
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of aluminum incorporation on the schwertmannite structure and surface properties†
Sergio
Carrero
 *a,
Alejandro
Fernandez-Martinez
b,
Rafael
Pérez-López
c,
Jordi
Cama
a,
Catherine
Dejoie
d and
José Miguel
Nieto
c
*a,
Alejandro
Fernandez-Martinez
b,
Rafael
Pérez-López
c,
Jordi
Cama
a,
Catherine
Dejoie
d and
José Miguel
Nieto
c
aInstitute of Environmental Assessment and Water Research (IDÆA-CSIC), 08034, Barcelona, Spain. E-mail: sergio.carrero@idaea.csic.es; Tel: +34 934 006 100 ext. 695
bUniversité Grenoble Alpes, Université Savoie Mont Blanc, CNRS, IRD, IFSTTAR, ISTerre, 38000 Grenoble, France
cDepartment of Earth Sciences & Research Center on Natural Resources, Health and the Environment, University of Huelva, Campus ‘El Carmen’, 21071, Huelva, Spain
dEuropean Synchrotron Radiation Facility, 71 avenue des Martyrs, Grenoble, 3800, France
First published on 15th June 2022
Abstract
Schwertmannite is a common nanomineral in acid sulfate environments such as Acid Mine Drainage (AMD) and Acid Sulfate Soils (ASS). Its high surface area and positively charged surface result in a strong affinity towards toxic oxyanions such as arsenate in solution. However, natural precipitation of schwertmannite also involves the accumulation of other impurities, in particular aluminum, an element that is often incorporated into the structure of Fe-oxide minerals, such as goethite and ferrihydrite, affecting their structural and surface properties. However, little is known about the effect of Al incorporation in schwertmannite on the removal capacity of toxic oxyanions found in AMD and ASS (e.g. arsenate). In this paper, schwertmannite samples with variable Al concentration were synthetized and employed in arsenate adsorption isotherm experiments at a constant pH of 3.5. Solid samples before and after arsenate adsorption were characterized using high energy X-ray diffraction and pair distribution function analyses in order to identify structural differences correlated with the Al content as well as variations in the coordination of arsenate adsorbed on the mineral surface. These analyses showed limited Al accumulation on schwertmannite (up to 5%) with a low effect on its structure. The maximum arsenate sorption capacity (258 mmolH2AsO4 molFe−1) was in the range of that with pure schwertmannite, but a higher proportion of inner-sphere coordination was observed. Finally, Al was found to desorb from schwertmannite, with adsorbed arsenate preventing this effect and increasing the stability of the mineral. These results are useful to interpret observations from the field, in particular from river water affected by AMD and ASS, where similar conditions are observed, and where aluminum incorporation is expected.
Environmental significanceSchwertmannite formation in areas affected by acid mine drainage and acid sulfate soils has been described as a natural attenuation process, where contaminants such as As and Fe are removed from solution. Historically, schwertmannite properties have been described in pure mineral phases, whereas impurities in schwertmannite, such as Al, are commonly observed in field samples. The identification of the surface properties in impure schwertmannite will shed light on the reaction, which takes place between schwertmannite and aqueous contaminants in critical areas such as river confluents, treatment plants and estuaries. |
1. Introduction
Schwertmannite is an Fe oxyhydroxysulfate nanomineral with a variable chemical composition (Fe8O8(OH)8−x(SO4)x·nH2O, where 1 < x < 1.75).1 Usually, it precipitates in acid sulfate-rich environments, such as acid mine drainage (AMD) and acid sulfate soils (ASS) and plays an important role as a scavenger of iron, sulfate and other aqueous contaminants (e.g., As, Cr and Se).2–5 The structure of schwertmannite is represented as a distorted akaganeite-like structure with double chains of edge-sharing FeO6 octahedra forming 2 × 2 tunnels, where sulfate ions are located in inner or outer-sphere coordination depending on the level of hydration.6,7The accumulation of structural defects makes the coherent domain size of schwertmannite in the order of a few nanometers, yielding a nanomineral with a high surface area and sorption capacity.8 Under acidic conditions in AMD and ASS (pH between 2.5 and 3.5), the surface of schwertmannite is positively charged resulting in a strong affinity for oxyanions, which increases its environmental importance.9–11 Two main mechanisms control the adsorption of oxyanions: 1) surface complexation and 2) ion-exchange with structural sulfate.12 Experimental results obtained using synthetic phases have shown that surface complexation is the main mechanism of As adsorption on schwertmannite. The adsorption capacity for As(V) is higher than that for As(III) at pH 3.5–4.5, via the formation of strong covalent bonds (inner-sphere complexes) between As and Fe.13,14 Other oxyanions such as selenate, chromate or molybdate are more prone to be adsorbed via anion exchange with structural sulfate, mostly forming outer-sphere complexes.6,12,14 Wang et al.15 reported that sulfate in schwertmannite brings about structural stability that could be compromised during desorption or exchange reactions. However, other authors showed that oxyanion incorporation does not only help maintain the integrity of the schwertmannite structure, but also increases its kinetic persistence against dissolution.16
Aluminum is also a common cation in many acid sulfate environments and its incorporation into Fe-oxide mineral structures has been largely reported.5,17–20 The replacement of Fe in octahedral coordination by Al seems to affect the structural and surface properties. For example, the presence of Al in goethite does not affect its dissolution, but it retards its crystal growth.18 About 20 and 30% of Fe in ferrihydrite can be replaced by Al in octahedral coordination. The limit of Al3+ incorporation is likely controlled by the accumulated strain caused by the different ionic radii between Al and Fe, which leads to a decrease of the unit cell lattice parameters.21 However, this Al incorporation in ferrihydrite increases the sorption capacity of As(V) species but does not affect the binding mechanisms between Fe and As observed in Al-free both natural and synthetic ferrihydrite.22,23
Recent field and lab studies have shown that natural schwertmannite precipitation also controls the solubility of Al at pH <4.3,24 The Al incorporation into schwertmannite is pH-dependent with an increase in pH from 2.5 to 4 inducing a progressively higher Al3+ content in schwertmannite (up to 20%). In addition, schwertmannite with a typical pincushion morphology showed a strong chemical contrast between the core and needles, with higher Al3+ concentrations in outer zones than in inner zones.24 However, all the studies of crystal structure and sorption experiments with schwertmannite have been done using a pure mineral phase. The possible effects of Al incorporation into the structure were not taken into account. There is a lack of knowledge of a potential effect of Al incorporation on the schwertmannite adsorption capacity of oxyanions and the adsorption mechanism and structural binding between oxyanions and the mineral surface. Likewise, the structural stability could be affected by an additional distortion imposed by the incorporation of Al with an ionic radius smaller than that of Fe.
To fill this gap in our knowledge of the reactivity of schwertmannite with Al substitutions, this study examines the structure of synthetic schwertmannite coprecipitated with Al and its effect on the As(V) adsorption capacity under river conditions affected by acid drainage (e.g., pH 3.5 and ionic strength of 0.1 mol L−1).25 The adsorption isotherms of As(V) were recorded using synthetic schwertmannite with different Al contents (between 0 and 30 mmolAl gsch−1). The solids retrieved from these experiments were examined using high-energy X-ray diffraction (HEXD) and pair distribution function (PDF) analysis in order to identify structural differences correlated with the Al content and to decipher variations in the coordination of arsenate adsorbed on the mineral surface.
2. Materials and methods
2.1. Synthesis
As(V) adsorption experiments and structural characterization were carried out with synthetic schwertmannite precipitated using a modified procedure by Loan et al.26 that includes Al in solution. In pure schwertmannite (Al-Sch0.00) synthesis, 2.506 g of a previously dehydrated Fe2(SO4)3 salt were added to 1 L of Milli-Q water preheated at 85 °C and stirred for 1 h. For schwertmannite with coprecipitated Al (Al-Sch0.03–0.32), a variable amount of Al2(SO4)3·18H2O (0.199 g, 0.499 g and 1.333 g for Al-Sch0.03, Al-Sch0.08 and Al-Sch0.32, respectively, reproducing AMDs with low, medium and high Al concentrations) was added into the preheated Milli-Q water before Fe2(SO4)3 incorporation with the pH stabilized at 3.5 by the addition of NaOH 0.1 M Merk solution. The necessary amount of Fe2(SO4)3 was calculated to keep the ME concentration (ME = Fe + Al, mol) constant as proposed by Loan et al.26 The Fe2(SO4)3 reagent was added gradually under vigorous stirring, while the pH was kept at 3.5 ± 0.1 by continuous addition of 0.1 mol L−1 NaOH. A total of 3 Al-Sch solids were synthetized with a final ratio of Al/Fe = 0.0025 (Al-Sch0.03), 0.01 (Al-Sch0.08) and 0.04 (Al-Sch0.32). All precipitates were retrieved by filtering the suspension through a 0.45 μm nylon membrane filter, and the solids were washed several times with Milli-Q water to be dried by lyophilization. The pH was monitored with a portable multiparametric Crison Mm40+ equipment, previously calibrated with different buffers for pH 4, 7 and 9.2.2.2. Adsorption experiments
Adsorption isotherms were obtained following the procedure described by Carrero et al.14 As(V) uptake by all Al-Schs was characterized by calculating the adsorption capacities at different initial As(V) concentrations (6 × 10−2, 0.1, 0.5, 0.8, 1.0, 1.5, 3.0, 5.0, 7.0 and 10 mmol L−1). As(V) solutions were prepared by dissolving disodium hydrogen arsenate (Na2HAsO4·7H2O ≥ 98% of purity, from Sigma) in Milli-Q water. Finally, the pH was established at 3.5 ± 0.1 by the addition of HCl 0.1 mol L−1 (from concentrate HCl 36% Merck reactive), and the background ionic strength was adjusted to 0.1 mol L−1 with NaCl (≥99% of purity, Panreac). Batch experiments were performed by the addition of 20 mL of As(V)-doped solutions to 0.05 g of solid at pH 3.5 ± 0.1, in high-density polyethylene plastic vials. The suspensions were continuously stirred at room temperature (23 ± 2 °C) and in the dark for 72 h. The pH was monitored periodically and maintained at 3.5 ± 0.1 by the addition of HCl or NaOH 0.1 mol L−1.After the equilibrium period, the samples were centrifuged and the supernatant was filtered through 0.2 μm nylon membrane filters, acidified with HNO3 (65%) to pH < 1 and stored at 4 °C for further chemical analysis. The composition of the starting solid was calculated by acid digestion of 0.05 g of solid in HNO3 (65%) and recovered with 50 mL of Milli-Q water for further chemical analysis. After the adsorption experiments, each solid was washed several times with Milli-Q water and dried at 35 °C for 48 h for subsequent PDF analysis.
