
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel high-energy-density lithium-free anode dual-ion battery and in situ revealing the interface structure evolution†
Li-Na
Wu‡
ab,
Zheng-Rong
Wang‡
ab,
Peng
Dai
b,
Yu-Xiang
Xie
b,
Cheng
Hou
 a,
Wei-Chen
Zheng
b,
Fa-Ming
Han
c,
Ling
Huang
*b,
Wei
Chen
*a and
Shi-Gang
Sun
*b
a,
Wei-Chen
Zheng
b,
Fa-Ming
Han
c,
Ling
Huang
*b,
Wei
Chen
*a and
Shi-Gang
Sun
*b
aSchool of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin 541004, China. E-mail: weichen@mailbox.gxnu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: huangl@xmu.edu.cn
cGuangxi Key Laboratory of Electrochemical and Magneto-chemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, China. E-mail: sgsun@xmu.edu.cn
First published on 8th March 2022
Abstract
Lithium-free anode dual-ion batteries have attracted extensive studies due to their simple configuration, reduced cost, high safety and enhanced energy density. For the first time, a novel Li-free DIB based on a carbon paper anode (Li-free CGDIB) is reported in this paper. Carbon paper anodes usually have limited application in DIBs due to their poor electrochemical performance. Herein, by using a lithium bis(fluorosulfonyl)imide (LiFSI)-containing electrolyte, the battery shows outstanding electrochemical performance with a capacity retention of 96% after 300 cycles at 2C with a stable 98% coulombic efficiency and 89% capacity retention after 500 cycles at 5C with a stable coulombic efficiency of 98.5%. Moreover, the electrochemical properties of the CGDIB were investigated with a variety of in situ characterization techniques, such as in situ EIS, XRD and online differential electrochemical mass spectrometry (OEMS). The multifunctional effect of the LiFSI additive on the electrochemical properties of the Li-free CGDIB was also systematically analyzed, including generating a LiF-rich interfacial film, prohibiting Li dendrite growth effectively and forming a defective structure of graphite layers. This design strategy and fundamental analysis show great potential and lay a theoretical foundation for facilitating the further development of DIBs with high energy density.
1. Introduction
Dual-ion batteries (DIBs) have emerged as promising energy storage systems with the merits of high energy density, outstanding rate performance, simple design, low cost, and green and environmentally friendly characteristics.1,2 With regard to DIBs, the negative electrode materials greatly affect the energy density.3–5 In a typical DIB system, graphite acts as both the anode and cathode material, while an ionic liquid (IL) is utilized as the electrolyte.6,7 Nevertheless, the application of graphite anodes is restricted by the low capacity, high cost and high viscosity of ILs and even the insufficient compatibility of ILs and graphite anodes.5,6,8 In 2016, Prof. Yongbing Tang and coworkers reported transformational work in which they developed a novel aluminum–graphite DIB (AGDIB) with low-cost aluminum (Al) as the anode and an ethyl methyl carbonate (EMC) electrolyte as the electrolyte.9 Remarkably, owing to its high theoretical capacity (993 mA h g−1) and low potential (≈0.38 V vs. Li/Li+), Al foil instead of traditional graphite was used as the anode to greatly improve the specific energy density of the AGDIB. The battery delivers an energy density of ≈220 W h kg−1 at a power density of ≈130 W kg−1, which is significantly superior to that of most commercial lithium-ion batteries (≈200 W h kg−1 at ≈50 W kg−1). Subsequently, using the lithium metal anode, due to its highest theoretical specific capacity (3862 mA h g−1) and lowest electrochemical potential (−3.04 V vs. standard hydrogen electrode),10 our group reported a Li–graphite DIB, which exhibits an outstanding theoretical energy density of 243 W h kg−1 at 234 W kg−1.11 Therefore, anode materials play a crucial role in determining the energy density of DIBs. However, metallic anodes (Al, Sn, Li, etc.) were subjected to inevitable volume changes during the repetitive alloying/dealloying process (Al: ≈97%; Sn: ≈260%). In addition, uncontrolled growth of lithium dendrites during metal plating/stripping may cause internal short-circuits, fast capacity fading and other potential safety issues. Moreover, Li metal anodes are employed in most research, usually with excess lithium, which will reduce the actual specific capacity and volume specific capacity of the battery and cause pollution and waste. Moreover, the instability of Li metal will lead to repeated SEI generation/rupture, which could result in the decomposition of the electrolyte, an increase in the internal impedance and a shortened lifespan of the battery.12–14 To solve the above issues, many effective approaches have been explored to enhance the stability of anode materials, including interface modification,15,16 structural engineering17,18 and introducing electrolyte additives19,20 or solid-state electrolytes.21,22 Despite the aforementioned effective strategies, it is essential to further improve the performance of DIBs for practical applications.Currently, lithium-free anodes with a simplified assembly process, higher energy density, and superior safety14 are attracting researchers' interest. Nevertheless, the large volume changes, Li dendrite growth, constant electrolyte consumption, and formation of a thick accumulated SEI layer23 in a lithium-free anode also bring about poor coulombic efficiency and cycling performance. Generally, an ultrathin Li metal anode (e.g., thickness < 50 μm, ideally 20 μm or less) is a potential material for improving the safety and stability of the battery. However, in fact, the price of high-quality thin Li foil even exceeds US$1000 kg−1 (graphite-based anode: US$20 kg−1), and the manufacturing techniques are rather complex. Therefore, the high cost of ultrathin Li metal anodes hinders their practical application in DIBs.14 Based on the above, it is imperative to develop other anode materials that are cheaper, safer and more stable and benefit the performance improvement of DIBs.
Herein, we for the first time assembled a novel lithium-free anode dual-ion battery coupled with a carbon paper anode (named Li-free CGDIB). Compared with metal anodes, carbon paper anodes exhibit several benefits, including (1) satisfactory electronic conductivity, mature preparation technology, low cost, and environment friendliness;24 and (2) 3D matrix host structures that can relieve volume variation and structural degradation and play a positive role in dendrite growth inhibition.25 Nevertheless, the poor electrochemical performance of the carbon paper electrode is a restricting factor for its application in DIBs.26 Notably, as an electrolyte additive, lithium bis(fluorosulfonyl)imide (LiFSI) showed superiority in improving the interface stability and cycling performance of graphite substrates, which was conducted to suppress the destruction of graphite from metallic lithium and form a stable SEI film, showing the excellent electrochemical performance of graphite electrodes.27,28 Consequently, combining the Li-free CGDIB technique with a LiFSI-containing electrolyte, it is worth noting that the Li-free CGDIBs enable a reversible discharge capacity of ≈95 mA h g−1 at 2C (1C = 100 mA g−1) with a capacity retention of 96% after 300 charge/discharge cycles and a superior capacity retention of 89% after 500 cycles at 5C. According to the estimation, the theoretical specific energy density of the Li-free CGDIBs is calculated to be ≈387 W h kg−1 at a power density of 450 W kg−1 and ≈320 W h kg−1 at 970 W kg−1, which clearly exceed those of most DIBs. The outstanding properties of the Li-free CGDIB demonstrate that it is a potential energy storage device with the key metrics of low cost, rapid charging performance and high energy density. In addition, for the first time, with a suit of complementary electrochemical characterization techniques and in situ characterization techniques, we can intensively analyze in detail the function of the LiFSI additive in Li-free CGDIBs to understand the advantage of the LiFSI additive and develop its potential for large-scale DIB techniques.
