P. Makam, B. Redko, E. Haimov, Y. Wang, L. J. W. Shimon, S. Rencus-Lazar, M. Ju, P. Tamamis, H. Dong and E. Gazit, Angew. Chem., Int. Ed., 2021, 60, 17164–17170 CrossRef CAS PubMed.
- E. Delort, T. Darbre and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642–15643 CrossRef CAS PubMed.
- N. Singh, M. P. Conte, R. V. Ulijn, J. F. Miravet and B. Escuder, Chem. Commun., 2015, 51, 13213–13216 RSC.
- L. Pasquato, F. Rancan, P. Scrimin, F. Mancin and C. Frigeri, Chem. Commun., 2000, 2253–2254, 10.1039/B005244M.
- Applications of Ion Chromatography for Pharmaceutical and Biological Products, ed. L. Bhattacharyya and J. S. Rohrer, Wiley VCH, 2012, pp. 449–453, ISBN: 9780470467091 Search PubMed.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- P. J. Pearce and R. J. J. Simkins, Can. J. Chem., 1968, 46, 241–248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental details and other supporting figures. See DOI: 10.1039/d1sc07203j |
| This journal is © The Royal Society of Chemistry 2022 |