
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A block copolymer templated approach for the preparation of nanoporous polymer structures and cellulose fiber hybrids by ozone treatment†
Lea
Gemmer
a,
Qiwei
Hu
bc,
Bart-Jan
Niebuur
d,
Tobias
Kraus
 de,
Bizan N.
Balzer
bcf and
Markus
Gallei
*ag
de,
Bizan N.
Balzer
bcf and
Markus
Gallei
*ag
aChair in Polymer Chemistry, Universität des Saarlandes, Campus Saarbrücken C4 2, 66123 Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bInstitute of Physical Chemistry, University of Freiburg, Albertstr. 21, 79104 Freiburg, Germany
cCluster of Excellence livMatS @ FIT-Freiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
dINM – Leibniz-Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
eColloid and Interface Chemistry, Universität des Saarlandes, Campus D2 2, 66123 Saarbrücken, Germany
fFreiburg Materials Research Center (FMF), University of Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany
gSaarene, Saarland Center for Energy Materials and Sustainability, Campus Saarbrücken C4 2, 66123 Saarbrücken, Germany
First published on 14th June 2022
Abstract
Functional amphiphilic block copolymers (BCPs) are versatile, smart, and promising materials that are often used as soft templates in nanoscience. BCPs generally feature the capability of microphase-separation leading to various interesting morphologies at the nanometer length scale. Materials derived from BCPs can be converted into porous structures while retaining the underlying morphology of the matrix material. Here, a convenient and scalable approach for the fabrication of porous functional polyvinylpyridines (P2VP) is introduced. The BCP polyisoprene-block-P2VP (PI-b-P2VP) is obtained via sequential anionic polymerization of the respective monomers and used to form either BCP films in the bulk state or a soft template in a composite with cellulose fibers. Cross-linking of the BCPs with 1,4-diiodobutane is conducted and subsequently PI domains are selectively degraded inside the materials using ozone, while preserving the porous and tailor-made P2VP nanostructure. Insights into the feasibility of the herein presented strategy is supported by various polymer characterization methods comprising nuclear magnetic resonance (NMR), size exclusion chromatography (SEC), and differential scanning calorimetry (DSC). The resulting bulk- and composite materials are investigated regarding their morphology and pore formation by scanning electron microscopy (SEM), atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS). Furthermore, chemical conversions were examined by energy dispersive X-ray spectroscopy (EDS), attenuated total reflection Fourier-transformation infrared spectroscopy (ATR-FTIR) and water contact angle (WCA) measurements. By this convenient strategy the fabrication of functional porous P2VP in the bulk state and also within sustainable cellulose composite materials is shown, paving the synthetic strategy for the generation of a new family of stimuli-responsive sustainable materials.
Introduction
Amphiphilic block copolymers (BCP) have attracted enormous attention due to their unique possibility for self-organization capabilities in the bulk state or in block-selective solvents to form a manifold of microphase-separated structures, micelles or vesicles based on their hydrophobic and hydrophilic segments.1–4 Polyvinylpyridines are versatile hydrophilic polymers, which are frequently used in functional materials’ preparations derived from BCPs,5,6 for instance for smart capsules,7 coatings,8 and membranes.9,10 Due to the pyridinic moieties, polyvinylpyridines can feature stimuli-responsiveness and they are capable of reversible swelling under acidic or basic conditions.7,9,11–13 Moreover, swelling could be made permanent by quaternization of the pyridinic moiety by treatment with alkyliodides.10,14–16 In general, polyvinylpyridine-containing BCPs are accessible via multiple synthetic routes such as controlled radical polymerization techniques (e.g., RAFT, NMP and ATRP)17–22 and sequential anionic polymerization.3 For the latter, the syntheses are carried out in polar-aprotic solvents such as tetrahydrofurane (THF) at low temperature.6 The polystyrene-b-polyvinylpyridine BCPs are one of the most-studied BCPs in functional materials research for example for filtration applications,9,10,23 or as polymeric particles.13,15 Poly-2-vinylpyridine (P2VP) based BCPs with polyisoprene (PI) as a non-polar flexible polymer segment are less reported, presumably due to solubility issues of the amphiphilic BCPs, which are even more pronounced for P4VP-based amphiphilic BCPs. Furthermore, there the sequential anionic polymerization is more complex, wherein the microstructure of the PI segment depends on the polymerization conditions.24 The anionic polymerization of isoprene can result in three different microstructures for the final PI. Vinylic linkages (1,2- and 3,4-linkages) exhibit a saturated polymeric backbone and a vinyl group as a side chain, whereas 1,4-linkages result in an unsaturated PI backbone, which influences the chemical properties for post-modification strategies and the glass transition temperature (Tg). PI featuring more than 90 mol% 1,4-linkages exhibit a Tg of about −70 °C, and with increasing vinylic content the Tg increases.8,25,26 The content of vinylic linkages can be controlled by reaction parameters.24 Vinylic linkages are kinetically preferred and polar-aprotic solvents such as THF and higher reaction temperatures enable the formation of high vinylic contents of about 75 mol% in the final polymer.8,27,28 By contrast, when conducting polymerization in cyclohexane at room temperature only 10 mol% of vinylic linkages can be found.25,29,30 For the efficient degradation of PI, a 1,4-PI content of 16 to 24 mol% turned out to be sufficient, as previously reported by Rüttiger et al. and Okumura et al.8,31 It is assumed that smaller degradation fragments are advantageous because they are more easily transported through the P2VP matrix. Therefore, in this work only BCPs with a high 1,4-content in the PI-block are used and the isoprene polymerization in non-polar solvents is preferred. Watanabe and Tirrell described a synthetic route for PI-b-P2VP BCPs via sequential anionic polymerization by taking advantage of a solvent exchange.30 BCPs with well-defined molar mass distribution and molar masses between 80 and 250 kg mol−1 were obtained, while the weight percentage of P2VP segment was varied from 12 to 64 wt%.The self-assembly of BCP and the formation of microphase-separated structures relies on many different parameters, and the χ of the underlying block segments is one of the most important.32–34 Amphiphilic BCPs typically tend to feature higher χ parameters and therefore exhibit microphase separation with well-defined structures.35 Additionally, the microstructures depend on the volume fractions of the block segments and the domain sizes correlate with molar masses.33,34 It is well-known that also the parameters for preparing the BCP films have a major influence on phase separation behavior. For PI-b-P2VP, an increasing phase expansion of P2VP-phase was observed when shifting from non-polar and more PI-selective solvent like tetrachloromethane (Hildebrandt and Hansen total solubility parameter of δT = 17.8 MPa0.5) to more polar and P2VP-selective solvent like THF (δT = 20.3 MPa0.5) or 1,4-dioxane (δT = 20.5 MPa0.5).36,37 In literature Hildebrandt and Hansen solubility parameters δT of 16.6 MPa0.5 for 1,4-cis-PI36,38 and multiple parameters for P2VP can be found, namely 19.8 MPa0.5, 21.3 MPa0.5, and 21.7 MPa0.5 (pyridine).5,36,37,39 As a result, a non-selective solvent should have a Hildebrandt and Hansen solubility parameter between 18.2 and 19.2 MPa0.5, as for example chloroform (δT = 19.0 MPa0.5). Though BCPs are frequently used as soft templates in various procedures, the sensitivity of morphology regarding solvents and swelling agents can cause complications. Free-standing or supported mesoporous functional materials can be fabricated from BCP containing a sacrificial block segment, which can be removed upon thermal or chemical treatment, while the matrix block segment remains unaffected.40 Chemical degradation of sacrificial segments from BCPs demands sufficient material transport and adequate chemical resistance, and therefore the degradation conditions for the sacrificial block segment have to be matched with care. Moreover, to avoid collapse of the matrix polymer during the degradation procedure and to maintain the templated pore structure, cross-linking of the residual domains is a common practice.16,40–43 Vinylpyridines can be cross-linked using 1,4-diiodobutane (DIB) from solution or from the vapor phase.16,41,42 Ozone is a small molecule that is capable of cleaving double bonds into various soluble carboxylic products. Since most polydienes contain double bonds in their polymeric backbone they can be degraded into smaller fragments by ozone treatment and washed away using suitable solvents.44 As a result, mesoporous BCP-templated materials with high pore order can be obtained.8,31 However, to the best of our knowledge, ozone-mediated PI degradation with PI rendering the minor block segment as a spherical BCP morphology has not been investigated in more detail yet, and there are no investigations on polydiene-based BCP templates as coatings within a cellulose fiber matrix. These insights would pave the way to a new route for the preparation of porous cellulose-based materials, stimuli-responsive coatings and smart membranes. Additionally, the hierarchical pore design and BCP coating at the cellulose surface should provide a direct and technologically straight-forward control over the wettability and functionality for sustainable microfluidics or sensing applications. For this purpose, within the present study amphiphilic BCPs, consisting of PI-b-P2VP with a high content of unsaturated moieties within the polyisoprene backbone, were synthesized via sequential anionic polymerization and investigated with respect to their self-assembly to a microphase-separated structure. Cross-linking with DIB was investigated and protocols for the generation of porous structures by ozone treatment were established. Finally, cellulose fiber materials were combined with the PI-b-P2VP-system to gain insights into the influence on cross-linking, pore formation and stability of BCP inside a cellulose fiber substrate.
Experimental section
Materials
Solvents and reagents were purchased from Alfa Aesar, Sigma-Aldrich (Merck), Fisher Scientific, VWR or Acros Organics and used as received unless otherwise stated. For anionic polymerization the following purification steps were carried out: dried cyclohexane was obtained by adding sec-butyllithium (s-BuLi, 1.3 M in hexane) and 1,1-diphenylethylene (DPE) to cyclohexane (Fisher, analytical reagent grade >99,8%) in a round-bottom glass-ampule until a dark red color was maintained (diphenylhexyllithium). The liquid was degassed via freeze–pump–thaw cycles and stored under vacuum until use. LiCl was dispersed in dry THF, treated with s-BuLi, and stirred overnight. The solvent was removed under reduced pressure and the LiCl was stored in a glovebox until use. Dried THF was prepared by adding n-butyllithium (n-BuLi, 1.6 M in hexane) and DPE to THF (Fisher, HPLC grade) in the same procedure. Before use, THF and cyclohexane were cryo-transferred to an ampule, which was transferred to a glovebox. In the case of THF, LiCl was added, followed by an additional treatment with s-BuLi. The solution was stirred for at least two hours before use. DPE (97%, Sigma Aldrich) was treated with n-BuLi until a dark red color was maintained, then cryo-transferred and stored in a glovebox until use. Methanol was dried using molecular sieve (3 Å), degassed via freeze–pump–thaw, then cryo-transferred and stored in a glovebox until use. Isoprene was pre-dried by stirring with calcium hydride. Before use, isoprene was freshly cryo-transferred two times, the first time to an ampule containing trioctylaluminium (25 wt% in hexane, Sigma Aldrich) for PI28-b-P2VP7263 or dibutylmagnesium (0.5 M in heptane, Acros Organics) for PI35-b-P2VP65186. 2-Vinylpyridine (2VP) was also pre-dried by stirring with calcium hydride. Before use, 2VP was freshly cryo-transferred three times, the first two times to an ampule containing trioctylaluminium for PI28-b-P2VP7263 or triethylaluminium (1 M in hexane, Sigma Aldrich) for PI35-b-P2VP65186. All purifying steps of reagents used in synthetic procedure were conducted using common Schlenk-technique at a full-glass high-vacuum line or, when stated, in a glovebox (MBRAUN, UNIlabplus ECO) working with nitrogen gas (5.0) and equipped with a Cryostat (Julabo FP89). Commercial Munktell cellulose-based filter discs were used as a paper substrate (Ahlstrom Munksjö, grade 3hw 65 g cm−3, 3.303.070).Methods
Size-exclusion chromatography (SEC) was performed with a 1260 Infinity II (Agilent Technologies) system and THF was used as the mobile phase (HPLC grade, flow rate 1 mL min−1) on an SDV column set from polymer standard service (PSS, SDV 103 Å, SDV 105 A, SDV 106 Å, 5 μm) with a PSS SECurity2 RI/UV detector. Calibration was carried out using polystyrene (PS) standards from PSS. For Multi-angle laser light scattering (SEC-MALLS) a PSS SLD 7000 detector was used. PSS WinGPC UniChrom V 8.31 was used for data acquisition and evaluation of the measurements. Differential scanning calorimetry (DSC) was carried out using a NETZSCH DSC 214 Polyma in aluminum crucibles with a heating rate of 10 K min−1 and nitrogen as both protective and purge gas in flow rates of 60 mL min−1 and 40 mL min−1, respectively. Thermogravimetric analysis (TGA) was conducted on a NETZSCH TG Libra in a crucible made of aluminum oxide with nitrogen flow of 20 mL min−1 and heating rate of 10 K min−1. Both TGA and DSC data were evaluated using NETZSCH Proteus Thermal Analysis 8.0.1. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II 400 spectrometer with a 9.4 T Ultrashield Plus Magnet, a BBFO probe, and referenced by using the solvent signals.45 For processing and evaluation of the sprectra MestReNova 14.2.0 was used. Fourier-transformed infrared (FTIR) spectra were collected on a BRUKER ALPHA II FTIR setup in attenuated total reflection mode (ATR) with spectrum output in transmittance. All spectra were processed with OPUS 8.5 (SP1) software (baseline correction) and Origin2020b (normalized). Ozonolysis experiments were carried out using an Anseros Ozomat COM CD HF4 (Ozone generator from Anseros Klaus Nonnenmacher GmbH) on generation level 100% at a gas flow of 50 NL h−1. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) was carried out on a Zeiss Sigma VP device (GeminiSEM 500) using the software SmartSEM Version 6.07. The samples were mounted on an aluminum stud using adhesive copper tape and sputter-coated with approximately 6 nm platinum using an Automatic Turbo Coater PLASMATOOL 125 SIN 2020_131 from Ingenieurbüro Peter Liebscher. High-resolution micrographs were collected via in-lens detector with acceleration voltages between 1 and 3 kV in high-current mode, 20 μm aperture, EDS-spectra were collected using acceleration voltage of 8 kV. Static water contact angle (WCA) measurements were performed using Hamilton syringe 100 μL in a syringe pump by KD Scientific adjusted to 10 μL and a custom-made xyz positioning table. In standard measurements deionized water was used, for measurements in acid a pH = 4 buffer solution consisting of citric acid, NaOH and NaCl purchased by Carl Roth was used. Photographs were collected using a Nikon D54000 and digiCamControl 2.1.2.0, OpenDrop 3.3.1 was used for evaluation.46 AFM imaging was performed with a Cypher ES (Asylum Research, an Oxford Instruments Company) in intermittent-contact mode. The following parameters were chosen for all measurements: pixel number of 1024 × 1024, scan area of 1 μm × 1 μm and scan rate of 1.95 Hz. First, all samples were scanned in air at 25 °C with AC240TS cantilevers (Olympus, Japan) having a tip with a radius of approx. 7 nm. In addition, sample Bs10b was also imaged in pure H2O (Purelab Chorus 1, Elga LabWater, Celle, Germany, with conductivity of 0.055 μS). Prior to the measurements in liquid environment, the samples were immersed in the respective solutions for 30 minutes for swelling and protected against light. AC40TS cantilevers (Olympus, Japan) with a tip radius of approx. 7 nm were utilized for experiments in liquid environment at 25 °C. The retrace images gained from AFM imaging were processed with the Gwyddion Free SPM analysis software (Petr Klapetek, version 2.59) for further evaluation as described in Friess et al.47 Small-angle X-ray scattering (SAXS) experiments were performed on a Xeuss 2.0 system (Xenocs SAS, Grenoble, France). The incident beam from a Copper Kα source with a wavelength λ = 0.154 nm was collimated and focused on the sample with a spot size of 0.25 mm2. 2D scattering intensity patterns were collected using a Pilatus 300 K detector (pixel size 172 × 172 μm2) located at a sample-detector distance of ∼2500 mm, calibrated using a silver behenate standard. For each measurement, the acquisition time was 3600 s. As no signs of anisotropic scattering were observed, the scattering patterns were azimuthally integrated to obtain the scattered intensity I in dependence on momentum transfer q = 4π × sin(θ/2)/λ, with θ being the scattering angle.Block copolymer synthesis
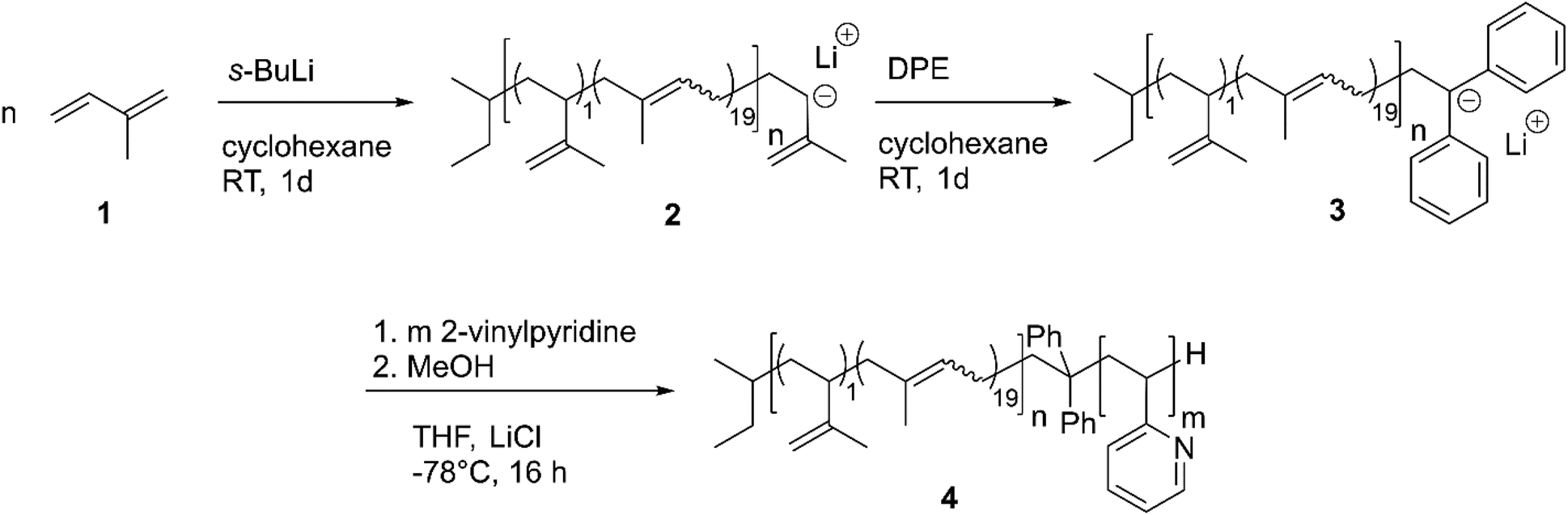 | ||
Scheme 1 Synthetic route leading to polyisoprene-b-poly(2-vinylpyridine) (PI-b-P2VP) via sequential anionic polymerization. Subscripts refer to molar microstructure composition of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 for 3,4- and 1,4-linkages, respectively. :19 for 3,4- and 1,4-linkages, respectively. | ||
SEC PI (vs. PS): Mn = 12500 g mol−1; Mw = 13800 g mol−1; Đ = 1.11.
SEC PI-b-P2VP (vs. PS): Mn = 39600 g mol−1; Mw = 42900 g mol−1; Đ = 1.08.
SEC-MALLS: Mw = 62501 g mol−1 (refractive index increment dn/dc: 0.140).
1
H NMR (300 MHz, 300 K, CDCl3, δ in ppm): 8.4–8.1 (pyr-H 2VP, 1H); 7.3–7.0 (pyr-H 2VP, 1H); 6.9–6.6 (pyr-H 2VP, 1H); 6.5–6.2 (pyr-H 2VP, 1H); 5.1 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-1,4-PI, 1H); 4.7 (CH2 3,4-PI, 2H); 2.7 (P2VP, bb); 2.3 (P2VP, bb); 2.0 (PI, bb); 1.8 (P2VP, bb); 1.7–1.6 (PI, bb); 1.3–0.9 (P2VP, bb).
CH-1,4-PI, 1H); 4.7 (CH2 3,4-PI, 2H); 2.7 (P2VP, bb); 2.3 (P2VP, bb); 2.0 (PI, bb); 1.8 (P2VP, bb); 1.7–1.6 (PI, bb); 1.3–0.9 (P2VP, bb).
Block copolymer film preparation and cross-linking with DIB
Polyisoprene degradation by ozonolysis
A three-necked round bottom flask featuring a reflux apparatus was filled with 40 mL heptane (GPR Rectapur, 99.8%), equipped with a gas inlet for ozone, and the flask was placed in an ice bath. Sample pieces of 10 to 50 mg in weight and edge lengths of 2 to 10 mm, often two or more sample pieces, were used per experiment. The sample was dispersed in heptane, the three-necked round bottom flask was closed with a stopper. The oxygen flow was set to 50 NL h−1 and the ozone generator was turned on. The former colorless and clear solvent turned light blue due to saturation with ozone and haze occurred. After 10 minutes, the ozone generator was turned off and oxygen was bubbled for two minutes to ensure complete displacement of residual ozone in the solvent. The treated sample was taken out, washed several times with fresh heptane (HPLC grade) and then soaked or stirred in deionized water for 30 minutes. The sample pieces were dried in vacuum at 40 °C for three days.Results and discussion
Synthesis and characterization of PI-b-P2VP
The desired block copolymer PI-b-P2VP was prepared by sequential anionic polymerization as shown in Scheme 1. For the intended post modification and degradation procedure of the PI segment, the presence of double bonds in the polymer backbone was favored. Therefore, in order to obtain a high content of 1,4-linked PI moieties, synthesis was conducted in cyclohexane as nonpolar solvent.24 As already described in the introduction, solvent exchange was necessary to enable 2VP polymerization in THF in the presence of LiCl at low temperatures. Following this synthetic route, PI-b-P2VP block copolymers with well-defined molar mass distribution, differing polyisoprene contents and high 1,4-PI contents were obtained (Fig. 1 and Table 1). In PI-b-P2VP BCPs subscripts refer to molar percentages of respective moieties and the superscript refers to the total molar mass.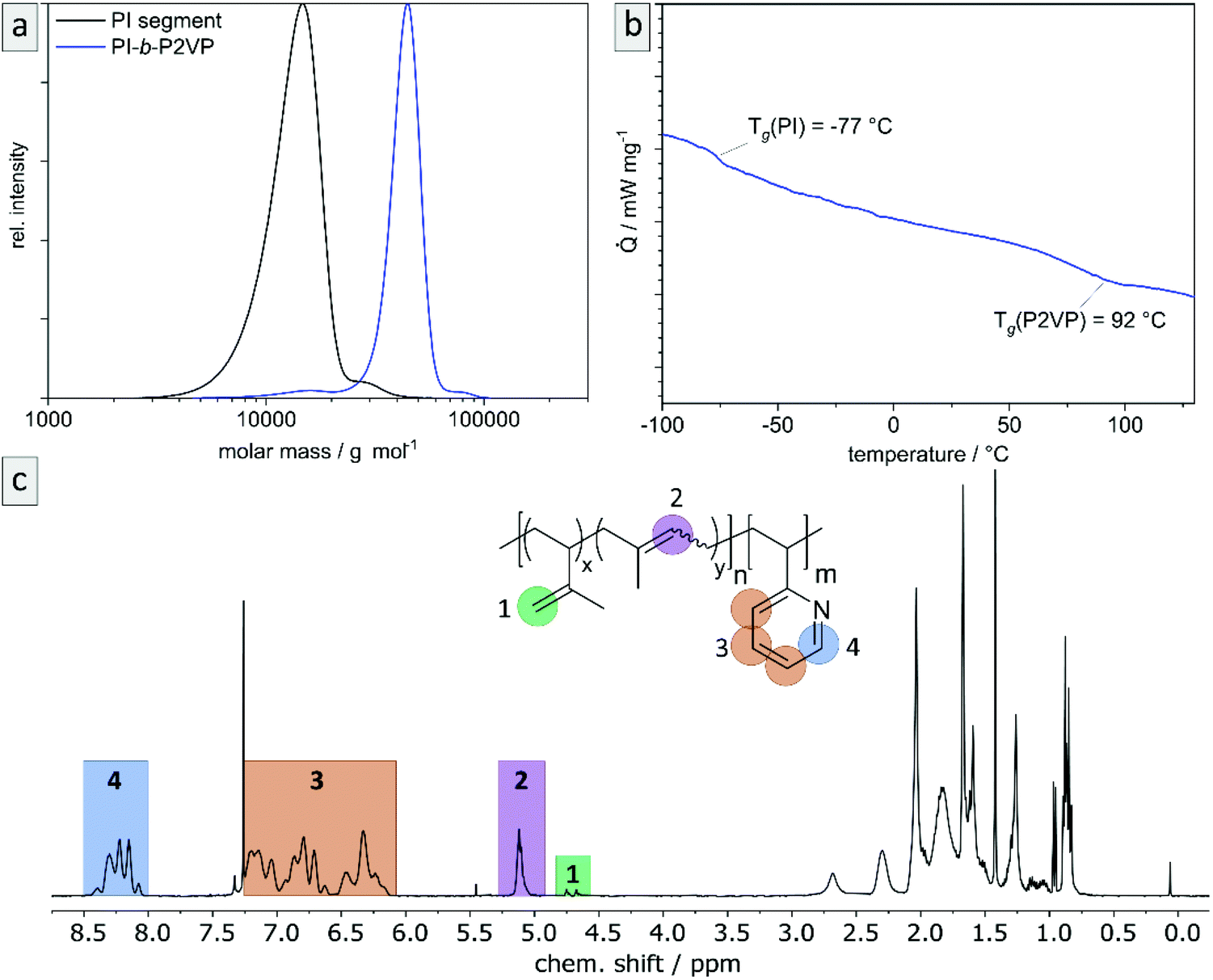 | ||
| Fig. 1 Exemplary characterization of PI28-b-P2VP7263 using (a) SEC to determine relative molar masses (vs. PS-standard) and molar mass distributions, (b) thermograms by DSC measurements to determine glass transition temperatures, and (c) 1H NMR spectrum in CDCl3 to determine chemical composition of BCP and microstructure composition of polyisoprene moieties. For evaluated data see Table 1. | ||
| Polymer | M SEC | Đ | mol%1,4-PId | mol%PIe | Φ PI | T g, PI | T g, P2VP |
|---|---|---|---|---|---|---|---|
| a Molar mass Mn in g mol−1 determined by SEC (PS standards, THF). b Molar mass Mw in g mol−1 determined by MALLS in SEC in THF. c Đ values determined by SEC (PS standards, THF). d Molar fractions of 1,4-PI microstructure in PI-segment in % determined from 1H NMR. e Molar fractions of PI segments in BCP in % calculated using molar fractions determined from 1H NMR (see Fig. 1). f Volume fractions of PI segments in % estimated from weight fractions using polymer densities (0.903 g cm−3 for PI48 and 1.153 g cm−3 for P2VP49). g T g in °C determined by DSC measurement. | |||||||
| PI | 12500a |
1.11 | 93 | ||||
| PI28-b-P2VP7263 | 62501b |
1.08 | 28 | 24 | −77 | 92 | |
| PI | 95 | ||||||
| PI35-b-P2VP65186 | 185720b |
1.15 | 35 | 31 | −78 | 99 | |
The exemplary molar mass distribution of PI as first block and corresponding PI28-b-P2VP7263 BCP is shown in Fig. 1, exhibiting a monomodal and narrow molar mass distribution both for the PI homopolymer precursor as well as the final BCP (1.11 and 1.08, respectively). The Tg of the PI segment is −77.6 °C, which is in accordance with a PI featuring a high content of 1,4-linkages.25 The 1H NMR spectrum of the BCP (Fig. 1) reveals the expected signals, while signals at 4.8 and 4.7 ppm correspond to the vinylic protons in the 3,4-PI unit and the signal at 5.1 ppm corresponds to the olefinic proton in the 1,4-PI unit. Signals marked in orange in the range from 6.1 to 7.3 ppm correspond to aromatic protons of 2VP (3 H, H3) and the signal at 8.2 ppm (1 H, H4) refers to the proton of 2VP-repeating unit next to the nitrogen atom. The composition of PI and the BCP were calculated from the relative integrals of H1, H2 and H4, summarized in Table 1.
Microphase separation of PI-b-P2VP and cross-linking reaction with DIB
For fabrication of PI-b-P2VP films chloroform was chosen as solvent.36 Films were solvent-cast and solvent-annealed for several days. Prior to their analysis, the BCP films were freeze-fractured in liquid nitrogen using a scalpel and a selection of samples was cross-linked using 1,4 diiodobutane (DIB) vapor (Experimental section). All polymer films were about 1 mm in thickness, flexible and non-brittle. Cross-linked films exhibited a yellow coloration (Fig. S2†). To gain insights into the influence on the morphology and pore formation, the BCP films derived from PI28-b-P2VP7263 were investigated by EDS, ATR-FTIR, SEM and SAXS before and after cross-linking, as described in the following. In Fig. 2a and b, SEM micrographs of cross-sections of the untreated film and the DIB-treated film are given. Both films exhibit PI spheres in a P2VP matrix as a morphology. In DIB-treated films highly ordered cylindrical domains can be found close to the edge of the cross-section, which is within 3 μm below the film surface, as it is shown in Fig. 2d.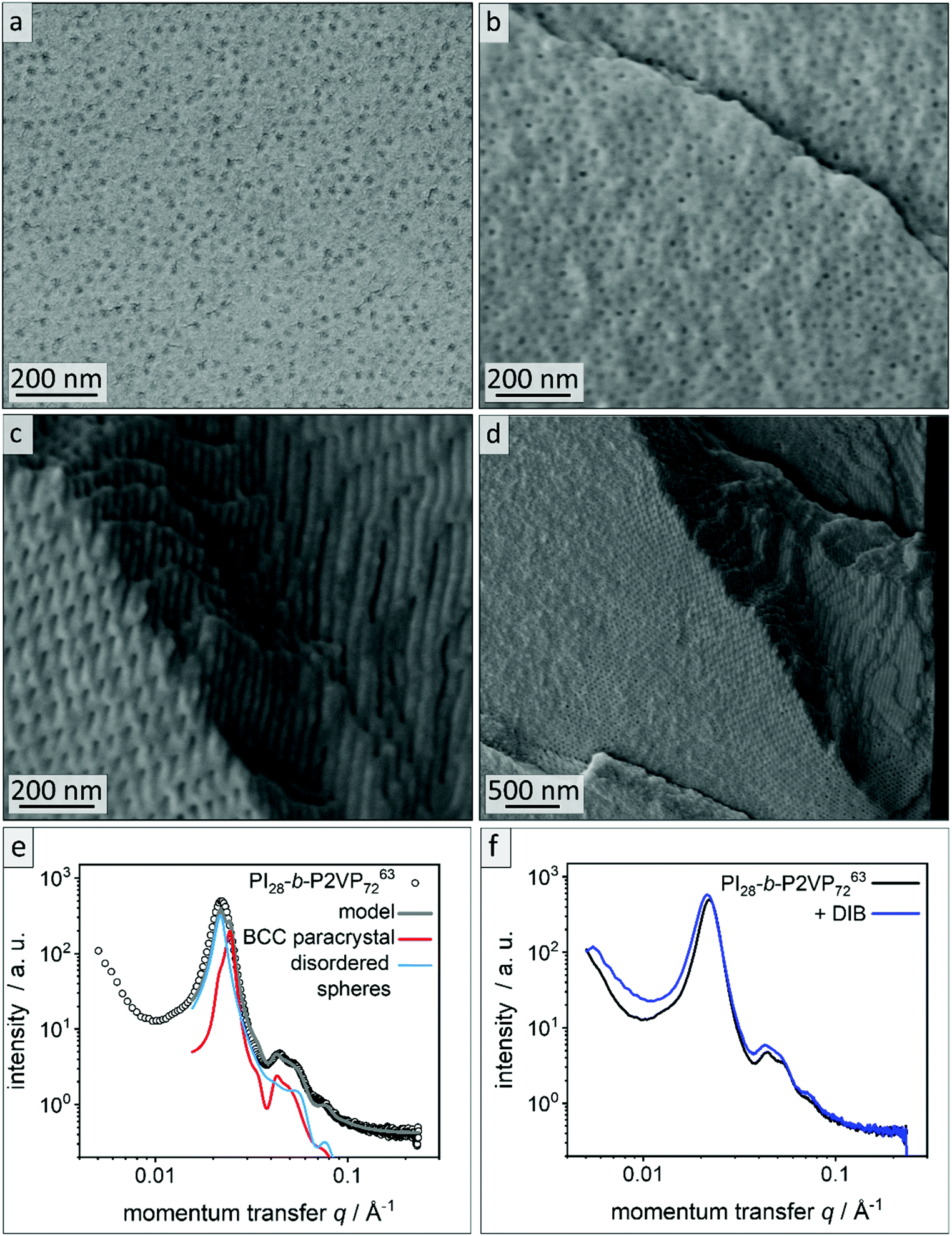 | ||
| Fig. 2 Morphological analysis of films derived from PI28-b-P2VP7263. (a) Cross-sectional SEM images of untreated bulk polymer, (b) cross-sectional SEM images of DIB-crosslinked bulk polymer, (c) area within 2 μm from surface of bulk morphology of DIB-crosslinked PI28-b-P2VP7263 and (d) overview image of the border area. (e) SAXS pattern of untreated PI28-b-P2VP7263 bulk polymer. The lines are the calculated theoretical model given by eqn (1) (grey) and its individual contributions from a BCC paracrystal (red) and disordered spheres (blue). (f) Untreated PI28-b-P2VP7263 bulk polymer compared to DIB stabilized sample. | ||
Fig. 2e shows the SAXS pattern obtained for PI28-b-P2VP7263 before DIB treatment. Besides a prominent primary Bragg peak at ∼0.022 Å−1, several shallow secondary peaks are present, indicating a well-ordered structure. The SEM images recorded for the untreated film (Fig. 2a) suggest a spherical morphology that may be well-ordered. As a qualitative examination of the peak positions did not allow for an unambiguous determination of the ordering state, the SAXS pattern was compared with a theoretical model. The positions of the secondary Bragg peaks at 0.045 and 0.055 Å−1 match those expected for a body centered cubic (BCC) structure. However, an additional contribution from disordered spheres is required to satisfactorily describe the full scattering pattern. A combination of BCC and disordered spheres have been observed previously in BCP morphologies.50–52 Theoretically, the scattering pattern of such a mixed structure can be described by:
| I(q) = SBCC(q)PS(q) + SHS(q)PS(q) + Ibkg | (1) |
Here, PS(q) is a sphere form factor with a Gaussian size distribution that depends on the radius of the spheres, RS, and the width of the distribution, σ.53SBCC(q) denotes the structure factor describing a BCC paracrystal,54,55 which is a function of the nearest neighbor distance a. The disordered arrangement of spheres was described by a hard-sphere structure factor, SHS(q).56 It is defined by the hard sphere radius, RHS, i.e., half of the center-to-center distance between the spheres. Ibkg accounts for background scattering.
The model captures all features of the scattering curve, which suggest that it qualitatively describes the structure of the sample. The analysis yields a sphere radius of 11–12 nm, and a nearest neighbor distance between spheres of 32–36 nm, which is in good agreement with the results from SEM. Slight deviations between the model and the scattering pattern point to small quantitative differences or the presence of minor amounts of additional structures that are not considered by the model.
Fig. 2f compares the scattering patterns of PI28-b-P2VP7263 before and after DIB treatment. The overall appearance of both scattering patterns is very similar, which implies that the DIB treatment did not lead to morphology changes in the sample. In comparison to the untreated sample, the DIB-treated sample has slightly broader Bragg peaks, which indicates a weaker long-range order. Furthermore, no signs of hexagonally packed cylinders (HPC), as observed by SEM (Fig. 2d), were found by SAXS. Therefore, this is most likely a surface effect only.
According to common microphase separation theory, volume fractions of PI between 21 and 31% in a PI-b-P2VP BCP (Table 1) should result in a morphology composed of PI cylinders in a P2VP matrix.32,33 When operating at BCP compositions close to phase transition, film casting conditions can cause changes in the observed morphology. The microphase separation is influenced by many factors, most importantly solvent selectivity, the presence of traces of water or underlying substrates like glass or paper.35 The sample shown in Fig. 2 clearly exhibits a spherical morphology. The deviation indicates that the P2VP phase is favored during casting and annealing process. As stated in the introduction, solubility parameters are essential when considering a solvent for a film casting process. Chloroform featuring a Hildebrandt and Hansen total solubility parameter of δT = 19.0 MPa0.5 can be considered as non-selective solvent. TGA of the polymer, however, exhibit mass losses of 10 wt% between 90 °C and 160 °C, which can be attributed to some water content (see Fig. S3†). However, polvinylpyridines feature polar and hydrophilic moieties that can adsorb water.57 To remove the bound water elevated temperatures are necessary, which can cause degrading or crosslinking processes in the polymer. We therefore avoided extensive drying procedures. In BCPs featuring a polar and hydrophilic segment those traces of water can shift the equilibrium morphology significantly,33,35 and according to analytical data this is the case in the present work. It is assumed that morphological changes occurred at the surface because of the DIB-mediated cross-linking reaction leading to a cylindrical morphology. Therefore, the cross-linking procedure was investigated in more detail and will be discussed further.
In literature, effective DIB-crosslinking is only described in films with a thickness below 60 μm,41,42 in solution,16 or in highly porous systems.58 In the present study, BCP films with a thickness up to 1 mm were used. It is assumed that cross-linking protocols could only affect the film's surface, because DIB is considered a bulky molecule with heavy iodide atoms that is introduced via vapor phase at atmospheric pressure. During the cross-linking procedure, iodide was incorporated into the polymer (chemical structure see Scheme S1†) and therefore EDS-data should give information on the locations of cross-linking reactions. In Fig. 3 cross-sectional SEM images of bulk sample PI28-b-P2VP7263, EDS-point-spectra and locations thereof are shown. Additionally, EDS-mapping of iodine in the cross-section is displayed with relative iodine concentration depicted as red dots. In locations c and d more iodine was found than in location e (Fig. 3c: 9.6 at% N and 2.4 at% I; Fig. 3d: 10.2 at% N and 1.8 at% I; Fig. 3e: 9.6 at% N and 0.4 at% I) and further away from the surface (Fig. 3f) no iodine could be detected. Mean values and standard deviations of these measurements can be found in the ESI (Table S1†), as well as a plot of iodine concentration in dependence of the distance from the surface (Fig. S4†). Considering the EDS-mapping (Fig. 3b) as well as the plot (Fig. S4†) it is self-evident that there is a gradient from higher iodine concentrations of 2 at% within 3 μm from the surface to no iodine content at 25 μm perpendicular to the surface, which can be directly correlated to the cross-linking density. Using the atomic ratios of iodine and nitrogen it can be estimated that within 3 μm from the surface (locations c and d) 25% of vinylpyridine units were quaternized with DIB and therefore covalently cross-linked (details and calculation in Table S2†).
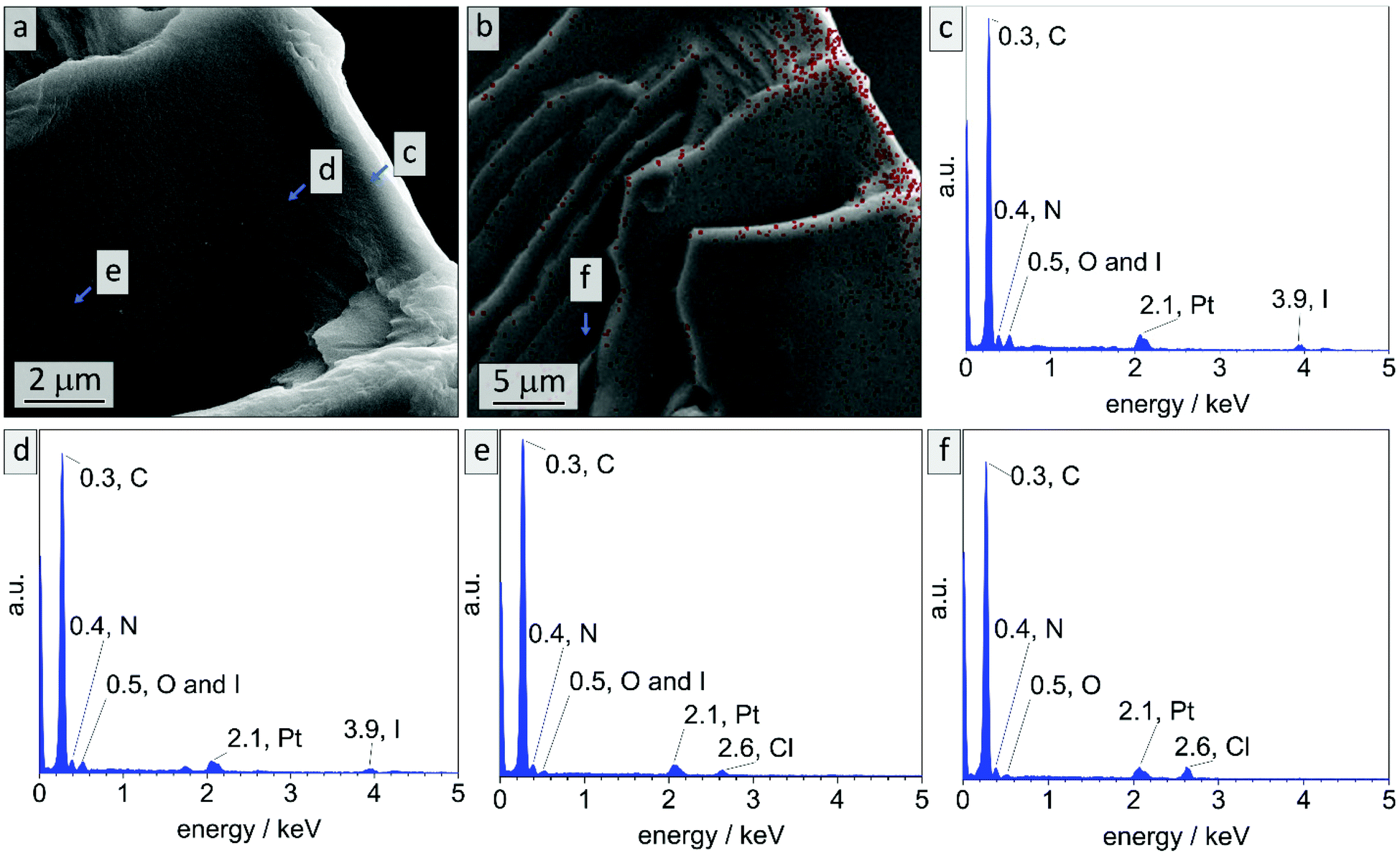 | ||
| Fig. 3 EDS data collected on bulk sample PI28-b-P2VP7263 after stabilization with DIB. (a) Cross-sectional SEM image, wherein the locations of exemplary EDS point spectra (c–e) are marked. (b) EDS mapping, wherein the location of an exemplary spectrum is shown in (f). The density of red dots refers to relative amount of iodine atoms. | ||
Locations c and d cover the area in which a cylindrical morphology is observed, as visible in Fig. 2. At spots e and f (8–10 μm and 25 μm from surface, respectively) solely spherical morphology is found. As a result, it can be stated that cross-linking is highly efficient in surface-near areas, considering the protocol used. There are two possible mechanisms for the morphological change at the surface: due to the change in chemical structure, the Flory–Huggins interaction parameter χ was changed by increasing the polarity of the 2VP moieties. Also, the volume fraction of the P2VP domains was affected by cross-linking. Moreover, DIB is expected to additionally soften the P2VP-phase during cross-linking and therefore enabling the rearrangement from spheres to cylinders. As a conclusion to these findings, the shift in morphology from spheres to cylinders in surface-near areas can be attributed to the reaction of the polymer with DIB, while the spherical morphology in the bulk state is preserved.
The untreated and cross-linked BCP films were furthermore analyzed by ATR-FTIR spectroscopy in order to detect characteristic changes within the chemical structure attributed to the reaction of pyridinic moieties with DIB. The corresponding chemical structure is given in the ESI (Scheme S1†). Peaks found at 1589 and 1567 cm−1 can be attributed to vibrations of pyridine rings and peaks at 1472 and 1433 cm−1 correspond to CH2-deformation vibrations of the polymer backbone. Therefore, these two double-peaks are characteristic signals for P2VP and are explicitly shown in Fig. 4b (partial spectrum Fig. 4a, PI28-b-P2VP7263, blue). The peak at 743 cm−1 (dominant signal, Fig. 4a) corresponds to aromatic C–H bonds. Changes after crosslinking with DIB are dominant in transmission spectrum at 1626 cm−1, 1509 cm−1 and 1450 cm−1 (Fig. 4b, PI28-b-P2VP7263 + DIB, gray curve). The two additional transmission peaks at 1626 and 1509 cm−1 are known to be characteristic for quaternized pyridium,41,57,59 whereas the signal at 1450 cm−1 can be attributed to additional CH2-groups that were introduced with the cross-linking agent.59 As a result, the iodine found in EDS measurements can be associated with quaternized pyridinium moieties.
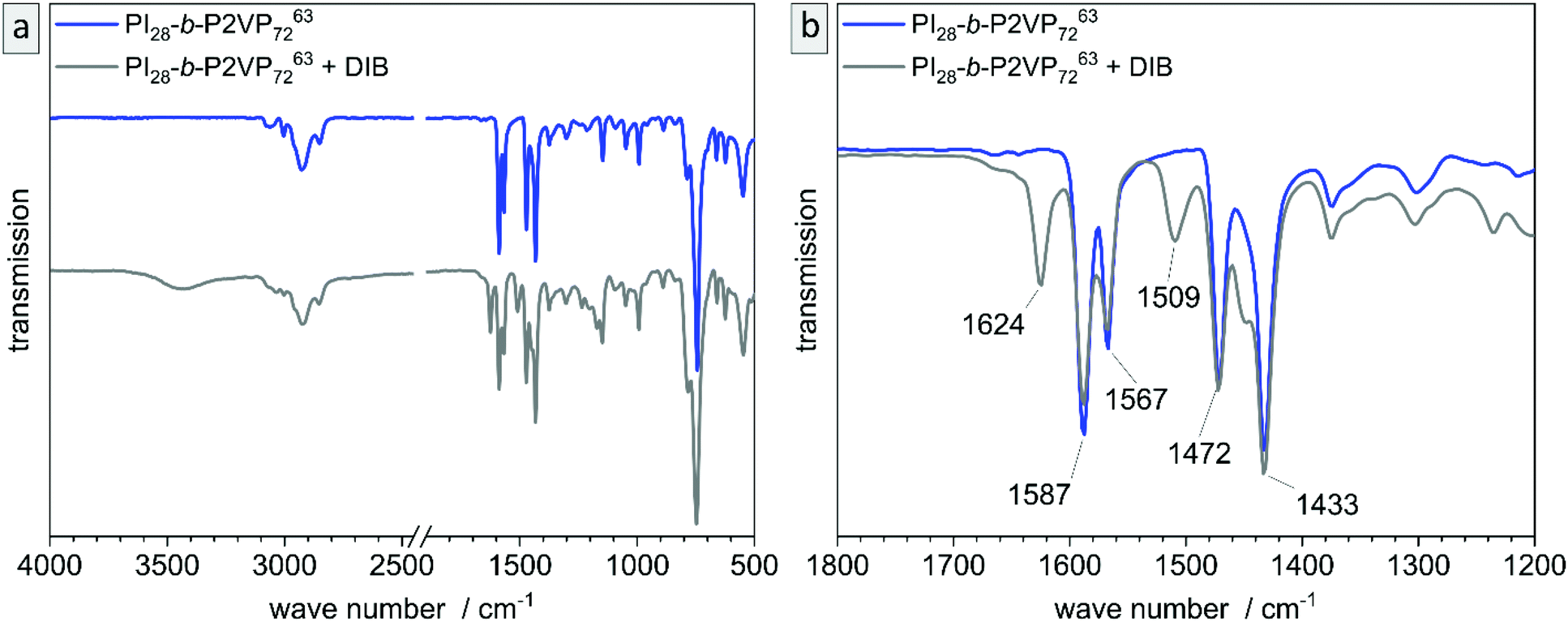 | ||
| Fig. 4 Analysis of untreated film (PI28-b-P2VP7263, blue) and DIB-crosslinked film (gray) by ATR-FTIR transmission spectra. (a) overview, whole spectrum, (b) partial spectrum with changes in P2VP signals. | ||
As a conclusion, the BCP in the bulk state self-assembled to a mixture of BCC and non-ordered spheres, whereas cross-linking with DIB shifted the morphology to cylinders in surface-near areas. As a next step, bulk samples were treated with ozone and the resulting materials were further investigated and will be discussed with respect to morphology and pore sizes.
Removal of PI domains by ozonolysis in the bulk material
In order to study the PI degradation in PI-b-P2VP BCPs, samples were treated with ozone. Here, a short treatment duration (10 min) and long treatment duration (90 min) were compared and the effect of cross-linking on microphase separation and pore formation was examined (Experimental section). The double bonds of the PI segments were cleaved by ozone and corresponding degradation products were removed during the washing procedure. Heptane is a non-solvent for the two block segments and the solvent properties of water regarding P2VP are pH-dependent. In general, P2VP and its solubility is very sensitive to the pH value. For pH ≥4.9, the polymer is partially soluble in water, and below this value, the good solvent condition is reached.7,60,61 Deionized water with a pH value of 5–7 was used for the rinsing steps. Therefore, the P2VP segments were not dissolved. It is noticed that after the introduction of charges by application of the DIB-mediated cross-linking protocols, swelling of the surface-near domains can occur. Nevertheless, a dissolution of the porous structure was impeded by the covalent linkages and no significant effect on the bulk morphology was observed after conversion into a mesoporous structure.For pore analysis, cross-sections were investigated by SEM, AFM and SAXS as obtained by freeze-fracturing of the BCP films. Bulk samples derived from PI28-b-P2VP7263 (termed B, numbers: duration of ozone-treatment in minutes) and DIB-stabilized bulk samples (termed Bs, numbers: duration of ozone-treatment in minutes) exhibited spherical pores with diameters between 15 and 18 nm according to SEM images and between 11 and 12 nm according to AFM measurements.
In Fig. 5, the topographies of the samples B90, Bs90, B10 and Bs10b are shown (for Bs10a see Fig. S5†). Fig. 5a–d show the aforementioned spherical pores derived from ozonolytic degradation of former PI domains with pore diameters inscribed. Close to the surface (Fig. 5e and f) the highly ordered cylindrical domains were retained and converted to hollow cylinders in a P2VP matrix. Diameters were determined using a visual image evaluation program (ImageJ) to measure 130 to 200 pores of the images shown in Fig. 5 and Fig. S5,† corresponding statistics are compiled in Table 2. The samples exhibit pore sizes between 15 and 18 nm, which is within the error values (±3) and therefore, no changes regarding the morphology with respect to varying ozonolysis protocols could be observed. However, sphere diameters determined from cross-sectional topography are assumed to be statistically distributed, since spheres will unlikely be exactly cut into two halves. All investigated film samples featuring a thickness of 1 mm revealed pores throughout the entire cross-sections.
 | ||
| Fig. 5 SEM images (mean pore diameters are indicated at the top right) of cross-sections of bulk polymer from PI28-b-P2VP7263 after ozone treatment. (a) non-stabilized and ozone treated (90 min) sample B90, (b) DIB-stabilized and ozone treated (90 min) sample Bs90, (c) non-stabilized and ozone treated (10 min) sample B10, (d) DIB-stabilized and ozone treated (10 min) sample Bs10b in the middle of the cross-section, (e) same sample Bs10b at the edge of the cross-section (near-surface area) and (f) overview thereof. | ||
| Sample | Stabilizeda | t /min | Pore-ø SEMc/nm | Pore-ø AFMd/nm |
|---|---|---|---|---|
| a 24 h treatment with DIB vapor. b Time span of ozone treatment. c Measured visually using Image-J on at least 132 pores. d Obtained from AFM image processed with Gwyddion Free SPM analysis software as twice the maximum inscribed radius rm (Fig. 6). e This sample was not washed with water. | ||||
| B90 | No | 90 | 15 ± 3 | — |
| Bs90 | Yes | 90 | 18 ± 3 | 10.7 ± 0.9 |
| B10 | No | 10 | 17 ± 2 | 11.1 ± 1.4 |
| Bs10ae | Yes | 10 | 15 ± 2 | 10.6 ± 0.9 |
| Bs10b | Yes | 10 | 16 ± 3 | 12.4 ± 2.2 |
AFM imaging revealed a porous surface structure for all samples (Bs90, B10, Bs10a and Bs10b, see Fig. 6), wherein the pore size is characterized by the maximum inscribed radius rm (see Table 2). As can be concluded from the height scale of Fig. 6b and d, Bs10b shows a flatter and more homogeneous surface than B10. The pore sizes of øB10 = (11.1 ± 1.4) nm and øBs10b = (12.4 ± 2.2) nm do not differ much. Bs10a was stabilized and underwent an ozonolysis for 10 minutes and also shows a similar pore size as B10 (øBs10a = (10.6 ± 0.9) nm). At the same time, with an ozonolysis for a longer time duration leads to a similar pore size of øBs90 = (10.7 ± 0.9) nm. The diameters of the pores of all samples from AFM data are smaller than those from SEM data, which could arise from cantilever tip broadening.62 In summary, the morphology is retained for stabilization with DIB and ozonolysis. An overview of samples examined in this chapter and results from SEM and AFM are compiled in Table 2.
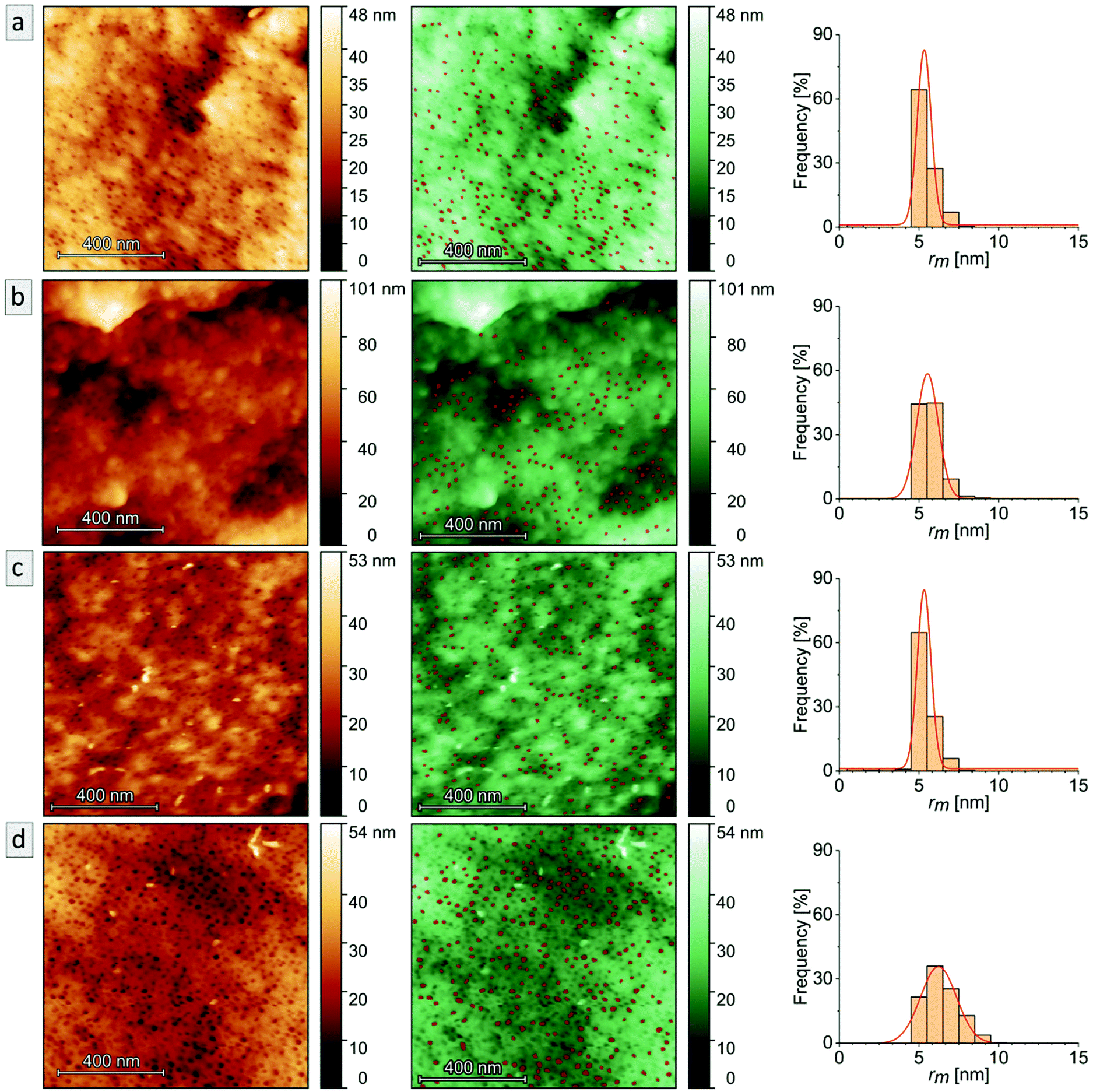 | ||
| Fig. 6 AFM images (left, in copper), AFM images masked pores (middle, in green) and the respective histograms (right) of the maximum inscribed radius rm with a Gaussian fit (red curve) of each sample: (a) Bs90, (b) B10, (c) Bs10a and (d) Bs10b. All AFM images were obtained in air at 25 °C. | ||
SAXS measurements of the samples B90, B10, Bs90 and Bs10 are shown in Fig. 7 in comparison to corresponding samples before ozone treatment. The non-stabilized bulk-sample and resulting porous materials (Fig. 7a) are compared to stabilized bulk-sample and resulting porous materials (Fig. 7b). All samples exhibit the same characteristic peaks as well as the same overall appearance as the non-treated sample. Therefore, SAXS confirms the results from SEM and AFM.
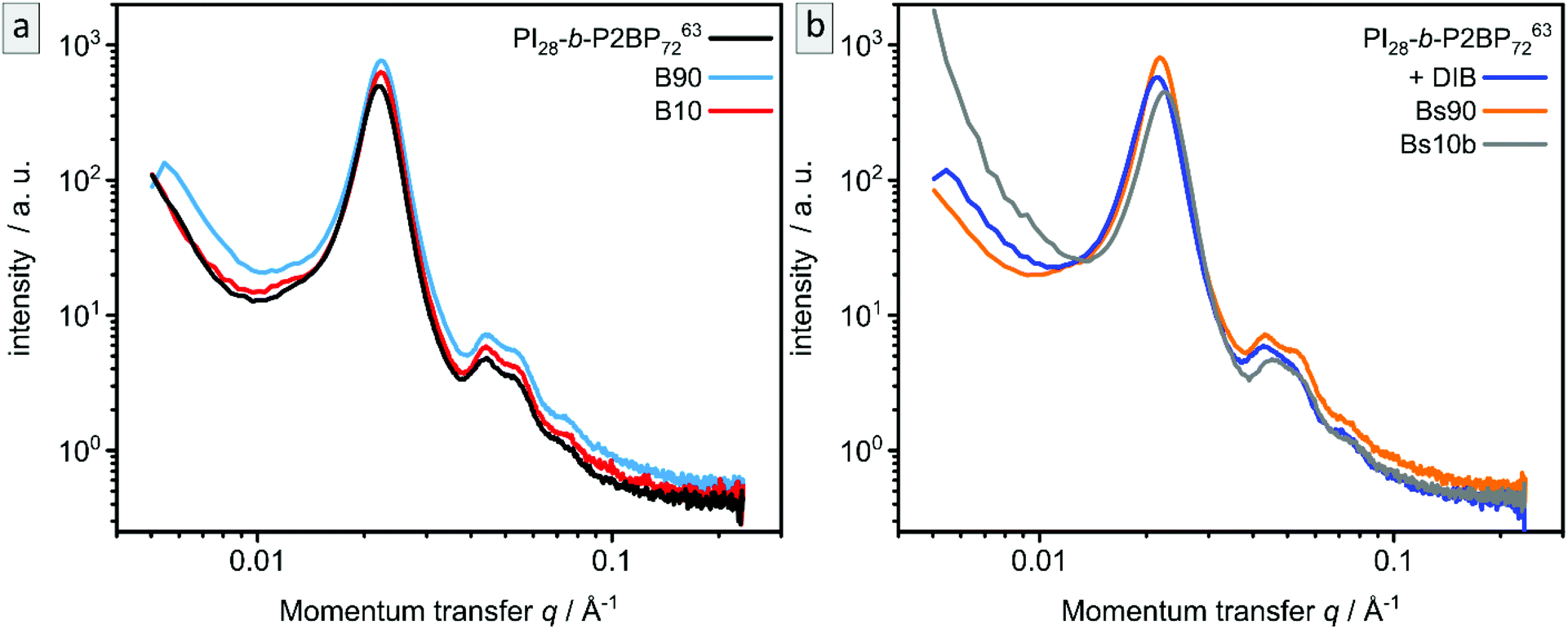 | ||
| Fig. 7 SAXS of bulk polymer PI28-b-P2VP7263 before and after ozone treatment. (a) non-stabilized (PI28-b-P2VP7263) and ozone treated samples B90 and B10. (b) DIB-stabilized (+DIB) and ozone treated samples Bs90 and Bs10b. | ||
Besides morphological and topographical analysis, conversion of functional groups was investigated by ATR-FTIR spectroscopy (Fig. S6†). The 2VP moieties stayed intact and are clearly detectible at 1587 and 1433 cm−1, as well as the characteristic peaks for pyridinium after DIB-crosslinking at 1626 and 1509 cm−1. After ozone treatment another signal occurs at 1729 cm−1, which corresponds to CO valence vibrations of carboxylic acids, aldehydes and ketones. That results from ozonolytic degradation of double bonds. Carboxylic derivatives are predominantly extracted from the pores by water, as can be seen in additional ATR-FTIR measurements shown in the ESI (Fig. S7†).
All experiments presented so far led to P2VP bulk material with hollow spheres, wherein the sphere sizes seemed to be independent of the ozone-treatment duration. Stabilizing the sample with DIB had no influence on the obtained morphology. Degradation of the PI in a sphere morphology still indicated a good diffusion of gases and degraded products of the PI segment through the P2VP matrix, while it retained its morphology. That underlines a high chemical stability of P2VP. Analysis of DIB-stabilized bulk samples indicated that accessibility and material transport can be an issue, since the penetration of the stabilizing agent is detectable for only a few micrometers. As long as the bulk substrates were relatively thick, stabilization was not necessary. To further highlight the application of the herein investigated ozone treatment and cross-linking protocols, these findings will be applied to a composite material consisting of cellulose fibers and amphiphilic BCP in order to gain access to a porous coating on a sustainable material.
PI-b-P2VP coatings on cellulose substrate and removal of PI domains
Within the next step, a BCP with a higher PI content was coated on cellulose fiber substrate as a model for more complex structures for tailored material and liquid transport. The composite materials were generated using commercially available cellulose-based filter discs (Munktell grade 3hw), which were infiltrated with a BCP solution. For this purpose, the polymer PI35-b-P2VP65186 was dissolved in chloroform and the paper was coated in a procedure similar to the vertical deposition method (Experimental section).63,64 In this procedure the interaction between the amphiphilic BCP and the cellulose fiber surface relies only on physical interactions such as hydrogen-bonding due to the presence of pyridinic moieties in the polymer and hydroxyl moieties at the cellulose fibers’ surface.35 Cross-linking with 1,4-diiodobutane and ozonolysis experiment were conducted in an analogous manner to the procedure used for the bulk BCP samples. The coated cellulose substrates as well as the ozone-treated substrates were examined using SEM, EDS, ATR-FTIR spectroscopy and WCA measurements.SEM analysis of untreated composite materials reveals that the paper-fibers are fully covered with polymer, as can be concluded from Fig. 8a and b, while the macroporous fiber-structure is retained (Fig. S8†). SEM images of untreated filter paper and overview images can be found in the ESI in Fig. S8 and S9.† When examining higher magnifications, BCP microphase separation can be observed, while the structure varies between lamellar structure or lying cylinders (Fig. S11a†) and spheres or standing cylinders (Fig. S11b†). In Fig. S11b,† few single spheres or standing cylinders are open, which may be due to sample preparation or air inclusions inside the film, while major parts of the BCP represent a closed polymer film.
 | ||
| Fig. 8 SEM analysis of (a and b) the surface of untreated and (c–f) ozone treated composite material using PI35-b-P2VP65186 on Munktell cellulose-based filter discs with increasing magnifications. (c) and (d) show the surfaces of the ozone-treated sample Cs10 (DIB-stabilized), (e) and (f) show surfaces of sample C10 (not stabilized). | ||
The as-prepared composite materials were treated with ozone for 10 minutes (Experimental section). While for the DIB-stabilized sample a dense polymer coating is retained (Cs10, Fig. 8c and d), the coating is mostly washed off in the non-stabilized sample while the remaining structure of the polymer matrix is mostly collapsed (C10, Fig. 8e and f). Consequently, cross-linking had a major influence on the samples’ stability when treating polymer-coated filter papers with ozone for only 10 minutes.
SEM images of the cross-sections of the untreated polymer-coating on cellulose substrates are shown in Fig. 9a and b. Fig. 9a gives indication on the microstructure of the cross-section of the coating. The thickness of the polymer layer varies between roughly 0.5 and 5 μm, as can be seen by comparing Fig. 9a and b. In Fig. 9b and c the border area between polymer and fiber is highlighted with arrows indicating the direction of the fibers. More SEM images of the cross-sections along with overview images can be found in the ESI in Fig. S12.† After 10 minutes of ozone treatment of sample Cs10 (Fig. 9c and d), a porous structure can be found. In particular, in Fig. 9d highly ordered domains of hexagonal hollow cylinders are visible, which is in agreement with the molar composition, i.e., PI content of 35% (see Table 1), according to BCP microphase separation theory. A combination of smaller and larger pore sizes on the nanometer scale could be observed in Fig. 9d, while the larger pores were already visible in the untreated samples (Fig. 9b and S11b†).
 | ||
| Fig. 9 SEM analysis of the (a and b) cross-sections of untreated and (c and d) ozone treated composite material (sample Cs10) using PI35-b-P2VP65186 on Munktell cellulose-based filter discs. Blue arrows indicate cellulose fibers. | ||
While in bulk non-stabilized samples could be treated with ozone for up to 90 min and were still intact (sample B90), non-stabilized coatings collapse already after 10 min of ozone treatment. To the contrary in DIB-stabilized samples (Cs10) highly meso- and macroporous composite materials could be obtained. It is assumed that the major difference between the two materials lies in the reduced film thickness while macropores are added. Meanwhile, the surface area increases and the effect of cross-linking with DIB is noticeable. EDS measurements were conducted on cross-sections of the cellulose composite material before treatment with ozone in order to investigate the limitation of cross-linking. In Fig. 10 exemplary EDS-point spectra are shown that were randomly collected in polymeric layers throughout the cross-section. It is noticeable that in all collected spectra characteristic emission energies for iodine can be detected. All locations of data points, mean values and standard deviations can be found in Fig. S14 and Table S3.†
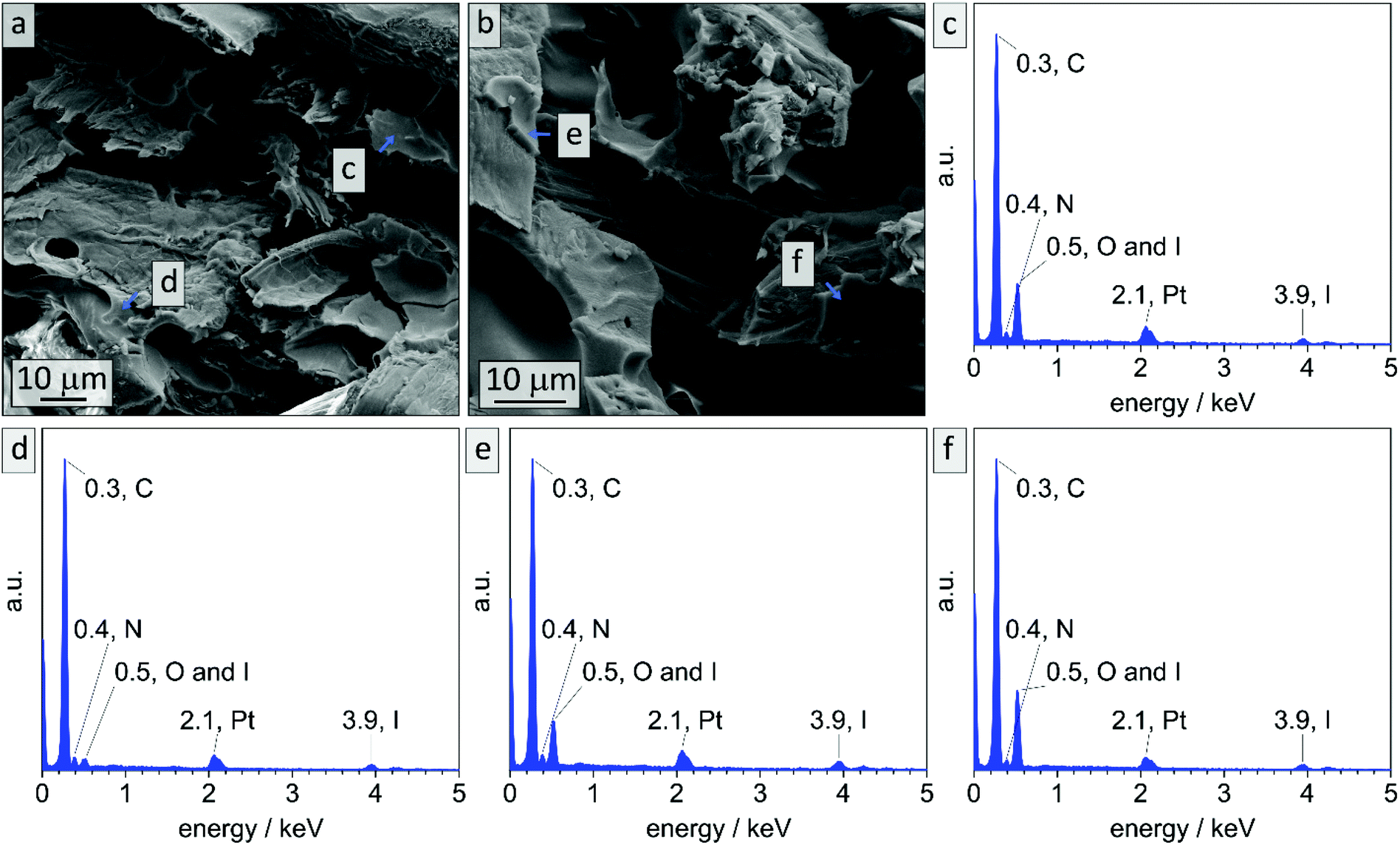 | ||
| Fig. 10 EDS data collected on composite sample made of PI35-b-P2VP65186 on cellulose substrate via vertical deposition after stabilization with DIB. (a and b) Cross-sectional SEM images and exemplary locations of EDS point spectra (c–f) are marked. | ||
As before, the chemical conversions in the process of coating, stabilizing and degrading PI is validated by ATR-FTIR measurements. In Fig. 11a and b IR-spectra of the untreated cellulose fiber substrate and the polymer-coated paper are shown. Characteristic signals of P2VP (compare to Fig. 4) are detected and highlighted in the partial spectrum (Fig. 11b). In Fig. 11c and d, the corresponding spectra of samples C10 and Cs10 are depicted. It is noticeable that P2VP-signals become less intensive, but are still clearly visible within both samples. In the DIB-stabilized sample Cs10 the characteristic signals for quaternization of pyridinium are visible as well as the signal for degrading products of PI (also compare to Fig. S6†). As a result, the chemical changing during the process of coating, stabilizing with DIB and PI degradation by ozone in the composite materials was followed and confirmed by ATR-FTIR measurements.
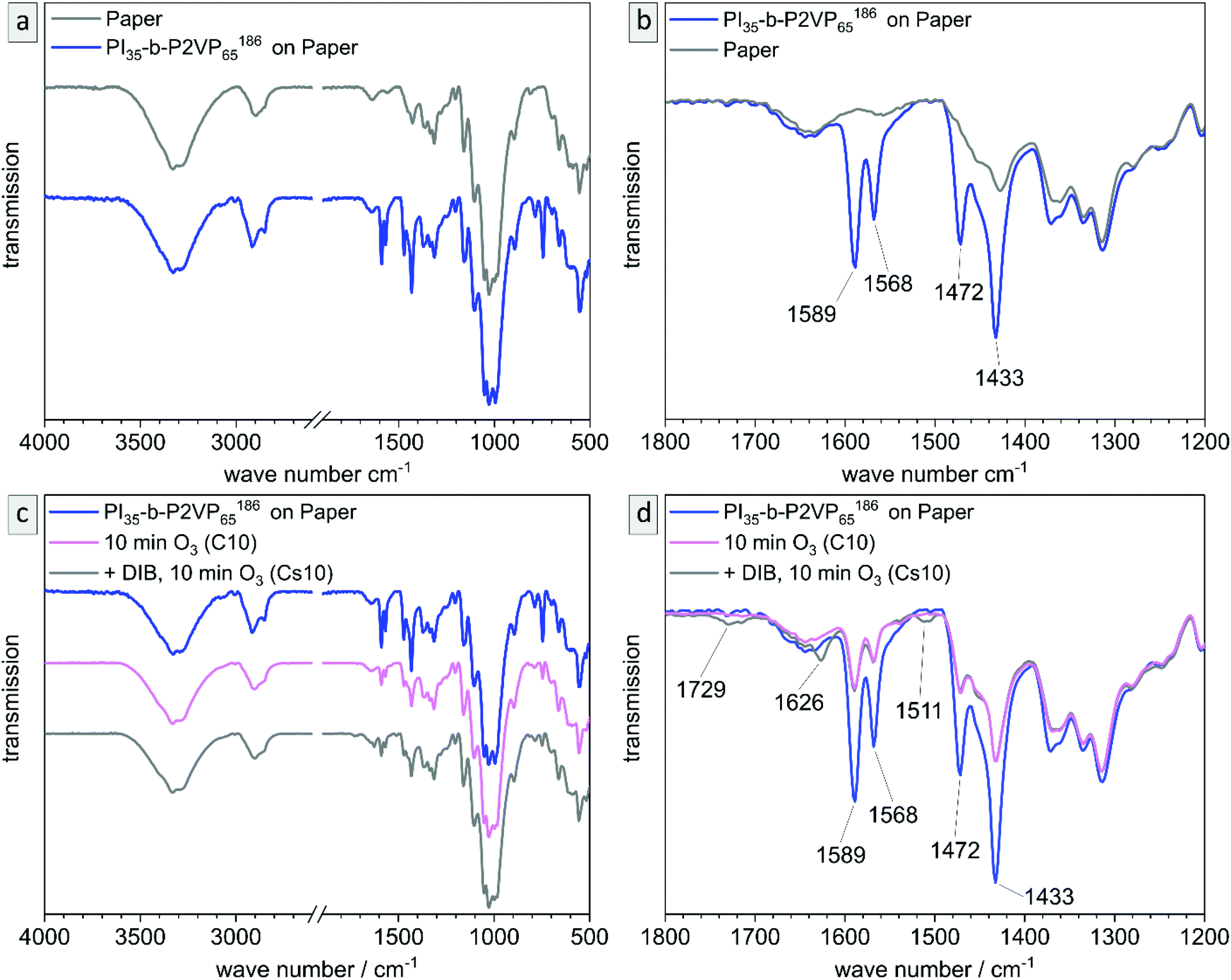 | ||
| Fig. 11 ATR-FTIR transmission spectra of (a) blank paper and polymer coated paper (PI35-b-P2VP65186via vertical deposition), (b) partial spectrum of polymer signals of (a). (c) polymer-coated paper (PI35-b-P2VP65186) before and after treating with ozone for 10 min, red curve without DIB-stabilization and blue curve with DIB-stabilization and (d) partial spectrum of polymer signals of (c). All IR-spectra are normalized to the most intensive peak (paper). | ||
The WCA (Θ) is a measure of surface hydrophilicity that is commonly used for surface characterization, which can give good indications on chemical transformations at the surface and their connection to surface properties.46,65 In this work, static WCA is used to follow the procedure of coating paper (Θ = 0°) with a polymer, stabilization with DIB and ozonolytic degradation. Fig. S15a–c† reveals a decrease to smaller contact angles, i.e., an increase of hydrophilicity: the untreated polymer coating features a mean-WCA of 114° ± 7°, while the DIB-stabilized polymer coating exhibits a mean-WCA of 109° ± 7° and the ozone treated sample (Cs10) is most hydrophilic with a WCA of 102° ± 9°. Mean values and errors were calculated taking six single values into account (see Table S4†). The difference between untreated polymer-coating and DIB-stabilized coating is within the standard deviation and rather indicates a tendency, while the absence of hydrophobic PI domains due to ozonolytic degradation causes a decrease in WCA.
In this section, it has been shown that a thin BCP-coating on cellulose substrate could be successfully retained during ozone treatment, while degrading PI led to spherical and cylindrical mesopores in a P2VP matrix. These samples featured only a fraction of the films thickness (∼0.5 to 5 μm) of the bulk samples investigated before (∼1 mm) and therefore, cross-linking with DIB became essential for retaining the films stability.
Stimuli-responsive behavior of porous P2VP films and coatings
Polyvinylpyridines are stimuli-responsive polymers that are sensitive to aqueous and acidic solutions because they contain pyridinic moieties which are Lewis bases. When treating polyvinylpyridines with aqueous or acidic aqueous solution, swelling can be observed.9,11–13 When applying this to porous materials, pores can shrink or close while the matrix swells.The cross-section of sample Bs10b was studied upon its swelling behavior in aqueous solution by AFM imaging (Fig. 12b). As a reference, an image of the sample measured in air at 25 °C is included (Fig. 12a). The root mean square (RMS) roughness of the whole images (1 × 1 μm2) and more local areas (100 × 100 nm2, as indicated by the red masks) are calculated to account for the amount of swelling of the sample (Table 3). The RMS roughness of the Bs10b surface in H2O increases due to the swelling of P2VP. Also, the pores appear even closed, which leads to a lower number of observable pores (Fig. 12b).
 | ||
| Fig. 12 Overview of masked areas (in red) for obtaining local RMS roughness in AFM images of sample Bs10b measured in (a) air and (b) in pure H2O. Photographs of WCA (1 minute) using an acidic buffer solution (pH = 4) on (c) polymer-coated and DIB-stabilized cellulose disc (Θ = 111°) and (d) corresponding ozone-treated sample Cs10 (Θ = 86°). SEM analysis of (e and f) the surface and (g and h) cross-section of ozone treated and acid-swollen sample Cs10 after washing and drying. | ||
| Sample | Measurement condition | RMS roughness for the whole scanned area/nm | RMS roughness for the masked area/nm |
|---|---|---|---|
| Bs10b | Air | 3.5 | 1.5 |
| H2O | 19.1 | 4.2 |
The stimuli-responsiveness of P2VP can also be observed via surface wettability. Therefore, WCA measurements were conducted on a polymer-coated and stabilized substrate versus the corresponding ozone-treated sample Cs10 with an acidic buffer solution at pH 4 (Fig. 12). After 1 minute, the droplet on sample Cs10 (Fig. 12d) exhibits a WCA of 86°, while the corresponding sample before ozone-treatment (Fig. 12c) exhibits a WCA of 111°. Hence, after ozonolytic treatment the wettability with aqueous acidic solution is increased, which can be attributed to the absence of the hydrophobic PI. Also, the acidic WCA of the ozone treated sample is smaller than the aqueous WCA (86° and 103°, respectively), which indicates that P2VP moieties in these materials are addressable by pH variations.
AFM images in water revealed that the P2VP matrix swells and WCA measurements showed that surface properties respond to change of pH value. In order to evaluate the possibility to use the herein presented cellulose based composite substrates as stimuli-responsive materials in future studies, the samples were stored in acid (HClaq., pH = 4.0) for 30 min, washed, dried and examined by SEM. Fig. 12e–h shows that the polymer coating is not delaminated from the cellulose fibers and therefore the macroporous fiber structure of the cellulose substrate is retained, as can be seen as well in Fig. S13.† Additionally, small open pores can be seen on the surface of the polymeric coating. The cylindrical pores (compare Fig. 12h to Fig. 9d) are less dominant and followingly changes in matrix structure cannot be excluded.
Conclusion
In this work we report the fabrication of a nanoporous functional polymeric bulk material featuring spherical pores with diameters below 20 nm. This was achieved by using a BCP-templated approach in which the BCP contained a sacrificial block segment that was degraded using ozone. For this purpose, a PI-b-P2VP BCP featuring high content of 1,4-linkages in the PI-segment (≥93 mol%) and high overall P2VP content (≥65 mol%) was synthesized via sequential anionic polymerization. The obtained BCP PI28-b-P2VP7263 exhibited spherical morphology in the bulk state, which was confirmed by SEM and SAXS measurements. Cross-linking of bulk-samples with DIB was conducted and the chemical conversion of 2VP moieties was confirmed by ATR-FTIR measurements. The cross-sections were additionally investigated using SEM-EDS to confirm the incorporation of iodine. A shift in morphology to cylinders was observed in locations where iodine was introduced during cross-linking, which is in the outer 3 μm of the cross-section. Ozonolytic degradation of PI-spheres in the bulk sample was then investigated using SEM, AFM and SAXS. The spherical morphology in the cross-section and the cylindrical morphology near the surface was retained. Hollow spheres and cylinders in a P2VP matrix were obtained throughout the whole cross-section with 1 mm thickness after only 10 minutes of ozone treatment. This result shows the potential of the applied concept and the efficiency of ozone treatment and removal of small molecules. Furthermore, the block copolymer PI35-b-P2VP65186 was coated on a cellulose fiber disc in a way that a macroporous fiber structure was obtained, which was shown in SEM images. Iodine was found throughout the whole cross-section in EDS spectra and after ozone treatment hollow cylinders are observed in SEM. Therefore, in this work the successful fabrication of hierarchically porous cellulose-based composite materials with BCPs is introduced. The tailored pore design at the nanometer length scale and at the cellulose fiber surface can enable the control over the wettability and functionality paving the way to applications in the field of sustainable microfluidics, membranes or sensors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was supported by the DFG project GA2169/7-1 in association with the DFG-funded consortium for advanced paper research (DFG-PAK 962/1) at TU Darmstadt (M. G. and L. G.). This work is also supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy − EXC-2193/1 − 390951807 (Q. H. and B. N. B.). The authors acknowledge Prof. Volker Presser and the EMG at the INM (Leibniz-Institute for New Materials Saarbrücken) for support. The authors thank Prof. Uli Kazmaier and his working group for providing the ozone generator, as well as Devid Hero and Blandine Boßmann of the Smart Polymers Group for their help and ideas with practical issues.References
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- H. Feng, X. Lu, W. Wang, N.-G. Kang and J. W. Mays, Polymers, 2017, 9, 494 CrossRef PubMed.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429 CrossRef CAS.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- N.-G. Kang, B.-G. Kang, H.-D. Koh, M. Changez and J.-S. Lee, React. Funct. Polym., 2009, 69, 470–479 CrossRef CAS.
- U. Borchert, U. Lipprandt, M. Bilang, A. Kimpfler, A. Rank, R. Peschka-Suss, R. Schubert, P. Lindner and S. Forster, Langmuir, 2006, 22, 5843–5847 CrossRef CAS PubMed.
- C. Rüttiger, H. Hübner, S. Schöttner, T. Winter, G. Cherkashinin, B. Kuttich, B. Stühn and M. Gallei, ACS Appl. Mater. Interfaces, 2018, 10, 4018–4030 CrossRef PubMed.
- A. Jung, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Macromol. Mater. Eng., 2012, 297, 790–798 CrossRef CAS.
- Z. Zhang, M. M. Rahman, C. Abetz, B. Bajer, J. Wang and V. Abetz, Macromol. Rapid Commun., 2019, 40, 1800729 CrossRef PubMed.
- M. Appold, C. Rüttiger, B. Kuttich, B. Stühn and M. Gallei, Macromol. Chem. Phys., 2018, 219, 1700187 CrossRef.
- N. Herzog, H. Hubner, C. Ruttiger, M. Gallei and A. Andrieu-Brunsen, Langmuir, 2020, 36, 4015–4024 CrossRef CAS PubMed.
- Z. Chen, C. He, F. Li, L. Tong, X. Liao and Y. Wang, Langmuir, 2010, 26, 8869–8874 CrossRef CAS PubMed.
- D. V. Pergushov, E. V. Remizova, M. Gradzielski, P. Lindner, J. Feldthusen, A. B. Zezin, A. H. E. Müller and V. A. Kabanov, Polymer, 2004, 45, 367–378 CrossRef CAS.
- Y.-H. Cho, J.-E. Yang and J.-S. Lee, Mater. Sci. Eng., C, 2004, 24, 293–295 CrossRef.
- M. Orlov, I. Tokarev, A. Scholl, A. Doran and S. Minko, Macromolecules, 2007, 40, 2086–2091 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, S. J. Brooks and K. Matyjaszewski, Macromolecules, 2006, 39, 6817–6824 CrossRef CAS.
- K. R. M. Vidts and F. E. Du Prez, Eur. Polym. J., 2006, 42, 43–50 CrossRef CAS.
- J. Zeng, K. Shi, Y. Zhang, X. Sun, L. Deng, X. Guo, Z. Du and B. Zhang, J. Colloid Interface Sci., 2008, 322, 654–659 CrossRef CAS PubMed.
- L. Volkmann, M. Köhler, F. H. Sobotta, M. T. Enke, J. C. Brendel and F. H. Schacher, Macromolecules, 2018, 51, 7284–7294 CrossRef CAS.
- M. Mazurowski, K. Sondergeld, J. Elbert, C. J. Kim, J. Li, H. Frielinghaus, M. Gallei, B. Stühn and M. Rehahn, Macromol. Chem. Phys., 2013, 214, 1094–1106 CrossRef CAS.
- T. Diaz, A. Fischer, A. Jonquieres, A. Brembilla and P. Lochon, Macromolecules, 2003, 36, 2235–2241 CrossRef CAS.
- K. V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- K. M. Axel and H. E. Muller, Controlled and Living Polymerizations, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed.
- J. M. Widmaier and G. C. Meyer, Macromolecules, 1981, 14, 450–452 CrossRef CAS.
- W.-l. Hsu, A. F. Halasa and B. A. Matrana, US Pat, US5336739, 1994 Search PubMed.
- C. Rüttiger, M. Appold, H. Didzoleit, A. Eils, C. Dietz, R. W. Stark, B. Stühn and M. Gallei, Macromolecules, 2016, 49, 3415–3426 CrossRef.
- M. Appold, J. Bareuther and M. Gallei, Macromol. Chem. Phys., 2019, 220, 1800548 CrossRef CAS.
- R. P. Quirk and S. Corona-Galvan, Macromolecules, 2001, 34, 1192–1197 CrossRef CAS.
- H. Watanabe and M. Tirrell, Macromolecules, 1993, 26, 6455–6466 CrossRef CAS.
- A. Okumura, Y. Nishikawa and T. Hashimoto, Polymer, 2006, 47, 7805–7812 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- M. Plank, F. Hartmann, B. Kuttich, T. Kraus and M. Gallei, Eur. Polym. J., 2020, 141, 110059 CrossRef CAS.
- Y. Funaki, K. Kumano, T. Nakao, H. Jinnai, H. Yoshida, K. Kimishima, K. Tsutsumi, Y. Hirokawa and T. Hashimoto, Polymer, 1999, 40, 7147–7156 CrossRef CAS.
- C. M. Hansen, J. Paint Technol., 1967, 39, 505 CAS.
- C. Hansen and K. Skaarup, J. Paint Technol., 1967, 39, 551 Search PubMed.
- S. Arichi, H. Matsuura, Y. Tanimoto and H. Murata, Bull. Chem. Soc. Jpn., 1966, 39, 434–439 CrossRef CAS.
- D. A. Olson, L. Chen and M. A. Hillmyer, Chem. Mater., 2007, 20, 869–890 CrossRef.
- R. C. Hayward, B. F. Chmelka and E. J. Kramer, Adv. Mater., 2005, 17, 2591–2595 CrossRef CAS.
- R. Saito, S.-I. Okamura and K. Ishizu, Polymer, 1992, 33, 1099–1101 CrossRef CAS.
- C. G. Schäfer, T. Winter, S. Heidt, C. Dietz, T. Ding, J. J. Baumberg and M. Gallei, J. Mater. Chem. C, 2015, 3, 2204–2214 RSC.
- M. P. Anachkov, S. K. Rakovski and R. V. Stefanova, Polym. Degrad. Stab., 2000, 67, 355–363 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- E. Huang, A. Skoufis, T. Denning, J. Qi, R. Dagastine, R. Tabor and J. Berry, J. Open Source Software, 2021, 6, 2604 CrossRef.
- F. V. Friess, Q. Hu, J. Mayer, L. Gemmer, V. Presser, B. N. Balzer and M. Gallei, Macromol. Rapid Commun., 2022, 43, 2100632 CrossRef CAS PubMed.
- D. Ferri and F. Greco, Macromolecules, 2006, 39, 5931–5938 CrossRef CAS.
- J. I. Brandrup, E. H. Grulke, E. A. Abe and A. Bloch, Polymer Handbook, John Wiley & Sons, 2003 Search PubMed.
- L.-Y. Shi, W.-W. Lei, F. Liao, J. Chen, M. Wu, Y.-Y. Zhang, C.-X. Hu, L. Xing, Y.-L. Zhang and R. Ran, Polymer, 2018, 153, 277–286 CrossRef CAS.
- N. Sota, N. Sakamoto, K. Saijo and T. Hashimoto, Polymer, 2006, 47, 3636–3649 CrossRef CAS.
- N. Sota, N. Sakamoto, K. Saijo and T. Hashimoto, Macromolecules, 2003, 36, 4534–4543 CrossRef CAS.
- T. Zemb and P. Lindner, Neutron, X-rays and light. Scattering methods applied to soft condensed matter, North Holland, 2002 Search PubMed.
- H. Matsuoka, H. Tanaka, N. Iizuka, T. Hashimoto and N. Ise, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 3854–3856 CrossRef PubMed.
- H. Matsuoka, H. Tanaka, T. Hashimoto and N. Ise, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 1754–1765 CrossRef PubMed.
- J. K. Percus and G. J. Yevick, Phys. Rev., 1958, 110, 1 CrossRef CAS.
- K. Mavronasou, A. Zamboulis, P. Klonos, A. Kyritsis, D. N. Bikiaris, R. Papadakis and I. Deligkiozi, Polymers, 2022, 14, 804 CrossRef CAS PubMed.
- Z. Zhang, M. M. Rahman, B. Bajer, N. Scharnagl and V. Abetz, J. Membr. Sci., 2022, 646, 120266 CrossRef CAS.
- J. Wu, Z. Lan, D. Wang, S. Hao, J. Lin, Y. Huang, S. Yin and T. Sato, Electrochim. Acta, 2006, 51, 4243–4249 CrossRef CAS.
- M. Kraska, M. Gallei, B. Stühn and M. Rehahn, Langmuir, 2013, 29, 8284–8291 CrossRef CAS PubMed.
- S. Behzadi, M. Gallei, J. Elbert, M. Appold, G. Glasser, K. Landfester and D. Crespy, Polym. Chem., 2016, 7, 3434–3443 RSC.
- J. Canet-Ferrer, E. Coronado, A. Forment-Aliaga and E. Pinilla-Cienfuegos, Nanotechnology, 2014, 25, 395703 CrossRef PubMed.
- Z. Zhou and X. S. Zhao, Langmuir, 2004, 20, 1524–1526 CrossRef CAS PubMed.
- D. Scheid, D. Stock, T. Winter, T. Gutmann, C. Dietz and M. Gallei, J. Mater. Chem. C, 2016, 4, 2187–2196 RSC.
- H.-J. Butt, K. Graf and M. Kappl, Physics and chemistry of interfaces, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00562j |
| This journal is © The Royal Society of Chemistry 2022 |