2. Results and discussion
2.1 Selection of anode material and optimization of LiFSI additive content
The choice of anode material is crucial for the performance of DIBs. Fig. S1† compares the cycling properties of DIBs with different anodes (Cu, graphite, and carbon paper) at 2C in the 3.0–5.0 V working voltage range. Obviously, the battery based on the Cu anode shows a “diving” phenomenon of discharge capacity after the activation process, which is most likely related to the short circuit triggered by the growth of Li dendrites on the Cu anode. It is worth noting that a rapid capacity decay phenomenon occurred for the DIB based on the graphite anode, which means that severe exfoliation ensued on the graphite anode during the cointercalation of the solvated Li+. Surprisingly, based on the carbon paper anode, the favorable cycling performance of the battery indicates that the carbon paper anode shows great potential for ensuring the electrochemical performance in DIBs. Therefore, carbon paper is a promising anode material for Li-free DIBs.Although a Li-free CGDIB coupled with a carbon paper anode based on an electrolyte (4 M LiPF6 + EMC + VC + FEC + LiFSI) has the potential to exhibit impressive electrochemical performance, the electrolyte composition plays a significant role in enhancing the electrochemical properties of the Li-free CGDIB. The conventional composition of 4 M LiPF6 + EMC + VC + FEC has been widely used in DIBs due to its superior performance arising from combining the merits of the electrolyte components. In this work, after screening and matching different electrolyte additives, it can be found that combining conventional DIB electrolytes (4 M LiPF6 + EMC + VC + FEC) with LiFSI is appropriate and useful in promoting the cycling ability of Li-free CGDIBs, and LiFSI greatly affects the electrochemical properties of Li-free CGDIBs. Naturally, we first investigated the correlation between the cycling performance of Li-free CGDIBs and the content of LiFSI in electrolytes with different molar contents. As shown in Fig. S2,† the capacity of the battery with only 0.02 M LiFSI additive decayed after 50 cycles. With increasing amount of LiFSI, the cycling ability of the battery is also enhanced. However, the capacity of the battery decreased with 0.12 M LiFSI additive. These results imply that 0.02 M LiFSI additive is not enough to participate in the decomposition reaction to form a stable interphase film, but a thick interphase film is generated with 0.12 M LiFSI additive due to its excess decomposition, which possibly weakens the diffusion of ions and leads to a decrease in the specific capacity. It is worth noting that a higher capacity of 90 mA h g−1 is obtained in the battery with 0.1 M LiFSI additive after activating cycles and can be retained in the remaining cycles. That is, 0.1 M LiFSI additive is the optimized amount for obtaining the outstanding performance of Li-free CGDIBs.
To further analyze the influence of the amount of LiFSI on the performance of the battery, the corresponding XPS experiments were employed to illustrate the performance of the battery at higher/lower concentrations of LiFSI based on the specific compositions of the SEI. It was reported that an inorganic SEI film is favorable for a high Li+ transference number and for effectively prohibiting the growth of Li dendrites, enhancing the stability of the interface and reducing the impedance, and thus leading to improved electrochemical performance.29,30 Meanwhile, the organic component is beneficial for enhancing the flexibility of the SEI films31 but unfavorable for Li+ transport. Fig. S3† displays the atomic percentages of C, P, O, F, and Li in anodes from cells after cycling in CDE with different amounts of LiFSI for ten cycles at 0.5C rate. Compared with higher/lower concentrations of LiFSI (0.02 M/0.12 M), the inorganic component content of LiF species increased with 0.1 M LiFSI. On the other hand, the content of the dominant C–O–C species, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species and oxygen amount distinctly decreased, which could be ascribed to the decreased organic component (possibly ROCOOLi),27 which may be due to the suppression of electrolyte decomposition.32 The XPS results indicate that the addition of 0.1 M LiFSI can result in the formation of an inorganic-rich SEI layer. At higher/lower concentrations of LiFSI (0.02 M/0.12 M), the decreased inorganic component seems incapable of imparting a satisfactory Li+ interfacial transfer ability and a stable interface, leading to a high interface resistance.29 Hence, a lower capacity and shorter lifespan of the battery was obtained at higher/lower concentrations of LiFSI. In contrast, the SEI component with a higher inorganic LiF component and lower organic component was achieved after the addition of 0.1 M LiFSI. Maybe this configuration is favorable for the high Li+ transference number. Meanwhile, the anions easily coordinate with Li+, creating a cross-linked network to inhibit the growth of Li dendrites, suppress the decomposition of electrolyte and improve the stability of the SEI layer,30,31 which eventually enhances the capacity and prolongs the lifespan of the battery.
O species and oxygen amount distinctly decreased, which could be ascribed to the decreased organic component (possibly ROCOOLi),27 which may be due to the suppression of electrolyte decomposition.32 The XPS results indicate that the addition of 0.1 M LiFSI can result in the formation of an inorganic-rich SEI layer. At higher/lower concentrations of LiFSI (0.02 M/0.12 M), the decreased inorganic component seems incapable of imparting a satisfactory Li+ interfacial transfer ability and a stable interface, leading to a high interface resistance.29 Hence, a lower capacity and shorter lifespan of the battery was obtained at higher/lower concentrations of LiFSI. In contrast, the SEI component with a higher inorganic LiF component and lower organic component was achieved after the addition of 0.1 M LiFSI. Maybe this configuration is favorable for the high Li+ transference number. Meanwhile, the anions easily coordinate with Li+, creating a cross-linked network to inhibit the growth of Li dendrites, suppress the decomposition of electrolyte and improve the stability of the SEI layer,30,31 which eventually enhances the capacity and prolongs the lifespan of the battery.
Furthermore, the influencing factors on the electrochemical performance of DIBs from electrolyte features, such as solvation structure, ion conductivity, and viscosity, were also studied. Fig. S4† shows the 7Li NMR spectra of the electrolyte with different amounts of LiFSI. The peak of 7Li in CDE + 0.02 M LiFSI electrolyte shifts from −0.600 ppm to −0.605 ppm in CDE + 0.1 M LiFSI and −0.614 ppm in CDE + 0.12 M LiFSI. The decreased chemical shifts can be attributed to the participation of more FSI− anions in the solvation sheath with increasing LiFSI concentration. Increased Li+–FSI− coordination may result in a weaker solvation between Li+ and PF6−, which can affect the composition of the SEI film and improve the conductivity of the SEI film, enabling a robust and uniform interface for improved cycling stability of the battery.31 However, the weaker solvation may lead to the decrease of inferior ionic conductivity of the electrolyte.33 As shown in Table S1,† the ionic conductivity (σ) of the CDE electrolyte is 2.63 mS cm−1. As the concentration of LiFSI increased from 0.02 M to 0.12 M, the ionic conductivity decreased from 2.50 to 2.26 mS cm−1. Meanwhile, the addition of FSI− can contribute to the increase of the viscosity (η). The viscosity of the CDE electrolyte is 30.01 mPa s. As the concentration of LiFSI increased from 0.02 to 0.12 M, the viscosity of the electrolyte increased from 31.82 mPa s to 36.12 mPa s. The decrease in inferior ionic conductivity and increase in viscosity of electrolyte with increasing concentration of LiFSI can be explained by the stronger Li+–FSI− interaction and poorer mobility of the charge carriers.33–35 Therefore, it is crucial to find the balance between electrolyte properties (higher viscosity and lower ionic conductivity of electrolyte) and high electrochemical performance. In the case of poor electrolytes, it is worth noting that the effect of the LiFSI additive on solvation structure, SEI composition, and Li ion deposition behaviors may become more dominant factors in the electrochemical performance of the DIB.
To supply strong evidence of the effect of the LiFSI additive, we mainly discuss the characterization results for conventional DIB electrolytes (CDEs) and 0.1 M LiFSI-containing CDE electrolyte (abbreviated as CDE + LiFSI).
The relationship between the electrochemical performances of the Li-free CGDIBs and LiFSI additive was investigated in the 3.0–5.0 V voltage window. Fig. 1a shows the cycling performance comparison of the battery at 2C. In CDE + LiFSI, the Li-free CGDIB exhibits a satisfactory reversible discharge capacity of 90 mA h g−1 after activating cycles at 0.5C and a capacity retention of 96% after 300 cycles at 2C with a corresponding coulombic efficiency of 98%. Conversely, a “fluctuation” phenomenon of capacity and coulombic efficiency occurred during cycling in CDE, possibly related to the instability of the battery caused by Li dendrite formation. Fig. S5† shows the galvanostatic voltage curves of the Li-free CGDIB in CDE + LiFSI for different cycles. Except for the 10th charge/discharge curve at 0.5C, other curves overlap well, and there is a small polarization after 10 cycles in the battery, indicating that LiFSI enables the cycling properties of the battery to improve considerably. Fig. 1b compares the average discharge middle voltage of the Li-free CGDIB. The inset shows the voltage profiles in CDE + LiFSI from 160 h to 200 h. It is clear that a ∼4.3 V average discharge middle voltage is obtained with LiFSI and that it shows 96% retention after 300 cycles. However, the “fluctuation” phenomenon also presents in the discharge middle voltage in CDE within the first few activating cycles, which could be due to the instability of the battery. Fig. 1c and d show the galvanostatic voltage curves of the Li-free CGDIB in CDE and CDE + LiFSI from 0.5 to 8C, respectively. The battery shows a large polarization and a decreased reversibility in CDE with increasing current rate (Fig. 1c), while the curves are basically identical to those with CDE + LiFSI (Fig. 1d), indicating decreased polarization and thereby improved rate capability of the battery. Fig. 1e further compares the rate capacities of the Li-free CGDIB. Impressively, the discharge capacity still holds high stability for each of the 5 cycles, even after the high current cycles. In particular, the 8C/0.5C capacity ratio increased to 92% from 51% compared with the battery in CDE. The above results further demonstrate that LiFSI can help improve the capacity reversibility and thus ensure the high rate ability of Li-free CGDIBs. The long-duration cycling stabilities of the Li-free CGDIBs at 5C are compared in Fig. 1f. The CGDIB with LiFSI exhibits an outstanding capacity retention of 89% after 500 cycles, and the corresponding coulombic efficiency is maintained at 98.5%. In contrast, the capacity of the CGDIB in CDE decays dramatically after 200 cycles, and the corresponding coulombic efficiency also shows the “fluctuation” phenomenon, which might be due to the unstable interfacial film during cycling in CDE. The comparison result indicates that LiFSI plays a significant role in enhancing the cycling ability of the CGDIB. Even more remarkably, the calculated energy density of the Li-free CGDIB is ≈387 W h kg−1 at a power density of 450 W kg−1 and ≈320 W h kg−1 at 970 W kg−1, which is competitive with most reported DIBs (Table S2†).
 | ||
| Fig. 1 (a) The cyclability of Li-free CGDIBs cycled in different electrolytes in the voltage range of 3–5 V at 0.5C (1–10 cycles) and 2C (11–300 cycles). (b) The discharge medium voltage of Li-free CGDIBs cycled in different electrolytes during cycling tests. The inset shows the galvanostatic charge/discharge curves cycled in the electrolyte with LiFSI during the period of 160–200 h. The corresponding galvanostatic charge/discharge curves of selected cycles for the Li-free CGDIBs (c) in CDE and (d) in CDE + LiFSI. (e) Rate performance of Li-free CGDIBs in the voltage range of 3–5 V. (f) Cyclability of Li-free CGDIBs cycled in different electrolytes in the voltage range of 3–5 V at a high current density of 5C (0.5C for 1–10 cycles and 5C for 11–500 cycles). | ||
2.2 Electrochemical testing and mechanism analysis by in situ techniques
The superior electrochemical performance of the Li-free CGDIB guides us to discuss the strong effect of the LiFSI additive on the electrochemical properties. Therefore, systematic characterization was carried out. Fig. 2a displays the typical cyclic voltammograms of the Li-free CGDIB at 3.0–5.0 V. Generally, each current peak position in the profile corresponds to the PF6− intercalation and deintercalation stages. Compared to CDE, upon charging, PF6− intercalation shows an obvious shift to the lower potential region in CDE + LiFSI. In particular, the onset potential of PF6− intercalation changes from 4.05 V (vs. Li/Li+) (in CDE) to 3.94 V (vs. Li/Li+) (in CDE + LiFSI). In addition, the main intercalation process is located at 4.05–5.0 V (vs. Li/Li+) in CDE, while ranging from 3.94 V to 4.9 V (vs. Li/Li+) with LiFSI. Upon discharging, the anion de-intercalation potential drops to about 4.38 and 3.89 V (vs. Li/Li+) with LiFSI compared to 4.45 and 4.00 V (vs. Li/Li+) observed in CDE. This result suggests that the LiFSI additive can possibly weaken the coordination ability between Li+ and PF6−, which further benefits PF6− prevailing over the initial intercalation kinetic process. Fig. 2b compares the nuclear magnetic resonance (NMR) spectra. Notably, the 7Li signal of lithium salts in CDE moves from −0.55 to −0.61 ppm after the addition of LiFSI, which may be the result of an introduction of electron-rich FSI− and the weakened coordination ability between Li+ and PF6−. In the following, we also studied the effect of LiFSI on the properties of electrodeposited Li nanoparticles. Comparing the Li electrodeposition behavior on the Pt electrode between −0.4 V and 0.2 V at a current density of 0.5 mV s−1 (Fig. 2c), it is apparent that the overpotential of Li electrodeposition increased with the addition of LiFSI. Furthermore, similar results can also be obtained from the exchange current density derived from the Tafel tests. Fig. 2d presents the Tafel curves based on the carbon paper‖Li battery at 1 mV s−1. By fitting the Tafel curves, the exchange current density decreases from 3.31 mA cm−2 in CDE to 2.21 mA cm−2 in the presence of LiFSI. Hence, the addition of LiFSI to CDE could increase the nucleation overpotential, which induces the smooth deposition of Li+ and thus controls the growth of Li dendrites. | ||
| Fig. 2 (a) The CV curves of Li-free CGDIBs cycled in different electrolytes. (b) 7Li NMR spectra of different electrolytes. (c) LSV tests of Li deposition on a Pt electrode. (d) Tafel plots of different electrolytes. | ||
To further demystify the effect of LiFSI on the initial interfacial reactions, the evolved gas resulting from the decomposition of electrolyte was monitored in the first two cycles by online differential electrochemical mass spectrometry (OEMS), and thus the electrolyte/electrode interface reaction was inferred. As shown in Fig. 3, only varied CO2 gas signals were detected. For CDE (Fig. 3a), the CO2 gas signal peak rises steeply starting from 3.9 V to 4.2 V and continues to slightly increase up to the end of the first charging process, suggesting the continued decomposition reaction of the electrolyte during interfacial film formation in the first charging cycle. It should be noted that a slight amount of CO2 gas is generated at the end of the first discharging stage, signifying the decomposition reaction associated with the repair of the interfacial film. During the second charging/discharging process, although the repeated variation trend of CO2 gas is detected, the intensity of the peak obviously decreases. This indicates the decomposition of the electrolyte and formation of the interfacial film again. For CDE + LiFSI (Fig. 3b), similar to CDE, the CO2 gas signal peak rises steeply and reaches a peak value within 3.9–4.2 V in the first charging process, indicating the decomposition of electrolyte and the formation of the interfacial film only in the potential range of 3.9–4.2 V. In contrast, less CO2 production can be observed during the end charging stage and the discharging process due to the mitigation of electrolyte decomposition. In addition, the repeated variation of the CO2 gas signal peak during the second charging/discharging process exhibits a weaker peak intensity compared to that in CDE. Therefore, it can be concluded that the LiFSI additive can suppress the excessive decomposition of electrolyte and benefit the formation of a stable interfacial film. In addition, DFT theoretical calculations indicate that the adsorption energy of the FSI− anion on graphene is −5.2 eV, while the adsorption energy of the PF6− anion on graphene is −4.5 eV (Fig. 3c). The result shows that there is a stronger interaction between the FSI− anion and carbon substrate and suggests that FSI− preferentially decomposes and participates in the formation of the interface film.
