
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirecting the H2-driven selective regeneration of NADH via Sn-doped Pt/SiO2†
Joseph W. H.
Burnett‡
ab,
Jianwei
Li‡
a,
Alan J.
McCue
 c,
Panagiotis N.
Kechagiopoulos
b,
Russell F.
Howe
c and
Xiaodong
Wang
*a
c,
Panagiotis N.
Kechagiopoulos
b,
Russell F.
Howe
c and
Xiaodong
Wang
*a
aChemical Engineering, Department of Engineering, Lancaster University, Lancaster LA1 4YW, UK. E-mail: xiaodong.wang@lancaster.ac.uk
bChemical and Materials Engineering, School of Engineering, University of Aberdeen, Aberdeen AB24 3UE, UK
cChemistry Department, University of Aberdeen, Aberdeen AB24 3UE, UK
First published on 31st January 2022
Abstract
H2-Driven NADH regeneration has long suffered from low selectivity when non-enzymatic, particularly heterogeneous catalysts, are used. In addition to the unselective nature of the catalysts, the typically unmeasured NAD+ conversion has inevitably hindered catalyst development. Here we report Sn-doped Pt/SiO2 catalysts for the selective regeneration of NADH and show that doping Pt/SiO2 with 10 at% Sn can deliver a selectivity of 90% (at full conversion) using H2. We propose that this is a result of Sn disturbing the Pt ensemble, altering the mode of NAD+ adsorption and directing the reduction to the 1,4-position of the nicotinamide ring.
Oxidoreductases constitute the largest group of the six different functional classes of enzymes and are capable of performing a wide range of industrially significant reactions under environmentally benign conditions with unparalleled selectivity.1–3 They have been utilised for a broad scope of applications, ranging from the production of chiral compounds for the pharmaceutical sector to atmospheric CO2 reduction to green fuels.4–6 To perform these reactions, the addition of the reduced form of the redox cofactor, nicotinamide adenine dinucleotide (1,4-NAD(P)H), is required. During the course of an NAD-dependant enzymatic reduction, hydride transfer occurs to the substrate from 1,4-NAD(P)H, with the latter concurrently oxidised to NAD(P)+ (Fig. 1). Due its high price and stoichiometric consumption, regeneration of the cofactor is essential to make these reactions commercially viable.7
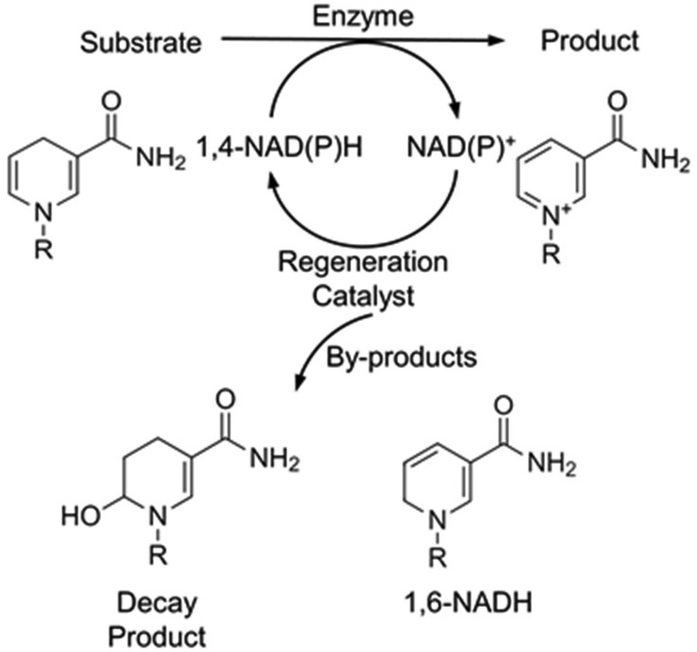 | ||
| Fig. 1 Schematic of an NAD-dependent bioreduction with in situ 1,4-NAD(P)H regeneration and the by-products that are formed in this study, R: adenine dinucleotide. | ||
Various approaches have been taken to the regeneration of 1,4-NAD(P)H (i.e. the reduction of NAD(P)+ back to 1,4-NAD(P)H), including the use of both enzymatic and non-enzymatic systems.8,9 Enzymatic regeneration has been a popular approach and due to the inherent selectivity of enzymes, exclusive formation of 1,4-NAD(P)H is achieved.10,11 However, because of the high cost and instability of the enzymes, along with the complexity of product separation due to the need for sacrificial substrates/products/enzymes, research into non-enzymatic regeneration is becoming more appealing.12 Non-enzymatic regeneration has spread across most types of catalysis, including photocatalysis, electrocatalysis, homogeneous and more recently, heterogeneous catalysis.13–25 However, process sustainability is still a major challenge, e.g. use of water-soluble electron mediators, photosensitisers, sacrificial electron donors or organometallic complexes, with subsequent cost and energy intensive separation.
The capability of employing H2 as a clean reducing agent and the ease of separation/recyclability make recently developed supported metal systems an attractive option. However, selectivity has been a crucial challenge. For example, we have studied Pt on a range of supports (SiO2, C, MgO and Fe3O4), achieving a maximum selectivity to 1,4-NADH of 25% when NAD+ is fully consumed.17,26 Recently, Yang et al. assessed a group of Pt/TiO2 catalysts in H2 and achieved a selectivity (and yield) of 63%.27 To date, supported metal catalysts have failed to deliver any further improvements in selectivity/yield. To overcome this, Vincent et al. have borrowed the natural performance of enzymes and used a commercial Pd/C (and carbon) to adsorb NAD+ reductase, which then exhibits exclusive selectivity to 1,4-NADH.28 Nevertheless, developing effective strategies to improve the intrinsic selectivity of supported metals alone has not been achieved, constituting a challenge that remains unsolved. Bimetallic alloy catalysts, as excellent candidates for selective hydrogenation reactions,29–31 may potentially contribute also to selective NADH regeneration. In this work, we propose to direct the selectivity by controlling the mode of NAD+ adsorption on Pt and therefore have designed a group of Sn-doped Pt/SiO2 catalysts intended to contain interrupted Pt ensembles. The results demonstrate that PtSn alloys enhance the selectivity of 1,4-NADH from 30 to 90%, the highest yet achieved with a heterogeneous catalyst to our knowledge.
We envisioned that by doping Pt/SiO2 catalysts with Sn, we could direct the selectivity by controlling the mode of NAD+ adsorption on Pt. With that in mind, a series of PtSn/SiO2 catalysts were prepared by wet impregnation whereby SiO2 was co-impregnated with aqueous solutions of H2PtCl6 and SnCl2 (full details in the ESI†). In all cases, the Pt loading was fixed at 1 wt% and the amount of Sn added is expressed in terms of nominal atomic percent (at%). For example, Pt90 refers to a catalyst containing 90 at% Pt and 10 at% Sn supported on SiO2. A 1 wt% Pt/SiO2 (labelled as Pt100) was also prepared by wet impregnation for comparison purposes. TEM images of Pt100 reveal well dispersed spherical Pt nanoparticles with a narrow size distribution and an average particle diameter of 2.2 nm (Fig. 2a). Sn doping results in an increase in the average particle size to 11.1, 10.9 and 11.1 nm for Pt90, Pt75 and Pt50, respectively (Table 1 and Fig. 2b–d) and is consistent with the literature for this type of preparation.32,33 The actual Pt and Sn loadings were determined by ICP analysis (see Table 1), which are in excellent agreement with the nominal ones. STEM/EDX results for the Sn-doped catalysts are shown in Fig. 2e–g. It can be seen that although there are some areas of isolated Pt and Sn, the majority of the Pt and Sn is located within the same regions, showing that bimetallic PtSn nanoparticles have been formed. Elemental composition determined by EDX of Pt90 is very close to the nominal composition (Pt91Sn9 vs. Pt90Sn10, respectively), indicating a homogeneous distribution of elements across the support. However, upon increasing Sn addition, the elemental composition determined by EDX differs from the nominal composition. The elemental composition determined by EDX of Pt75 is Pt84Sn16 and Pt50 is Pt39Sn61, showing a more heterogeneous distribution of elements as the amount of Sn-doping increases, which is not uncommon with the higher degrees of Sn-doping.34,35
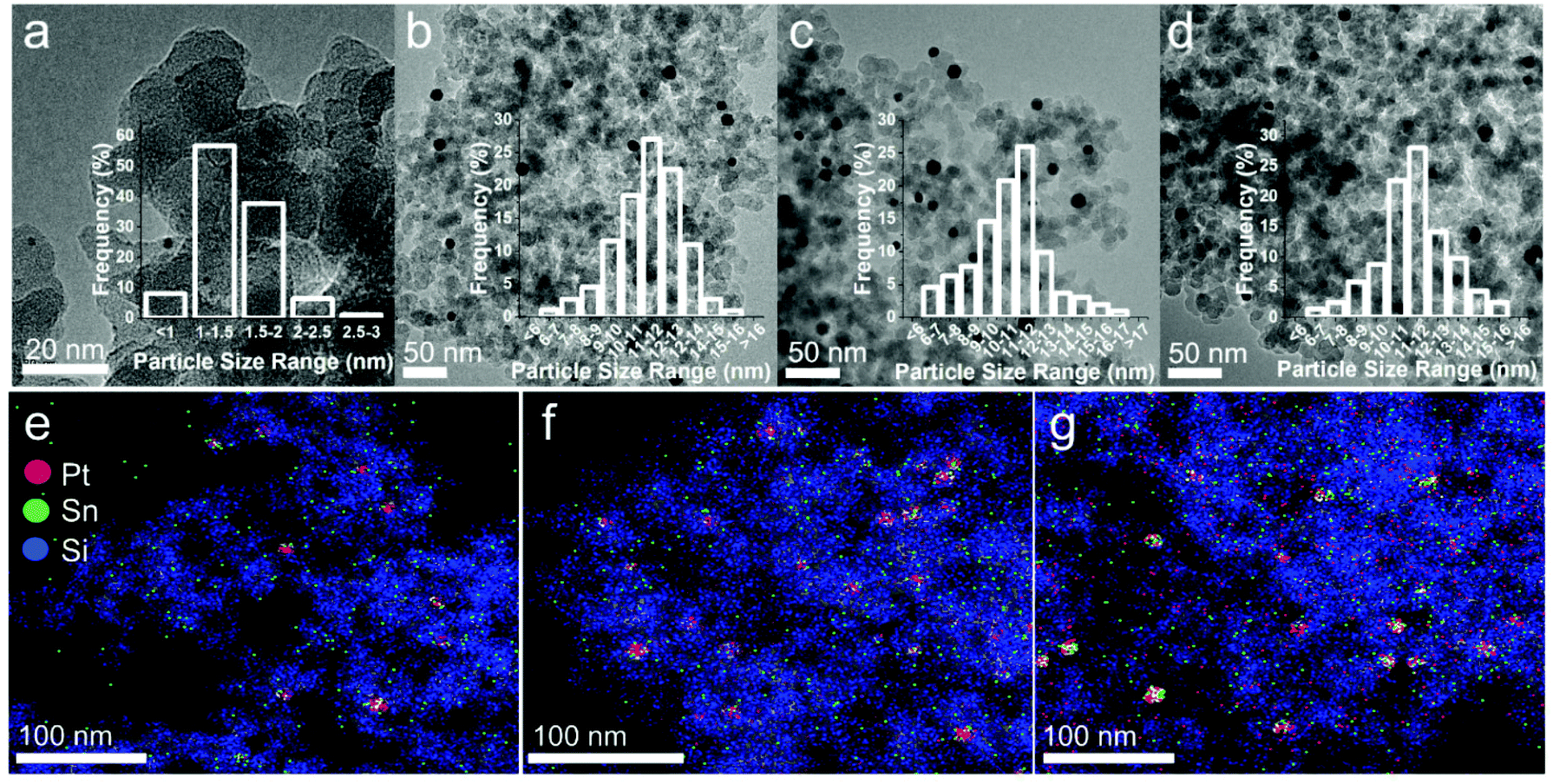 | ||
| Fig. 2 Representative TEM images of (a) Pt100, (b) Pt90, (c) Pt75 and (d) Pt50, with the associated particle size distribution histograms. STEM/EDX images of (e) Pt90, (f) Pt75 and (g) Pt50. | ||
| Pt100 | Pt90 | Pt75 | Pt50 | |
|---|---|---|---|---|
| a Determined by ICP-OES. b Determined by XPS. c Determined by XRD. d At full conversions except Pt50. | ||||
| Pt (at%) | 100 | 90 | 75 | 50 |
| Sn (at%) | 0 | 10 | 25 | 50 |
| CO/Pt | 0.35 | 0.09 | 0.07 | 0.05 |
| d TEM (nm) | 2.2 | 11.1 | 10.9 | 11.1 |
| Pt (wt%)a | 1.01 | 0.98 | 1.00 | 0.98 |
| Sn (wt%)a | - | 0.07 | 0.21 | 0.62 |
| Sn/Pt (atomic)a | 0 | 10/85 | 25/73 | 50/48 |
| Surface area (m2 g−1) | 165 | 145 | 152 | 151 |
| Pt0 (%)b | 72 | 75 | 77 | 80 |
| Pt2+ (%)b | 28 | 25 | 23 | 20 |
| 2θmax (°) | 67.7 | 67.5 | 67.1 | 66.7 |
| Lattice parameter (Å)c | 3.913 | 3.924 | 3.942 | 3.965 |
| TOF (h−1) | 1462 | 514 | 309 | 93 |
| S 1,4-NADH (%)d | 30 | 90 | 82 | 71 |
Fig. 3a shows the XRD patterns of the catalysts with Pt100 exhibiting peaks at 39.7, 46.2, 67.7 and 81.5°, corresponding to the (111), (200), (220) and (311) reflections typical of the face-centred cubic (fcc) crystallite structure of Pt (JCPDS-ICDD 00-004-0802). Upon Sn addition (Pt90, Pt75 and Pt50), the peaks corresponding to fcc Pt decrease and peaks at 30.1, 41.8, 44.1 and 62.5° appear, which can be attributed to (101), (102), (110) and (202) reflections, indicative of a hexagonal PtSn alloy (JCPDS-ICDD 00-025-0614). The diffraction peaks of the Pt (100) reflections are consistently shifted to lower 2θ values with increasing Sn addition, indicating enhanced Sn incorporation into the fcc structure of Pt.36,37 Due to the large ionic radius of Sn relative to Pt, examination of the (220) reflections reveals an expansion of the lattice parameter (from 3.915 to 3.965 Å) as Sn addition increases (Table 1), providing further evidence of the extent of alloy formation. N2 physisorption experiments reveal that all catalysts exhibit type IV isotherms with a hysteresis loop characteristic of mesoporous materials (Fig. S1a–d†). The surface area, pore volume and pore sizes of the catalysts are very similar (154 m2 g−1 (±16%), 0.86 cm3 g−1 (±6%) and 27.4 nm (±3%), respectively).
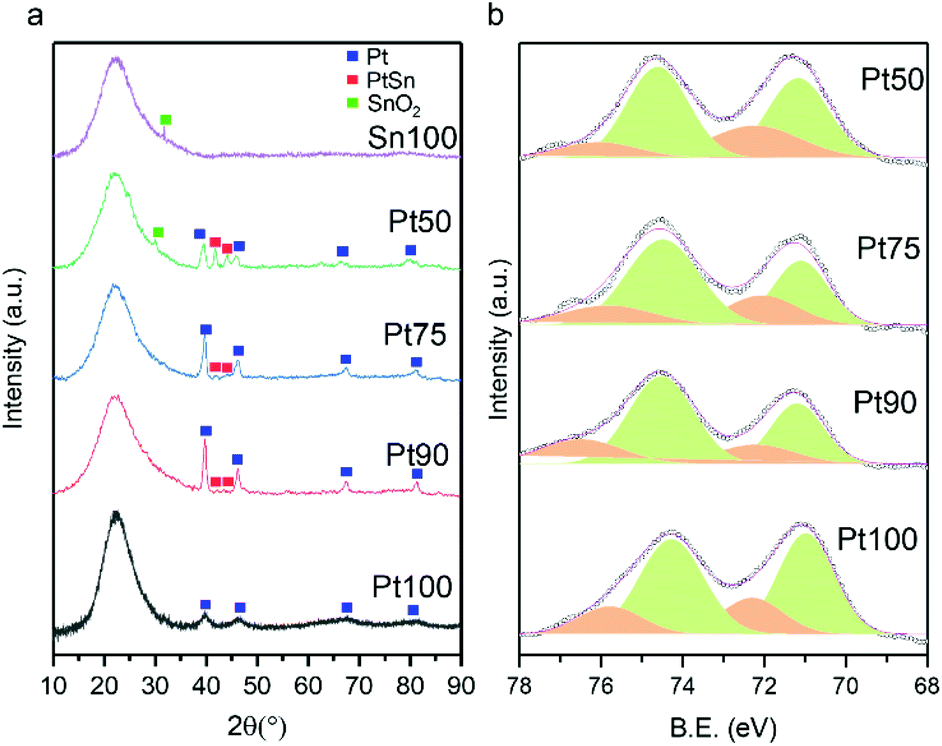 | ||
| Fig. 3 (a) XRD patterns of the Pt and Sn-doped Pt catalysts, with Sn/SiO2 as a reference. (b) Pt 4f XPS spectra of the set of catalysts showing Pt0 (Green), Pt2+ (orange) and experimental data (circles). | ||
The CO chemisorption capacities of the catalysts are summarised in Table 1. The irreversible chemisorption of CO is observed in all catalysts and the amount of CO chemisorption decreased with increasing Sn incorporation. Since the particle sizes of the Sn doped catalysts are similar, the addition of Sn will dilute the surface Pt ensembles and thus result in less CO chemisorption as the amount of Sn incorporation increases. XPS was used to probe the surface chemical composition of the catalysts and is shown in Fig. 3b. The binding energies of Pt 4f5/2 and Pt 4f7/2 in the undoped catalyst (Pt100) are centred around 71.1 and 74.3 eV but deconvolution reveals the presence of small amounts of Pt2+. The composition of Pt oxidation states in all catalysts is summarised in Table 1 and it can be observed that the amount of metallic Pt increases (72 to 80%) as the amount of Sn increases. This is in line with the Sn 3d spectra shown in Fig. S2,† where a clear positive shift can be observed with increasing Sn, evidencing the electron transfer from Sn to Pt. As the extent of Sn doping increases, there is an observed positive shift in binding energies (+0.5 eV in Pt50) of the Pt 4f5/2 and Pt 4f7/2 peaks, indicating PtSn alloy formation consistent with previous literature.38–40 For example, Chen et al. observed a shift of +0.6 eV and Li et al. a shift of +0.4 eV with their supported bimetallic PtSn catalysts.39,40
The catalytic performance of the Sn doped Pt/SiO2 catalysts for NADH regeneration with hydrogen was evaluated as described in the ESI,† and the time on stream profiles are shown in Fig. 4. Although NAD+ conversion can reveal fundamental and important characteristics of the regeneration reaction, due to the difficulty of its experimental examination, it is hardly reported in the literature.41 Here we employed a laboratory-developed analytical method based on enzymatic assays and measured the conversion data for each experimental points.42 The Pt100, Pt90 and Pt75 catalysts all show an NAD+ conversion approaching 100% after >80 minutes reaction time, whereas the Pt50 catalyst plateaus at around 45% conversion. Taking account of the surface Pt concentrations determined by CO chemisorption shows TOF values increasing with decreasing Sn concentration, with Pt50, Pt75, Pt90 and Pt100 achieving TOFs of 93, 309, 514 and 1462 h−1, respectively. The cause of the reduction in activity is that increasing the extent of Sn doping will cause a dilution of Pt adsorption ensembles. It was suggested by Yang et al. that the hydrogenation of NAD+ on Pt (supported on TiO2) occurs by the end-on adsorption of the nicotinamide ring via the carbonyl group,27 and since less surface Pt is available to accommodate CO adsorption, this is likely to contribute to the reduction in activity.
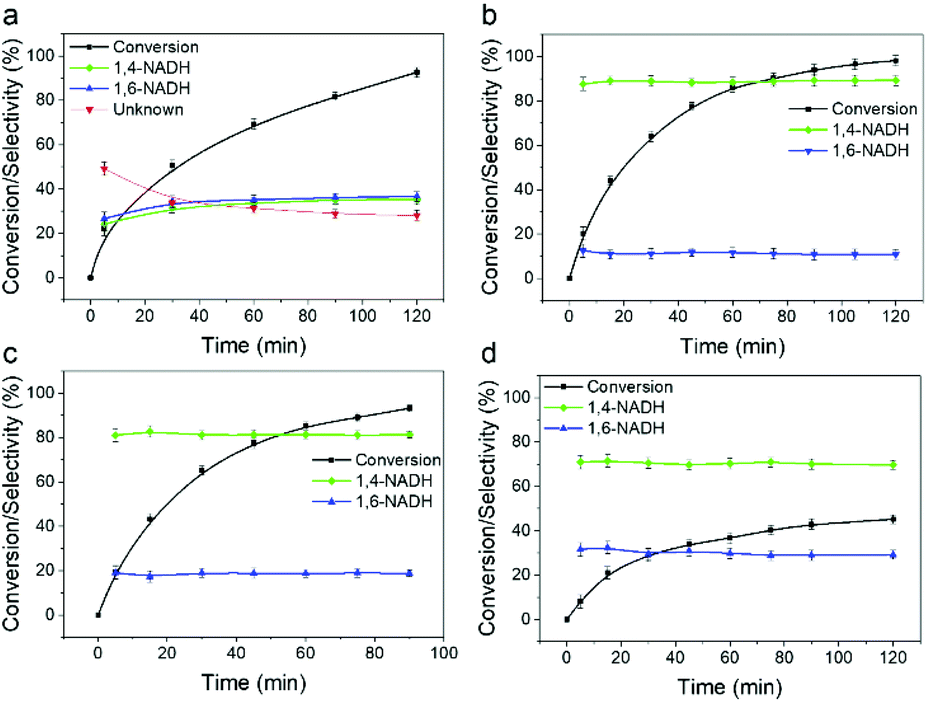 | ||
| Fig. 4 Conversion and selectivity as a function of time for (a) Pt100, (b) Pt90, (c) Pt75 and (d) Pt50. Reaction conditions: 1.5 mM NAD+ (a) or 100 μM NAD+ (b–d), 0.005 g (a and b)/0.010 g (c)/0.020 g (d) of catalyst, 22 °C, 9 bar H2 in a 0.1 M, pH 7 phosphate buffer. | ||
Enzymatic assays42 were first used to systematically examine the selectivity data shown in Fig. 4. Sn doping has a clear effect on the selectivity to 1,4-NADH. The only products detected by the assays for the Sn doped catalysts are 1,4- and 1,6-NADH (with already closed mass balance), i.e. no production of the 6-hydroxytetrahydropyridine previously reported to be formed by Pt/SiO2 catalysed decay of 1,4-NADH.17 The optimum catalyst is the Pt90, which gives a selectivity to 1,4-NADH of ∼90% at all conversions; this decreases as the Sn content is further increased. The maximum yields of 1,4-NADH of 88% and 76% for the Pt90 and Pt75 catalysts are significantly improved from those previously reported for Pt/TiO2 (up to 63%).27 The selectivity of the reaction was then investigated by analysing products with 1H NMR spectroscopy. Fig. 5 shows the 1H NMR spectra of the pure reactant (NAD+), desired product (1,4-NADH) and the reaction mixture obtained from Pt100 and Pt90 catalysts at ∼50% conversion. The 1H NMR spectra of NAD+ (Fig. 5a) and 1,4-NADH (Fig. 5d) shows the main peaks of interest that are characteristic of these molecules. These are singlets located at 9.20 ppm and 6.86 ppm, which correspond to the 2-H of the nicotinamide ring of NAD+ and 1,4-NADH, respectively. Fig. 5b shows the resulting 1H NMR spectrum obtained from the regeneration reaction using Pt100. It can be seen that the spectrum contains the characteristic singlets at 9.20 ppm and 6.86 ppm of NAD+ and 1,4-NADH, but also contains two new peaks at 7.01 and 7.13 ppm. The appearance of the two new peaks are attributed to the formation of 1,6-NADH and the decay product (6-hydroxytetrahydropyridine), respectively.17 The NMR spectrum obtained from the Pt90 catalyst (Fig. 5c) shows only the signals of 1,4-NADH with a trace of 1,6 NADH, indicating a much more selective response and consistent with the enzymatic assay results.
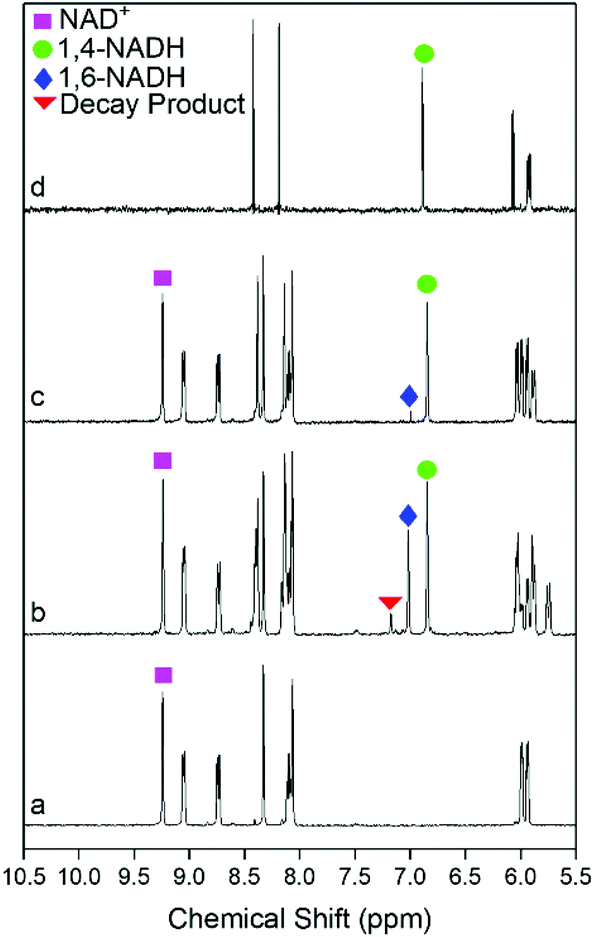 | ||
| Fig. 5 1H NMR spectra of (a) NAD+ (6 mM, 0.1 M pH 7 phosphate buffer in D2O), (b) reaction mixture of Pt100 (6 mM NAD+, 0.1 M pH 7 phosphate buffer in D2O, 40 mg of catalyst, 22 °C and 9 bar of H2), (c) reaction mixture of Pt90 (6 mM NAD+, 0.1 M pH 7 phosphate buffer in D2O, 40 mg of catalyst, 22 °C and 9 bar of H2) and (d) 1,4-NADH (6 mM, 0.1 M pH 7 phosphate buffer in D2O). | ||
The independence of selectivity from conversion of the Sn-doped catalysts is another important finding, which suggests a parallel reaction pathway/mechanism with the relative rates of 1,4- and 1,6-NADH formation determined by the amount of Sn. On the pure Pt surface of Pt100, adsorption of the nicotinamide ring is likely to occur in a planar fashion, resulting in unselective reduction of the nicotinamide ring. However, Sn incorporation will hinder the planar adsorption of the ring and end-on adsorption (via the 1,4 position) is likely to be preferred, especially since this is the least sterically hindered position of the ring, increasing the selectivity to 1,4-NADH. Although a precise mechanistic understanding has yet to be realised, the results show a clear trend between the degree of PtSn alloying and selectivity, attributable to differing adsorption modes of NAD+.
Conclusions
In summary, a series of Sn doped Pt/SiO2 catalysts were prepared and tested for the regeneration of 1,4-NADH using H2. It was found that the catalyst doped with the lowest amount of Sn (Pt90) gave rise to the highest selectivity to 1,4-NADH, achieving 90%. This is a significant improvement on the previously reported heterogeneous catalysts (which attained a maximum selectivity of 63%27). The improved selectivity can be attributed to directing the adsorption mode of NAD+ by disturbing the Pt ensemble. We hope these results will not only show the feasibility of heterogeneous catalysts for NADH regeneration but also provide a strategy for designing selective regeneration catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC New Horizons scheme (EP/V048635/1) and The Royal Society (ICA\R1\180317). We are also grateful for support from the UK Catalysis Hub funded by EPSRC grant reference EP/R026645/1.Notes and references
- H. Chen, F. Y. Dong and S. D. Minteer, Nat. Catal., 2020, 3, 225–244 CrossRef.
- C. K. Prier and B. Kosjek, Curr. Opin. Chem. Biol., 2019, 49, 105–112 CrossRef CAS PubMed.
- R. N. Patel, ACS Catal., 2011, 1, 1056–1074 CrossRef CAS.
- H. H. Sun, H. F. Zhang, E. L. Ang and H. M. Zhao, Bioorg. Med. Chem., 2018, 26, 1275–1284 CrossRef CAS PubMed.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
- A. T. Martinez, F. J. Ruiz-Duenas, S. Camarero, A. Serrano, D. Linde, H. Lund, J. Vind, M. Tovborg, O. M. Herold-Majumdar, M. Hofrichter, C. Liers, R. Ullrich, K. Scheibner, G. Sannia, A. Piscitelli, C. Pezzella, M. E. Sener, S. Kilic, W. J. H. van Berkel, V. Guallar, M. F. Lucas, R. Zuhse, R. Ludwig, F. Hollmann, E. Fernandez-Fueyo, E. Record, C. B. Faulds, M. Tortajada, I. Winckelmann, J. A. Rasmussen, M. Gelo-Pujic, A. Gutierre, J. C. del Rio, J. Rencoret and M. Alcalde, Biotechnol. Adv., 2017, 35, 815–831 CrossRef CAS PubMed.
- W. F. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef CAS PubMed.
- T. Quinto, V. Kohler and T. R. Ward, Top. Catal., 2014, 57, 321–331 CrossRef CAS.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- J. Preissler, H. A. Reeve, T. Z. Zhu, J. Nicholson, K. Urata, L. Lauterbach, L. L. Wong, K. A. Vincent and O. Lenz, ChemCatChem, 2020, 12, 4853–4861 CrossRef CAS.
- X. D. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. F. Shi, Chem, 2017, 2, 621–654 CAS.
- L. Tensi and A. Macchioni, ACS Catal., 2020, 10, 7945–7949 CrossRef CAS.
- M. W. Yuan, M. J. Kummer, R. D. Milton, T. Quah and S. D. Minteer, ACS Catal., 2019, 9, 5486–5495 CrossRef CAS.
- D. Yang, H. J. Zou, Y. Z. Wu, J. F. Shi, S. H. Zhang, X. D. Wang, P. P. Han, Z. W. Tong and Z. Y. Jiang, Ind. Eng. Chem. Res., 2017, 56, 6247–6255 CrossRef CAS.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880–1886 CrossRef CAS.
- T. Saba, J. Li, J. W. H. Burnett, R. F. Howe, P. N. Kechagiopoulos and X. Wang, ACS Catal., 2021, 11, 283–289 CrossRef CAS.
- A. Marrone and R. H. Fish, J. Organomet. Chem., 2021, 943, 121810 CrossRef CAS.
- Z. B. Zhang, J. J. Li, M. B. Ji, Y. R. Liu, N. Wang, X. P. Zhang, S. J. Zhang and X. Y. Ji, Green Chem., 2021, 23, 2362–2371 RSC.
- Y. Y. Zhang, Y. J. Zhao, R. Li and J. Liu, Sol. RRL, 2021, 5, 2000339 CrossRef CAS.
- H. Li, J. Liu, M. Wang, X. Ren, C. Li, Y. Ren and Q. Yang, Sol. RRL, 2021, 5, 2000641 CrossRef CAS.
- D. Ju, G. Lin, H. Xiao, Y. Zhang, S. Su and J. Liu, Sol. RRL, 2020, 4, 2000559 CrossRef CAS.
- L.-J. Zhao, Z. Yin, Y. Shi, W. Sun, L. Sun, H. Su, X. Sun, W. Zhang, L. Xia and C. Qi, Catal. Sci. Technol., 2021, 11, 7982–7991 RSC.
- P. Singh, R. K. Yadav, K. Kumar, Y. Lee, A. K. Gupta, K. Kumar, B. C. Yadav, S. N. Singh, D. K. Dwivedi, S.-H. Nam, A. P. Singh and T. W. Kim, Catal. Sci. Technol., 2021, 11, 6401–6410 RSC.
- D. Yadav, A. Kumar, J. Y. Kim, N.-J. Park and J.-O. Baeg, J. Mater. Chem. A, 2021, 9, 9573–9580 RSC.
- T. Saba, J. W. H. Burnett, J. W. Li, X. N. Wang, J. A. Anderson, P. N. Kechagiopoulos and X. Wang, Catal. Today, 2020, 339, 281–288 CrossRef CAS.
- M. D. Wang, X. M. Ren, M. Guo, J. L. Liu, H. Li and Q. H. Yang, ACS Sustainable Chem. Eng., 2021, 9, 6499–6506 CrossRef CAS.
- X. Zhao, S. E. Cleary, C. Zor, N. Grobert, H. A. Reeve and K. A. Vincent, Chem. Sci., 2021, 12, 8105–8114 RSC.
- M. Wang, M. Guo, X. Ren, X. Liu and Q. Yang, J. Phys. Chem. C, 2021, 125, 15275–15282 CrossRef CAS.
- X. Ren, J. Lu, M. Wang, M. Guo, H. Li, X. Pan, L. Li, A. Munyentwali and Q. Yang, ACS Catal., 2020, 10, 13701–13709 CrossRef CAS.
- M. Cardona-Farreny, P. Lecante, J. Esvan, C. Dinoi, I. del Rosal, R. Poteau, K. Philippot and M. R. Axet, Green Chem., 2021, 23, 8480–8500 RSC.
- F. Colmati, E. Antolini and E. R. Gonzalez, J. Solid State Electrochem., 2008, 12, 591–599 CrossRef CAS.
- G. Neri, C. Milone, S. Galvagno, A. P. J. Pijpers and J. Schwank, Appl. Catal., A, 2002, 227, 105–115 CrossRef CAS.
- X. D. Wang, J. Stover, V. Zielasek, L. Altmann, K. Thiel, K. Al-Shamery, M. Baumer, H. Borchert, J. Parisi and J. Kolny-Olesiak, Langmuir, 2011, 27, 11052–11061 CrossRef CAS PubMed.
- H. Q. Li, G. Q. Sun, L. Cao, L. H. Jiang and Q. Xin, Electrochim. Acta, 2007, 52, 6622–6629 CrossRef CAS.
- S. Stevanovic, D. Tripkovic, V. Tripkovic, D. Minic, A. Gavrilovic, A. Tripkovic and V. M. Jovanovic, J. Phys. Chem. C, 2014, 118, 278–289 CrossRef CAS.
- M. M. Magalhaes, J. F. Gomes, G. Tremiliosi, P. B. S. de Figueiredo, R. B. de Lima and F. Colmati, J. Appl. Electrochem., 2021, 51, 173–181 CrossRef CAS.
- J. Zhu, X. Zheng, J. Wang, Z. Wu, L. Han, R. Lin, H. L. Xin and D. Wang, J. Mater. Chem. A, 2015, 3, 22129–22135 RSC.
- W. Chen, Z. Lei, T. Zeng, L. Wang, N. C. Cheng, Y. Y. Tan and S. C. Mu, Nanoscale, 2019, 11, 19895–19902 RSC.
- H. P. Rong, Z. Q. Niu, Y. F. Zhao, H. Cheng, Z. Li, L. Ma, J. Li, S. Q. Wei and Y. D. Li, Chem. – Eur. J., 2015, 21, 12034–12041 CrossRef CAS PubMed.
- J. W. H. Burnett, R. F. Howe and X. Wang, Trends Chem., 2020, 2, 488–492 CrossRef CAS.
- T. Saba, J. W. H. Burnett, J. Li, P. Kechagiopoulos and X. Wang, Chem. Commun., 2020, 56, 1231 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc04414a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |