
Recharging upconversion: revealing rubrene's replacement
Colette M.
Sullivan
and
Lea
Nienhaus
 *
*
Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA. E-mail: lnienhaus@fsu.edu
First published on 10th November 2022
Abstract
One of the major limitations of solid-state perovskite-sensitized photon upconversion to date is that the only annihilator successfully paired with the perovskite sensitizer has been rubrene, raising the question of whether this appraoch of triplet sensitization is universal or limited in scope. Additionally, the inherent energetic mismatch between the perovskite bandgap and the rubrene triplet energy has restricted the apparent anti-Stokes shift achievable in the upconversion process. To increase the apparent anti-Stokes shift for upconversion processes, anthracene derivates are of particular interest due to their higher triplet energies. Here, we demonstrate successful sensitization of the triplet state of 1-chloro-9,10-bis(phenylethynyl)anthracene using the established formamidinium methylammonium lead triiodide perovskite FA0.85MA0.15PbI3, resulting in upconverted emission at 550 nm under 780 nm excitation. We draw a direct comparison to rubrene to unravel the underlying differences in the upconversion processes.
 Lea Nienhaus | Dr. Lea Nienhaus received her B.Sc. from the Universität Ulm, Germany in 2010. In 2015, she obtained her Ph.D. from the University of Illinois at Urbana-Champaign working with Professor Gruebele on optical scanning tunneling microscopy. Following her postdoctoral work with Professor Bawendi at MIT, she began her independent career at Florida State University in the Fall of 2018. Her current research focuses on utilizing bulk perovskite materials as triplet sensitizers in photon upconversion and understanding the complex photophysical processes occurring in these systems by using a combination of optical spectroscopy and scanning probe microscopy. |
Introduction
Perovskite-sensitized triplet fusion upconversion (UC) via triplet–triplet annihilation (TTA) first emerged in 2019.1,2 Since its inception, focus has been placed primarily on understanding the mechanism of triplet generation and role of the fabrication conditions on the device performance.3–11 High absorption cross sections12 and long charge carrier diffusion lengths in the perovskite sensitizer13–15 in addition to the underlying triplet sensitization mechanism based on free charge carrier injection to the bound triplet state of the annihilator1–3 promise a path towards the realization of efficient solid-state UC devices.16 However, to date, the only triplet annihilator that has been successfully paired with bulk perovskites in the solid state and reported in bulk lead halide perovskite-sensitized UC has been rubrene, allowing for near-infrared-to-yellow UC.17,18Rubrene has long been the ‘gold standard’ of solid-state near-infrared-to-visible UC due to its known high UC efficiencies in solution as well as solid state configurations.19–23 However, the inherent ∼0.4 eV energy loss caused by the mismatched energy alignment between the perovskite bandgap (1.55 eV) and triplet energy (T1 = 1.14 eV)24,25 negates the benefits of using a sensitizer that does not rely on intersystem crossing to generate the prerequisite triplet states. The resulting small apparent anti-Stokes shift from 800 nm (1.55 eV) to 605 nm (2.05 eV) in the case of rubrene doped with ∼1% dibenzotetraphenylperiflanthene (DBP) is far from optimized. In the ideal case, the TTA-UC process should be able to double the sensitizer bandgap energy due to minimal losses when generating the triplet state Ebandgap = E(T1) and achieve an apparent anti-Stokes shift equal to the triplet energy E(T1) resulting in an emitted singlet energy E(S1) = 2E(T1). Hence, even when allowing for small energy losses in the TTA processes, achieving upconverted emission around 450 nm (2.8 eV) should be targeted when using the established formamidinium methylammonium lead triiodide (FA0.85MA0.15PbI3, FAMA) perovskite with a bandgap of Ebandgap = 1.55 eV as the triplet sensitizer.
In this desired triplet energy regime, anthracene derivatives are of general interest26–30 as these allow an increase in triplet energy and reduced energy losses during hole extraction due to generally deeper highest occupied molecular orbital (HOMO) energy levels.31,32 While 9,10-diphenylanthracene (DPA) is the common workhorse in solution-phase visible-to-blue UC,33–38 it is incompatible with the FAMA sensitizer used here due to its high triplet energy level (T1 = 1.77 eV) and strong excimer formation reduces the achievable UC yield and apparent anti-Stokes shift in solid state.39 However, the green dye 1-chloro-9,10-bis(phenylethynyl)anthracene (1-CBPEA), which has been commercially used in glowsticks, has been reported as an efficient triplet annihilator with peak emission at 490 nm (2.53 eV) in solution.40–43 Similarly, Castellano and co-workers have successfully demonstrated TTA-UC using 2-chloro-9,10-bis(phenylethynyl)anthracene (2-CBPEA) which has similar optical properties as 1-CBPEA and a FAMA-compatible upper bound of the triplet energy level set at T1 < 1.61 eV.44,45 A previous study has given a triplet energy for 1-CBPEA of T1 = 1.20 eV in solution,40 which appears to be at odds with TTA-UC occurring successfully as the requirement of E(S1) ≤ E(2T1) is not fulfilled.40–43 While slightly endothermic UC is in principle possible, we suggest a marginally higher native triplet energy level in the range of T1 = 1.3 − 1.2 eV in solution, estimated at roughly half of the singlet energy S1 (vide infra). However, additional effects on the singlet and triplet energy surfaces caused by intermolecular coupling and different rotational conformations upon condensation into the solid state cannot be ruled out; this effect has previously been observed for the parent molecule 9,10-bis(phenylethynyl)anthracene (BPEA), where different molecular arrangements and rotational conformations influence the singlet and triplet energy surfaces and therefore the rates of both TTA and singlet fission (SF).46–49
In this contribution, we investigate the triplet sensitization at the FAMA/1-CBPEA interface and the resulting UC process comparing the performance to the established FAMA/rubrene/1%DBP (FAMA/Rub) device. We find that FAMA perovskites can act as triplet sensitizers for 1-CBPEA resulting in upconverted emission at 550 nm (2.4 eV) highlighting the fact that triplet generation at the perovskite/organic semiconductor (OSC) interface is a general phenomenon and not limited to rubrene. However, despite the larger apparent anti-Stokes shift of the FAMA/1-CBPEA UC device, the overall performance is slightly diminished and the threshold intensity for efficient UC is increased. These effects can in large part be traced back to the significantly reduced triplet lifetime of 1-CBPEA in comparison to rubrene and a reduced OSC quantum yield. However, further optimization of the energetic driving force for charge extraction by compositional8 and interfacial tuning4 or addition of a dopant dye to improve the OSC quantum yield21 bear promise in further advancing this next-generation perovskite-sensitized UC.
Results and discussion
The general optical characterization and comparison of 1-CBPEA and rubrene doped with ∼1% DBP (RubDBP) OSC thin films are depicted in Fig. 1. Fig. 1a shows the absorption spectra of 1-CBPEA and RubDBP. In the latter case, the expected vibronic progression of rubrene is visible, as well as the vibronic feature corresponding to DBP at 600 nm. For 1-CBPEA, in addition to the expected vibronic progression, we find a broad red shifted feature at 520 nm (2.4 eV), which we attribute to an aggregation-induced redshift due to intermolecular coupling in the solid state.47 As reported previously, the emission profile of RubDBP (Fig. 1b) is governed by the dopant dye DBP (605 nm), indicating efficient Förster resonance energy transfer (FRET) between rubrene and DBP. The 1-CBPEA emission does not show the vibronic progression observed in solution, rather is dominated by the red-shifted aggregate emission peaking at 550 nm. A comparison of the photoluminescence (PL) intensity normalized by the optical density of the OSC at the excitation wavelength (405 nm) indicates that the PL quantum yield of the RubDBP film is ∼1.5 times that of 1-CBPEA.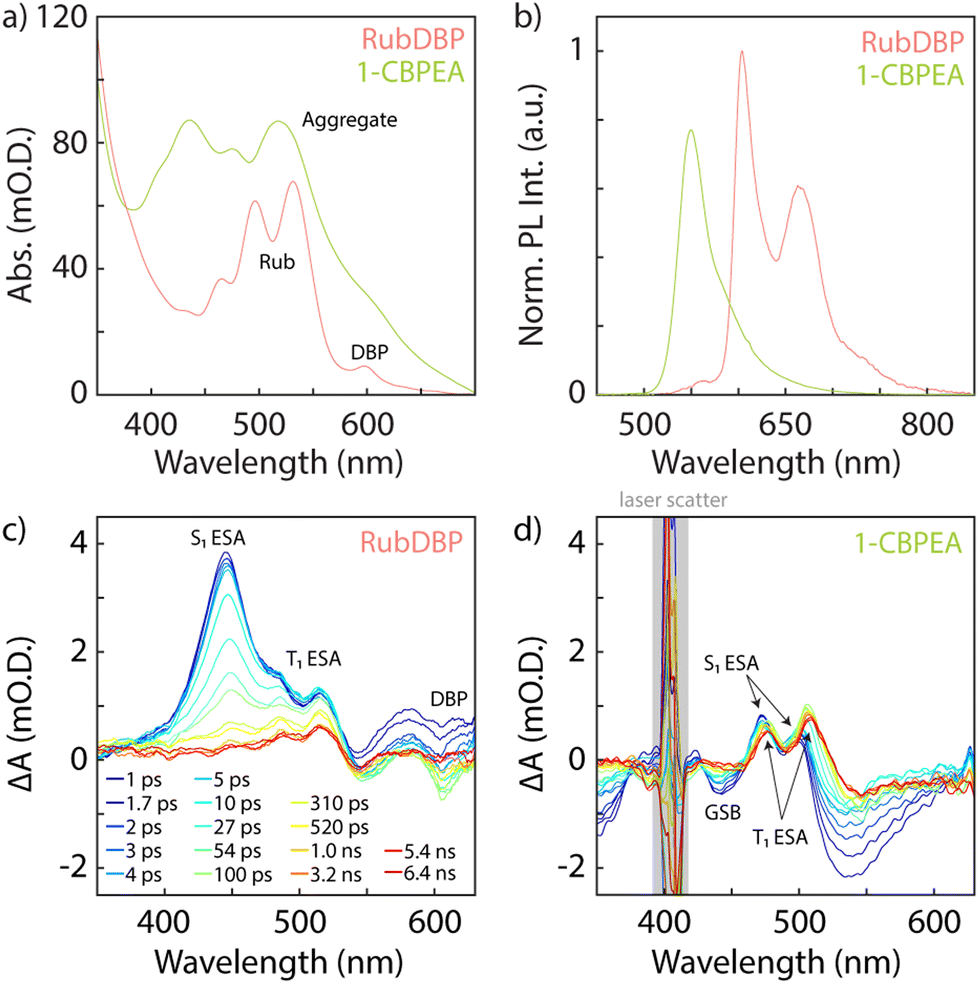 | ||
| Fig. 1 Comparison of the RubDBP and 1-CBPEA OSC thin films. (a) UV-visible absorbance spectra of RubDBP and 1-CBPEA, where a redshifted aggregation-induced absorption feature can be seen at 520 nm. (b) Emission spectra of the OSC films normalized to their optical density at the excitation wavelength of 405 nm. 1-CBPEA emits at 550 nm and RubDBP is dominated by DBP emission at 605 nm. Absorption spectra at selected delay times for RubDBP (c) and 1-CBPEA (d) thin films. The relevant spectral features are identified for both OSC films. Rubrene exhibits the characteristic S1 excited state absorption (ESA) at 445 nm and T1 ESA features at 485 nm and 515 nm and a bleach corresponding to DBP is observed at 605 nm. Two bleach features are found for 1-CBPEA, which we attribute to the ground state bleach (GSB) at 440 nm and 530 nm, respectively. The S1 ESA is found at 470 nm and 500 nm, T1 ESA features emerge at 475 nm and 510 nm. Excess pump laser scatter is denoted by the gray box. | ||
Transient absorption (TA) spectroscopy gives further insight into the spectral signatures of the singlet and triplet states under direct excitation. Rubrene is known to undergo both SF and TTA, due to its triplet energy T1 at half of the singlet energy S1. Therefore, direct excitation of the RubDBP OSC thin film at 400 nm yields the singlet signature dominated by the S1 excited state absorption (ESA) at 445 nm which rapidly evolves to reveal the T1 → Tn ESA features characteristic of rubrene triplet state at 485 nm and 515 nm.24,25 Furthermore, a DBP-related bleach feature can be observed at 605 nm which we attribute to stimulated emission under direct excitation or to the DBP ground state bleach (GSB). To the best of our knowledge, SF has not been previously reported in 1-CBPEA; however, has been shown in the parent molecule BPEA.47 Due to the similarity of the TA spectra observed here for 1-CBPEA and previous reports for BPEA, we suggest that SF also occurs in the 1-CBPEA OSC thin films under direct excitation at 400 nm.47 In particular, we find bleach features at 440 nm and 530 nm which we attribute to the GSB or stimulated emission of 1-CBPEA. Overlapping ESA features corresponding to the S1 state are found at 470 nm and 500 nm. Additional redshifted ESA features rapidly emerge at 475 nm and 510 nm, which can be attributed to the spin-allowed T1 → Tn optical transitions of triplet states generated by SF on an ultrafast timescale.44,47
To investigate the charge extraction at the perovskite/OSC interface and resulting TTA-UC process, bilayer devices consisting of a ∼30 nm thin FAMA sensitizer film and either RubDBP or 1-CBPEA as the annihilator were fabricated. The anticipated energy flow resulting in UC in each device is highlighted in Fig. 2. Upon photoexcitation of the perovskite at 780 nm, free charge carriers are generated which transfer to the OSC and recombine to populate the triplet state of either 1-CBPEA or RubDBP. Following triplet generation, two triplets can combine via TTA to yield the desired higher energy singlet state. After creation of the singlet state, the photon can be emitted directly from the S1 state (1-CBPEA) or the higher energy state can be harvested by the DBP dopant (RubDBP) with subsequent emission from the S1 state of DBP.
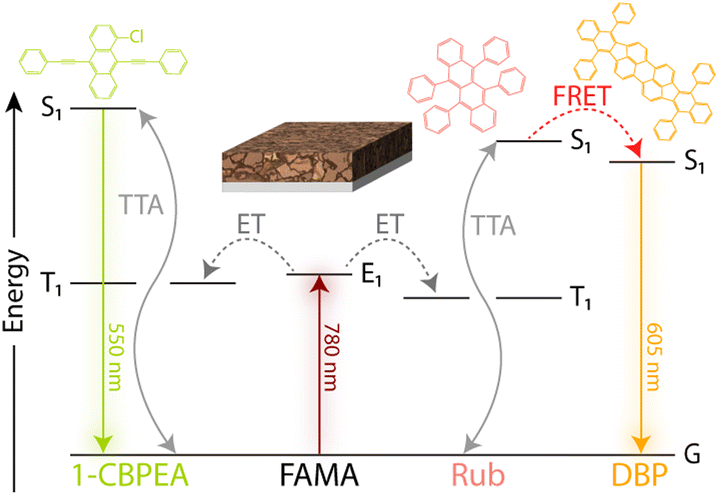 | ||
| Fig. 2 Energy flow schematic for UC process in the bilayer devices with the corresponding chemical structures for the OSC molecules. Incident 780 nm photons are absorbed by the perovskite, where charge transfer (ET) populates the triplet state T1 of 1-CBPEA (left) or Rub (right). Interaction of two triplet states results in TTA, yielding the higher energy S1 state. For Rub, an additional energy transfer (FRET) occurs to the DBP S1 energy level. Resulting UC emission wavelengths are 550 nm for 1-CBPEA and 605 nm for RubDBP. | ||
The absorption spectra of the bilayer devices and a FAMA control film are shown in Fig. 3a. The absorption onset of the perovskite is unchanged at 800 nm, and the OSC-related vibronic progression near 500 nm is observed indicating successful fabrication of the bilayers. Further investigation by time-correlated single-photon counting yields information into the charge carrier extraction at the FAMA/OSC interface where we monitor the perovskite PL decay dynamics (>800 nm) under 780 nm excitation (Fig. 3b). In comparison to the FAMA control, both the RubDBP and 1-CBPEA show quenched PL decay dynamics, indicating successful charge extraction. Anthracene derivatives generally have deeper highest occupied molecular orbital (HOMO) levels in comparison to their tetracene analogues.31,32,50 The HOMO of the 1-CBPEA parent molecule BPEA has been reported at −5.49 eV,51 deeper with respect to the HOMO level of rubrene at −5.4 eV.50 Electron withdrawing groups such as the chlorine in 1-CBPEA, further deepen the HOMO level and simultaneously reduce the bandgap.52–54 As a result, we expect the driving force for initial hole extraction from the perovskite valence band to the OSC HOMO to be inherently reduced in 1-CBPEA in contrast to RubDBP when utilizing the same FAMA perovskite sensitizer composition. Hence, the increased amount of quenching and more rapid PL decay for RubDBP over 1-CBPEA can be attributed to a higher driving force for charge extraction for RubDBP.
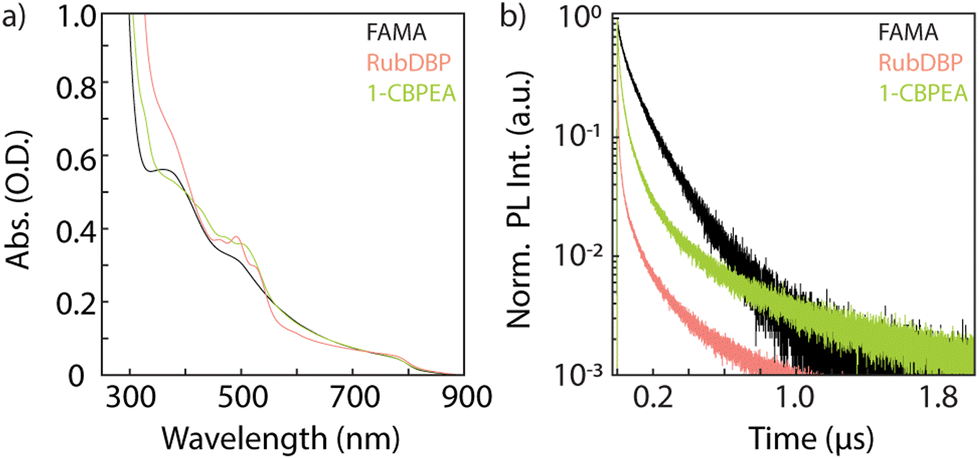 | ||
| Fig. 3 (a) Absorption spectra for the FAMA perovskite thin film (black) and bilayer devices (RubDBP: pink, 1-CBPEA: green). (b) Perovskite PL decays for the FAMA and bilayer devices under 780 nm excitation at an excitation density of 44.2 mW cm−2. A 780 nm notch filter and an 800 nm long pass filter were used to remove laser scatter and isolate the perovskite emission. | ||
Thus far, we have shown that charge extraction occurs at the perovskite/OSC interface. However, the observation of charge extraction at the perovskite/OSC interface is not sufficient for triplet generation since single charge transfer would similarly reduce the PL lifetime of the perovskite. To investigate whether the triplet of 1-CBPEA is indeed populated following the hole extraction process at the perovskite/OSC interface, we turn to TA spectroscopy to probe the ESA corresponding to the T1 → Tn transition.9Fig. 4a shows the TA spectra for the underlying FAMA sensitizer at selected delay times under 700 nm excitation in the relevant spectral region from 440–650 nm for the triplet-related ESA. The two characteristic bleach features of the perovskite are denoted as PB1 and PB2 respectively, while the perovskite photoinduced absorption feature centred at 550 is labelled as PIA. An additional overlapping bleach and photoinduced absorption attributed to PbI2 is found at 510 and 520 nm, respectively. This excess PbI2 is caused by a post-fabrication acetonitrile treatment of the perovskite surface. We have previously shown that this solvent treatment increases triplet generation in rubrene due to n-type doping of the perovskite, which increases the hole density at the surface due to interfacial band bending.55 Once interfaced with the OSCs, additional spectral features corresponding to the T1 → Tn ESA transitions previously shown in Fig. 1 for both OSCs emerge and can be detected on a sub-nanosecond timescale for both RubDBP (Fig. 4b) and 1-CBPEA (Fig. 4c). Hence, we confirm that the charge extraction observed by PL spectroscopy indeed yields triplet excitons in the OSC layers.
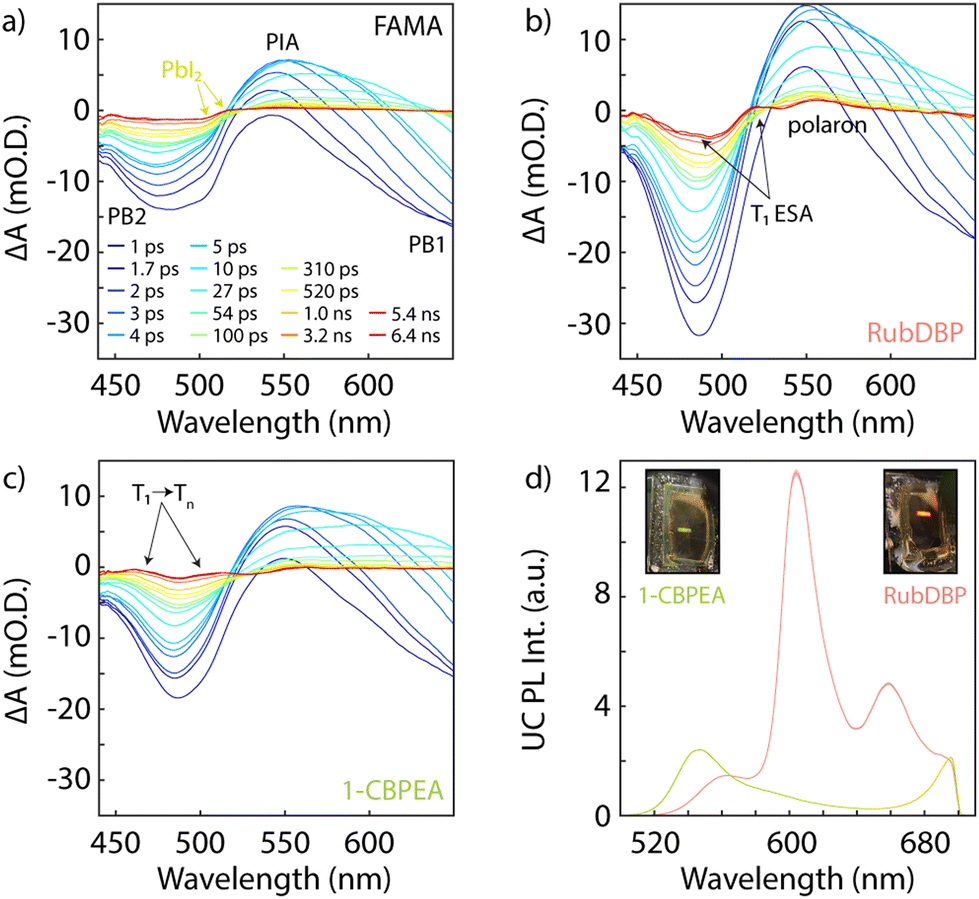 | ||
| Fig. 4 Absorbance spectra at selected delay times for (a) FAMA, (b) RubDBP, and (c) 1-CBPEA in the spectral range of 440–650 nm showcasing the higher energy perovskite photobleach (PB2) at 495 nm and photoinduced absorption (PIA) at 550 nm. The onset of the ground state bleach PB1 can be seen at 650 nm. Residual PbI2 signals at 510 nm and 520 nm are present in the FAMA only spectra due to surface treatment with acetonitrile to increase interfacial charge extraction. In (b), both the Rub T1 and polaron signatures are present at 520 nm and 550 nm signifying successful triplet sensitization. The T1 → Tn transitions at 460 nm and 510 nm for 1-CBPEA are highlighted in (c). All TA measurements were taken under 700 nm pump. (d) UC emission for the bilayer films with photos of the corresponding bilayer films under 780 nm excitation. | ||
To validate that the triplet generation in the OSC layer results in upconverted emission from the OSC singlet state, we probe the emission of the bilayer devices under 780 nm excitation (Fig. 4d). The RubDBP UC device yields the expected strong emission with a peak emission at 605 nm (Fig. 4d, pink).2 The 1-CBPEA UC device yields an upconverted emission spectrum peaking at 550 nm, which increases the apparent anti-Stokes shift achievable by perovskite-sensitized UC by 0.2 eV (Fig. 4d, green) relative to the FAMA/RubDBP UC device. The insets show photographs of the corresponding devices under 780 nm excitation where the strong upconverted emission is visible for both UC devices. A stronger intensity of upconverted emission is found for RubDBP than for 1-CBPEA which can be traced back to two factors: (i) a higher triplet generation in rubrene than in 1-CBPEA as indicated by the stronger quenching of the perovskite PL in the case of RubDBP. (ii) A higher native OSC quantum yield: the observed upconverted emission is directly dependent on the underlying OSC PL quantum yield which dictates the fraction of singlet states generated in the TTA-UC process which undergo radiative recombination.2,21
Lastly, we investigate the properties of the upconverted PL. TTA-UC exhibits a characteristic power dependence: at low incident powers, the upconverted PL intensity increases quadratically. In this regime, the triplets decay primarily through other decay pathways. However, above the threshold intensity Ith for efficient UC, the upconverted PL intensity increases linearly with incident power.35 Here, TTA is the predominant triplet decay pathway and the upconversion process becomes efficient. Increasing the excitation power further results in a saturation regime, where the UC process becomes less efficient. To include TTA-UC devices into photovoltaics, an Ith significantly lower than the integrated solar spectrum at the wavelength region of interest is required, on the order of Ith < 10 mW cm−2.
Mathematically, the power threshold Ith can be expressed by the following equation as described by Monguzzi et al.,35
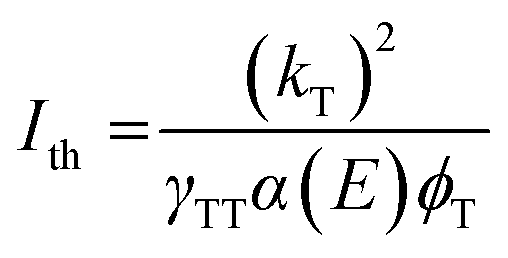
Here, kT is the annihilator triplet decay rate, γTT is the second order rate constant characterizing the TTA process for a given annihilator, α(E) is the absorption coefficient of the perovskite, and ϕT represents the efficiency of the generation of the OSC triplet state.35,56,57
For both annihilators investigated here, we find the characteristic turnover from a slope α = 2β to α = β, where β is the inherent power dependency of the underlying perovskite PL (β = 1.3). We extract a threshold intensity Ith = 18 mW cm−2 for RubDBP and Ith = 195 mW cm−2 for 1-CBPEA. As a note, in this study, the underlying perovskite sensitizer films are kept significantly thinner than the 100 nm optimal thickness we have previously determined1 to allow for sufficiently low optical density for TA studies.9 Upon increasing the perovskite thickness, we have previously shown that the Ith is further reduced due to the increase of α(E).1 Hence, increasing the sensitizer film thickness will push the Ith of the 1-CBPEA UC device towards viable intensity thresholds for device applications.
A back-of-the-envelope calculation allows for further insight into the UC process for the two annihilators and the cause of the discrepancy in the Ith. The optical density α(E) at 780 nm for both devices is similar and therefore, is not a determining factor. We estimate a slightly lower triplet population yield ϕT for 1-CBPEA in comparison to RubDBP based on the magnitude of perovskite PL quenching, which in part accounts for a higher threshold intensity Ith of 1-CBPEA. However, the effects of the annihilator properties on the efficiency threshold cannot be overlooked and kT or γTT must be the underlying cause of the differences in the Ith.
To determine the underlying cause of the difference in the Ith, we investigate the dynamics of the UC PL (Fig. 5b). The UC PL dynamics are generally governed by two separate processes: (i) the initial rise time which gives the characteristic time of singlet generation. This time scale is a convolution of the triplet generation, TTA-UC and S1 decay. (ii) The long-lived decay, which is rate-limited by the triplet decay. In both cases, we find a rapid rise of the UC PL. RubDBP exhibits two rise times τrise,1 = 50 ns and τrise,2 = 400 ns, due to rapid TTA-UC occurring close to the interface and diffusion-mediated TTA-UC far from the interface.6 1-CBPEA on the other hand shows a single characteristic rise of τrise = 28 ns, indicating rapid generation of the emissive singlet state. However, the dynamics of the UC PL shown in Fig. 5b also clearly highlight a major difference between the annihilators. Within the 6 μs time window shown, the UC PL of RubDBP has barely started decaying, while the UC PL of 1-CBPEA has already fully decayed. Estimating the triplet lifetime in the solid-state devices based on a tail fit results in a long-lived decay for RubDBP (τRubDBP > 10 μs) and a much more rapid decay for 1-CBPEA (τ1-CBPEA = 1 μs). Therefore, based on the UC PL dynamics alone (kT,1-CBPEA = 106 s−1, while kT,RubDBP < 105 s−1), we would expect the Ith of RubDBP to be a minimum of two orders of magnitude lower than that of 1-CBPEA.
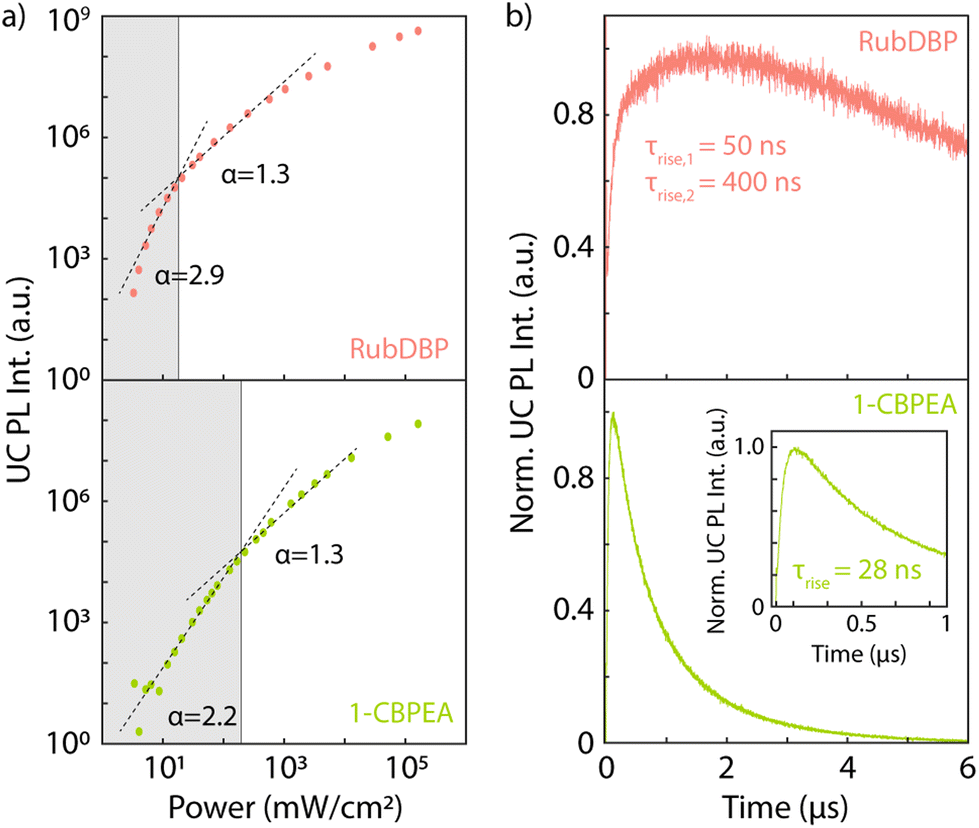 | ||
| Fig. 5 (a) Power dependent upconverted emission for the RubDBP (top, pink) and 1-CBPEA (bottom, green) bilayer devices. The intersect of the dashed lines indicating the different TTA-UC regimes yields the Ith (vertical grey lines). Calculated intensity threshold Ith values for RubDBP and 1-CBPEA films are 18.2 mW cm−2 and 195 mW cm−2, respectively. (b) UC PL dynamics for the RubDBP (top) and 1-CBPEA (bottom) films (repetition rate: 50 kHz, power density: 120 mW cm−2). A magnification of the early time rise for 1-CBPEA is shown in the inset. | ||
To summarize thus far, the trends for kT and ϕT predict a significantly larger difference in the Ith than we observe experimentally. The last key factor in the calculation of the Ith is the second order rate constant γTT, which is unique to each individual annihilator. According to Monguzzi et al.,35γTT is dependent on the triplet exciton diffusion length, the probability of TTA generating a singlet state, as well as the exciton interaction distance. To counterbalance the decrease in the Ith caused by the significantly longer-lived triplets in RubDBP and higher yield of triplets ϕT, 1-CBPEA must have a much larger γTT to account for the relative differences in the Ith. Hence, the triplet diffusion rate or probability of TTA-UC occurring in 1-CBPEA must be higher than in the current state-of-the-art solid-state annihilator rubrene. This further solidifies the promise of this new annihilator 1-CBPEA, despite the currently superior performance of RubDBP.
Circling back to the previous discussion of the triplet energy T1 of 1-CBPEA, due to the observation of both TTA-UC and SF, we acknowledge that the previously reported triplet energy of T1 ≈ 1.2 eV is indeed accurate in the solid-state devices investigated here.40 This amounts to half of the singlet energy S1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E(T1) = 0.5E(S1) – the fundamental requirement for both SF and TTA-UC to occur in the same material. However, this result begs the question whether the TTA-UC process here occurs through the true singlet state S1 of 1-CBPEA (∼2.6 eV in solution) and which then relaxes to the aggregate-induced redshifted singlet state at (2.4 eV) or, whether the lower singlet energy of the aggregated state in solid state enables TTA-UC in this material in the first place. As a result, we emphasize that the translation of solution-phase annihilator properties to the solid state is not straight-forward, as the effect of intermolecular coupling cannot be ignored. In fact, we suggest that the properties of possible annihilators must first be investigated in their aggregated form, prior to making an educated decision on whether they are viable candidates for TTA-UC.
:E(T1) = 0.5E(S1) – the fundamental requirement for both SF and TTA-UC to occur in the same material. However, this result begs the question whether the TTA-UC process here occurs through the true singlet state S1 of 1-CBPEA (∼2.6 eV in solution) and which then relaxes to the aggregate-induced redshifted singlet state at (2.4 eV) or, whether the lower singlet energy of the aggregated state in solid state enables TTA-UC in this material in the first place. As a result, we emphasize that the translation of solution-phase annihilator properties to the solid state is not straight-forward, as the effect of intermolecular coupling cannot be ignored. In fact, we suggest that the properties of possible annihilators must first be investigated in their aggregated form, prior to making an educated decision on whether they are viable candidates for TTA-UC.
Conclusions
We have demonstrated successful perovskite-sensitized solid-state TTA-UC with a novel annihilator 1-CBPEA in the solid state, enabling near-infrared-to-green UC. While we acknowledge that the direct comparison between the RubDBP and 1-CBPEA UC devices does highlight that RubDBP at present still outperforms its replacement 1-CBPEA in terms of efficiency and intensity threshold, the 0.2 eV increase in apparent anti-Stokes shift cannot be ignored. Further device improvements by compositional tuning of the perovskite to better match the promise increased triplet quantum yields to boost the UC efficiency.Additionally, improvements to the OCS film itself may yield increased TTA-UC efficiencies. Wasielewski and co-workers have shown that for the parent molecule BPEA, the fabrication method directly influences the crystal arrangement, and hence the OSC PL quantum yield due to a change in rates of SF.47 Work by Moth-Poulsen and co-workers highlight the role of the molecular conformation on the singlet and triplet energy surfaces,46 which dictate the rates of SF and TTA. Together, these results emphasize that a clear fundamental understanding of the molecular conformation at the nanoscale and the effects of the resulting intermolecular interactions are required to further advance this technology. Since 1-CBPEA is also capable of both SF and TTA-UC, the balance between these two processes must be shifted towards TTA. Hence, tailoring the fabrication conditions to favour TTA over SF will be necessary.
In conclusion, we have demonstrated that triplet generation at the perovskite/OSC interface by charge injection is a universal process and is not inherently limited to the perovskite/rubrene interface. Moving forward, focus should be placed on developing new sensitizer/annihilator pairs to expand the library of viable partners.
Experimental
Device synthesis
Glass substrates were cleaned via sonication for 15 min in each of the respective solutions: 2% Hellmanex, deionized water, and acetone. Following sonication, the substrates were cleaned by UV-ozone (Ossila) treatment for 15 min. Precursor solutions of PbI2 (1.2 M, TCI), MAI (1.2 M, Dyenamo), and FAI (1.2 M, Dyenamo) were prepared in anhydrous DMF:DMSO (9:1, v/v, Sigma-Aldrich) in a 1:1.09 ratio. The precursor solution was then diluted to 0.24 M (∼30 nm) and spin-coated at 1000 rpm for 10 s and 5000 rpm for 30 s. Anhydrous chlorobenzene (Sigma-Aldrich) was used as the antisolvent. The films were then annealed at 120 °C for 30 min prior to solvent treatment with acetonitrile (Sigma-Aldrich).
Rubrene (Rub 99.99%, Sigma-Aldrich), dibenzotetraphenyl-periflanthene (DBP, 98%, Sigma-Aldrich), and 1-chloro-9,10-bis(phenylethynyl)anthracene (1-CBPEA, 99%, Sigma-Aldrich) were used without further purification. A 10 mg mL−1 solution of Rub in anhydrous toluene (Sigma-Aldrich) was prepared and doped with ∼1% DBP, similarly a 10 mg mL−1 solution of 1-CBPEA in anhydrous toluene (Sigma-Aldrich) was prepared. All annihilator solutions were spin-coated onto the perovskite substrates at 6000 rpm for 20 s then annealed at 100 °C for 1 min. Films were encapsulated with a cover slip using a two-part epoxy (Devcon) under an inert nitrogen atmosphere (<0.5 ppm O2) prior to removal from the glovebox.
Steady-state absorption spectroscopy
A Thermo Scientific Evolution 220 Spectrophotometer was used to collect the steady-state absorption spectra.Steady-state emission spectroscopy
Direct excitation emission spectra were collected with a 405 nm continuous wave (CW) laser (LDH-D-C-405, PicoQuant) at a power density of 92 W cm−2 using a 425 nm long-pass filter (Chroma Tech) to remove excess laser scattering. Upconverted emission spectra were taken with a 780 nm CW laser (LDH-D-C-780, PicoQuant) at a power density of 150 W cm−2 using a 700 nm short-pass laser (ThorLabs) to remove laser scatter. An Ocean Insight emission spectrometer (HR2000+ES) was used to collect all spectra.Time-resolved emission spectroscopy
Perovskite PL decays were measured through time-correlated single photon counting (TCSPC) with a 780 nm picosecond pulsed laser (LDH-D-C-780, PicoQuant) with a 125 kHz repetition frequency at a power density of 44.2 mW cm−2. An 800 nm long-pass filter (ThorLabs) and 780 nm notch (ThorLabs) were used to remove laser scatter as well as isolate the perovskite emission. OSC PL decays were taken under 405 nm picosecond pulsed excitation (LDH-D-C-405, PicoQuant) at a repetition frequency of 1 MHz and power density of 4.97 W cm−2. A 425 nm long pass filter (ThorLabs) was used to remove laser scattering. Upconverted PL decays were measured under a 780 nm picosecond pulsed laser (LDH-D-C-780, PicoQuant) at a repetition frequency of 50 kHz at a power density of 120 mW cm−2. A combination of a 650 nm short pass (ThorLabs), 700 nm short pass (ThorLabs), and 780 nm notch (ThorLabs) were used to isolate the upconverted PL emission and remove scattering. A MultiHarp 150 event timer (PicoQuant) connected to a single-photon avalanche photodiode (Micro Photon Devices) was used to collect photon arrival times for all measurements. Laser powers were measured with a silicon power meter (PM100-D, ThorLabs), and spot sizes were determined using the razor blade method (90:10).
PL power dependencies
All power dependent measurements were taken using a 780 nm CW laser (LDH-D-C-780, PicoQuant). Laser powers were measured using a silicon power meter (PM100-D, ThorLabs) and arriving photons were counted for 25 s by a MultiHarp 150 event timer (PicoQuant) with a single-photon photodiode (Micro Photon Devices) connected. For the UC PL power dependencies, both a 780 nm notch filter (ThorLabs) and 600/40 nm (centre/width) band pass filter (ThorLabs) were used to isolate the UC signal and remove laser scattering. Laser powers were attenuated with neutral density filters (ThorLabs).Transient absorption spectroscopy
A HELIOS Fire transient absorption spectrometer (Ultrafast Systems) was used for all transient absorption measurements. Femtosecond laser pulses were generated by an Astrella-V-F-1K amplifier where the Vitara-S Coherent Ti:Sapphire laser used was amplified using a 1 kHz Coherent Revolution-50 pump laser. All resulting laser pulses were 5 mJ with a full width half max of 100 fs at 800 nm. Pump and probe beams were directed through an optical parametric amplifier (OperaA Solo, Coherent) and delay stage, respectively. The visible probe (400 nm to 780 nm) was generated via a sapphire crystal for bilayer and perovskite only measurements while a CaF2 crystal generated the ultraviolet probe for the OSC-only measurements (320 nm to 650 nm). Excess laser scattering was minimized through a dual chopper system, and neutral density filters were used to attenuate pump power. For all measurements, 3 spectra were collected with a 2 seconds integration time for each delay position with an exponential point collection method starting at 0.01 ps, resulting in a total of 150 points. Perovskite only measurements were taken under 700 nm pump at 4.7 mW, and bilayer films were taken under 700 nm pump with a power of 6.0 mW. A 400 nm pump was used for the OSC measurements at a power of 0.6 mW for both. The TA maps were processed through the Surface Xplorer software package from Ultrafast Systems in addition to MATLAB.Author contributions
L. N. conceptualized the study. C. M. S. and L. N. fabricated the samples. C. M. S. performed the experiments. C. M. S. and L. N. analysed the data. L. N. wrote the manuscript with contributions by C. M. S.Conflicts of interest
FSU has filed a provisional application for a US patent based on this technology that names L. N. as an inventor.Acknowledgements
The authors acknowledge funding by Florida State University. This project used resources provided by the Materials Characterization Laboratory (FSU075000MAC) at the FSU Department of Chemistry and Biochemistry. Ultrafast transient absorption measurements were performed on a spectrometer supported by the National Science Foundation under Grant No. CHE-1919633.References
- S. Wieghold, A. S. Bieber, Z. A. VanOrman, L. Daley, M. Leger, J.-P. Correa-Baena and L. Nienhaus, Matter, 2019, 1, 705–719 CrossRef CAS.
- L. Nienhaus, J.-P. Correa-Baena, S. Wieghold, M. Einzinger, T.-A. Lin, K. E. Shulenberger, N. D. Klein, M. Wu, V. Bulović, T. Buonassisi, M. A. Baldo and M. G. Bawendi, ACS Energy Lett., 2019, 888–895 CrossRef CAS.
- S. Wieghold and L. Nienhaus, J. Phys. Chem. Lett., 2020, 11, 601–607 CrossRef CAS PubMed.
- L. Wang, J. J. Yoo, T.-A. Lin, C. F. Perkinson, Y. Lu, M. A. Baldo and M. G. Bawendi, Adv. Mater., 2021, 33, 2100854 CrossRef CAS PubMed.
- K. Prashanthan, B. Naydenov, K. Lips, E. Unger and R. W. MacQueen, J. Chem. Phys., 2020, 153, 164711 CrossRef CAS.
- S. Wieghold, A. S. Bieber, Z. A. VanOrman and L. Nienhaus, J. Phys. Chem. Lett., 2019, 3806–3811 CrossRef CAS.
- S. Wieghold, A. S. Bieber, J. Lackner, K. Nienhaus, G. U. Nienhaus and L. Nienhaus, ChemPhotoChem, 2020, 4, 704–712 CAS.
- A. S. Bieber, Z. A. VanOrman, H. K. Drozdick, R. Weiss, S. Wieghold and L. Nienhaus, J. Chem. Phys., 2021, 155, 234706 CrossRef CAS PubMed.
- C. R. Conti, A. S. Bieber, Z. A. VanOrman, G. Moller, S. Wieghold, R. D. Schaller, G. F. Strouse and L. Nienhaus, ACS Energy Lett., 2022, 7, 617–623 CrossRef CAS.
- A. S. Bieber, Z. A. VanOrman, S. Wieghold and L. Nienhaus, J. Chem. Phys., 2020, 153, 084703 CrossRef CAS PubMed.
- Z. A. VanOrman, J. Lackner, S. Wieghold, K. Nienhaus, G. U. Nienhaus and L. Nienhaus, Appl. Phys. Lett., 2021, 118, 203903 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- W. Tress, N. Marinova, O. Inganäs, M. K. Nazeeruddin, S. M. Zakeeruddin and M. Graetzel, Adv. Energy Mater., 2015, 5, 1400812 CrossRef.
- J. Alves, J. Feng, L. Nienhaus and T. W. Schmidt, J. Mater. Chem. C, 2022, 10, 7783–7798 RSC.
- Z. A. VanOrman and L. Nienhaus, ACS Energy Lett., 2021, 6, 3686–3694 CrossRef CAS.
- S. Wieghold, Z. A. VanOrman and L. Nienhaus, Adv. Opt. Mater., 2021, 9, 2001470 CrossRef CAS.
- M. Mahboub, Z. Huang and M. L. Tang, Nano Lett., 2016, 16, 7169–7175 CrossRef CAS PubMed.
- L. Nienhaus, M. Wu, N. Geva, J. J. Shepherd, M. W. B. Wilson, V. Bulović, T. Van Voorhis, M. A. Baldo and M. G. Bawendi, ACS Nano, 2017, 11, 7848–7857 CrossRef CAS PubMed.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS.
- M. Wu, J. Jean, V. Bulović and M. A. Baldo, Appl. Phys. Lett., 2017, 110, 211101 CrossRef.
- N. Geva, L. Nienhaus, M. Wu, V. Bulović, M. A. Baldo, T. Van Voorhis and M. G. Bawendi, J. Phys. Chem. Lett., 2019, 10, 3147–3152 CrossRef CAS.
- D. G. Bossanyi, Y. Sasaki, S. Wang, D. Chekulaev, N. Kimizuka, N. Yanai and J. Clark, J. Mater. Chem. C, 2022, 10, 4684–4696 RSC.
- L. Ma, K. Zhang, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Phys. Chem. Chem. Phys., 2012, 14, 8307–8312 RSC.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171–1174 CrossRef CAS.
- K. Wang, S. Huang, P. Ding, Z. Liang, S. Chen, L. Li, C. Ye and X. Wang, RSC Adv., 2021, 11, 17755–17759 RSC.
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, ACS Photonics, 2014, 1, 382–388 CrossRef CAS.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef CAS.
- M. Häring, R. Pérez-Ruiz, A. J. von Wangelin and D. D. Díaz, Chem. Commun., 2015, 51, 16848–16851 RSC.
- M. Y. Vorona, N. J. Yutronkie, O. A. Melville, A. J. Daszczynski, J. S. Ovens, J. L. Brusso and B. H. Lessard, Materials, 2020, 13, 1961 CrossRef CAS PubMed.
- Y.-Y. Lyu, J. Kwak, O. Kwon, S.-H. Lee, D. Kim, C. Lee and K. Char, Adv. Mater., 2008, 20, 2720–2729 CrossRef CAS PubMed.
- Z. A. VanOrman, A. S. Bieber, S. Wieghold and L. Nienhaus, Chem. Mater., 2020, 32, 4734–4742 CrossRef CAS.
- C. J. Imperiale, P. B. Green, M. Hasham and M. W. B. Wilson, Chem. Sci., 2021, 12, 14111–14120 RSC.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B, 2008, 78, 195112 CrossRef.
- T. Miyashita, P. Jaimes, T. Lian, M. L. Tang and Z. Xu, J. Phys. Chem. Lett., 2022, 13, 3002–3007 CrossRef CAS PubMed.
- E. M. Rigsby, T. Miyashita, P. Jaimes, D. A. Fishman and M. L. Tang, J. Chem. Phys., 2020, 153, 114702 CrossRef CAS PubMed.
- Z. Huang and M. L. Tang, J. Phys. Chem. Lett., 2018, 9, 6198–6206 CrossRef CAS PubMed.
- A. Nandi, B. Manna and R. Ghosh, Phys. Chem. Chem. Phys., 2019, 21, 11193–11202 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS.
- S. Guo, L. Xu, K. Xu, J. Zhao, B. Küçüköz, A. Karatay, H. G. Yaglioglu, M. Hayvali and A. Elmali, Chem. Sci., 2015, 6, 3724–3737 RSC.
- F. Zhong and J. Zhao, Dyes Pigm., 2017, 136, 909–918 CrossRef CAS.
- S. Guo, J. Sun, L. Ma, W. You, P. Yang and J. Zhao, Dyes Pigm., 2013, 96, 449–458 CrossRef CAS.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Inorg. Chem., 2009, 48, 2541–2548 CrossRef CAS PubMed.
- V. Gray, A. Dreos, P. Erhart, B. Albinsson, K. Moth-Poulsen and M. Abrahamsson, Phys. Chem. Chem. Phys., 2017, 19, 10931–10939 RSC.
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- B. Manna, A. Nandi and R. Ghosh, J. Phys. Chem. C, 2018, 122, 21047–21055 CrossRef CAS.
- Y. J. Bae, J. A. Christensen, G. Kang, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Chem. Phys., 2019, 151, 044501 CrossRef PubMed.
- G. Ji, G. Zheng, B. Zhao, F. Song, X. Zhang, K. Shen, Y. Yang, Y. Xiong, X. Gao, L. Cao and D.-C. Qi, Phys. Chem. Chem. Phys., 2017, 19, 6546–6553 RSC.
- B. vander Zee, S. Paulus, R.-Q. Png, P. K. H. Ho, L.-L. Chua, G.-J. A. H. Wetzelaer and P. W. M. Blom, Adv. Electron. Mater., 2020, 6, 2000367 CrossRef CAS.
- J. F. Tannaci, M. Noji, J. McBee and T. D. Tilley, J. Org. Chem., 2007, 72, 5567–5573 CrossRef CAS PubMed.
- S. Liu, L. Zheng, M. Chen, Y. Sun, P. Wang, S. Li, H. Wu, X. Zhang and W. Hu, J. Mater. Chem. C, 2021, 9, 4217–4222 RSC.
- M. Aydemir, G. Haykır, A. Battal, V. Jankus, S. K. Sugunan, F. B. Dias, H. Al-Attar, F. Türksoy, M. Tavaslı and A. P. Monkman, Org. Electron., 2016, 30, 149–157 CrossRef CAS.
- C. M. Sullivan, A. S. Bieber, H. K. Drozdick, G. Moller, J. E. Kuszynski, Z. A. VanOrman, S. Wieghold, G. F. Strouse and L. Nienhaus, Adv. Opt. Mater., 2022, n/a, 2201921 CrossRef.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- A. Ronchi, P. Brazzo, M. Sassi, L. Beverina, J. Pedrini, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2019, 21, 12353–12359 RSC.
| This journal is © The Royal Society of Chemistry 2022 |