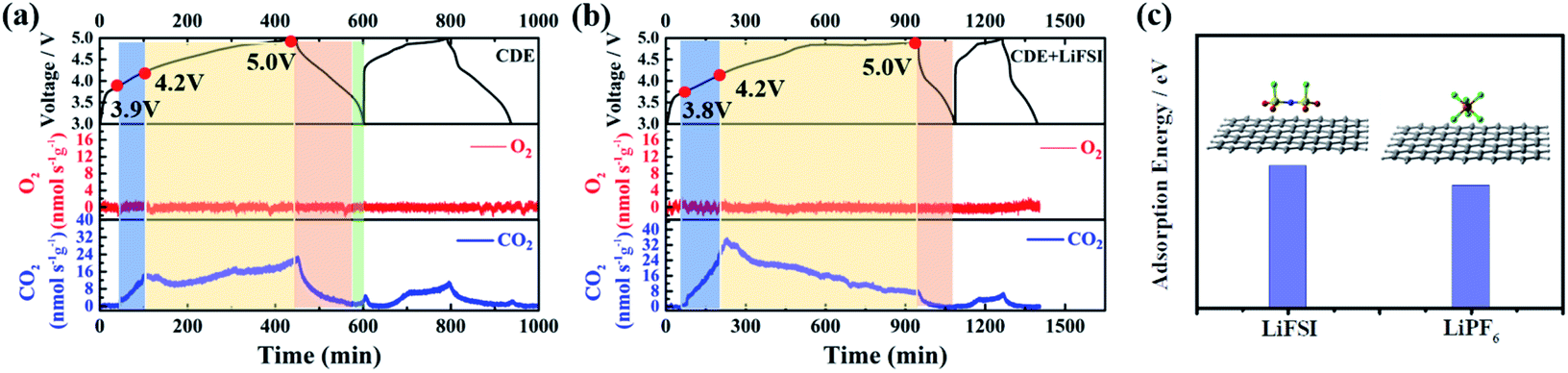 | ||
| Fig. 3 OEMS tests of Li-free CGDIBs cycled in (a) CDE and (b) CDE + LiFSI at 0.5C for two cycles. (c) The DFT calculated structure and adsorption energy at the M06-L/(def2-SVP) level of theory. | ||
To gain further insights into the effect of LiFSI on CEI/SEI composition, the surface compositions of the carbon paper anode and graphite cathode after cycling were investigated by X-ray photoelectron spectroscopy (XPS). The bar graph in Fig. S6† displays the atomic percentages of C, P, O, F and Li in the cycled cathode and anode from cells after cycling in CDE and CDE + LiFSI for ten cycles at 0.5C rate, respectively. It can be seen that the cathode and anode cycled in CDE + LiFSI have lower oxygen content and C–O–C content, demonstrating that the LiFSI additive is effective in suppressing the decomposition of solvent.36 Moreover, a negligible variation of phosphorus content was detected in CDE + LiFSI. This phenomenon indicates that the addition of LiFSI can effectively inhibit the decomposition of LiPF6,27 and result in more PF6− anions participating in the intercalation/de-intercalation process in the cathode reaction, which will alleviate the corrosion reactions from decomposed PF5 and ensure the stability of the CEI/SEI film.31 In addition, the higher LiF content of the electrode in CDE + LiFSI compared to that in CDE suggests that FSI− is more labile to be reduced and participates in the formation of a stable LiF-rich interfacial film.37 This can improve the diffusion of Li+ and thus decrease the charge transfer resistance, thereby ensuring a smooth charge transfer reaction and suppressing the growth of Li dendrites.29
The chemical environment of dominant species C, F, and P is identified by the XPS spectra, and the results are compared in Fig. 4. Fig. 4a–c compare the effect of LiFSI on the CEI composition. In the C 1s spectra (Fig. 4a), the peaks at ∼284.4, ∼285.7 and ∼291.4 eV belong to C–H/C–C, C–O–C and C–F (PVDF binder), respectively.38 The F 1s spectra show two peaks at ∼685.7 eV (Li–F) and ∼691.4 eV (organic fluorides) (Fig. 4b).39 It is obvious that the ratio of Li–F on the CEI increases in CDE + LiFSI. The increment in Li–F content is most likely responsible for the decomposition of LiFSI. Moreover, the LiF- rich layer is beneficial to improving the diffusion ability of Li+ and suppressing the electrolyte decomposition.40 In the P 2p spectra (Fig. 4c), the peaks at ∼133.9 eV and ∼134.9 eV correspond to P–O and LixPOyFz, respectively. The existence of P–O can enhance the interfacial transfer kinetics of Li+,41 while LixPOyFz can improve the stability of the interfacial film.31 In CDE + LiFSI, the increased peak intensity of LixPOyFz indicates that LiFSI can contribute to building a more stable CEI on the graphite cathode.
 | ||
| Fig. 4 The XPS deconvoluted spectra of C 1s, F 1s, and P 2p for the cathode (a–c) and anode (d–f) after cycling in CDE and CDE + LiFSI for ten cycles. | ||
Furthermore, the surface composition of the SEI is also compared in Fig. 4d–f. Obviously, the LiFSI additive has a noticeable effect on the composition of the SEI film. For the C 1s spectra (Fig. 4d), observably, CO shows a weaker peak intensity in CDE + LiFSI than in CDE, signifying that LiFSI is beneficial for forming a uniform SEI and prevents the side reaction of the electrolyte. From the F1 s spectra (Fig. 4e), although the composition of the SEI is similar to that of the CEI, the content ratio is different. Notably, the increased proportion of the Li–F peak in the SEI than in the CEI indicates that electrolyte decomposition mainly participates in the formation of the SEI. The higher peak intensity of Li–F favors the improvement of Li+ interfacial transfer ability, thus effectively prohibiting the growth of Li dendrites and further electrolyte decomposition. From the spectra of P 2p shown in Fig. 4f, it is clear that the peak at ∼133.63 eV (LixPFy) disappears in CDE + LiFSI, while LixPFy is probably residual LiPF6 according to the reported work.42 The present result confirms that LiFSI prevents the decomposition of LiPF6. Altogether, combined with the results of NMR and DFT theoretical calculations, the LiFSI can decompose prior to the electrolyte and LiPF6 and form a more stable and homogeneous LiF-rich SEI. In addition, the LiF-rich SEI can enhance the interfacial diffusion of Li+, induce a homogeneous deposition of Li+ and effectively inhibit Li dendrite growth.
Electrochemical impedance spectroscopy (EIS) is a powerful technique to identify the electrode/electrolyte interphase properties and kinetic processes. To further reveal the influence of LiFSI on the interfacial changes, in situ EIS tests were performed during the initial cycling process with a three-electrode system, which recorded the impedance spectra of the anode, cathode and full cell. In general, the high-frequency region of EIS is related to interphase resistance (Rf), while the intermediate-frequency region is related to charge transfer resistance (Rct) through the electrode/electrolyte interface. The measured interfacial resistance and charge transfer resistance can reflect the effect of LiFSI on the interface stability. Obviously, the Nyquist plots show an apparent radius variation of the semicircle during the charging/discharging process (Fig. 5a–h and S7a–d†). From the equivalent circuit shown in Fig. S8,† the correlated variations of Rf and Rct are achieved by fitting the Nyquist plots using ZView software.
 | ||
| Fig. 5 In situ EIS profiles of Li-free CGDIBs on the cathode and anode at the first two cycles cycled in CDE (a–d) and in CDE + LiFSI (e–h). Variation trend plots of Rf and Rct on the cathode and anode at the first two cycles cycled in CDE and CDE + LiFSI (i–l). | ||
As shown in Fig. 5i, upon introducing the LiFSI additive, a significant difference in the variation tendency of Rf was observed in the first cycle. In CDE, Rf begins to increase from 4.3 V at the first charging stage and stepwise increases up to 4.8 V, which is associated with the formation of an SEI and excessive decomposition of the electrolyte.43 A decreasing tendency in Rf is observed at higher voltages (>4.8 V), which is related to the unstable SEI caused by possible inner microstress or corrosion of the electrolyte.44 Moreover, combined with the OEMS results and the working mechanism of Li-free CGDIBs, it can be inferred that SEI formation is preferentially associated with the decomposition of the electrolyte and interphase change on the anode surface. During the discharging process, Rf restarts to increase from 4.5 V, suggesting the decomposition of the electrolyte and its main responsibility for repairing the unstable SEI.43 After adding the LiFSI additive, the Rf at 4.3 V in the charged state and 4.5 V in the discharged state can be observed due to electrolyte decomposition and SEI formation and repair. Remarkably, the value of Rf in the presence of LiFSI can maintain a relatively high stability, suggesting that less electrolyte decomposed. The difference in the variation tendencies of Rf is clear evidence that LiFSI plays a vital role in suppressing electrolyte decomposition and favoring stable SEI formation.
The variation tendency of Rct is shown in Fig. 5i. In CDE, a slight increase of Rct at 4.35 V can be observed during the first charging process. This result may be associated with the concentration polarization of the Li+ embedding process on the anode followed by an increase in Rct.44 On the other hand, Rct decreases with potentials higher than 4.4 V, which represents the insertion of Li+ into the anode accompanied by reduced concentration polarization, thereby resulting in an increased content of Li on the anode along with a decrease in Rct.43 At the end of the discharging process, the minor increase in Rct indicates the end of the deinsertion process of Li+ on the anode. After the addition of the LiFSI additive, there was a minimum at 4.3 V, which corresponds to the insertion of Li+ into the anode. In addition, LiF on the anode is helpful for improving the diffusion of Li+ and thus for decreasing the concentration polarization. At 4.2 V during the discharging process, the smaller increase in Rct is possibly because of the decreased concentration polarization caused by the reduced Li+ content at the discharging state.
Meanwhile, compared to the variation tendencies of Rf on the SEI, the value of Rf on the CEI decreases significantly (Fig. 5j), which strongly confirms that the SEI plays a decisive role in determining the interphase properties. In CDE, a slight increase in Rf at higher voltages (>4.8 V) correlates with the construction of a new phase and the continuous decomposition of the electrolyte. After the addition of the LiFSI additive, the stability of Rf suggests that the electrolyte decomposition reaction is effectively suppressed and CEI formation is controlled. Notably, a smooth variation tendency of Rct is reflected in both CDE and CDE + LiFSI. Similarly, the increase of Rct at 4.4 V is related to the concentration polarization during the intercalation reaction of PF6−, and the decrease in Rct at 4.8 V is associated with the completion of the PF6− intercalation reaction. At the same time, after the addition of the LiFSI additive, the Rct remains stable during the charging/discharging process, signifying the unobstructed intercalation/deintercalation reaction of PF6−.
Fig. S9a† compares the corresponding effect of LiFSI on the variation tendencies of Rf and Rct in the full cell. In CDE, similar to the variation tendencies of Rf during the charging process, at 4.8 V, the value of Rf decreases sharply and reaches a minimum at 5 V, which signifies the decomposition of the interfacial film at a higher voltage. Meanwhile, the increase of Rct at 4.3 V is due to the concentration polarization caused by the electrode/electrolyte interphase reaction. Upon charging to 4.4 V, the value of Rf starts to decrease because of the end of the electrode/electrolyte interphase reaction. During the discharging process, Rf slightly increases until 3.9 V suggesting the formation of interfacial film again. Simultaneously, the corresponding Rct begins to increase, showing the completion of the electrode/electrolyte interphase reaction. After the addition of the LiFSI additive, the stable variation tendencies of Rf and Rct indicate that LiFSI can effectively inhibit the decomposition of the electrolyte and is beneficial for building a stable interfacial film during the charging/discharging process. Furthermore, the LiFSI additive can facilitate the diffusion of Li+ and thus decrease the charge transfer resistance, thereby ensuring a smooth charge transfer reaction.
Fig. 5k compares the variation tendencies of Rf and Rct on the anode in the second cycle. Obviously, the value of Rf in CDE is higher than that in CDE + LiFSI. Combined with the OEMS results, the decomposition reaction of the electrolyte is significantly reduced in the presence of LiFSI, indicating that the formed SEI film is stable enough in the first cycle, and no more SEI film is reformed in the second cycle. For the variation tendencies of Rct, a slight increase in Rct starts at 4.4 V in CDE, which suggests the existence of concentration polarization during the insertion reaction of Li+. After the addition of the LiFSI additive, an increase in Rct occurs at the discharging state, which means the end of the deinsertion reaction of Li+.
Fig. 5l compares the variation tendencies of Rf and Rct on the cathode in the second cycle. Remarkably, much difference can be observed for CDE and CDE + LiFSI. In CDE, the irregular increases in Rf and Rct starting at the end of the charging process are clear evidence that an unstable CEI forms, which could be due to the excessive decomposition of the electrolyte which in turn affects the intercalation/deintercalation reaction of PF6−. After the addition of the LiFSI additive, the stable Rf and Rct are correlated with the stable CEI and intercalation/deintercalation reaction of PF6−.
Fig. S9b† compares the variation tendencies of Rf and Rct in the full cell in the second cycle. In CDE, the observed irregular increasing trends of Rf and Rct during discharging are similar to those for the cathode. This abnormal increasing trend could be ascribed to two reasons: the change in the graphite structure and uncontrolled growth of the CEI. After the addition of the LiFSI additive, the stable trends of Rf and Rct can be understood by the fact that LiFSI suppresses the excessive decomposition of electrolyte and facilitates a stable interfacial film, thus ensuring the completion of the electrode reaction.
In the following, EIS was employed to elucidate the influence of LiFSI on the interphase properties between electrodes and the electrolyte after 10, 50, 100 and 200 cycles at 2C. From the Nyquist impedance spectra shown in Fig. S10a and b,† the battery resistance in CDE experiences a significant increase from 10 to 200 cycles (Fig. S10a†), which indicates that an unstable interphase is formed and leads to continuous electrolyte decomposition. In contrast, the battery resistance in CDE + LiFSI remains relatively stable from 10 to 200 cycles (Fig. S10b†). The results verify that LiFSI has a positive effect on the interfacial stability, and the LiF-rich interface also facilitates charge transport at the interphase to indirectly protect the electrode materials and prevent side reactions during long-term cycling.
Subsequently, explicit evidence for the formation of the SEI induced by LiFSI can be further observed with SEM. Fig. 6 presents the SEM images of the carbon paper anode after 10, 50, 100 and 200 cycles in Li-free CGDIBs at 2C. Meanwhile, as shown in Fig. S11,† pure carbon paper has a 3D structure and consists of randomly interlaced carbon fibers with a smooth surface. After 10 cycles in CDE, it is obvious that the inhomogeneous SEI on the surface of carbon fibers with small deposits (Fig. 6a) is probably associated with nonuniform Li deposition. However, in the presence of the LiFSI additive, a more uniform SEI forms without any deposits (Fig. 6e). Moreover, in CDE, after 50 cycles, the SEI was still not completely coated on the carbon paper, and Li deposition with spherical structures was generated (Fig. 6b). It is worth noting that after 50 cycles, a homogeneous SEI has already been formed on carbon paper in the presence of LiFSI (Fig. 6f). Moreover, from Fig. 6c, exposed carbon fibers can be clearly observed after 100 cycles in CDE due to the generated porous and incomplete SEI. In contrast, a stable and complete SEI is formed after cycling in CDE + LiFSI. As expected, in the 200th cycle, the carbon paper cycled in CDE shows an incomplete SEI and uneven Li deposition (Fig. 6d). In comparison, the SEI formed in CDE + LiFSI is significantly homogeneous and complete without any deposits. Altogether, the SEM images prove that the addition of LiFSI is beneficial for generating a uniform and complete SEI, and the LiF component of the SEI is favorable to facilitate the diffusion ability of Li+ and suppress the growth of Li dendrites.
 | ||
| Fig. 6 SEM images of the carbon paper anode after cycling in CDE and CDE + LiFSI at the 10th (a and e), 50th (b and f), 100th (c and g) and 200th (d and h) cycles at 2C. | ||
In general, X-ray diffraction (XRD) is an authoritative technique to recognize the structural change of graphite material during the anion intercalation/deintercalation process. Here, to further confirm the impact mechanism of LiFSI on the PF6− intercalation/deintercalation behavior, in situ XRD was performed for the Li-free CGDIBs in CDE and CDE + LiFSI during the galvanostatic charging/discharging process. The structural parameters obtained from XRD spectra are summarized in Table S3.†
As shown in Fig. 7a, obviously, a dominant (002) peak of graphite appears as expected at 26.4°, which will be analyzed in detail. Once charging starts, the intensity of the (002) peak begins to decline. With charging to 4.61 V, the intensity of the (002) peak continuously decreases, and (00n + 1) and (00n + 2) act as two new peaks appearing on each side of the (002) peak. Here, stage 5 is first observed, and the corresponding periodic repeat distance (Ic) is calculated to be 21.14 Å and the PF6− gallery height (di) is 7.74 Å (Fig. 7c). Stage 3 can be reached at 4.68 V. Subsequently, a voltage drop step occurs from 4.68 to 4.66 V, which corresponds to a staging transition from stage 3 to 2. During the interval of voltage, the (002) peak gradually disappeared, and the (00n + 1) and (00n + 2) peaks continued to move sideways with an increased intensity. By charging from 4.66 to 4.85 V, it can be observed that the (00n + 1) and (00n + 2) peak positions remain stable with a corresponding stage number of 2. The stable stage number and the increased voltage are presumably related to the decomposition of the electrolyte.45 The staging transition from stage 2 to 1 is finished between 4.85 V and 4.89 V, where the (00n + 1) and (00n + 2) peaks continually move sideways, while Ic is equal to di, indicating the maximum stage number of 1 attained in this stage. At higher voltage (>4.89 V), for the (00n + 1) and (00n + 2) peaks, the stage number of 1 remains, but there is a slight shift of the peak position to higher 2θ values and the distance remains unchanged, which is associated with the structural rearrangement of the embedded PF6− anions.46
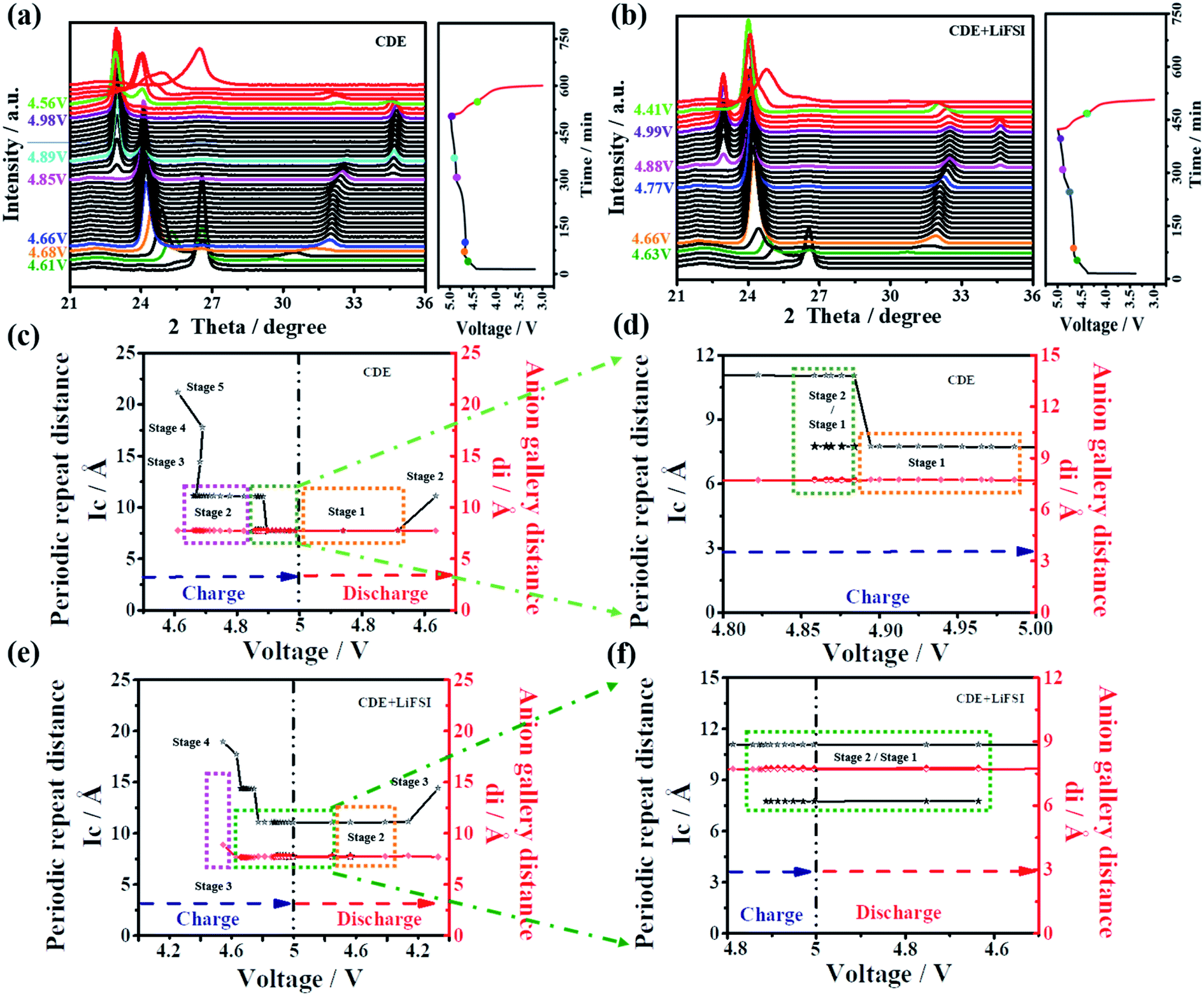 | ||
| Fig. 7 In situ XRD spectra and the corresponding charge–discharge curves of Li-free CGDIBs during the first cycle in (a) CDE and (b) CDE + LiFSI. (c and e) The relevant change of periodic repeat distance Ic, and the intercalated PF6− gallery height di. (d and f) The amplified plots of (c) and (e), respectively. | ||
As soon as the discharge reaction starts, the distance between the (00n + 1) and (00n + 2) peaks gradually shortens, and the intensity of the peaks gradually weakens, signifying that PF6− undergoes a deintercalation reaction. At 4.56 V, the staging transition from 1 to 2 finishes. Meanwhile, after the (00n + 1) and (00n + 2) peaks disappeared, the (002) peak rearose with a relatively low intensity at the end of the discharging process, which may be correlated with the fact that the embedded PF6− is incompletely deintercalated from the graphite cathode.
With further analysis of Fig. 7b, the variation tendency of the in situ XRD spectra in CDE + LiFSI is similar to that in CDE. At approximately 4.63 V, the first observable intercalation stage is 4, the corresponding Ic is calculated to be 17.69 Å, and di is 7.65 Å. The stage number of 3 is gained at 4.66 V and remains unchanged until 4.77 V. The staging transition from stage 3 to 2 finishes between 4.74 V and 4.87 V. The XRD spectra at higher voltages (>4.88 V) show that the (00n + 1) and (00n + 2) peaks remain and two new peaks appear on either side of the (00n + 1) and (00n + 2) peaks, which are named (00n) and (00n + 3), respectively. The simultaneous presence of four peaks indicates the coexistence of stage 2 and stage 1 in the voltage range (Fig. 7f). During the discharging process, the staging transition from 1 to 2 also finishes at 4.41 V. However, the reappeared (002) peak in fact does not return to the original position. Such a result would be related to the increased complexity because more PF6− anions participate in the intercalation process in CDE + LiFSI, thereby causing an irreversible change in the graphite layers and forming a defective structure of the graphite cathode.
According to the above results, the addition of LiFSI can effectively inhibit the decomposition of the electrolyte and LiPF6, which will alleviate the corrosion reactions on the SEI from decomposed PF5, and more PF6− anions will participate in the intercalation/deintercalation process. Meanwhile, the presence of LiFSI can increase the coexisting composition of stage 2 and stage 1, and the defective structure favors offering more embedded spacing to receive future PF6− anions, which ensure high-rate capacitive charge storage. Furthermore, the PF6− gallery height (di) was calculated to be approximately 7.75 Å, and gallery expansion (Δd) was calculated to be approximately 4.4 Å at stage 1. These results further demonstrate that FSI− does not participate in the PF6− intercalation/deintercalation reaction on the graphite cathode.
3. Conclusion
In summary, based on the carbon paper anode and LiFSI-containing electrolyte, we first successfully constructed a novel Li-free CGDIB with superior electrochemical performance. The key advantages of using carbon paper as an anode are the ability to effectively avoid the instability caused by the volume expansion of the metal anode and the safety hazard caused by lithium dendrites and to eliminate the structural collapse of the graphite anode. Moreover, by combining electrochemical characterization and a variety of in situ characterization techniques, the work here for the first time systematically analyzes the multifunctional effect of the LiFSI additive on the Li-free CGDIB, including the following: (1) it can restrain the excessive decomposition of the electrolyte and participates in the formation of a stable and homogeneous LiF-rich interfacial film; (2) it can improve the diffusion properties of Li+, induce the uniform deposition of Li+ and further prohibit the growth of lithium dendrites; and (3) it can contribute to the formation of a defective structure of graphite layers when more PF6− anions participate in the intercalation process, which is beneficial for increasing the rate capability of the Li-free CGDIB. As a consequence, with the carbon paper anode and LiFSI-containing electrolyte, the theoretical specific energy density of the Li-free DIB reaches ≈387 W h kg−1 at a power density of 450 W kg−1 and ≈320 W h kg−1 at 970 W kg−1. Furthermore, the Li-free CGDIB presents an excellent rate ability with a 96% 8C/0.5C capacity ratio. Consequently, the design of Li-free CGDIBs and the understanding obtained from the multifunctional effect of the LiFSI additive provide theoretical support for enabling the future development of high-energy-density Li-free DIBs.Data availability
All relevant data are presented in the main text and ESI.†Author contributions
Li-Na Wu and Zheng-Rong Wang contributed equally to this work. Li-Na Wu, Ling Huang, Wei Chen and Shi-Gang Sun conceived the project and designed the experiments. Li-Na Wu, Zheng-Rong Wang, Peng Dai, Yu-Xiang Xie, Wei-Chen Zheng and Fa-Ming Han conducted the material synthesis, characterization and electrochemical measurements. Cheng Hou performed DFT calculations. Li-Na Wu wrote the manuscript. The manuscript was discussed and revised by all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (22005068) and the Natural Science Foundation of Guangxi Province (2019GXNSFGA245003).References
- L. Zhang, H. Wang, X. Zhang and Y. Tang, Adv. Funct. Mater., 2021, 31, 2010958 CrossRef CAS
.
- X. Zhou, Q. Liu, C. Jiang, B. Ji, X. Ji, Y. Tang and H. M. Cheng, Angew. Chem., Int. Ed., 2020, 59, 3802–3832 CrossRef CAS PubMed
- Z. Y. Tang, Sci. China Mater., 2017, 60, 368–370 CrossRef CAS
- M. Wang, F. Zhang, C. S. Lee and Y. B. Tang, Adv. Energy Mater., 2017, 7, 1700536 CrossRef
- T. Placke, A. Heckmann, R. Schmuch, P. Meister, K. Beltrop and M. Winter, Joule, 2018, 2, 2528–2550 CrossRef CAS
- S. Rothermel, P. Meister, G. Schmuelling, O. Fromm, H. W. Meyer, S. Nowak, M. Winter and T. Placke, Energy Environ. Sci., 2014, 7, 3412–3423 RSC
- T. Placke, P. Bieker, S. F. Lux, O. Fromm, H. W. Meyer, S. Passerini and M. Winter, Z. Phys. Chem., 2012, 226, 391–407 CrossRef CAS
- M. Wang and Y. B. Tang, Adv. Energy Mater., 2018, 8, 1703320 CrossRef
- X. L. Zhang, Y. B. Tang, F. Zhang and C. S. Lee, Adv. Energy Mater., 2016, 6, 1502588 CrossRef
- Y. Chen, M. Yue, C. Liu, H. Zhang, Y. Yu, X. Li and H. Zhang, Adv. Funct. Mater., 2019, 29, 1806752 CrossRef
- L. N. Wu, J. Peng, Y. K. Sun, F. M. Han, Y. F. Wen, C. G. Shi, J. J. Fan, L. Huang, J. T. Li and S. G. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 18504–18510 CrossRef CAS PubMed
- L. N. Wu, J. Peng, F. M. Han, Y. K. Sun, T. Sheng, Y. Y. Li, Y. Zhou, L. Huang, J. T. Li and S. G. Sun, J. Mater. Chem. A, 2020, 8, 4300–4307 RSC
- P. P. Paul, E. J. McShane, A. M. Colclasure, N. Balsara, D. E. Brown, C. Cao, B. R. Chen, P. R. Chinnam, Y. Cui, E. J. Dufek, D. P. Finegan, S. Gillard, W. Huang, Z. M. Konz, R. Kostecki, F. Liu, S. Lubner, R. Prasher, M. B. Preefer, J. Qian, M. T. F. Rodrigues, M. Schnabel, S. B. Son, V. Srinivasan, H. G. Steinrück, T. R. Tanim, M. F. Toney, W. Tong, F. Usseglio-Viretta, J. Wan, M. Yusuf, B. D. McCloskey and J. Nelson Weker, Adv. Energy Mater., 2021, 11, 2100372 CrossRef CAS
- Z. Xie, Z. Wu, X. An, X. Yue, J. Wang, A. Abudula and G. Guan, Energy Storage Mater., 2020, 32, 386–401 CrossRef
- R. Xu, X. B. Cheng, C. Yan, X. Q. Zhang, Y. Xiao, C. Z. Zhao, J. Q. Huang and Q. Zhang, Matter, 2019, 1, 317–344 CrossRef
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2020, 60, 3661–3671 CrossRef PubMed
- S. S. Chi, Y. Liu, W. L. Song, L. Z. Fan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1700348 CrossRef
- W. D. Zhang, Z. Y. Shen, S. Y. Li, L. Fan, X. Y. Wang, F. Chen, X. X. Zang, T. Wu, F. Y. Ma and Y. Y. Lu, Adv. Funct. Mater., 2020, 30, 2003800 CrossRef CAS
- Y. Liu, D. Lin, Y. Li, G. Chen, A. Pei, O. Nix, Y. Li and Y. Cui, Nat. Commun., 2018, 9, 3656 CrossRef PubMed
- C. Yan, Y. X. Yao, X. Chen, X. B. Cheng, X. Q. Zhang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14055–14059 CrossRef CAS PubMed
- X. Ji, S. Hou, P. Wang, X. He, N. Piao, J. Chen, X. Fan and C. Wang, Adv. Mater., 2020, e2002741 CrossRef PubMed
- X. Zhang, S. Wang, C. Xue, C. Xin, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Mater., 2019, 31, e1806082 CrossRef PubMed
- Y. Tian, Y.-L. An, C.-L. Wei, H.-Y. Jiang, S.-L. Xiong, J.-K. Feng and Y.-T. Qian, Nano Energy, 2020, 78, 105344 CrossRef CAS
- S. Waseem, N. Chandrasekaran, P. H. Maheshwari and S. R. Dhakate, J. Electrochem. Soc., 2020, 167, 160538 Search PubMed
- Y. Zhao, Q. Sun, X. Li, C. Wang, Y. Sun, K. R. Adair, R. Li and X. Sun, Nano Energy, 2018, 43, 368–375 CrossRef CAS
- B. Hou, X. Cui and Y. Chen, Solid State Ionics, 2018, 325, 148–156 CrossRef CAS
- G. Yang, S. Zhang, S. Weng, X. Li, X. Wang, Z. Wang and L. Chen, Nano Lett., 2021, 21, 5316–5323 CrossRef CAS PubMed
- G. J. Yang, Y. J. Li, S. Liu, S. M. Zhang, Z. X. Wang and L. Q. Chen, Energy Storage Mater., 2019, 23, 350–357 CrossRef
- T. Zheng, J. Xiong, X. Shi, B. Zhu, Y. J. Cheng, H. Zhao and Y. Xia, Energy Storage Mater., 2021, 38, 599–608 CrossRef
- W. Liu, P. C. Liu and D. Mitlin, Adv. Energy Mater., 2020, 10, 2070177 CrossRef CAS
- S. Zhang, G. Yang, Z. Liu, X. Li, X. Wang, R. Chen, F. Wu, Z. Wang and L. Chen, Nano Lett., 2021, 21, 3310–3317 CrossRef CAS PubMed
- J. D. Xie, J. Patra, P. Chandra Rath, W. J. Liu, C. Y. Su, S. W. Lee, C. J. Tseng, Y. A. Gandomi and J.-K. Chang, J. Power Sources, 2020, 450, 227657 CrossRef CAS
- S. C. Kim, X. Kong, R. A. Vila, W. Huang, Y. Chen, D. T. Boyle, Z. Yu, H. Wang, Z. Bao, J. Qin and Y. Cui, J. Am. Chem. Soc., 2021, 143, 10301–10308 CrossRef CAS PubMed
- J. Neuhaus, E. von Harbou and H. Hasse, J. Power Sources, 2018, 394, 148–159 CrossRef CAS
- J. Hao, X. Li, X. Song and Z. Guo, EnergyChem, 2019, 1, 100004 CrossRef
- P. Shi, H. Zheng, X. Liang, Y. Sun, S. Cheng, C. Chen and H. Xiang, Chem. Commun., 2018, 54, 4453–4456 RSC
- G. G. Eshetu, X. Judez, C. M. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS PubMed
- S. Li, W. Zhang, Q. Wu, L. Fan, X. Wang, X. Wang, Z. Shen, Y. He and Y. Lu, Angew. Chem., Int. Ed., 2020, 14935–14941 CrossRef CAS PubMed
- Z. Zhu, Y. Tang, Z. Lv, J. Wei, Y. Zhang, R. Wang, W. Zhang, H. Xia, M. Ge and X. Chen, Angew. Chem., Int. Ed., 2018, 130, 3718–3722 CrossRef
- X. Zhang, X. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef
- S. Liu, Q. Zhang, X. Wang, M. Xu, W. Li and B. L. Lucht, ACS Appl. Mater. Interfaces, 2020, 12, 33719–33728 CrossRef CAS PubMed
- X. Han and J. Sun, J. Power Sources, 2020, 474, 228664 CrossRef CAS
- Q. D. Li, L. Li, P. J. Wu, N. Xu, L. Wang, M. Li, A. Dai, K. Amine, L. Q. Mai and J. Lu, Adv. Energy Mater., 2019, 9, 1901153 CrossRef CAS
- C. H. Shen, Q. Wang, H. J. Chen, C. G. Shi, H. Y. Zhang, L. Huang, J. T. Li and S. G. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 35323–35335 CrossRef CAS PubMed
- L. N. Wu, S. Y. Shen, Y. H. Hong, C. H. Shen, F. M. Han, F. Fu, X. D. Zhou, L. Huang, J. T. Li and S. G. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 12570–12577 CrossRef CAS PubMed
- T. Placke, G. Schmuelling, R. Kloepsch, P. Meister, O. Fromm, P. Hilbig, H. W. Meyer and M. Winter, Z. Anorg. Allg. Chem., 2014, 640, 1996–2006 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, more electrochemical results, SEM characterization, and Tables S1–S3. See DOI: 10.1039/d2sc00244b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |