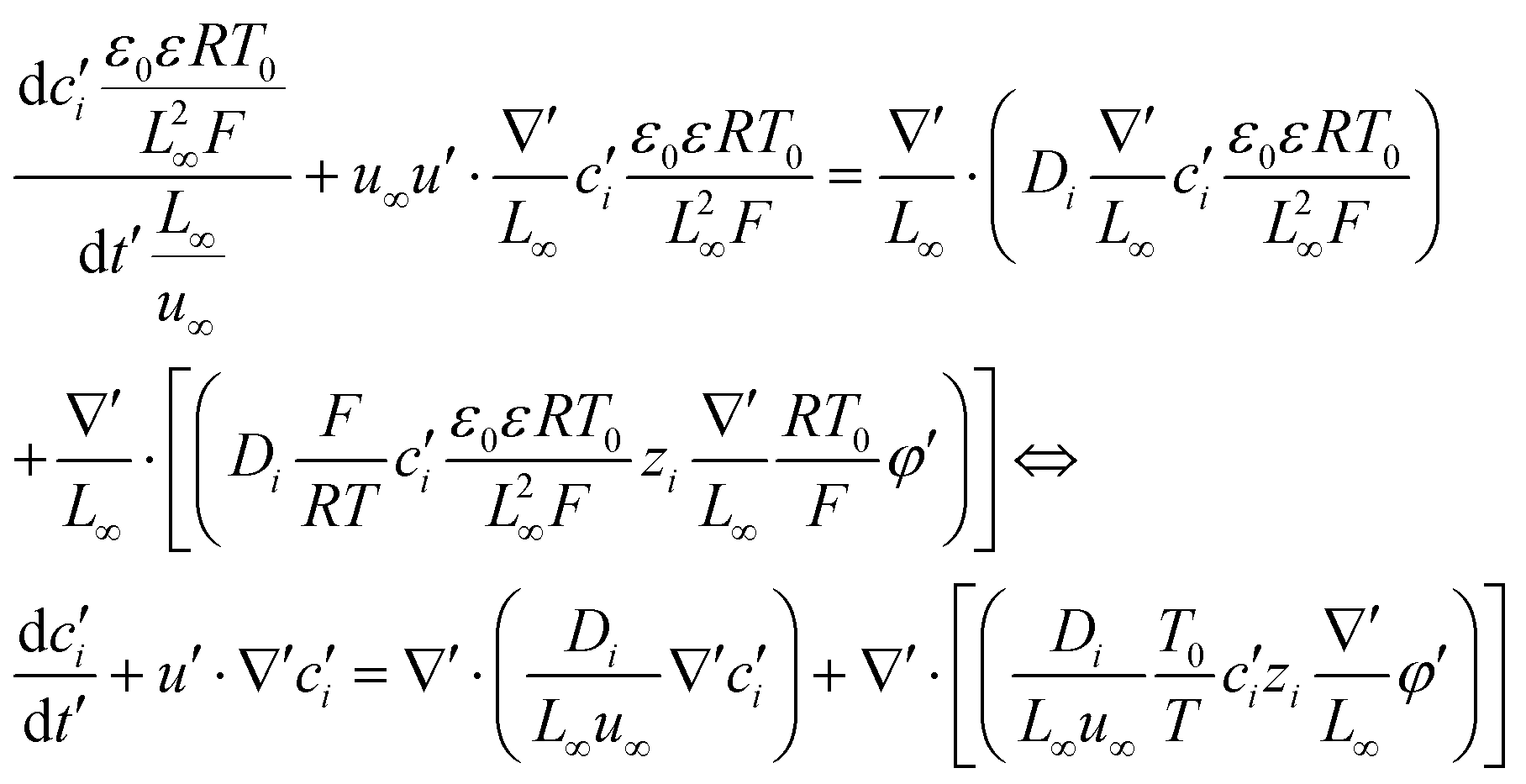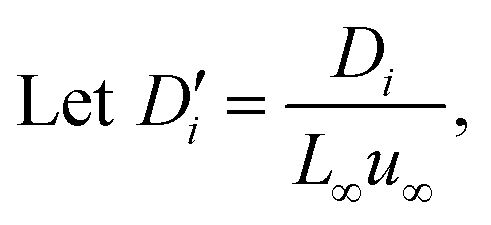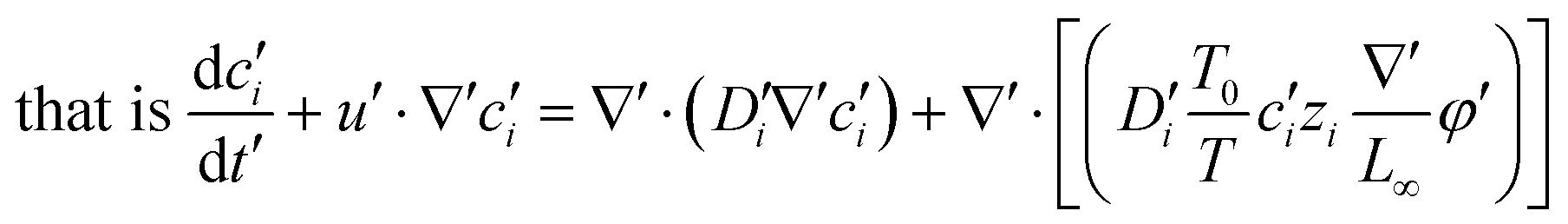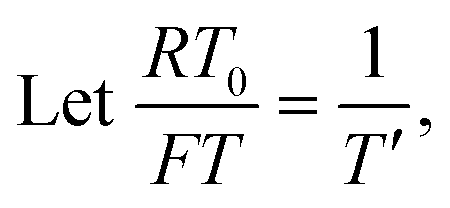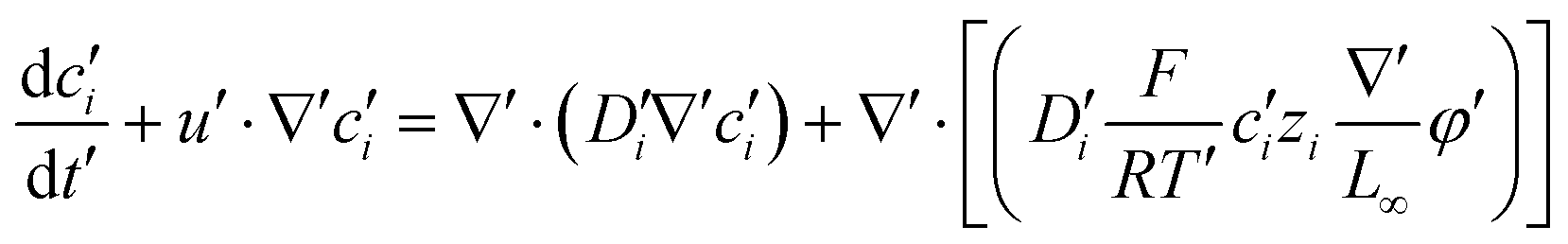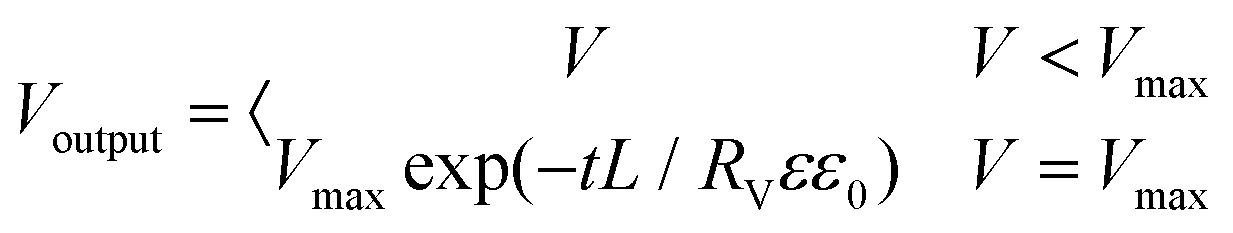DOI:
10.1039/D2NH00323F
(Communication)
Nanoscale Horiz., 2022,
7, 1523-1532
Magnetic-field-controlled counterion migration within polyionic liquid micropores enables nano-energy harvest†
Received
9th July 2022
, Accepted 13th October 2022
First published on 14th October 2022
Abstract
Efficient separation of positive and negative charges is essential for developing high-performance nanogenerators. In this article, we describe a method that was not previously demonstrated to separate charges which enables us to fabricate a magnetic energy harvesting device. The magnetic field induces the migration of the mobile magnetic counterions (Dy(NO3)4−) which establishes anion gradients within a layer of polyionic liquid micropores (PLM). The PLM is covalently cross-linked on which the positive charges are fixed on the matrix, that is, immobile. In a device with a structure of Au/dielectric//mag-PLM//dielectric/Au, the charge gradient is subsequently transformed into the output voltage through electrostatic induction. Removing the magnetic field leads to the backflow of magnetic anions which produces a voltage with a similar magnitude but reversed polarity. The parameters in fabricating the magnetic PLM such as photoinitiator concentration, UV irradiation time, water treatment time, and temperature are found to dramatically influence the size of micropores and the effective concentration of magnetic anions. Under optimized conditions, an output voltage with an amplitude of approximately 4 V is finally achieved. We expect this new method could find practical applications in further improving the output performance.
New concepts
Developing new techniques/methods and proposing new mechanisms are always important but challenging for high-performance nanogenerators. Herein, we describe a method that was not previously demonstrated to separate the charges which enables us to fabricate a magnetic energy harvesting device. Inspired by the electric-field-driven migration of ionic charges, the magnetic field is firstly used to separate charges by designing a new type of magnetic polyionic liquid micropore. Under a magnetic field, the migration of the mobile magnetic counterions establishes charge gradients within the PLM layer and then transforms into the output voltage through electrostatic induction. In addition, this new concept—separating charges with a magnetic field—is also applicable for switching on an electrolyte gate transistor.
|
Introduction
Nanogenerators, which can produce ∼milliwatts of electrical energy, have attracted considerable attention because of the increasing energy demand from small electronic devices such as smart homes1 and human–machine interfaces,2,3 and these require self-powering functions.4–6 Understanding the underlying mechanism of electricity generation is the key aspect of fabricating high-performance nanogenerators. In principle, it is closely related to charge separation which establishes effective charge gradients, unipolar and/or ambipolar within the ‘active’ materials. There are many techniques/methods that are developed to create such charge gradients.7–9 In one example, positive and negative charges are induced/separated by piezoelectric effects,10,11 triboelectric effects,12–14 and/or bringing two materials with different electron affinities into contact. The inductive potential difference has then driven the flow of electrons between two metal electrodes through an electrostatic induction process. Second, it could also be created by the synthesis of materials with intrinsic functional group gradient distributions.15,16 For example, with moisture-electric annealing and/or electrochemical polarization techniques, oxygen-containing group (e.g., OH, COOH) gradients within carbon materials are produced.17–19 Upon exposure to moisture, the diffusion of disassociated free protons (released from the oxygen-containing group) induces potential and free electron movement in the external circuit. In addition, employing an asymmetric external field could be another effective method.20,21 For example, the temperature difference on the two sides of a polyacid film could drive the migration of mobile counterions and output a thermal voltage referring to an ionic Seebeck effect.22 This concept is also applicable to an environment without an external asymmetric field. Due to the unique porous structure, humidity gradient is automatically generated within a film of protein nanowire covered by a small-area top electrode.23 This humidity gradient is then transformed into the gradient distribution of protons, leading to continuous electricity output under ambient conditions.
Currently, the research in this emerging field is mainly focused on improving the device performances and/or expanding the application of nanogenerators.24–26 Developing new techniques/methods and proposing new mechanisms are always challenging. Inspired by the electric-field-driven migration of ionic charges, the magnetic field could also be used to separate charges if these charges are magnetically active. Therefore, in this article, we design a new type of magnetic polyionic liquid micropore (mag-PLM) on which the fixed positive charges on the framework are balanced by the mobile magnetic counterions. Under a magnetic field, an anion gradient is created which enables us to fabricate a magnetic energy harvesting device.
Results and discussion
Design and fabrication of the magnetic energy harvesting device
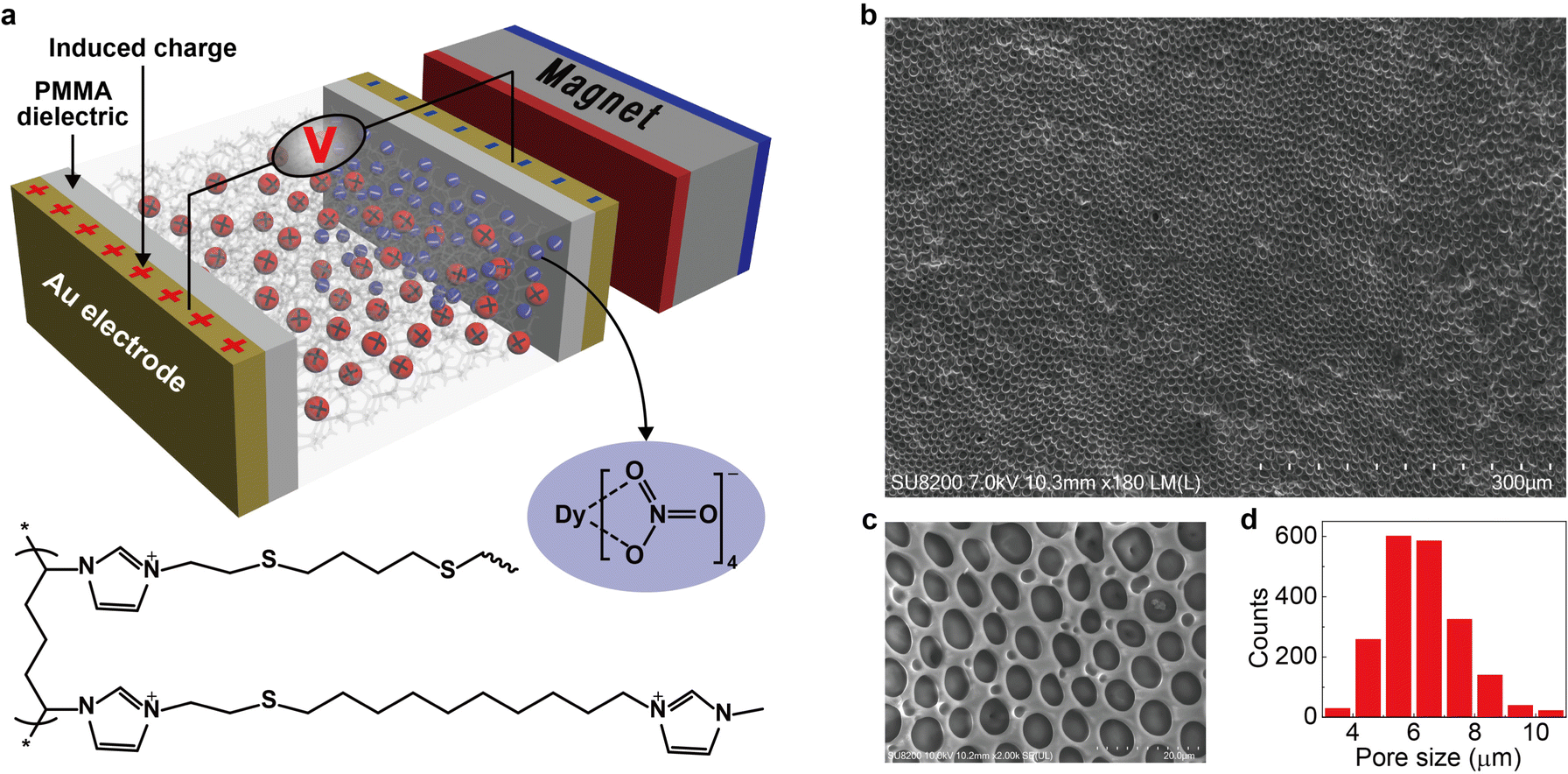
The key idea in designing the magnetic energy harvesting device is to separate positive and negative charges efficiently under an external magnetic field. This can be achieved by combining the porous framework structure and the magnetic ionic liquid. The non-magnetic positive charges on the porous matrix are immobile (confined), whereas the magnetic negative counterions (free) could respond to the magnetic field and establish an anion gradient within the magnetically responsive layer. In addition, the porous framework should be highly-uniform with sufficient micropores (as a free channel for ions) to ensure efficient movement of magnetic anions. Based on this proposal, we design a new type of magnetic polyionic liquid micropore in which the porous structure is created by covalently cross-linking a polyionic liquid material (Fig. 1a, bottom half). The structural characterization indicates highly-uniform micropores distributed on the whole layer (Fig. 1a–d). A paramagnetic negative counterion (Dy(NO3)4− is selected because of its sufficient magnetic susceptibility (χg ∼ 10−6 emu g−1) and ease of synthesis.
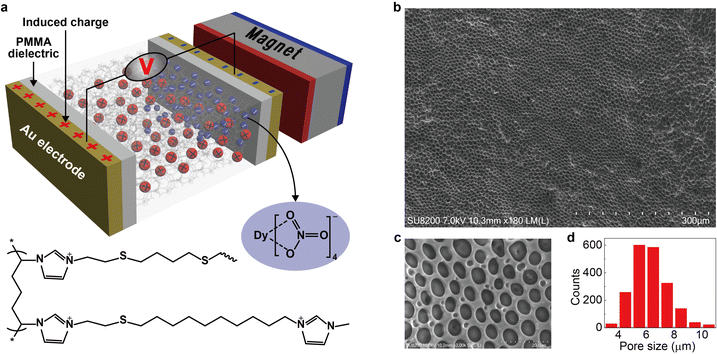 |
| | Fig. 1 Scheme of the device and typical polyionic liquid micropores. (a) The scheme of a magnetic energy harvesting device consisting of two Au electrodes (50 nm), two PMMA dielectric layers (550 nm), one magnet, and a layer of magnetically responsive polyionic liquid micropores (100 μm). The bottom half is the molecular structure of the polyionic liquid in which the negative counterions (Dy(NO3)4−, blue ball) are paramagnetic and could migrate under a magnetic field. The positive charge (red ball) is immobile. (b) Typical magnetically responsive polyionic liquid micropores prepared by using a 5 mg mL−1 photoinitiator, 3 h UV irradiation, and 72 h water treatment at room temperature. (c) Magnified SEM image and (d) size distribution of micropores in (b). The statistic histogram is based on 2000 micropores. | |
With mag-PLM in hand, the magnetic energy harvesting device with the Au/dielectric//mag-PLM//dielectric/Au structure (Fig. 1a, upper half) is then fabricated, in which the dielectric layer is 550 nm thick polymethyl methacrylate (PMMA). By applying a magnetic field, the (Dy(NO3)4− anions migrate to the mag-PLM/PMMA interface which induces the dielectric layer to carry opposite charges. Subsequently, the electron flow occurs in the external circuit to balance the dielectric potential, leading to the electrical energy output. When the magnetic field is removed, the charge density within the dielectric layer has decreased due to the backflow of (Dy(NO3)4− anions. Consequently, the electron flow direction in the external circuit is also reversed, generating voltage with a similar magnitude but reversed polarity.
Synthesis of magnetically responsive polyionic liquid micropores
To synthesize magnetically responsive polyionic liquid micropores, the non-magnetic PLM is firstly prepared by using a covalent cross-linking method.27 Briefly, the acetone solution of poly(3-allylmethyl-1-vinylimidazole) bis(trifluoromethanesulfonyl)imide and the cross-linker 1,4-dithioerythritol is drop-casted on a glass substrate. The polymer film is then placed in an aqueous solution containing a photoinitiator (UV2959) to finish the cross-linking reaction under UV irradiation. The polyionic liquid micropores are finally produced by immersing the cross-linked film in water for tens of hours (see the details in the Experimental section).
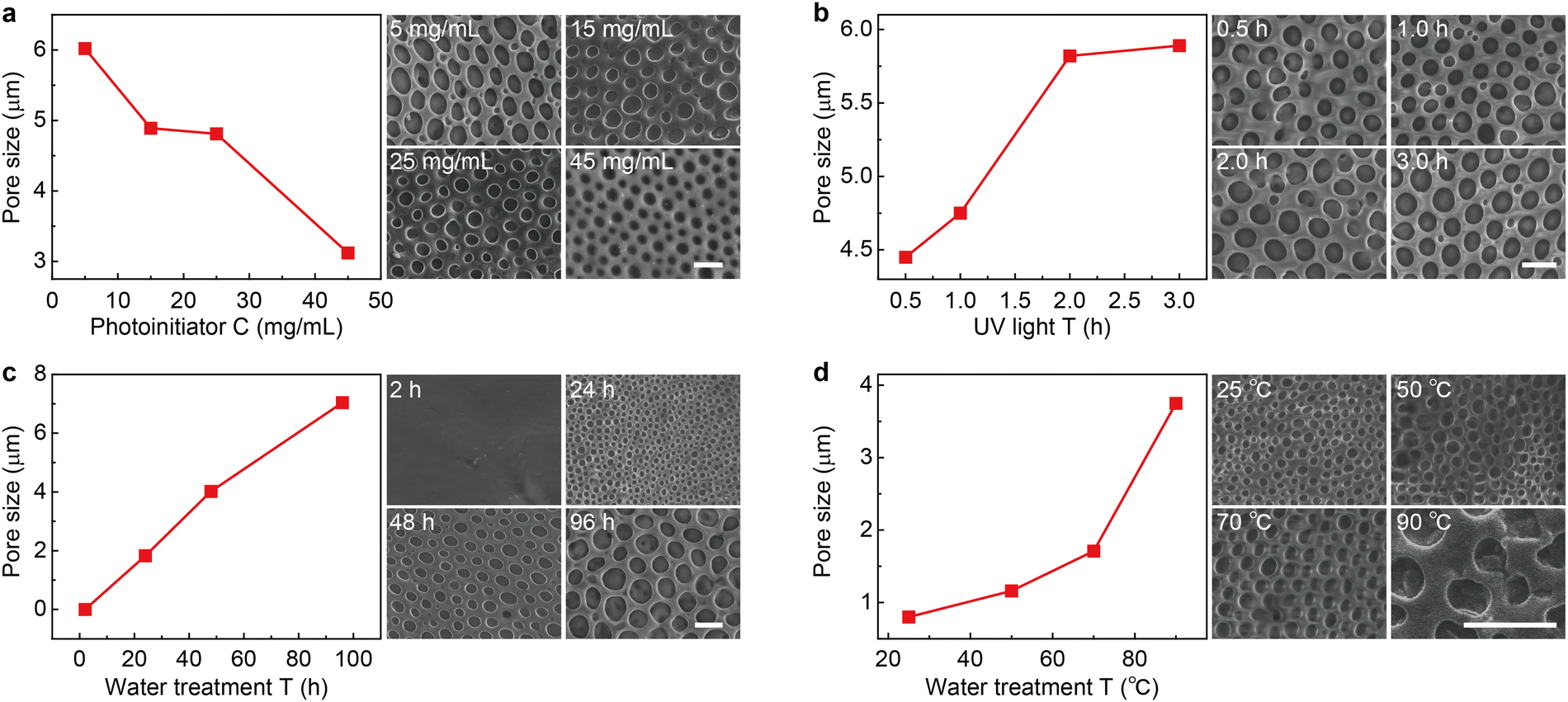
Since the counterion movement within the magnetically responsive layer is significantly influenced by the structure of micropores (e.g., size and uniformity), the synthesis conditions of PLM such as photoinitiator concentration, UV irradiation time, water treatment time, and temperature are comprehensively studied. With the increase of photoinitiator concentration from 5 mg mL−1 to 45 mg mL−1, the average micropore size gradually decreases from 5.98 μm to 3.42 μm (Fig. 2a and Fig. S1, ESI†). However, increasing the UV irradiation time leads to an increase of the micropore size from 4.45 μm (0.5 h) to 5.89 μm (3 h) (Fig. 2b and Fig. S2, ESI†). Both the photoinitiator concentration and UV irradiation time determine the degree of cross-linking of PLM. Lowering the dosage of the photoinitiator and extending the reaction time is useful for producing micropores with a larger size.
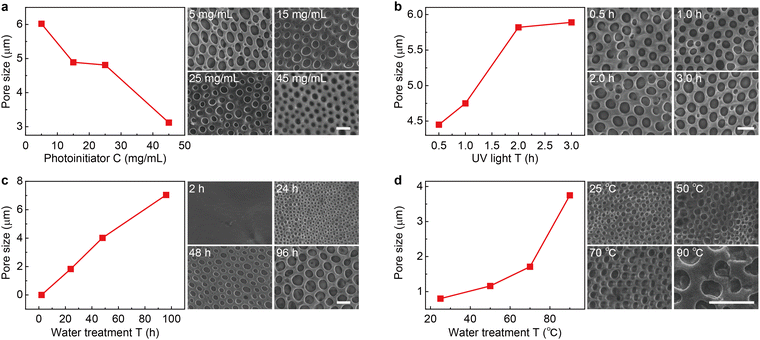 |
| | Fig. 2 Fabrication of the polyionic liquid micropores. (a) Micropore size as the function of photoinitiator concentration (left). The typical SEM image is given on the right. Other synthesis conditions are as follows: 3 h UV irradiation, 72 h water treatment at room temperature. Similar micropore size evolution with the increase of UV irradiation time (b), water treatment time (c), and water treatment temperature (d). The other fabrication conditions are 5 mg mL−1 photoinitiator, 72 h water treatment at room temperature for (b), 5 mg mL−1 photoinitiator, 3 h UV irradiation at room temperature for (c), and 5 mg mL−1 photoinitiator, 3 h UV irradiation, 12 h water treatment for (d). The size statistics for all conditions are based on 200 micropores (see the statistic histograms in Fig. S1 to S4, ESI†). The scale bars are 5 μm. | |
Interestingly, the water treatment time is found to dramatically change the PLM size. With a short treatment time (e.g., 2 h), no apparent micropore is produced. With the increase of treatment time from 24 h to 96 h, the size of micropores dramatically increased from 1.83 μm to 7.04 μm (Fig. 2c and Fig. S3, ESI†). In fact, this time determines the swelling process of a poor solvent (H2O) within hydrophobic polymers.28 Therefore, increasing this time is beneficial for expanding the porous structure. In addition, long-term interaction between water molecules and polyionic liquid chains is also favorable for building through-hole mesoporous structures (Fig. S5, ESI†). Another parameter in water treatment is temperature. Apparently, elevating temperatures could accelerate the diffusion of a poor solvent within PLM and produce larger size micropores (Fig. 2d and Fig. S4, ESI†). However, it also leads to less control and poor uniformity.
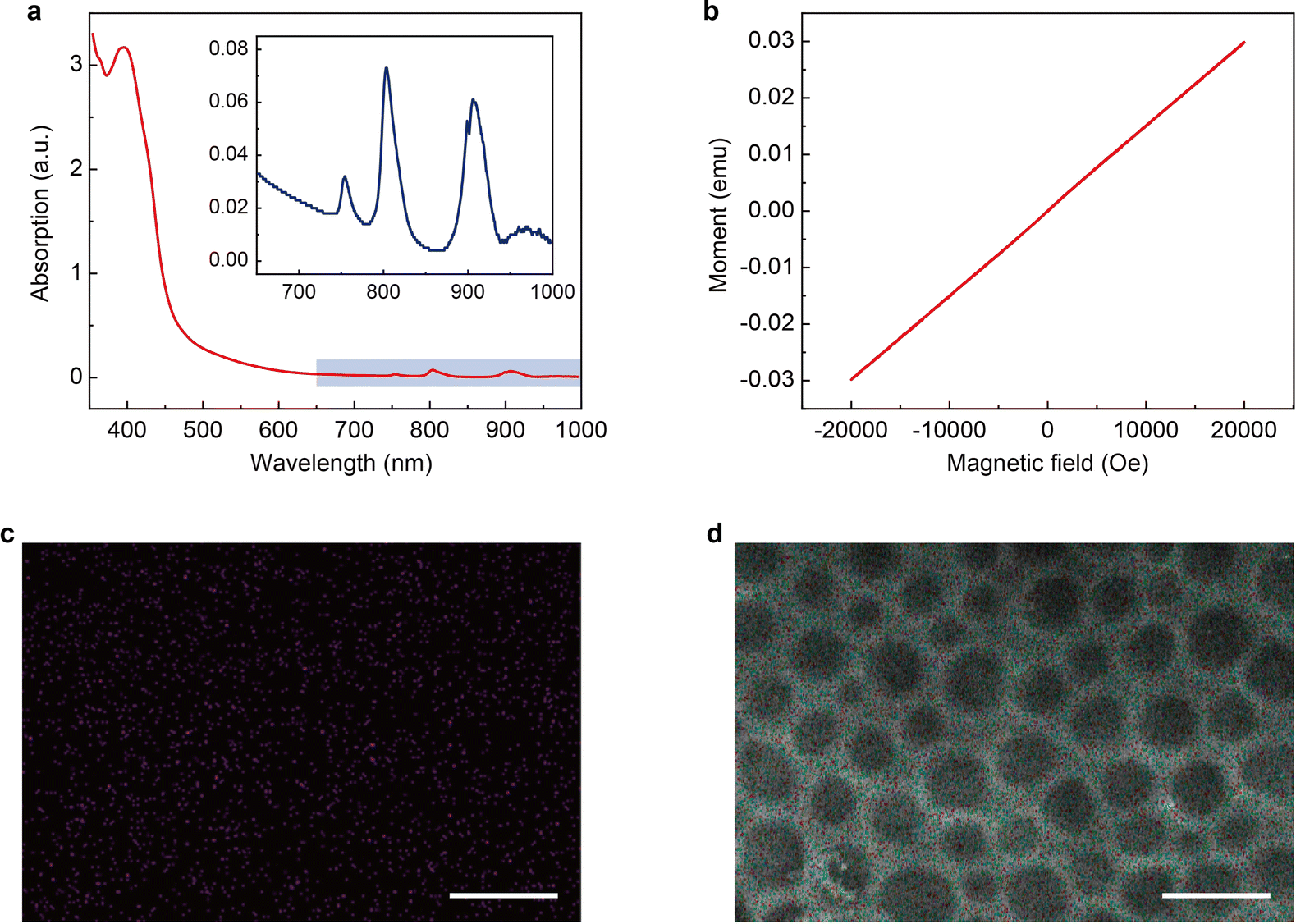
The magnetic PLM is then synthesized through a thiol-ene click reaction29–31 between the remaining C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds on PLM and the thiol-functionalized magnetic ionic liquid (1-10-mercaptodecyl-3-methylimidazolium dysprosium tetranitrate, [HS-C10mim] [Dy(NO3)4]). The details of the synthesis of [HS-C10mim] [Dy(NO3)4] can be found in the Experimental section. The UV-vis spectrum of the magnetic ionic liquid (MIL) shows an intense peak at 395 nm which corresponds to the absorption of imidazolium cations and several weak absorption peaks at 755 nm, 804 nm, and 907 nm which could be ascribed to the dysprosium ions (Fig. 3a).32 The M–H measurement performed at room temperature indicates that the [HS-C10mim] [Dy(NO3)4] MIL is paramagnetic. The magnetic susceptibility is approximately 10−6 emu g−1 (Fig. 3b) which is close to the value observed for other magnetic anions.33,34 The PLM is subsequently soaked into the [HS-C10mim] [Dy(NO3)4] MIL solution to complete the addition reaction under UV irradiation. The residual MIL is removed and the magnetic PLM is finally produced. Energy dispersive spectroscopy (EDS) confirms the presence of dysprosium elements (Fig. 3c). In addition, the porous structure of PLM is maintained after the addition reaction (Fig. 3d). The validity of this method is further verified by using two other MILs, 1-10-mercaptodecyl-3-methylimidazolium ferric chloride bromide (Fig. S6 and S7, ESI†) and 1-10-mercaptodecyl-3-methylimidazolium dysprosium chloride bromide (Fig. S8 and S9, ESI†).
C bonds on PLM and the thiol-functionalized magnetic ionic liquid (1-10-mercaptodecyl-3-methylimidazolium dysprosium tetranitrate, [HS-C10mim] [Dy(NO3)4]). The details of the synthesis of [HS-C10mim] [Dy(NO3)4] can be found in the Experimental section. The UV-vis spectrum of the magnetic ionic liquid (MIL) shows an intense peak at 395 nm which corresponds to the absorption of imidazolium cations and several weak absorption peaks at 755 nm, 804 nm, and 907 nm which could be ascribed to the dysprosium ions (Fig. 3a).32 The M–H measurement performed at room temperature indicates that the [HS-C10mim] [Dy(NO3)4] MIL is paramagnetic. The magnetic susceptibility is approximately 10−6 emu g−1 (Fig. 3b) which is close to the value observed for other magnetic anions.33,34 The PLM is subsequently soaked into the [HS-C10mim] [Dy(NO3)4] MIL solution to complete the addition reaction under UV irradiation. The residual MIL is removed and the magnetic PLM is finally produced. Energy dispersive spectroscopy (EDS) confirms the presence of dysprosium elements (Fig. 3c). In addition, the porous structure of PLM is maintained after the addition reaction (Fig. 3d). The validity of this method is further verified by using two other MILs, 1-10-mercaptodecyl-3-methylimidazolium ferric chloride bromide (Fig. S6 and S7, ESI†) and 1-10-mercaptodecyl-3-methylimidazolium dysprosium chloride bromide (Fig. S8 and S9, ESI†).
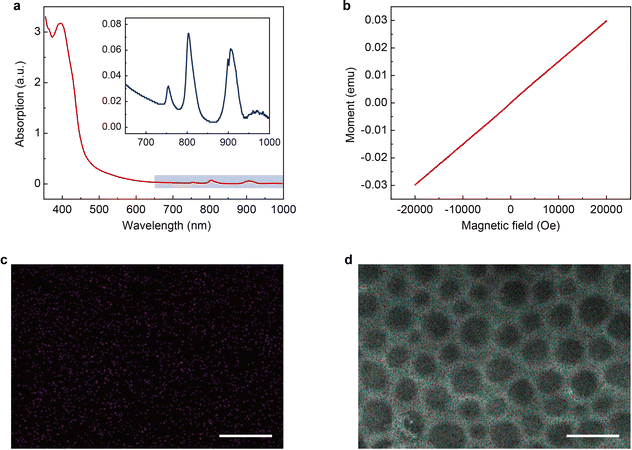 |
| | Fig. 3 Characterization of magnetic polyionic liquid micropores and the magnetic ionic liquid. (a) UV-Vis spectrum of the magnetic ionic liquid [HS-C10mim] [Dy(NO3)4]. The inset shows the magnified image of the 650–1000 nm region. (b) Magnetization curve of [HS-C10mim] [Dy(NO3)4]. The measurement was performed at room temperature. (c) Dy elemental distribution and (d) EDS results of magnetic polyionic liquid micropores. The scale bars in (c) and (d) are 5 μm. | |
Device output characteristics and performance optimization
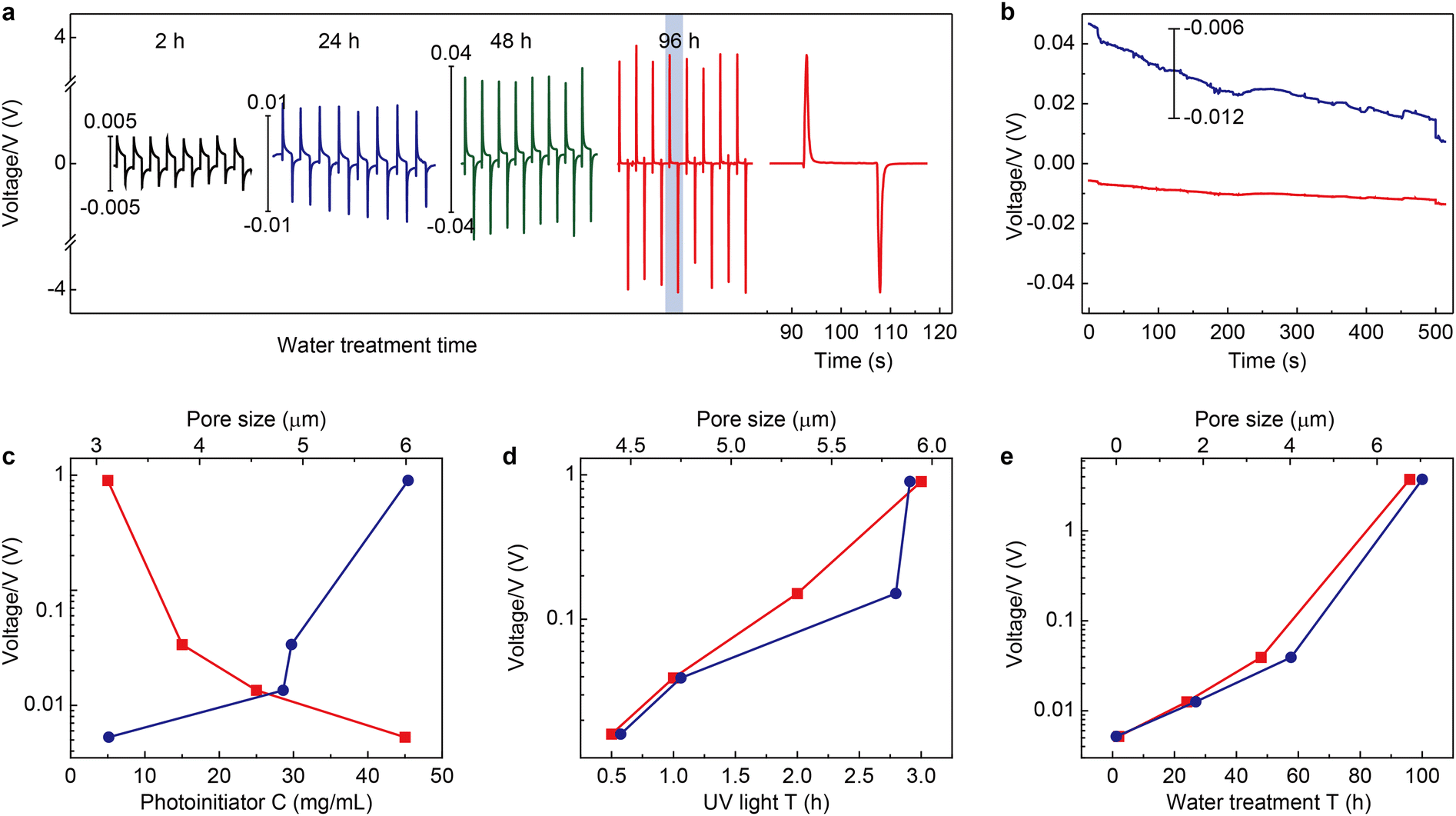
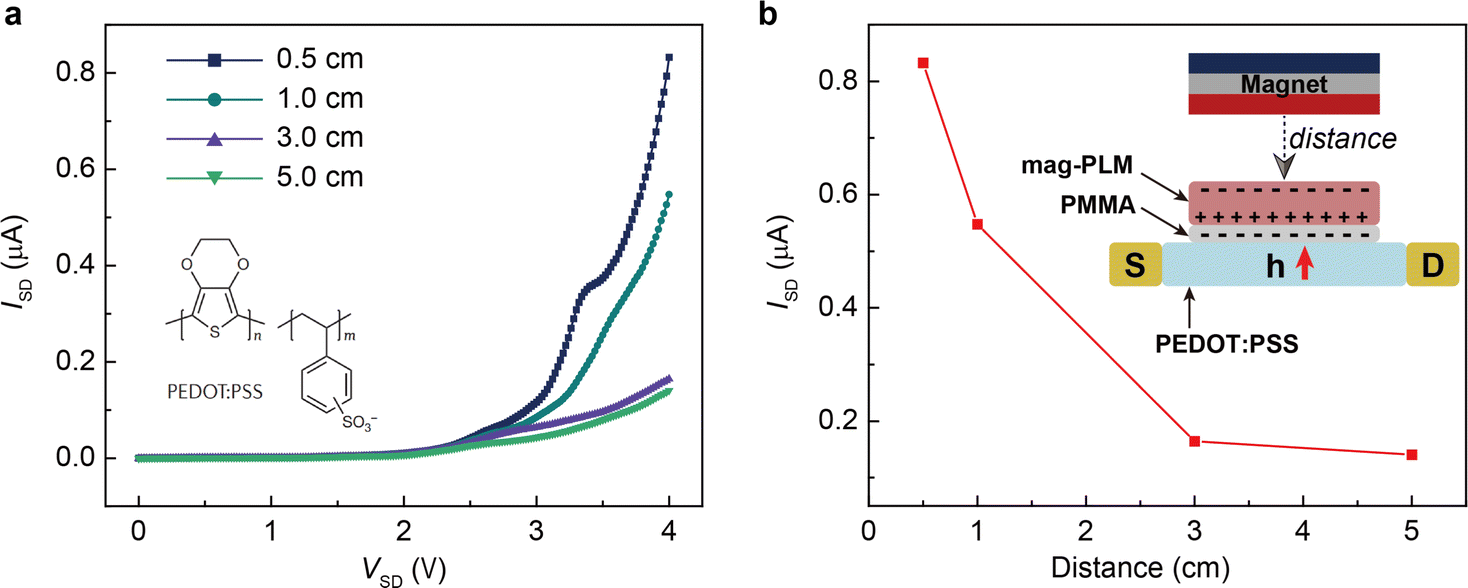
The electrical output of Au/dielectric//mag-PLM//dielectric/Au devices is measured by applying an external magnetic field. Fig. 4a shows the open circuit voltages of the devices using mag-PLMs prepared under different water treatment times. Alternating voltage outputs are recorded with the application and removal of the magnetic field, indicating the reversed movement of electrons in the external circuit. However, this switchable performance is not observed when the mag-PLM is replaced by a non-magnetic PLM ([HS-C10mim] NO3) (Fig. 4b). Remarkably, the magnitude of output voltage has increased with the water treatment time of PLM. The device with 2 h water-treated mag-PLM outputs only approximately 5 mV. This value has exponentially increased to around 4 V when using the 96 h water-treated mag-PLM (Fig. 4a and e).
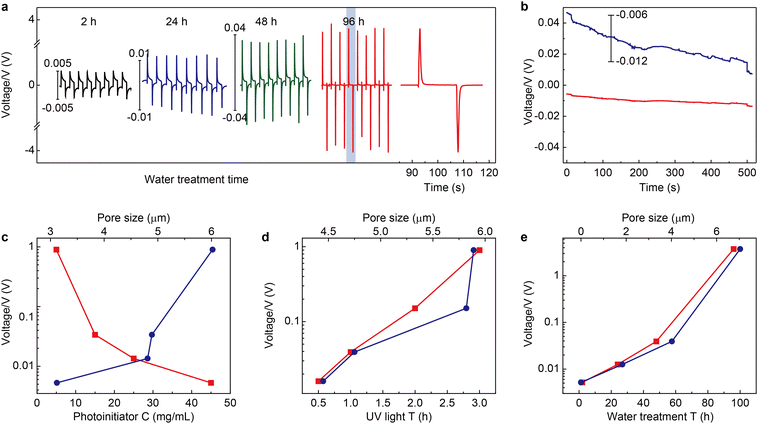 |
| | Fig. 4 Output characteristics of the magnetic energy harvesting device. (a) Time-dependent output voltages of the devices fabricated by using magnetic polyionic liquid micropores synthesized with different water treatment times. The voltage scales for 2 h (black curve), 24 h (blue curve), and 48 h (green curve) devices are given on the left side of the curves. The 96 h device (red curve) uses the left y-axis. The time interval of each cycle is 30 s. The far-right curve is the magnified one cycle in the 96 h device. Note: the distinct waveform and significant enhancement of output voltage between 96 h and others are probably due to the changes in the pore structure, including the pore size and through-hole channel structure. (b) A similar experiment is performed by using non-magnetic polyionic liquid micropores ([HS-C10mim] NO3). The blue curve is the magnified view of the time-dependent output curve. The output voltage amplitude of devices as a function of (c) photoinitiator concentration (bottom x-axis, red markers) and the corresponding pore size (top x-axis, blue markers), (d) UV irradiation time (bottom x-axis, red markers) and the corresponding pore size (top x-axis, blue markers), and (e) water treatment time (bottom x-axis, red markers) and the corresponding pore size (top x-axis, blue markers). | |
The output performances of the devices fabricated using mag-PLMs with the increase of photoinitiator concentration and UV irradiation time are also recorded. Increasing the photoinitiator concentration leads to a decrease in output voltage (Fig. 4c). However, with the increase of UV irradiation time, the recorded voltage has also increased (Fig. 4d). Interestingly, the change of output voltage is qualitatively consistent with the evolution of micropore size (Fig. 2vs.Fig. 4). Apparently, in a device with a larger micropore size, the ‘effective’ magnetic anion concentration is higher which generates a higher voltage. However, the connection between the output voltage and micropore size is found to be non-linear and probably exponential. This indicates the complexity of the system where the ‘effective’ magnetic anion concentration is dependent not only on magnetic anion concentration but also on its mobility within mag-PLM channels. The short-circuit current of the device is also measured (Fig. S10, ESI†); however, most devices output only nano-ampere current which is in the same magnitude as other electrostatic induction devices.35,36
Modeling of the magnetic energy harvesting device
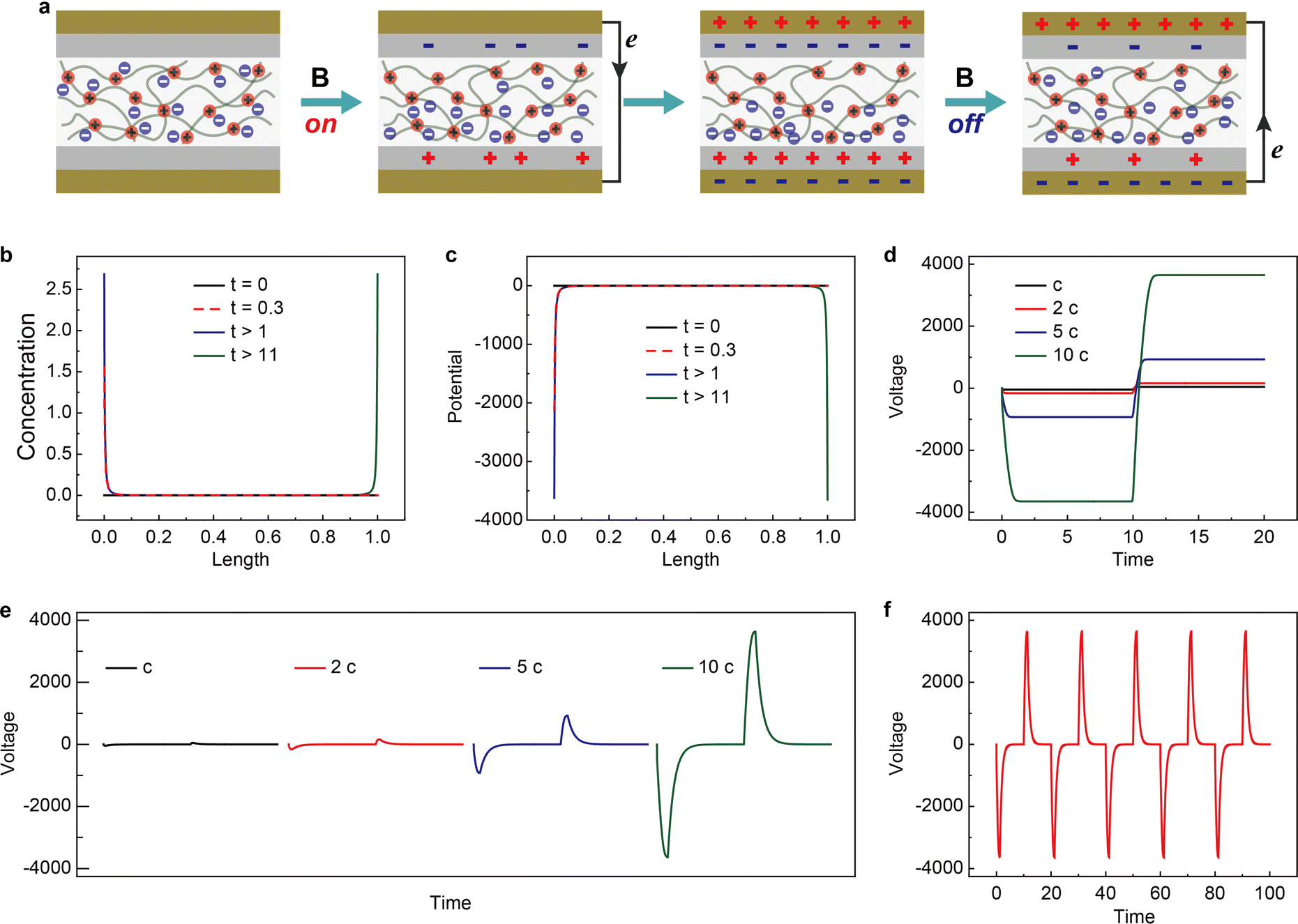
The working mechanism of the device is schematically shown in Fig. 5a. Similar to previously reported nanogenerators,37,38 the output originates from the coupling of charge transfer between dielectric layers and electrostatic induction.39,40 However, the charge separation method has not been demonstrated before. Applying and removing a magnetic field leads to the reversed migration of electrons in the external circuit. This is closely related to the magnetic-field-controlled magnetic anion migration and gradient distribution within the mag-PLM layer. To simulate the magnetic energy harvesting process, a continuum charge transport model combining Poisson and Nernst–Planck diffusion equations is developed (see the Experimental section for details).41,42 Since the movement of magnetic anions is driven by the magnetic field, we convert this magnetic force into an electric one in which only magnetic counterions could migrate in response to local electric fields and concentration gradients. The non-magnetic positive charges are however immobile. Due to the electrostatic induction effect, the dielectric layers and Au electrodes will induce the corresponding number of charges. In addition, once the potential is different between two gold electrodes, the electrons could flow through the external circuit to reach electric neutrality (Fig. 5a).
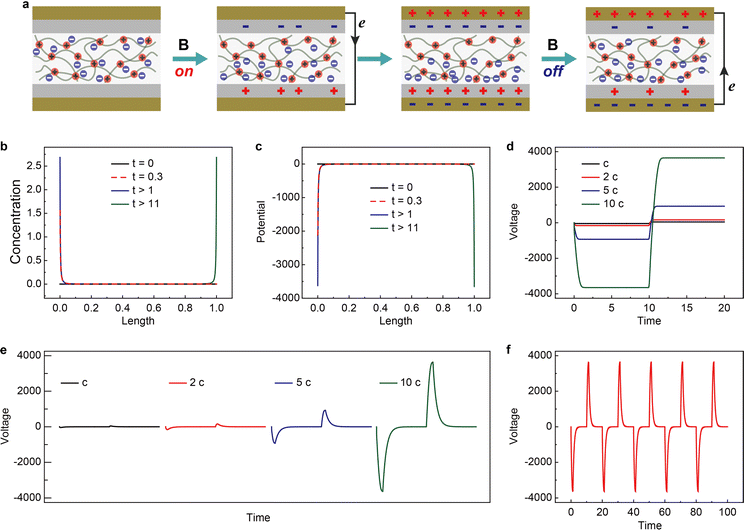 |
| | Fig. 5 Theoretical simulations of the magnetic energy harvesting device. (a) The schematic of the operating mechanism of the magnetic energy harvesting device. Simulated time-dependent distribution of Dy(NO3)4− anions (b) and potential (c) under applied magnetic field. (d) Dimensionless voltage–time characteristics within magnetically responsive polyionic liquid micropores. (e) Dimensionless output voltage collected between two Au electrodes. Four types of magnetic anion concentrations are simulated (c to 10 c). (f) Dimensionless cycling (magnetic field on/off) performance of the magnetic energy harvesting device. The concentration of magnetic anions is 10 c. | |
The magnetic counterion concentration distribution is then simulated. Under a magnetic field, the counterions accumulate on one side of mag-PLM and eventually reach equilibrium (Fig. 5b). The distribution of ionic charges directly affects the potential distribution within the mag-PLM layer (Fig. 5c). Since removing the magnetic field in the experiment has the same consequence as applying a magnetic field on the other side of the device, the simulated magnetic counterion concentration and potential distributions under reversed field display mirror images towards the forward field (blue curve vs. green curve in Fig. 5b and c). The gradient distribution of charges and potential is then transformed into electric potential within mag-PLM (Fig. 5d) and output voltage in the external circuit (Fig. 5e). In addition, with the increase of magnetic counterion concentration, the output voltage has also increased (Fig. 5e). Interestingly, it is non-linear which is consistent with the experimental results. The cycling performance of the device is also reproduced by simulation (Fig. 5fvs. Fig. S11, ESI†).
Apparently, these are only proof-of-concept demonstrations and future improvements – in various aspects – will be needed for practical applications. The first improvement is the output voltage. This is closely related to the ‘effective’ concentration of magnetic counterions migrating toward the PLM/dielectric interface within the mag-PLM layer. Except for further improvement of PLM, materials with high charge density and ion mobility can be employed such as porous carbon materials,43,44 metallic nanostructures,45–48 and even metal–organic frameworks.49,50 In addition, the induced charge density can also be increased by choosing an insulating layer with a higher dielectric constant. The second one regards the short-circuit current. Herein, we only measured nano-ampere current which is due to the slow and insufficient counterion migration within mag-PLM. Besides the increase of ‘effective’ counterion concentration, fabricating a through-hole structure to facilitate the diffusion of magnetic ions could be one solution. Synthesis of a magnetic ion with higher magnetic susceptibility and smaller size would be also effective.51 Another improvement should be the design of the whole system. Repeatedly applying and removing the magnetic field is far away from the realistic application. An optimized situation could be continuing rotation of the device while constantly applying the magnetic field.
Finally, we wish to emphasize the idea of separating charges with a magnetic field. Since the magnetic field has the same effect as the electric field, one can envision the use of mag-PLM beyond energy harvesting devices, for example, a magnetic field effect transistor in which the mag-PLM plays the role of the electrolyte gate.52,53 By applying a magnetic field, the movement of magnetic ions could induce the charge carriers within semiconductors. We have conceptually demonstrated such a device in which while approaching the magnet, holes accumulate within the PEDOT:PSS layer, leading to an increase of current between the source and drain (Fig. 6). Interestingly, on the other side of mag-PLM (Semiconductor 1//mag-PLM//Semiconductor 2), charge carriers with reversed polarity could be induced. In addition, when the magnetic field is applied parallelly to an ambipolar semiconductor plane, charge gradients and internal electric potential can be established to produce a pn junction. These works are principally demonstrated and will be published later.
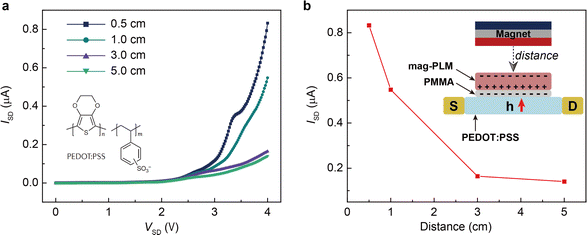 |
| | Fig. 6 Switching on a transistor with the mag-PLM. (a) Source to drain current–voltage characteristics of a PEDOT:PSS transistor in which the mag-PLM is used as the electrolyte gate. As the magnet approaches the mag-PLM, the current has increased. (b) Distance-dependent source to drain current. The inset shows the scheme of the device. | |
Conclusion
To conclude, we have demonstrated a magnetic energy harvesting device in which charge separation is achieved by applying a magnetic field. With a comprehensive study on the synthesis conditions of magnetic polyionic liquid micropores, the device finally outputs approximately 4 V voltage. A continuum charge transport model is developed which qualitatively reproduces the experimental results. Although the device performance is not competitive to other methods,54,55 we suggest an application in which our device could be placed close to devices generating strong magnetic fields (for example, NMR systems). In addition, we expect our concept could be applied beyond energy harvesting devices.
Experimental
The synthesis of polyionic liquids
In a typical procedure, 30 g of 1-vinylimidazole was firstly dissolved in 100 mL of N,N-dimethylformamide. Then 36 mg of the initiator 2,2′-azobis(2-methylpropionitrile) dissolved in 1 mL N,N-dimethylformamide solution was injected using a needle-tipped syringe under nitrogen protection at 90 °C. The reaction was carried out for 24 h. Poly(1-vinylimidazole) was precipitated by using 500 mL acetone and the resulting white solid was collected and dried under vacuum at 50 °C for 24 h. 6 g poly(1-vinylimidazole) and 0.627 g butylated hydroxytoluene were dissolved in 100 mL N,N-dimethylformamide. The mixture was heated to 90 °C, followed by dropwise addition of 15.5 g 1-allyl bromide. The reaction proceeded for 24 h and then cooled down to room temperature. The poly(3-allylmethyl-1-vinylimidazole)bromide was precipitated by using 500 mL diethyl ether and the resulting yellow-white solid was collected and dried under vacuum at 50 °C for 48 h. The structure was confirmed by 1H NMR (400 MHz, DMSO-d6) (Fig. S12, ESI†). Then 1 g of the product and 2 g lithium bis(trifluoromethanesulfonyl)imide were separately dissolved in 10 mL water. The lithium salt was then dropwise-added into the former solution, stirred for 6 h, and filtered, producing a yellow-white solid. The solid is then washed with deionized water to remove excess lithium salts, followed by vacuum drying at 50 °C for 48 h. The structure of the polyionic liquid was confirmed by 1H NMR (400 MHz, DMSO-d6) (Fig. S13, ESI†).
Fabrication of the PLM layer
The fabrication of the PLM layer was according to a covalent cross-linking method. 1 g of the polyionic liquid and 156 mg of the cross-linking agent dithiothreitol were dissolved in 10 mL acetone. 100 μL solution was dropped on the glass substrate and dried for 1 h under ambient conditions, followed by vacuum drying for 12 h at room temperature to remove the remaining acetone. The substrate was then immersed into an aqueous solution containing 50 mg of the photoinitiator UV2959 (10 mL). The reaction proceeded by UV irradiation for 4 h at ice bath temperature. The film was then washed with deionized water and soaked in deionized water for tens of hours to produce micropores. To control the size of the micropores, the photoinitiator concentration (5–45 mg mL−1), UV irradiation time (0.5–3 h), water treatment time (2–96 h), and water treatment temperature (25–90 °C) were all studied.
Synthesis of thiol-functionalized MILs
1-10-Mercaptodecyl-3-methylimidazole bromide was synthesized according to a previous procedure.56 Typically, 2 g 1,10-dibrominated dodecane and 14.63 g 1-methylimidazole were mixed and refluxed in 20 mL chloroform for 24 h. The chloroform was then removed by rotary evaporation at 50 °C to obtain a white viscous material. The prepared material was repeatedly centrifuged and washed by using n-hexane to remove the unreacted halogenated hydrocarbons. A small amount of deionized water (5 mL) was used to dissolve the product and repeatedly extracted by using n-hexane three times. The product was then collected by removing extra water. The transparent ionic liquid was then dried under vacuum at 50 °C for 48 h to obtain a transparent yellow bromide, 1-(10-bromodecyl)-3-methylimidazole. The prepared product (2.7 g) was then mixed with 1.08 g potassium thioacetate in 20 mL acetonitrile solvent and refluxed at 65 °C for 24 h. The unreacted potassium thioacetate was removed by filtration and the liquid solution was rotary-evaporated at 55 °C to remove acetonitrile. The product was vacuum-dried at 50 °C for 48 h to obtain 1-(10-thioethyl decyl)-3-methylimidazole bromide in the form of a dark reddish ionic liquid. Then 1 g IL was then dissolved in 6 mL ethanol, followed by a dropwise addition of 2 mL sodium hydroxide solution (10.6 M) at ice bath temperature. After 24 h hydrolysis, the pH of the solution was adjusted to around 2 by adding 8.6 M hydrobromic acid. Dichloromethane was used to extract the product, followed by addition of anhydrous magnesium sulfate to remove trace water. With filtration and rotary evaporation (at room temperature), a pungent viscous product was collected and dried under vacuum at 40 °C for 48 h to finally obtain 1-(10-mercaptodecyl)-3-methylimidazole bromide (see the 1H NMR spectra in Fig. S14–S16, ESI†).
A similar procedure was used to synthesize 1-(10-mercaptodecyl)-3-methylimidazole nitrate. 0.45 g silver nitrate and 1 g 1-(10-thioethyl decyl)-3-methylimidazole bromide were dissolved in 30 mL methanol. The reaction proceeded for 12 h in dark conditions and the product was collected by filtration and rotary evaporation. Then 0.95 g of the product was dissolved in 6 mL ethanol, followed by dropwise addition of 2 mL sodium hydroxide solution (10.6 M) at ice bath temperature. After 12 h of hydrolysis, the pH of the solution was adjusted to around 2 by adding 5 M nitric acid. The following procedures were the same as above. Finally, 1-(10-mercaptodecyl)-3-methylimidazole nitrate was obtained (see the ESI-MS results in Fig. S17, ESI†).
To synthesize the MIL, 0.25 g 1-(10-mercaptodecyl)-3-methylimidazole bromide and 0.12 g ferric chloride were mixed with 0.5 mL methanol and stirred for 24 h under nitrogen protection. The product was then repeatedly extracted with diethyl ether to remove the unreacted ferric chloride to produce 1-(10-mercaptodecyl)-3-methylimidazolium ferric chloride bromide (see the typical UV-vis spectrum in Fig. S6, ESI†). 0.33 g 1-(10-mercaptodecyl)-3-methylimidazole bromide and 0.37 g dysprosium chloride hydrate were mixed in 1 mL methanol and stirred for 24 h under nitrogen protection. By removing the remaining methanol, 1-(10-mercaptodecyl)-3-methylimidazolium dysprosium chloride bromide was produced (see the ESI-MS results in Fig. S18, ESI†). 0.26 g 1-(10-mercaptodecyl)-3-methylimidazole nitrate and 0.37 g dysprosium nitrate hydrate were mixed in 1 mL methanol and stirred for 24 h under nitrogen protection. The remaining methanol was removed and 1-10-mercaptodecyl-3-methylimidazolium dysprosium tetranitrate was finally produced as an orange-reddish viscous liquid (see the ESI-MS results in Fig. S19, ESI†).
Fabrication of mag-PLMs
The PLM was sandwiched between two glass slides and then immersed in an ethylene glycol solution of 30 wt% 1-(10-mercaptodecyl)-3-methylimidazolium dysprosium tetranitrate. The reaction was performed by UV irradiation for 4 h, followed by further soaking for 24 h at room temperature. The mag-PLM was finally produced by washing it with ethylene glycol. The other two mag-PLMs used 1-(10-mercaptodecyl)-3-methylimidazolium ferric chloride bromide and 1-(10-mercaptodecyl)-3-methylimidazolium dysprosium chloride bromide dissolved in the propylene carbonate solvent, and the rest of the steps were the same. To prepare the non-magnetic PLM for the control experiment, the non-magnetic IL 1-(10-mercaptodecyl)-3-methylimidazole nitrate was used.
Characterization and measurement
The structures of the non-magnetic ionic liquids were characterized by using a 400 MHz nuclear magnetic resonance spectrometer (AVANCE III HD 400, Bruker, USA). The UV-vis spectra were recorded using a spectrophotometer (Shimadzu, Japan, UV 2600). The ESI-MS was performed by using an LCMS/ESI/QTOF analyzer (UPLM-QTOF). The porous structures of the PLM and mag-PLM were characterized by using a scanning electron microscope (SEM, Hitachi-SU8220). Energy dispersive X-ray spectroscopy (EDS) was performed using a Horiba EMAX X-ray detector equipped with a SEM. The VSM measurement of the MIL was performed on a PPMS-ANC300 (Quantum Design, America; attocube systems AG, Germany).
Fabrication and electrical characteristics of the magnetic energy harvesting device
A sandwich structure was used to fabricate the device, Au/dielectric//mag-PLM//dielectric/Au. The gold electrode was prepared by ion sputtering with a thickness of 50 nm. The PMMA dielectric layer was spin-coated (550 nm thick). The mag-PLM was then transferred onto a glass substrate with precoated Au/dielectric. Two glass slides with were then laminated in a face-to-face configuration and finally encapsulated with polydimethylsiloxane (PDMS). The electrical characteristics were recorded using a Keithley 6517 high-resistance electrometer, and the data were collected using an acquisition card. The magnetic field was provided by a 0.3 T neodymium magnet. The time interval between the application and removal of the magnetic field was 30 s.
Theoretical calculations
According to Maxwell's equations, the magnetic field generated by the magnetic anion and its movement was much smaller than the external magnetic field, so it could be ignored. The magnetic field applied to the system was considered to be a constant magnetic field. Since the magnetic anion has a negative charge, the applied magnetic field can be converted into an electric field by using the transformation of force; see eqn (1):where χm and V are the magnetic susceptibility and volume per unit mass; H and gradH are the magnetic field intensity and magnetic field gradient.
A coupled system of Poisson and Nernst–Planck equations was employed to model the nano-energy harvesting device. Under an electrostatic field, ions could be described using the Nernst–Planck equation:
| | 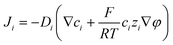 | (2) |
where index
i corresponds to the
ith ionic species;
Ji,
Di,
ci,
zi, and
ui are the flux, diffusion coefficient, concentration, charge, and velocity of the species
i, respectively;
φ is the electrostatic potential. Considering the mass balance,
eqn (2) can be transformed to obtain the change of each ion concentration over time:
| | 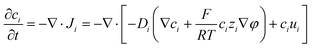 | (3) |
In addition, the electrostatic potential depends on the fixed charges on the polymer and the mobile charges, which could be described using the Poisson equation:
| | 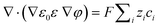 | (4) |
where
ε0 is the vacuum dielectric constant and
ε is the relative dielectric constant.
To facilitate the calculation, the equation is dimensionless. Here, we set the characteristic scale as follows: length scale L∞, potential scale 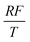 , and time scale
, and time scale 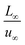 .
.
By the potential scale 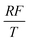 ,
, 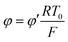 , eqn (4) can be written as
, eqn (4) can be written as
| | 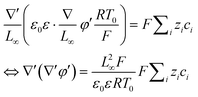 | (5) |
| | 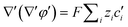 | (6) |
Substituting equation 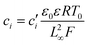 ,
, 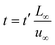 into eqn (3), we get
into eqn (3), we get
| | 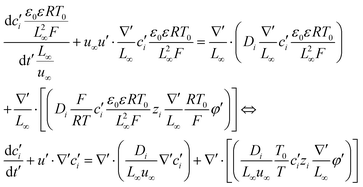 | (7) |
| | 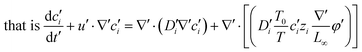 | (8) |
| | 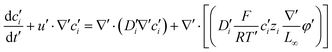 | (9) |
For simplicity, we considered a one-dimensional domain [0, L]. The initial and boundary conditions can be summarized as
| φ(x = 0, t) = 0; φ (x = L, t) = φ0; |
where
φ0 is given by the potential difference calculated using
eqn (1); the ion concentration
ci0 is set referring to the actual situation. The coupled equations
(eqn (6) and (9)) were then solved numerically by using a commercial finite element solver (COMSOL 5.6) to obtain the distribution of ion concentration and electrostatic potential. The potential distribution (
V) was then obtained from the ion concentration. The output voltage can be split into two parts, electrostatic induction and condenser discharge. So it can be finally integrated by
| | 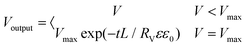 | (10) |
The parameters were as follows: L = 1; c0 = 10−3 mol m−3; Danion = 10−12 m2 s−1; ε = 8; ε0 = 8.85 × 10−12 F m−1; Rv = 104 Ω; T = 298.15 K; F = 96500 C mol−1; and R = 8.314 J mol−1 K−1.
Author contributions
T. X. carried out the experiments and, along with J. W., J. G., and X. Z., performed the data analysis. J. W. modeled the experimental results. All authors wrote the manuscript. Y. Y. conceived and supervised the project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21875053) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36000000). The authors thank B. Tu and B. Lu for their helpful discussions and support on modeling.
References
- Q. F. Shi, Z. X. Zhang, T. Y. Y. He, Z. D. Sun, B. J. Wang, Y. Q. Feng, X. C. Shan, B. Salam and C. Lee, Nat. Commun., 2020, 11, 4609 CrossRef CAS.
- Z. D. Sun, M. L. Zhu, X. C. Shan and C. K. Lee, Nat. Commun., 2022, 13, 5224 CrossRef CAS.
- M. L. Zhu, Z. D. Sun, Z. X. Zhang, Q. F. Shi, T. Y. Y. He, H. C. Liu, T. Chen and C. K. Lee, Sci. Adv., 2020, 6, eaaz8693 CrossRef CAS PubMed.
- Z. L. Wang, T. Jiang and L. Xu, Nano Energy, 2017, 39, 9–23 CrossRef CAS.
- L. Persano, C. Dagdeviren, Y. W. Su, Y. H. Zhang, S. Girardo, D. Pisignano, Y. G. Huang and J. A. Rogers, Nat. Commun., 2013, 4, 1633 CrossRef.
- Z. Q. Bai, T. Y. Y. He, Z. X. Zhang, Y. L. Xu, Z. Zhang, Q. F. Shi, Y. Q. Yang, B. G. Zhou, M. L. Zhu, J. S. Guo and C. Lee, Nano Energy, 2022, 94, 106956 CrossRef CAS.
- Y. Yan, J. V. I. Timonen and B. A. Grzybowski, Nat. Nanotechnol., 2014, 9, 901–906 CrossRef CAS.
- S. O. Krabbenborg and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 9152–9167 CrossRef CAS PubMed.
- Q. Q. Sun, D. H. Wang, Y. N. Li, J. H. Zhang, S. J. Ye, J. X. Cui, L. Q. Chen, Z. K. Wang, H. J. Butt, D. Vollmer and X. Deng, Nat. Mater., 2019, 18, 936–941 CrossRef CAS PubMed.
- Z. L. Wang and J. H. Song, Science, 2006, 312, 242–246 CrossRef CAS.
- C. Chang, V. H. Tran, J. Wang, Y.-K. Fuh and L. Lin, Nano Lett., 2010, 10, 726–731 CrossRef CAS PubMed.
- F. R. Fan, Z. Q. Tian and Z. L. Wang, Nano Energy, 2012, 1, 328–334 CrossRef CAS.
- W. Seung, M. K. Gupta, K. Y. Lee, K.-S. Shin, J.-H. Lee, T. Y. Kim, S. Kim, J. Lin, J. H. Kim and S.-W. Kim, ACS Nano, 2015, 9, 3501–3509 CrossRef CAS.
- Y. Q. Yang, Q. F. Shi, Z. X. Zhang, X. C. A. Shan, B. Salam and C. Lee, InfoMat, 2022, e12360 Search PubMed.
- S. Morgenthaler, C. Zink and N. D. Spencer, Soft Matter, 2008, 4, 419–434 RSC.
- S. Inagi, Y. Ishiguro, M. Atobe and T. Fuchigami, Angew. Chem., Int. Ed., 2010, 49, 10136–10139 CrossRef CAS.
- F. Zhao, H. H. Cheng, Z. P. Zhang, L. Jiang and L. T. Qu, Adv. Mater., 2015, 27, 4351–4357 CrossRef CAS.
- F. Zhao, Y. Liang, H. H. Cheng, L. Jiang and L. T. Qu, Energy Environ. Sci., 2016, 9, 912–916 RSC.
- H. Wang, Y. Sun, T. He, Y. Huang, H. Cheng, C. Li, D. Xie, P. Yang, Y. Zhang and L. Qu, Nat. Nanotechnol., 2021, 16, 811–819 CrossRef CAS.
- Y. Liang, F. Zhao, Z. Cheng, Y. Deng, Y. Xiao, H. Cheng, P. Zhang, Y. Huang, H. Shao and L. Qu, Energy Environ. Sci., 2018, 11, 1730–1735 RSC.
- H. Wang, U. Ail, R. Gabrielsson, M. Berggren and X. Crispin, Adv. Energy Mater., 2015, 5, 1500044 CrossRef.
- D. Zhao, S. Fabiano, M. Berggren and X. Crispin, Nat. Commun., 2017, 8, 14214 CrossRef CAS PubMed.
- X. M. Liu, H. Y. Gao, J. E. Ward, X. R. Liu, B. Yin, T. D. Fu, J. H. Chen, D. R. Lovley and J. Yao, Nature, 2020, 578, 550–554 CrossRef CAS.
- X. J. Li, J. J. Luo, K. Han, X. Shi, Z. W. Ren, Y. Xi, Y. B. Ying, J. F. Ping and Z. L. Wang, Nat. Food, 2022, 3, 133–142 CrossRef CAS.
- B. L. Cheng, Q. Xu, Y. Q. Ding, S. Bai, X. F. Jia, Y. D. C. Yu, J. Wen and Y. Qin, Nat. Commun., 2021, 12, 4782 CrossRef CAS.
- J.-H. Lee, K. Y. Lee, M. K. Gupta, T. Y. Kim, D.-Y. Lee, J. Oh, C. Ryu, W. J. Yoo, C.-Y. Kang, S.-J. Yoon, J.-B. Yoo and S.-W. Kim, Adv. Mater., 2014, 26, 765–769 CrossRef CAS PubMed.
- A. Dani, K. Tauber, W. Y. Zhang, H. Schlaad and J. Y. Yuan, Macromol. Rapid Commun., 2017, 38, 1700167 CrossRef.
- Q. Zhao, M. J. Yin, A. P. Zhang, S. Prescher, M. Antonietti and J. Y. Yuan, J. Am. Chem. Soc., 2013, 135, 5549–5552 CrossRef CAS.
- Y. Y. Ren, J. N. Guo, Z. Y. Liu, Z. Sun, Y. Q. Wu, L. L. Liu and F. Yang, Sci. Adv., 2019, 5, eaax0648 CrossRef CAS PubMed.
- Y. J. Song, J. H. He and Y. T. Zhang, Macromol. Rapid Commun., 2020, 41, 2000456 CrossRef CAS PubMed.
- B. Liu, X. R. Deng, Z. X. Xie, Z. Y. Cheng, P. P. Yang and J. Lin, Adv. Mater., 2017, 29, 1604878 CrossRef.
- H. B. Yuan, J. H. Zhang, R. J. Yu and Q. Su, J. Rare Earths, 2009, 27, 308–311 CrossRef.
- J. Wang, H. Yao, Y. Nie, X. Zhang and J. Li, J. Mol. Liq., 2012, 169, 152–155 CrossRef CAS.
- Y. M. Tang, X. L. Hu, P. Guan, X. P. Lin and X. Q. Li, J. Phys. Org. Chem., 2014, 27, 498–503 CrossRef CAS.
- M. Zhang, T. Gao, J. S. Wang, J. J. Liao, Y. Q. Qiu, Q. Yang, H. Xue, Z. Shi, Y. Zhao, Z. X. Xiong and L. F. Chen, Nano Energy, 2015, 13, 298–305 CrossRef CAS.
- Z. Y. Pi, J. W. Zhang, C. Y. Wen, Z. B. Zhang and D. P. Wu, Nano Energy, 2014, 7, 33–41 CrossRef CAS.
- Z. Y. Wu, H.
Y. Guo, W. B. Ding, Y. C. Wang, L. Zhang and Z. L. Wang, ACS Nano, 2019, 13, 2349–2356 CAS.
- Z. Y. Wu, B. B. Zhang, H. Y. Zou, Z. M. Lin, G. L. Liu and Z. L. Wang, Adv. Energy Mater., 2019, 9, 1901124 CrossRef.
- Z. L. Wang and A. C. Wang, Mater. Today, 2019, 30, 34–51 CrossRef CAS.
- J. Briscoe and S. Dunn, Nano Energy, 2015, 14, 15–29 CrossRef CAS.
- X. Zhao, B. Tu, M. Y. Li, X. J. Feng, Y. C. Zhang, Q. J. Fang, T. H. Li, B. A. Grzybowski and Y. Yan, Sci. Adv., 2018, 4, eaau3546 CrossRef CAS PubMed.
- X. Zhao, L. Yang, J. H. Guo, T. Xiao, Y. Zhou, Y. C. Zhang, B. Tu, T. H. Li, B. A. Grzybowski and Y. Yan, Nat. Electron., 2021, 4, 109–115 CrossRef CAS.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- X. Wu, Y. L. Chen, Z. Xing, C. W. K. Lam, S. S. Pang, W. Zhang and Z. C. Ju, Adv. Energy Mater., 2019, 9, 1900343 CrossRef.
- H. W. Huang, J. W. Wang, X. F. Yang, R. Z. Hu, J. L. Liu, L. Zhang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 14504–14510 CrossRef CAS.
- M. Liu, H. Fan, O. Zhuo, J. C. Chen, Q. Wu, L. J. Yang, L. M. Peng, X. Z. Wang, R. C. Che and Z. Hu, Nano Energy, 2020, 68, 104368 CrossRef.
- W. Yang, Z. P. Luo, W. K. Bao, H. Xie, Z. S. You and H. J. Jin, Sci. Adv., 2021, 7, eabb9471 CrossRef PubMed.
- Z. P. Su and T. H. Chen, Small, 2021, 17, 2005354 CrossRef.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. Xiao, L. L. Zou, H. Pang and Q. Xu, Chem. Soc. Rev., 2020, 49, 301–331 RSC.
- B. Mallick, B. Balke, C. Felser and A. V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7635–7638 CrossRef PubMed.
- J. Lenz, F. del Giudice, F. R. Geisenhof, F. Winterer and R. T. Weitz, Nat. Nanotechnol., 2019, 14, 579–585 CrossRef.
- S. H. Kim, K. Hong, W. Xie, K. H. Lee, S. P. Zhang, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2013, 25, 1822–1846 CrossRef CAS PubMed.
- W. H. Xu, H. X. Zheng, Y. Liu, X. F. Zhou, C. Zhang, Y. X. Song, X. Deng, M. Leung, Z. B. Yang, R. X. Xu, Z. L. Wang, X. C. Zeng and Z. K. Wang, Nature, 2020, 578, 392–396 CrossRef CAS.
- L. Yin, J. M. Moon, J. R. Sempionatto, M. Y. Lin, M. Z. Cao, A. Trifonov, F. Y. Zhang, Z. Y. Lou, J. M. Jeong, S. J. Lee, S. Xu and J. Wang, Joule, 2021, 5, 1888–1904 CrossRef CAS.
- H. Zhang, L. Ye, X. Wang, F. Li and J. Wang, Chem. Commun., 2014, 50, 2565–2568 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Jiahui
Guo
ab,
Xing
Zhao
ab,
Jiahui
Guo
ab,
Xing
Zhao
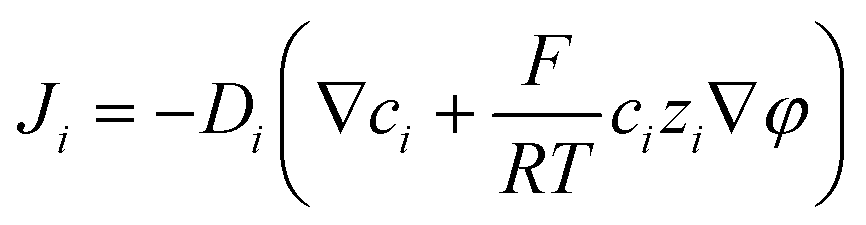
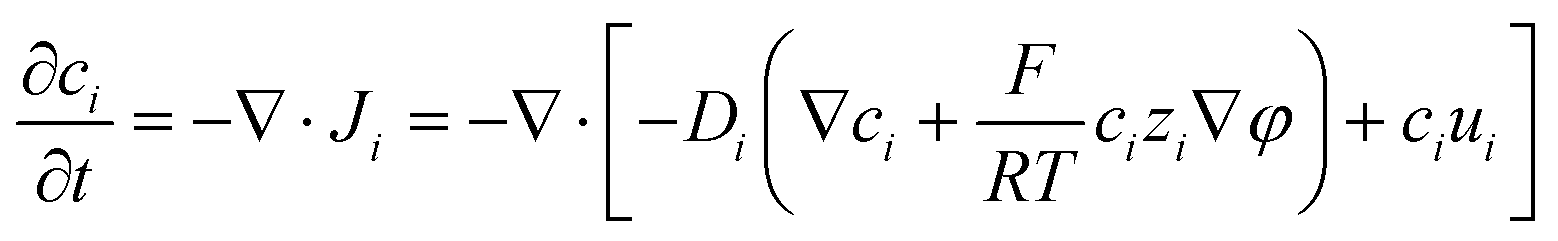
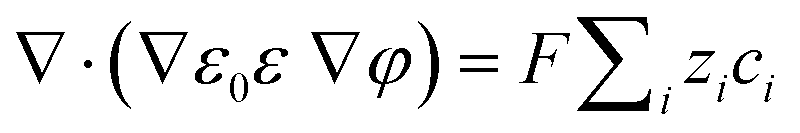
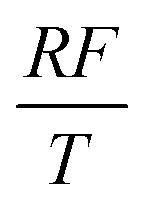 , and time scale
, and time scale 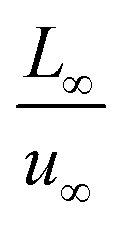 .
.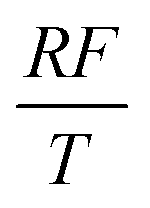 ,
, 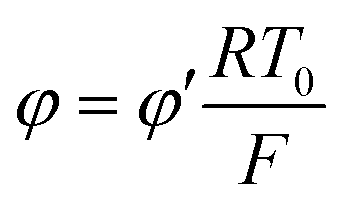 , eqn (4) can be written as
, eqn (4) can be written as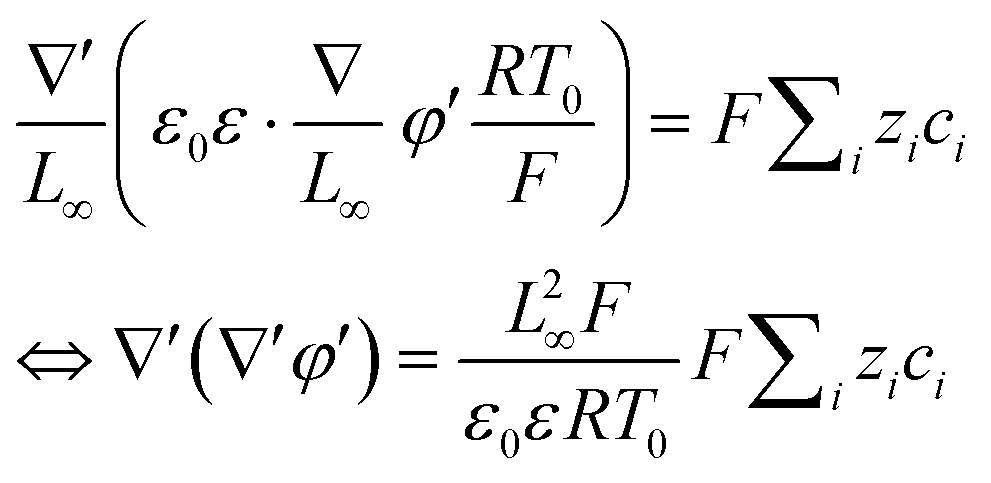
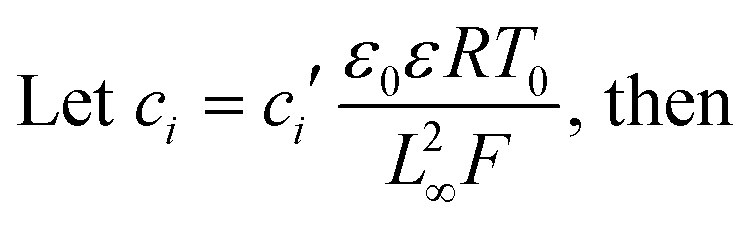
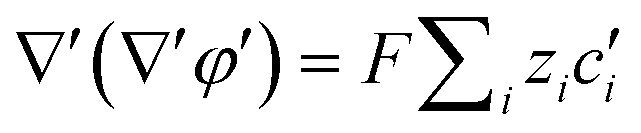
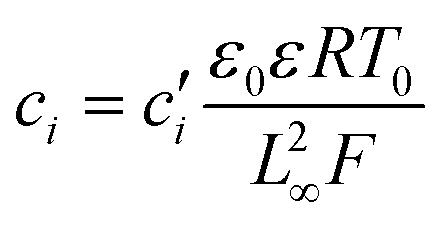 ,
, 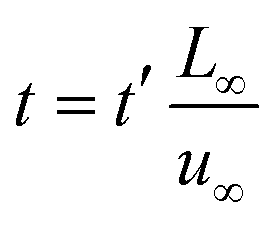 into eqn (3), we get
into eqn (3), we get