
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical-mediated thiol–ene ‘click’ reactions in deep eutectic solvents for bioconjugation†
Mark D.
Nolan
 a,
Andrea
Mezzetta
b,
Lorenzo
Guazzelli
*b and
Eoin M.
Scanlan
*a
a,
Andrea
Mezzetta
b,
Lorenzo
Guazzelli
*b and
Eoin M.
Scanlan
*a
aTrinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland. E-mail: eoin.scanlan@tcd.ie
bDepartment of Pharmacy, University of Pisa, Via Bonanno 6, 56126, Pisa, Italy
First published on 3rd February 2022
Abstract
Herein, we report the first application of deep eutectic solvents (DESs) in radical-mediated hydrothiolation reactions. Under UV and atmospheric-oxygen mediated conditions, the thiol–ene reaction was applied to amino acid and peptide ligation in DESs. The conditions facilitate highly-efficient synthesis of biomolecular targets using a ‘green’ methodology, with complete recycling of the reaction medium. Fluorescent labelling and glycosylation of a minimal sequence mimetic of angiotensin-converting enzyme 2 capable of binding the SARS-CoV-2 spike protein is demonstrated for applications in the study of inhibition of COVID-19 infectivity.
The United Nations Sustainable Development Goal 12 aims to substantially reduce production waste, including through recycling and reuse. The improvement of sustainability within pharmaceutical development manufacturing is particularly closely linked with the principles of ‘green’ chemistry.1 In recent years, the growing awareness in the public opinion of the impact caused by climate change and raw material depletion is undoubtedly rising.2 In this context, ‘green’ or sustainable chemistry can play a pivotal role on account of its potential contribution to reaching several of the 17 Sustainable Development Goals, established in 2016.3 According to the views expressed in this deal, chemical processes need to be redesigned with alternative targets in mind, certainly beyond yield and productivity. The most critical factor to consider when evaluating a chemical process from this perspective is the solvent. In the pharmaceutical industry solvents represent up to 80–90% of the chemicals employed, and it is estimated that less than 50% are reused.4
In this context, deep eutectic solvents (DESs), among other possible alternatives to volatile organic solvents (VOCs), have attracted an exponentially growing interest in the last five years. DESs usually have 2 constituting components which, through the establishment of strong interactions, cause a decrement of the eutectic point temperature below that of the ideal mixture.5 In the vast majority of cases, these interactions occur between a hydrogen bond acceptor (HBA), and a hydrogen bond donor (HBD). When both partners are present in nature, these media are called Natural DESs (NaDESs).6
The key favourable aspects of (Na)DESs are their ease of preparation, the possibility to tailor their physico-chemical properties by selecting suitable HBA-HBD combinations, and the reduced volatility and toxicity when compared to VOCs.14 DESs have found application in several areas of research spanning from biomass treatment15,16 to added value compounds for extraction,17,18 and from electrochemistry19,20 to biotechnology.21
Furthermore, the exploitation of DESs as solvents and/or catalysts for organic reactions has been a recent and intense area of study. Several archetypical reactions22–25 including Grignard and organolithium additions26–28 as well as the preparation of pharmaceutical active ingredients,29 have been optimised in these protic media.
However, to the best of our knowledge, the thiol–ene ‘click’ (TEC) reaction in DESs has not previously been explored. Radical thiol–ene chemistry in traditional solvents has found significant application in synthetic chemistry,30 including in carbohydrate31 and peptide science.32,33 The ‘click’ characteristics of the radical thiol–ene reaction,34 together with excellent atom economy, complement the development of ‘green’ syntheses, in particular bioconjugation reactions (Fig. 1). DESs have proven highly compatible media for protein processing as demonstrated by numerous biotechnological applications.21 We therefore hypothesised that the thiol–ene reaction in DES or DES–water mixtures would provide an ideal, green system for access to ligated amino acids and peptides via reaction at the cysteine thiol residue. Furthermore, the thiol–ene reaction is an efficient method for the formation of thioethers or thioesters, with advantageous application in pharmaceuticals synthesis due to the prevalence of such structures.35
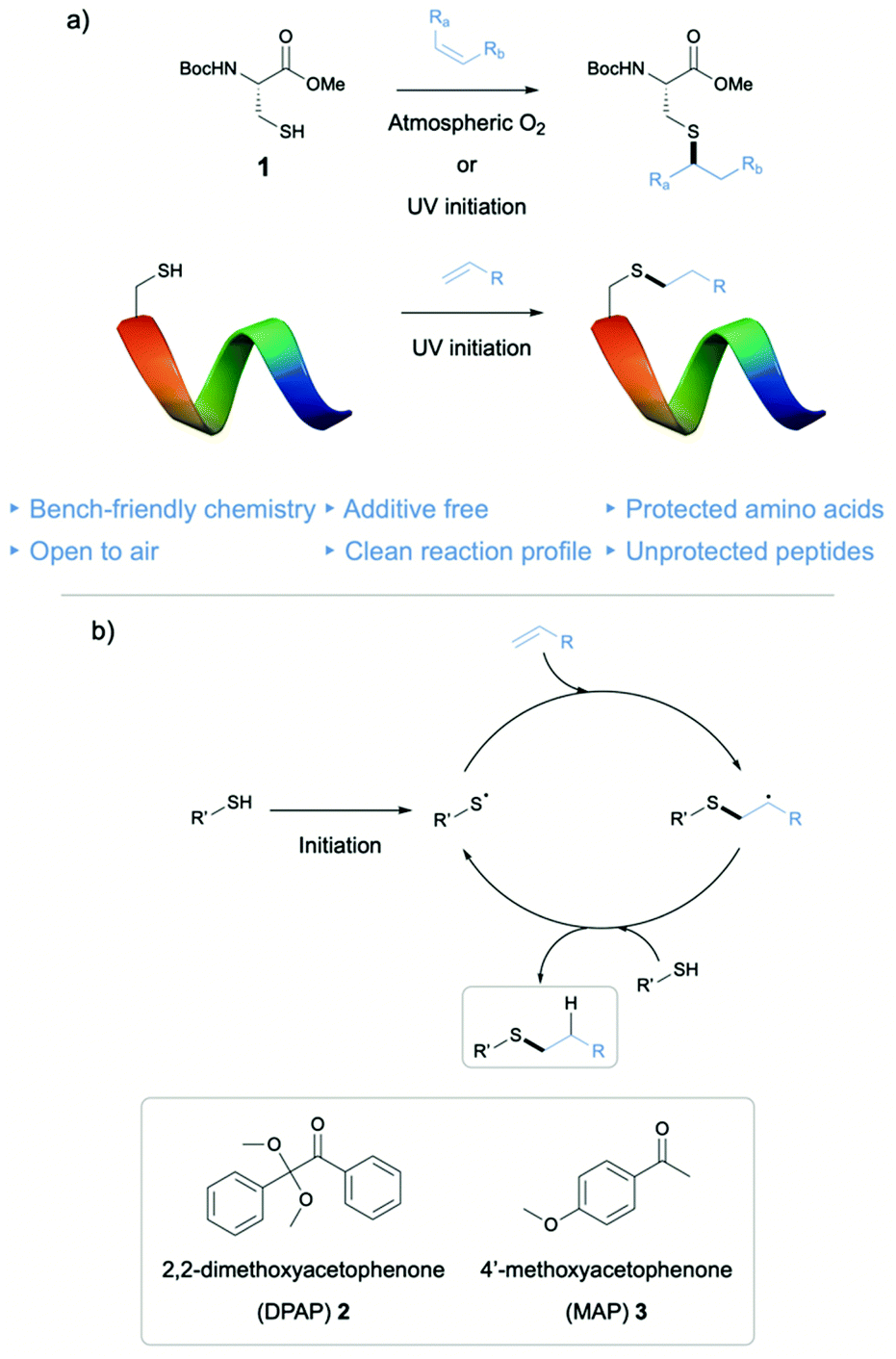 | ||
| Fig. 1 (a) Modification of protected Cys and of an unprotected peptide in DESs via either UV or atmospheric O2 initiated thiol–ene reaction. (b) Mechanism of the thiol–ene radical chain process. | ||
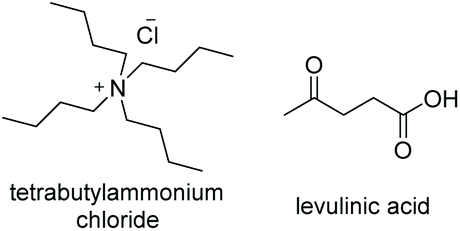
Initial studies into the compatibility of selected DESs as shown in Table 1 with the thiol–ene reaction commenced with a screen of a range of common DES solvents. Reaction mixtures underwent irradiation at 365 nm for 1 hour, after which time the reaction was diluted with H2O and extracted with EtOAc. Reaction products together with decomposed initiator 2,2-dimethoxy-2-phenylacetophenone (DPAP) and photosensitiser 2-methoxyacetophenone (MAP) were collected in the organic layer, whilst the DES could be completely recovered via concentration of the aqueous layer in vacuo. NMR analysis of the crude organic extract confirmed efficient product formation in all cases (see ESI†). Of the choline chloride (ChCl) based DESs, those containing glycerol (Gly) or ethylene glycol (EG) as hydrogen bond donors (HBDs) gave cleanest product extract (Fig. 2). The resulting extract from the reaction run with a urea (U) HBD gave small amounts of a solid urea impurity. The levulinic acid (LevA) HBD gave significant impurity in the extract, accounting for the majority of the crude mass. For both of the tetrabutylammonium chloride (TBACl) based DESs investigated, impurities resulting from the DES components was again observed by NMR. The betaine (Bet) containing DESs showed little impurity in the crude NMR, however over the course of the reaction these DESs underwent significant colour change and a precipitate was observed. Importantly, this indicates poor potential for recycling of these particular DESs, a critical aspect for their use in ‘green’ synthesis. Furthermore, the Bet DESs were the only solvents in which full conversion was not achieved. For this reason, the ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Gly (1:2) and ChCl:EG (1:2) DESs were considered for further study. Due to Gly displaying lower oral toxicity and higher boiling point than EG, together with very slightly improved profile of the extract, focus was directed to the ChCl:Gly DES for future investigation. However, it is important to note that the reaction proceeded to high or quantitative conversion in all DESs investigated. This highlights the potential for tailoring of the DES components to the individual reactants on a case-by-case basis if desired, and highlights the robustness of the reaction in a variety of DESs. To provide comparison, the reaction was also performed in Gly, but upon extraction a significant amount of the glycerol was obtained with the reaction products. Thus, it is necessary to use the DES for recyclability and efficient product isolation.
:Gly (1:2) and ChCl:EG (1:2) DESs were considered for further study. Due to Gly displaying lower oral toxicity and higher boiling point than EG, together with very slightly improved profile of the extract, focus was directed to the ChCl:Gly DES for future investigation. However, it is important to note that the reaction proceeded to high or quantitative conversion in all DESs investigated. This highlights the potential for tailoring of the DES components to the individual reactants on a case-by-case basis if desired, and highlights the robustness of the reaction in a variety of DESs. To provide comparison, the reaction was also performed in Gly, but upon extraction a significant amount of the glycerol was obtained with the reaction products. Thus, it is necessary to use the DES for recyclability and efficient product isolation.
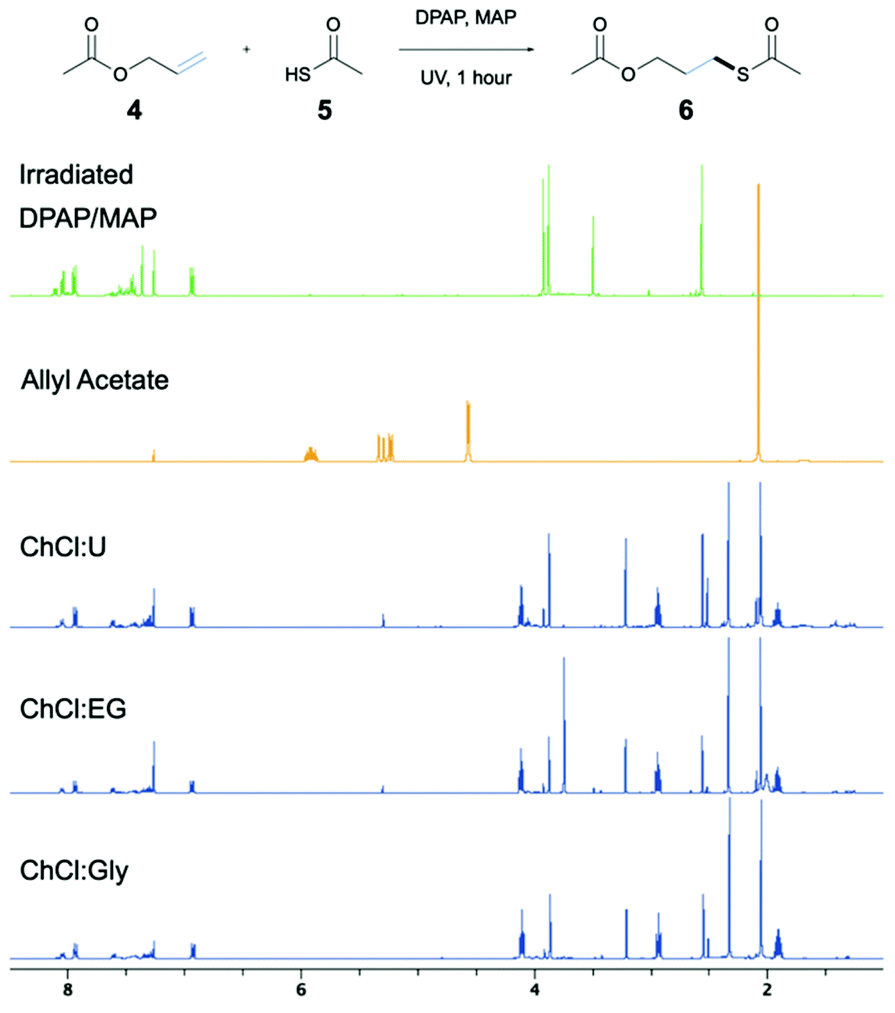 | ||
| Fig. 2 NMR spectra of crude products obtained from DES screen compared to allyl acetate starting material and DPAP/MAP irradiation products. | ||
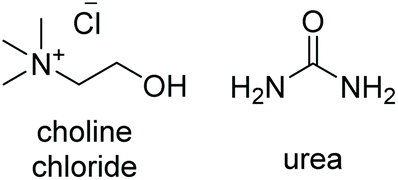
| DES | DES structure | Ratio | T m (°C) | T c (°C) | T deg (°C) | η @25 °C (cP) | ρ @25 °C (Kg m−3) |
|---|---|---|---|---|---|---|---|
| a Glass transition temperature (Tg). b Viscosity at 30 °C. | |||||||
| ChCl:U |
|
1:2 |
12 (ref. 7) | — | 211 (ref. 8) | 1398 (ref. 9) | 1197 (ref. 9) |
| ChCl:EG |
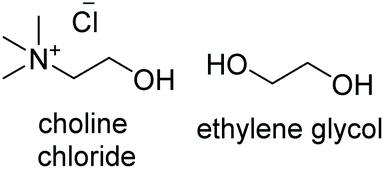
|
1:2 |
−25.8 | 0.0 | 199 (ref. 10) | 46 | 1116 |
| ChCl:LevA |
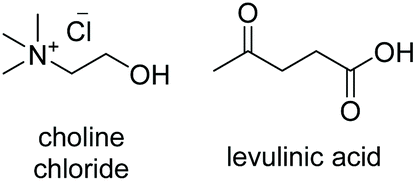
|
1:2 |
−76.1a | — | 180 (ref. 10) | 256 | 1138 |
| ChCl:Gly |
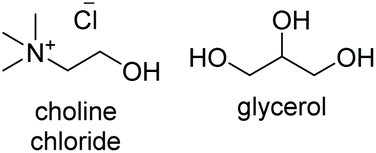
|
1:2 |
< −90 | — | 202 (ref. 10) | 375 | 1191 |
| Bet:EG |
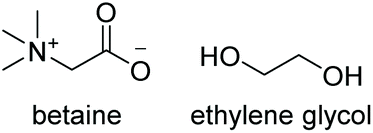
|
1:2 |
−17.9 | −11.9 | 183 | — | — |
| 59.6 | |||||||
| Bet:Gly |

|
1:2 |
−74.1a | — | 176 | 2102 | 1216 |
| TBACl:EG |
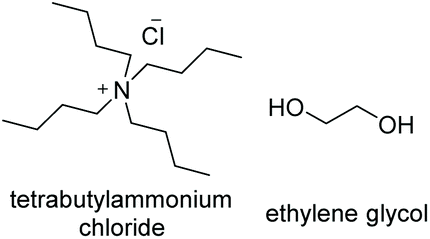
|
1:2 |
−60.8a | — | 154 (ref. 12) | 127b (ref. 11) | 1020 (ref. 13) |
| −30.1 (ref. 11) | |||||||
| TBACl:LevA |
|
1:2 |
−57.5a | — | 238 | 258 | 1019 |
Following identification of the optimum DES system, investigation into recycling of the DES solvent in multiple runs of the same reaction was investigated. The reaction between diethyl 2-allylmalonate and thioacetic acid, initiated using a DPAP/MAP photoinitiator/photosensitiser system was chosen for this study. The reaction in previously unused DES gave a yield of 71%. The DES solvent was subsequently collected by concentration of the aqueous layer following reaction workup and reused ×5 times for a total of 6 reactions. The 5th and final repeat gave an undiminished yield, showing no change due to reuse of the reaction solvent. NMR analysis of the DES in D2O prior to the initial reaction and after each subsequent reaction showed no change in the solvent. This result therefore demonstrates significant potential for the reuse of these solvents in the thiol–ene reaction, with considerable advantages for ‘green’ chemistry through reduction of waste produced. In addition to the DES, only a comparatively small amount of biodegradable EtOAc and H2O were used during reaction workup. Furthermore, only small, catalytic amounts (0.2 equivalents) of DPAP and MAP are required for the photoactivated process.
We next investigated the scope of the thiol–ene reaction in the ChCl:Gly DES system, with focus on demonstrating application in synthesis of biologically relevant or desirable products displaying a range of common functional and protecting groups (Scheme 1). Firstly, a small number of relatively simple thiol and alkene combinations were investigated, affording good yields (7–10). The reaction was next applied to the synthesis of modified amino acids (AAs), taking Boc-Cys-OMe as the thiol component of the reaction. Initial study of this reaction with cyclohexene gave a 67% yield of AA 11, demonstrating potential for the synthesis of further protected AAs, suitable for use in solid-phase peptide synthesis. To this end the reaction was demonstrated for synthesis of lipo-AA 13, protected diol-containing AA 14, and steroid-conjugated AA 15. Following from the success of the reaction with fully protected amino acids, the reaction was performed on Boc-Cys-OH (12). Furthermore, reaction of thioglucose showed potential for application to carbohydrate ligation (16). Further investigation into the reaction scope included use of thioacetic acid in an Acyl Thiol–Ene (ATE) reaction. Again, a general study into a number of simple examples gave promising yields (17–20). With these results in hand, the reaction was applied to synthesis of S-acetylated homocysteine 21. The reaction was then also applied to an ATE-initiated radical cyclisation cascade furnishing cyclopentane product (22).
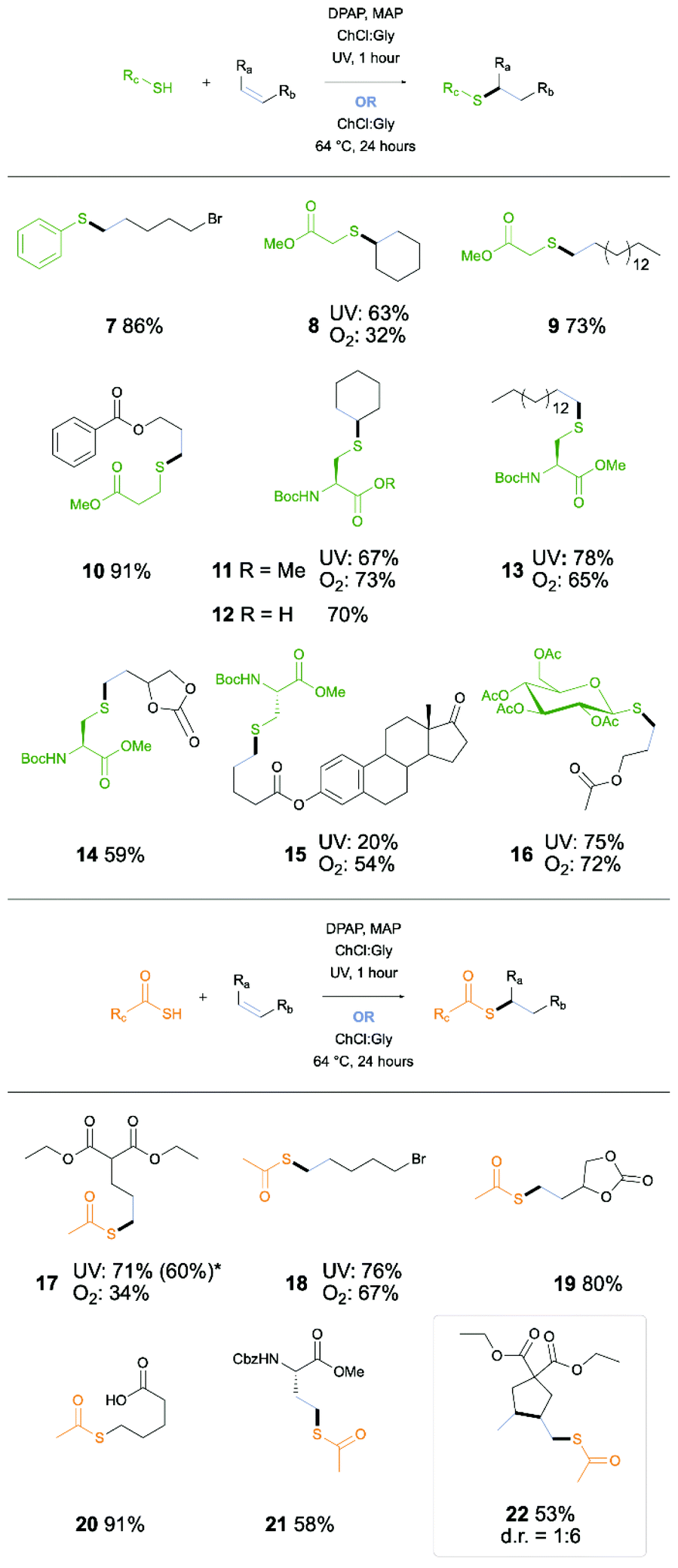 | ||
| Scheme 1 Scope for thiol–ene reaction in DESs under either UV or O2 initiation. *Reaction performed without MAP. | ||
Olefin-based radical syntheses in DESs in the presence of air have recently been reported in the form of Cu-catalysed polymerisation36 and Meyer–Schuster rearrangement.29 Having recently reported atmospheric oxygen mediated TEC and ATE reactions,37 we next investigated these conditions and their scope within the context of the DES solvent system (Scheme 1). Due to lack of requirement for an initiator or sensitiser, these initiation conditions further strengthen the ‘green’ chemistry characteristics of this methodology, with high atom economy and use of non-volatile, recyclable solvents. The non-volatility and non-toxicity of the solvents as well as lack of requirement for UV irradiation renders this chemistry highly suitable for benchtop practise. Oxygen mediated conditions do however trade ‘green’ chemistry advantages for extended reaction times. Whilst efficient oxygen incorporation into volatile solvents was previously achieved through refluxing of the reaction mix, this is not possible for the non-volatile DES system. Recently, the oxygen transfer rate for a number of DESs has been investigated, including study of the potential scope for tailoring reaction conditions to favour oxygen solvation.38 Of particular influence on this factor is the viscosity of the solvent, which can be readily influenced through heating or addition of water. Furthermore, increased stirring has been shown to improve yield in oxygen-dependent reactions for DESs.39 Through combination of these factors, the reaction proceeded with rapid stirring and gentle heating, demonstrating that the DES is capable of sufficient oxygen solvation for thiyl radical initiation. Furthermore, the addition of a small amount of water as co-solvent was applied to select examples (see ESI†), providing lower viscosity and also increasing the oxygen transfer rate.38 The oxygen-mediated conditions were first applied to simple thiol (8) and thioacid (17) examples for investigation of compatibility and efficiency. Of particular note is the result obtained for alkyl bromide example 18, in which bromide substitution products were not observed. Although, moderately reduced yields compared to the UV initiated process were afforded for these examples, (8, 32%; 17, 34%), the application to biologically relevant examples was undertaken. Demonstration of TEC via cysteinyl radical was investigated, coupling with cyclohexene (11), a hexadecene lipid alkene (9), and steroid alkene (13) as previously demonstrated in UV conditions. For both the cyclohexene and hexadecene examples, the yields of the O2 initiated reaction were comparable to UV (11, 73%, 13 63%). However, for the steroid example 15, the O2-initiated conditions gave a significant improvement yield, increasing from 20% to 54%. This is likely due to the improved solubility of the alkene-bearing steroid component of the reaction in the increased temperature of the oxygen mediated conditions, and may also be aided by the longer reaction time. Importantly, this demonstrates a further advantage of these conditions in addition to the green chemistry advantages previously mentioned, in tailoring the methodology to specific substrates that may exhibit difficult solubilities. O2 initiated conditions when applied to carbohydrate example 16 also gave a similar yield to that obtained under UV irradiation. Additionally, this exemplifies the potential for tailoring of both DES and reaction conditions to reactants or individual requirements (i.e. time constraints or green chemistry considerations).
The success of the Cys examples in the O2 initiated conditions gives an important mechanistic insight, as the postulated mechanism involves generation of H2O2,37 which has been shown to react with thiols to generate disulfides via the sulfenic acid,40 and also to oxidise thioethers to sulfones, including in DESs.41,42 The lack of side products of these types that are observed in this reaction implies that any H2O2 generated is only present in small quantities insufficient to cause side reactions. To further examine the potential for application of this methodology to large-scale synthesis of modified Cys residues, the O2 initiated synthesis of 11 was performed at a 6.5 mmol scale, providing the target in 47% yield. As the mechanism previously proposed for this reaction hypothesises the presence of peroxides,37 a potential hazard at larger scale, the peroxide concentration was monitored using MQuant peroxide strips. Throughout the reaction time, no peroxides were detected by this method. While this does not rule out the presence of any peroxide species, it further demonstrates that any peroxides formed are present at very low concentrations (<0.5 mg L−1). Due to the radical chain nature of the TEC reaction, only trace quantities of peroxide may be produced whilst still maintaining an efficient reaction. An additional important observation in these conditions is the direct reaction of highly lipophilic alkenes with relatively polar thiols. The DES provides sufficient solvation of both components for reaction, and results show that tailoring of reaction conditions can aid in solvation of more lipophilic, solid reagents.
Following from successful results involving protected Cys monomers, the application of the thiol–ene reaction in DES systems to bioconjugation of a peptide example was identified as a desirable further objective (Scheme 2). For this purpose, a portion of the N-terminal α-helix of human angiotensin-converting enzyme 2 (ACE-2) was selected. This human protein binds the SARS-CoV-2 spike protein in infection, and the EDLFYQ motif present in this protein has previously been shown to serve as the minimal sequence sufficient for binding of the spike protein.43 This interaction is an essential route of entry for SARS-CoV-2.44 For demonstration of the use of thiol–ene chemistry in DESs on an analogue of this peptide, the peptide sequence CEDLFYQ (23) was synthesised by Fmoc-SPPS, wherein the Cys residue replaces an alanine in the sequence of the human protein, and provides the thiol moiety for functionalisation of the peptide without alteration of the minimal sequence required for binding. The optimised UV-initiated conditions were applied to the peptide with excess cyclohexene, as this alkene is commercially available and had shown good yield in both UV- and O2-initiated TEC with Boc-Cys-OMe. However, only trace product was observed by HPLC and MS analysis, with the majority of the peptide starting material forming the disulfide dimer. To overcome this undesired reaction, the use of a reducing agent in the reaction mix was investigated. Further, it has been shown that, for selected choline based DES examples, the DES nanostructure is maintained up to 42 wt% H2O, while above 51 wt% the structure is disrupted.45 Thus, a DES/H2O (3:2) mix was used for efficient solvation of the peptide, with reduced viscosity when compared to the DES alone. This also serves as further demonstration of the potential for exploitation of DESs as designer solvents through adjusting of their ability to dissolve compounds of different hydrophobic or hydrophilic nature. The peptide starting material was dissolved in degassed DES/H2O (3:2) mix, along with 1 equivalent each of DPAP, MAP and TES and 5 equiv. of cyclohexene and irradiated under UV for 3 hours. An aliquot was taken and analysed by RP-HPLC, revealing 86% conversion of the starting material peak with retention time of 15.19 min and clean reaction profile. In turn, consistent with consumption of the free thiol and installation of the hydrophobic cyclohexyl group, a new peak with longer retention time of 16.93 min was observed, MS analysis of which confirmed the identity of the modified peptide product. Semi-preparative HPLC of an aliquot of the reaction mix provided the product in 56% isolated yield.
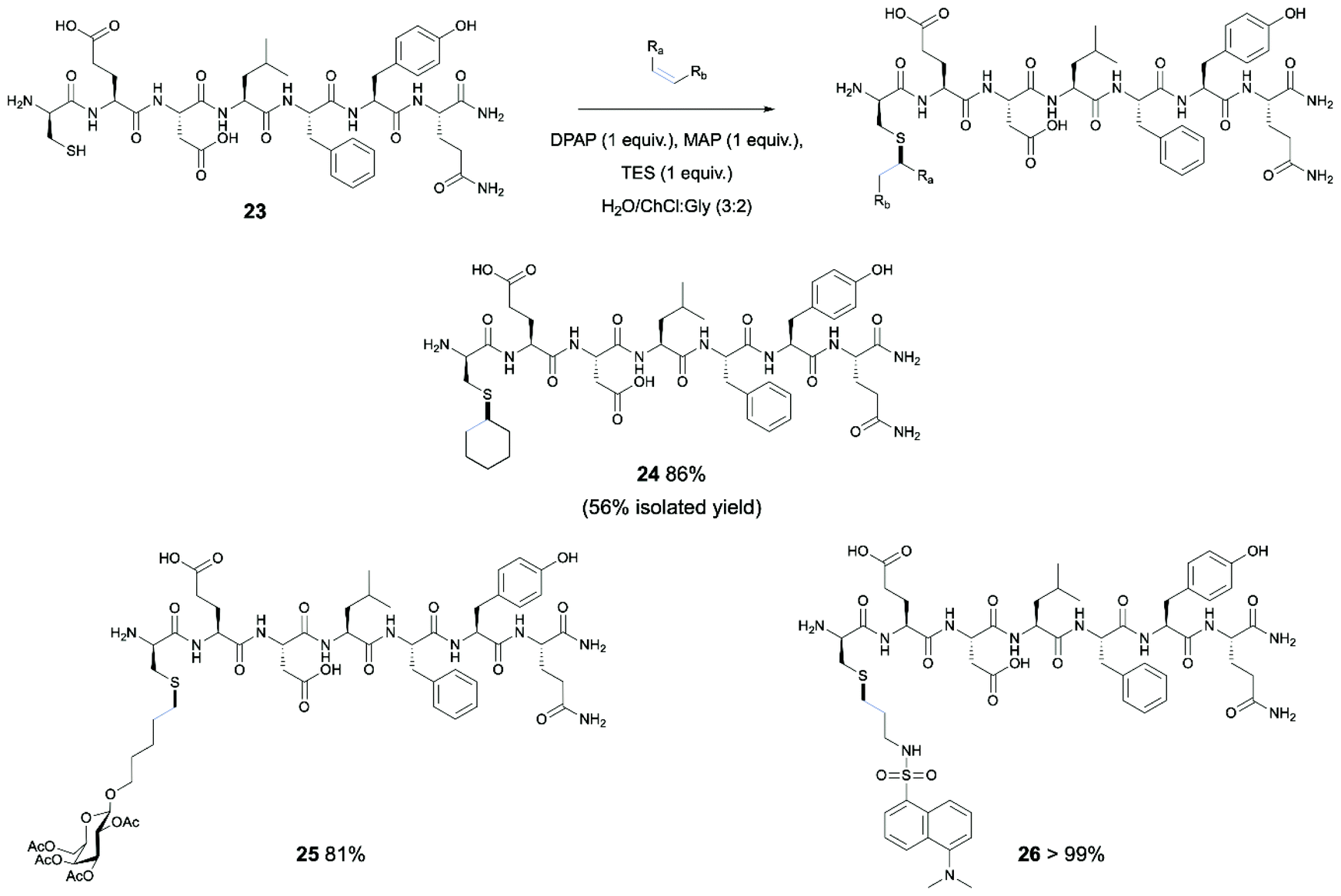 | ||
| Scheme 2 Modification of the CEDLFYQ helix via TEC in DES system. Conversions obtained via HPLC analysis. | ||
Furthermore, the oxygen-initiated conditions were investigated for conjugation of the cyclohexyl group to the CEDLFYQ sequence, again incorporating an equivalent of TES as a reducing agent due to the increased likelihood of disulfide formation when stirred in oxygenated conditions. Unfortunately, after 24 h, HPLC analysis showed no conversion of the starting material. After a further 24 h (48 h total), still no conversion was observed. Despite the presence of TES, small amounts of disulfide were observed.
For this reason, and with the success of the UV-initiated reaction in hand, we turned to synthesis of a carbohydrate-modified peptide, incorporating a galactosyl moiety onto the Cys residue of the CEDLFYQ sequence through UV-initiated TEC. The reaction proceeded with 81% conversion. Importantly, this demonstrates a “post-translational” approach to peptide glycosylation via TEC in the DES system.
Finally, we undertook the synthesis of a fluorophore-tagged peptide, with potential for application in study of SARS-CoV-2 infection. For this purpose, the previously utilised reaction conditions were applied to conjugation of the CEDLFYQ sequence with a dansyl alkene (Scheme 2). The UV-initiated conditions previously applied to this sequence were utilised, gratifyingly giving complete consumption of the peptide starting material with >99% conversion to 21 by HPLC.
Conclusions
We have developed a green system for formation of thioester and thioether linkages by TEC utilising DESs, with particular focus on synthesis of modified amino acids and peptide modification. The reaction showed compatibility with all DESs investigated and in the case of the ChCl:Gly DES, it was shown that the solvent can be reused without impact on reaction outcome. The reaction was first demonstrated for simple thiol and thioacid examples, and then utilised for synthesis of modified amino acids. Finally, the reaction was applied to modification of an unprotected peptide derived from the minimal sequence of human ACE-2, allowing synthesis of a lipopeptide, glycopeptide and fluorescently labelled peptide. We therefore envisage application of this methodology in facile synthesis of modified peptides or proteins, either using modified AA monomers or modification of unprotected sequences.
Author contributions
M. N., A. M., L. G. and E. M. S. devised the study and prepared the manuscript. M. N. performed experiments.Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Fetting, The European Green Deal, ESDN Report, December 2020, ESDN Office, Vienna.
- A European Green Deal European Commission, https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal, (accessed 7 May 2021).
- T. Welton, Curr. Opin. Green Sustain. Chem., 2018, 13, A7–A9 CrossRef.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- V. Migliorati, F. Sessa and P. D'Angelo, Chem. Phys. Lett., 2019, 737, 100001 CrossRef.
- W. Chen, Z. Xue, J. Wang, J. Jiang, X. Zhao and T. Mu, Acta Phys.–Chim. Sin., 2018, 34, 904–911 CAS.
- V. Agieienko and R. Buchner, J. Chem. Eng. Data, 2019, 64, 4763–4774 CrossRef CAS.
- J. González-Rivera, E. Husanu, A. Mero, C. Ferrari, C. Duce, M. R. Tinè, F. D'Andrea, C. S. Pomelli and L. Guazzelli, J. Mol. Liq., 2020, 300, 112357 CrossRef.
- F. S. Mjalli, J. Naser, B. Jibril, V. Alizadeh and Z. Gano, J. Chem. Eng. Data, 2014, 59, 2242–2251 CrossRef CAS.
- M. F. Majid, H. F. Mohd Zaid, C. F. Kait, N. A. Ghani and K. Jumbri, J. Mol. Liq., 2019, 294, 111588 CrossRef CAS.
- D. K. Panda and B. L. Bhargava, J. Mol. Liq., 2021, 335, 116139 CrossRef CAS.
- Y. Liu, J. B. Friesen, J. B. McAlpine, D. C. Lankin, S. N. Chen and G. F. Pauli, J. Nat. Prod., 2018, 81, 679–690 CrossRef CAS PubMed.
- G. Colombo Dugoni, A. Mezzetta, L. Guazzelli, C. Chiappe, M. Ferro and A. Mele, Green Chem., 2020, 22, 8680–8691 RSC.
- J. L. K. Mamilla, U. Novak, M. Grilc and B. Likozar, Biomass Bioenergy, 2019, 120, 417–425 CrossRef CAS.
- E. Husanu, A. Mero, J. G. Rivera, A. Mezzetta, J. C. Ruiz, F. D′andrea, C. S. Pomelli and L. Guazzelli, ACS Sustainable Chem. Eng., 2020, 8, 18386–18399 CrossRef.
- P. V. de Almeida Pontes, I. Ayumi Shiwaku, G. J. Maximo and E. A. Caldas Batista, Food Chem., 2021, 352, 129346 CrossRef CAS PubMed.
- J. Wu, Q. Liang, X. Yu, L. Qiu-Feng, L. Ma, X. Qin, G. Chen and B. Li, Adv. Funct. Mater., 2021, 2011102 CrossRef CAS.
- S. Arnaboldi, A. Mezzetta, S. Grecchi, M. Longhi, E. Emanuele, S. Rizzo, F. Arduini, L. Micheli, L. Guazzelli and P. R. Mussini, Electrochim. Acta, 2021, 380, 138189 CrossRef CAS.
- M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef PubMed.
- S. E. Hooshmand, R. Afshari, D. J. Ramón and R. S. Varma, Green Chem., 2020, 22, 3668–3692 RSC.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS.
- S. Nejrotti, M. Iannicelli, S. S. Jamil, D. Arnodo, M. Blangetti and C. Prandi, Green Chem., 2020, 22, 110–117 RSC.
- C. Y. Chien and S. S. Yu, Chem. Commun., 2020, 56, 11949–11952 RSC.
- C. Vidal, J. García-Álvarez, A. Hernán-Gõmez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969–5973 CrossRef CAS PubMed.
- J. García-Álvarez, E. Hevia and V. Capriati, Chem. – Eur. J., 2018, 24, 14854–14863 CrossRef PubMed.
- D. Arnodo, S. Ghinato, S. Nejrotti, M. Blangetti and C. Prandi, Chem. Commun., 2020, 56, 2391–2394 RSC.
- N. Rios-Lombardia, L. Cicco, B. Kota Yamamoto, J. A. Hernández-Fernández, F. Morí, V. Capriati, J. Garcia-Alvarez and J. Gonzalez-Sabin, Chem. Commun., 2020, 56, 15165–15168 Search PubMed.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- L. McSweeney, F. Dénès and E. M. Scanlan, Eur. J. Org. Chem., 2016, 2080–2095 CrossRef CAS.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 CrossRef CAS PubMed.
- J. T. McLean, A. Benny, M. D. Nolan, G. Swinand and E. M. Scanlan, Chem. Soc. Rev., 2021, 50, 10857–10894 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- L. Quirós-Montes, G. A. Carriedo, J. García-Álvarez and A. P. Soto, Green Chem., 2019, 21, 5865–5875 RSC.
- R. O. McCourt and E. M. Scanlan, Chem. – Eur. J., 2020, 26, 15804–15810 CrossRef CAS PubMed.
- N. Zhang, F. Steininger, L. E. Meyer, K. Koren and S. Kara, ACS Sustainable Chem. Eng., 2021, 9, 8347–8353 CrossRef CAS.
- L. Huang, J. P. Bittner, P. Domínguez de María, S. Jakobtorweihen and S. Kara, ChemBioChem, 2020, 21, 811–817 CrossRef CAS PubMed.
- D. Luo, S. W. Smith and B. D. Anderson, J. Pharm. Sci., 2005, 94, 304–316 CrossRef CAS PubMed.
- D. Y. Dai, L. Wang, Q. Chen and M. Y. He, J. Chem. Res., 2014, 38, 183–185 CrossRef CAS.
- F. G. Bordwell and P. J. Boutan, J. Am. Chem. Soc., 2002, 79, 717–722 CrossRef.
- R. C. Larue, E. Xing, A. D. Kenney, Y. Zhang, J. A. Tuazon, J. Li, J. S. Yount, P. K. Li and A. Sharma, Bioconjugate Chem., 2021, 32, 215–223 CrossRef CAS PubMed.
- A. Sadremomtaz, Z. M. Al-Dahmani, A. J. Ruiz-Moreno, A. Monti, C. Wang, T. Azad, J. C. Bell, N. Doti, M. A. Velasco-Velázquez, D. de Jong, J. de Jonge, J. Smit, A. Dömling, H. van Goor and M. R. Groves, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.1c00477.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03714e |
| This journal is © The Royal Society of Chemistry 2022 |