The amount of As(V) adsorbed by the solids, [XO4]s (mmol molFe−1), was calculated from the difference between the initial concentration of As, [XO4]0 in mmol L−1, and the equilibrium concentration in solution, [XO4]eq in mmol L−1, normalized by the Fe concentration in the solid (MC in molFe) to the volume (V in L) according to the equation:
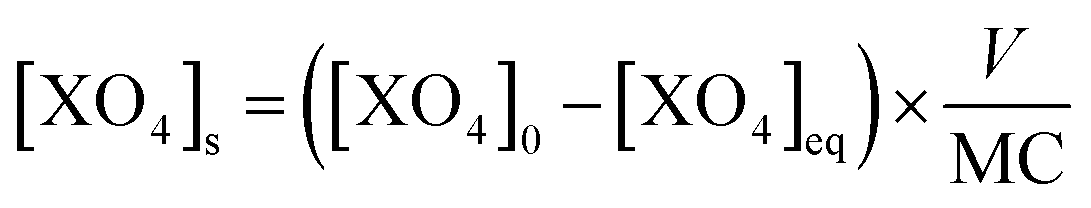 | (1) |
The non-competitive Langmuir isotherm used to model the results from the adsorption experiment is defined as:
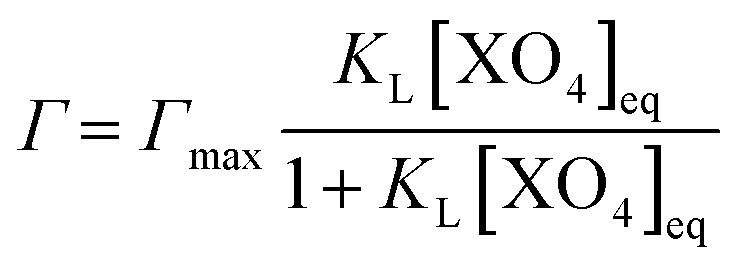 | (2) |
2.3. Analytical techniques
As(V) solutions, before and after each adsorption experiment, and acid digestions of the starting solids were analyzed for Fe, Al, As and S by inductively coupled plasma atomic emission spectrometry (ICP-AES Thermo Jarrel-Ash). Three blanks and three duplicates were analyzed every 20 samples to check the analytical accuracy. The detection limits were 140 μg L−1 for Al and Fe, 15 μg L−1 for As and 300 μg L−1 for S. The analytical error was lower than 5%. In addition, the saturation index of the solid phases and aqueous speciation of solutions were calculated using the PHREEQC code27 with the Mintq.v4 thermodynamic database,28 which was enlarged with data from Bigham et al.1 to account for schwertmannite solubility.Schwertmannite precipitates were lyophilized using a VirTis Benchtop freeze-dryer (Hucoa-Erlöss, Spain) in order to obtain a dry powder. The water proportion in each solid phase was calculated by thermogravimetric analysis (TGA) using a TGA-DSC3+ (Mettler Toledo®). A thermal treatment from 25 to 1000 °C was performed under a N2 flow of 20 mL min−1. HEXD and PDF analyses were performed at the beamline ID22 at the European Synchrotron Radiation Facility (ESRF) using a monochromatic X-ray beam with an energy of ∼70 keV (λ = 0.1784 Å) in Debye–Scherrer geometry. The energy was calibrated using a LaB6 standard (NIST SRM660). Samples were loaded in polyamide capillaries. The diffraction patterns were collected using a PerkinElmer flat-panel detector and were integrated using the program PyFAI.29 PDFs were obtained by Fourier transformation of the reduced structure factor, F(Q), using a Qmax = 24.5 Å−1 with a background scattering correction using the PDFgetX3 program.30
Differential pair distribution functions (d-PDF) were obtained by subtracting a reference PDF of the pure material (synthetic As(V)-free Al-Sch) from the PDFs of the samples recovered after adsorption experiments. Retention models of As(V) adsorbed onto schwertmannite structures were constructed from the structure proposed by Fernandez-Martinez et al.7 The partial pair distribution functions of As(V) located in different structural positions were calculated using PDFgui software.31
3. Results and discussion
3.1. Solid phase characterization
HEXD analysis confirms that the X-ray scattering patterns of the Al-Sch solids match well the diffraction pattern of schwertmannite (Fig. 1a).32 The chemical analyses and TGA data indicate their stoichiometry (Table 1, Table S1 and Fig. S1 of the ESI†). Aluminum incorporation into schwertmannite at pH 3.5 increases gradually with the Al concentration in solution up to an Fe/(Fe + Al) ratio of 0.95, which seems to be the limit at this pH (Table 1).24 All the Al-Sch samples display a sulfate content that is higher than in previously reported chemical formulas of synthetic schwertmannite,1 but remains within the range observed in natural samples.33,34 The high sulfate content in schwertmannite has been related to a significant amount of outer-sphere complexes occupying sorption positions on the mineral surface, compensating for the positive surface charges.6,7,35 The increment in structural Al barely affects the sulfate content, which shows a slightly decreasing trend in the presence of Al (Fig. S2†).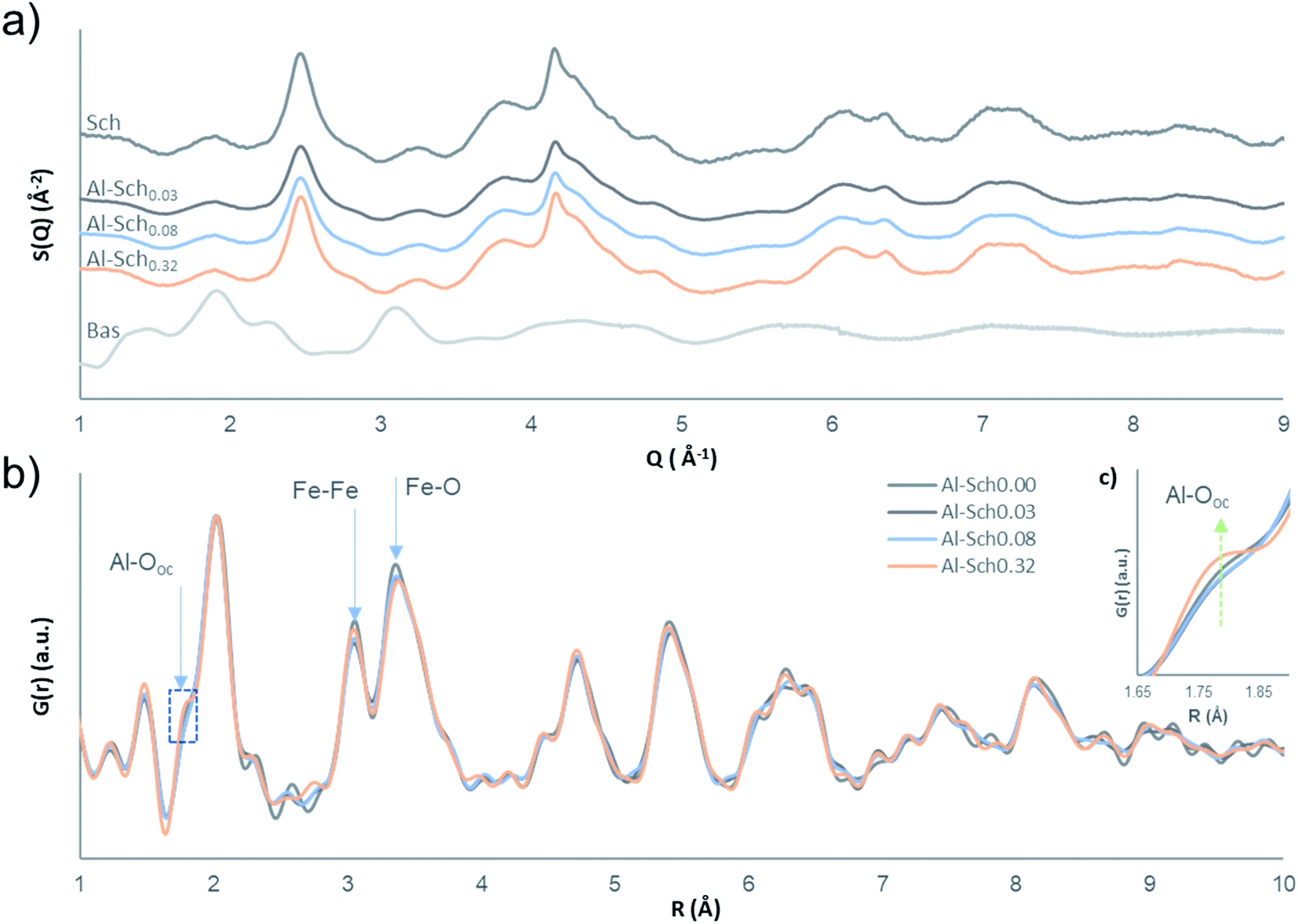 | ||
| Fig. 1 (a) Synchrotron-based HEXD patterns of the synthetic Al-Sch solid compared with the standards of schwertmannite (Sch) and basaluminite (Bas). The latter was included as a reference of a nanomineral Al-phase reported in an acidic environment. (b) PDFs of Al-Sch0.00 to Al-Sch0.32. Intensities were normalized to that of the Fe–O peak at ∼1.99 Å. Arrows indicate the distances affected by Al incorporation in the schwertmannite structure. (c) Zoomed-in image of the Al–O shoulder indicated with a dashed square. | ||
| Sample | Fe (mmol g−1) | Al (mmol g−1) | S (mmol g−1) | Stoichiometry |
|---|---|---|---|---|
| Al-Sch0.00 | 8.21 | 0.00 | 1.73 | Fe8O8(OH)4.62(SO4)1.69·8.84H2O |
| Al-Sch0.03 | 8.34 | 0.03 | 1.88 | Fe7.98Al0.02O8(OH)4.48(SO4)1.79·7.47 H2O |
| Al-Sch0.08 | 8.45 | 0.08 | 1.89 | Fe7.92Al0.08O8(OH)4.46(SO4)1.77·6.62H2O |
| Al-Sch0.32 | 7.78 | 0.32 | 1.62 | Fe7.69Al0.31O8(OH)4.8(SO4)1.60·10.4H2O |
The diffraction patterns of Al-bearing schwertmannite solids display broad peaks, which are coincident with those previously reported for schwertmannite.7,32,35,36 Although Al concentrations in Al-Sch samples increase (Table 1), secondary Al minerals, such as alunite, basaluminite or jurbanite, were not observed (Fig. 1). This fact suggests that Al is coprecipitated and included into the schwertmannite structure, as is the case in other Fe oxide minerals.20,37 The intensity of the diffraction patterns slightly decreases when increasing the Al content. This lower intensity in diffraction has been previously associated with an increase in the structural water content. Nevertheless, results from TGA do not show a clear correlation between the water content and Al in schwertmannite (Table S1 and Fig. S1†). The lower intensity of the X-ray patterns could thus be due to differences in the density of the final solid.
The PDFs of the Al-Sch0.03–0.32 solids compared with the pure Al-Sch0.00 sample reveal that the incorporation of Al does not result in strong structural differences with the pure schwertmannite sample, and the coherent domain of the nanoparticles remains constant (Fig. 1b). The peaks at 3.05 Å and 3.39 Å, corresponding to the respective Fe–Fe and Fe–Fe/Fe–O distances in the Fe framework,7 slightly decrease in intensity concomitantly with an increase in the intensity of a shoulder located at 1.88 Å, which is attributed to the Al–O distance of Al in octahedral coordination (Fig. 1b).38,39 These observations indicate that octahedral occupation by Fe decreases, whereas the Al octahedral coordination appears in the structure, evidencing that Fe is effectively replaced by Al. The small differences observed between the PDFs of the different samples at longer distances imply that the Al replacement (up to 5% of Fe) does not affect strongly the medium-range order of the schwertmannite structure.
3.2. As(V) uptake onto Al-Sch
Aqueous chemical speciation models with the PHREEQC code indicate that H2AsO4− (arsenate) is the dominant As species during adsorption experiments with a proportion higher than 95% at pH 3.5 (Table S2†). The time needed to reach equilibrium between schwertmannite and arsenate was determined to be 72 h as arsenate adsorption reaches a maximum and mineral dissolution is minimized.10,12,14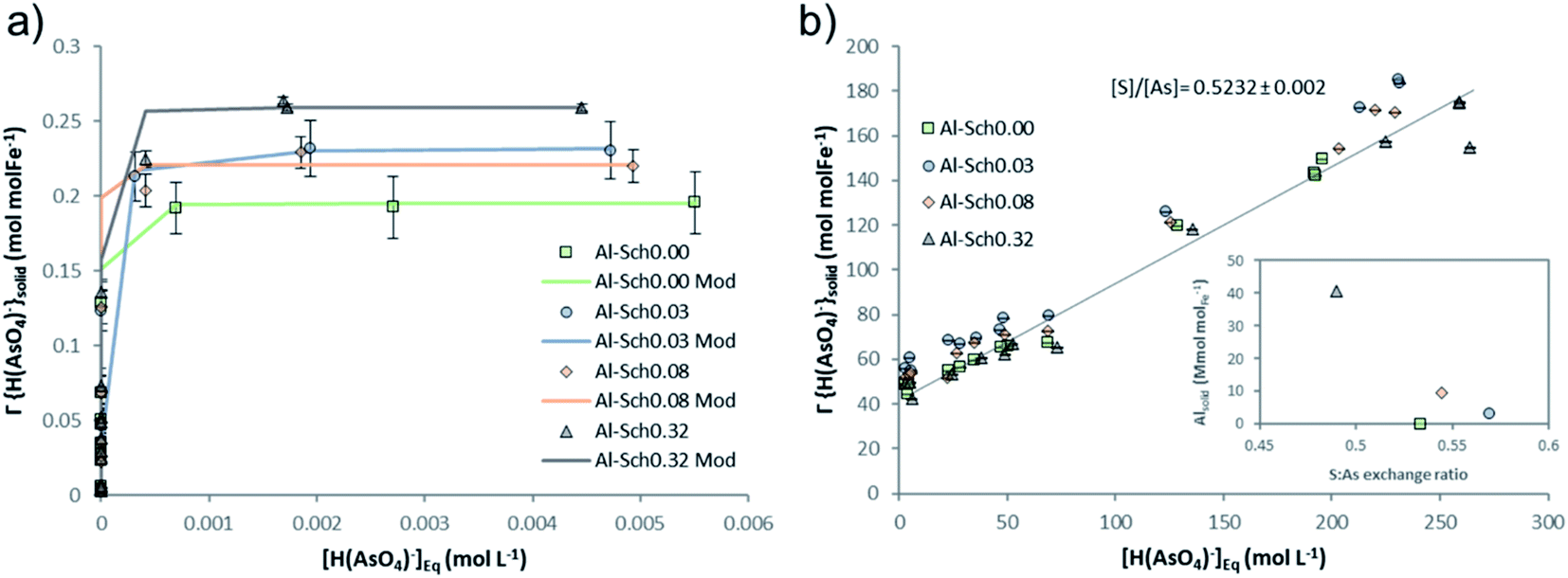 | ||
| Fig. 2 (a) Adsorption isotherm of arsenate (mmolAsO4 molFe−1) onto different Al-Sch solids. Curves obtained using the Langmuir equations are shown as continuous lines to compare with the experimental data. (b) Relationship between sulfate released from the solid phase (mmolSO4 molFe−1) and adsorbed aqueous arsenate (mmolAsO4 molFe−1). The trend line was calculated considering the values from all solids. The experiment was conducted at pH 3.5, an ionic strength of 0.1 mol L−1 and oxyanion concentration from 6 × 10−2 to 10 mmol L−1. | ||
The main process controlling the arsenate adsorption was the ion exchange with sulfate, where around 50–70% of the initial sulfate was replaced by arsenate in Al-Sch0.00 and Al-Sch0.32, respectively (Table S3, Fig. S3†), yielding a value that is higher than in previous studies.14 Moreover, exchange coefficients (Rex) were obtained as the slope of the linear regression of the relationship between desorbed sulfate (mmolSO4 molFe−1) and adsorbed arsenate (mmolH2AsO4 molFe−1) (Fig. 2b). A value of Rex close to 1 mmolSO4 mmolH2AsO4−1 can be interpreted as a complete substitution of the structural sulfate by an equivalently charged oxyanion through an ion exchange mechanism. Values higher than 1 mmolSO4 mmolH2AsO4−1 would indicate only a partial substitution. The Rex values for arsenate varied between 0.49 and 0.56 (Table S3†), indicating an exchange reaction where two arsenates replace one sulfate to balance the surface charge. The high sulfate content in schwertmannite observed in this experiment correlates with the higher proportion of exchangeable sulfate, indicating a greater presence of outer-sphere complexes onto the mineral surface that is easily replaced by oxyanions from the solution. Aluminum incorporation into the schwertmannite structure seems to increase the amount of exchangeable sulfate but not the total sulfate content in the Al-Sch0.03–0.32 sample compared with that of Al-Sch0.00. This fact suggests a reduction of structural sulfate in favor of surficial complexes with Al incorporation, accounting for the slight increase in the arsenate retention capacity.
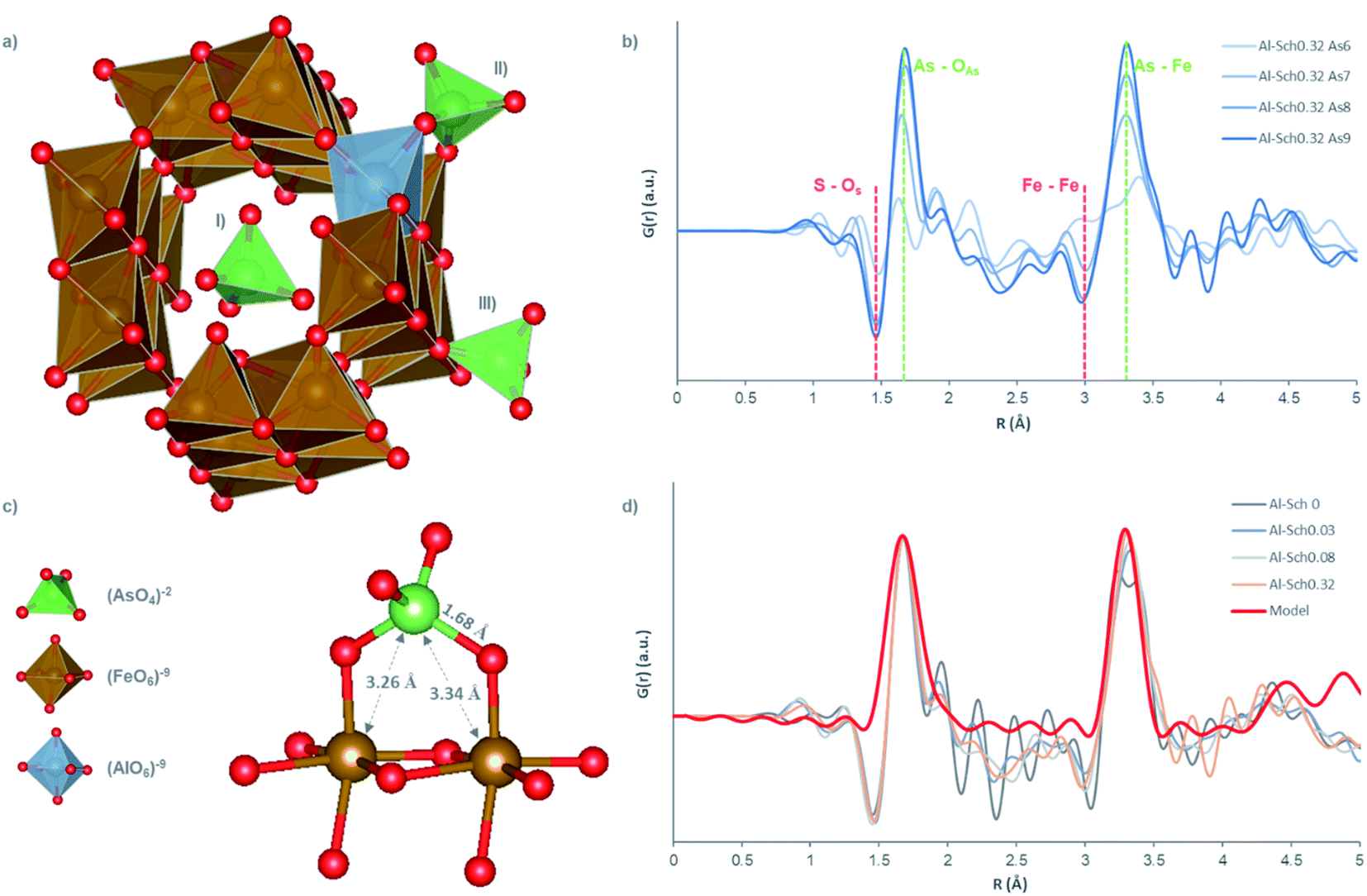 | ||
| Fig. 3 (a) Structural model of schwertmannite reported by Fernandez-Martinez et al. (2010) including Al replacement (hypothetical coordination) and doping with arsenate in three surface coordination: (i) outer-sphere, (ii) bidentate-binuclear inner-sphere and (iii) monodentate inner-sphere ligands. (b) d-PDFs of arsenate onto Al-Sch0.32 with 1.5, 3.0, 5.0, and 7.0 mmol L−1 of arsenate. (c) Local order of arsenate at the schwertmannite structure. (d) d-PDFs of the Al-bearing schwertmannite samples doped with 7.0 mmol L−1 of arsenate. The signal intensity in (b) and (d) was normalized with the maximum of the Fe–O distance. | ||
The d-PDF showed a peak at 1.67 Å corresponding to the As–O distance in the arsenate molecule.14,41,42 A second peak at 3.28 Å was observed with a similar distance to the As–Fe pair distance found in previous studies dealing with arsenate adsorption onto Fe-phases,41 and it is coincident with the interatomic distances reported by other studies about arsenate sorption onto Fe oxyhydroxides.9,14,42,43 Both peaks were found in all solids with arsenate concentration higher than 70 mmolH2AsO4 molFe−1 (Fig. 3b and S4†) at any Al concentration in schwertmannite. The position and relative intensity between As–O and As–Fe peaks perfectly match those of a model where 100% of arsenate is bound through a bidentate binuclear inner-sphere ligand (Fig. 3c and d). This result contrasts with previous findings reported by Carrero et al.,14 where arsenate retention in schwertmannite by bidentate-binuclear inner-sphere coordination was limited to 50% of the total at arsenate concentrations in solution higher than 1 mmolH2AsO4 L−1, whereas Al-phases (i.e., basaluminite and alunite) can reach 100%.44,45 This fact indicates that Al incorporation in the schwertmannite structure could favor the formation of covalent bonds between As and Fe, in detriment of outer-sphere and hydrogen bond complexes, increasing thus the stability of the adsorbed oxyanions.
Finally, the d-PDF showed two negative peaks at 1.46 Å and 3 Å whose negative intensity increases simultaneously with the arsenate concentration in the solid (Fig. 3b). The first peak can be attributed to the S–O distance in tetrahedral sulfate, confirming that the anion exchange is the main arsenate retention mechanism (Fig. 3b). The second peak is coincident with the Fe–Fe first shell distance and indicates that structural changes in the Fe-framework are taking place (e.g., formation of Fe vacancies). This type of structural distortion has been previously reported during adsorption reactions.14,46 However, the Fe-framework distortion observed in this experiment seems to be lower than the values reported by Carrero et al.14 (Fig. 3b). This latter study identifies secondary negative peaks associated with the distortions of Fe-frameworks (i.e., Fe–O first shell at 1.98 Å, Fe–O second shell at 4.73 Å and Fe–Fe second shell at 5.4 Å), which have not been found here, as well as a higher intensity of the Fe–Fe negative peak at 3 Å.14 This suggests that Al incorporation into the schwertmannite structure minimizes the structural defects during arsenate adsorption, although the mechanism controlling these effects remains unidentified.
3.3. Stability of Al in schwertmannite
As shown in Fig. 4, Al desorption from the schwertmannite structure correlates with the total arsenate concentration in the solids. For each solid, the proportional amount of Al released from schwertmannite dissolution (less than 5% of total solid) was subtracted from total Al in solution in order to show the exact desorbed amount. Al is desorbed from all the Al-Sch solid samples with a significant Al concentration (i.e., higher than 0.1 mmol gsch−1) during the arsenate adsorption experiments. Regardless of the Al content in the solids, around 30% of the initial Al was desorbed from the Al-Sch samples and displays an inverse correlation with the adsorbed arsenate, reaching around 5% of Al desorption when the solid reaches the arsenate Γmax (Fig. 4). Similar desorption reactions have been described by Antelo et al.47 for schwertmannite coprecipitated with copper during the first days after mineral formation likely due to the transformation of schwertmannite into goethite. Moreover, the aging schwertmannite reactions require more time than that elapsed for the current arsenate adsorption experiments.16 However, As adsorption seems to increase schwertmannite stability, delaying Al desorption and, subsequently, the aging of schwertmannite to goethite as previously described in both natural and synthetic schwertmannite samples.16,36,48,49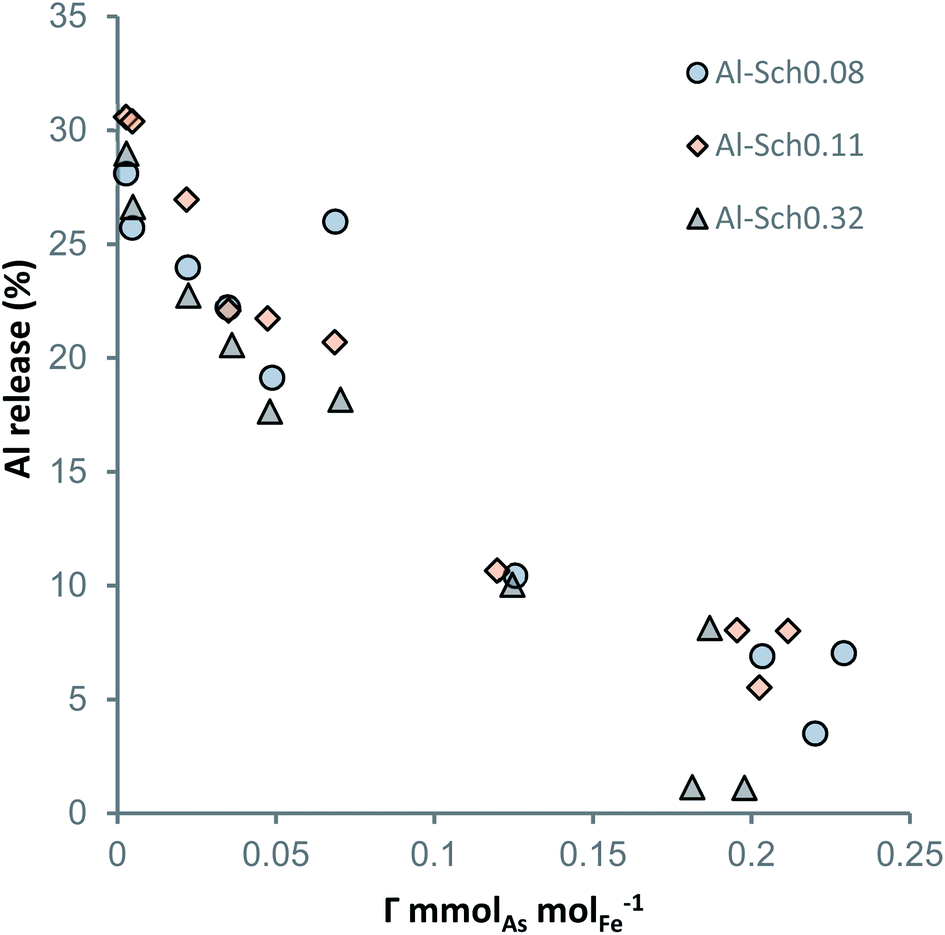 | ||
| Fig. 4 Al released from Al-Sch after 72 h equilibration with the arsenate solution as a function of the adsorbed aqueous arsenate (mmolAsO4 molFe−1). | ||
The results from structural and chemical analyses shown here cannot be used to identify the exact structural position at which Al is included in schwertmannite. The only piece of data suggesting the substitution by an octahedral iron is the fact that the Fe–O and Fe–Fe peaks in the PDFs of Al-Sch show a lower intensity than that of pure schwertmannite. However, this decrease is not monotonic, and hence is not a strong evidence of Fe substitution by octahedral Al (Fig. S5†). The fact that 30% of Al in Al-Sch is released to solution without schwertmannite dissolution suggests that schwertmannite was not a monomineralic precipitate, although thermodynamic calculations show that acid water solutions are undersaturated with respect to Al-phases at pH below 4.5, so no secondary Al-phase would be stable during the adsorption experiments (Table S4†).17,33,38,39 The possibility that an Al-rich domain may form within the schwertmannite structure, in a sort of ordered nanoscopic solid solution, cannot be ruled out. Experiments probing the structure with a nm-resolution would be needed to ascertain the nature of the substitution. Therefore, our study shows evidence of Fe replacement in octahedral coordination by Al; although the existence of Al-oxyhydroxide nano-domains within schwertmannite cannot be ruled out.
4. Conclusions
Aluminum incorporation in schwertmannite, a process that is frequently observed in iron oxide minerals, has negligible effects on its structure at least at low pH (i.e., 3.5). Only up to 5% of Fe octahedral positions were occupied by Al in different schwertmannite samples synthetized with Al/Fe ratios ranging from 0.03 to 0.32. In addition, the structural Al incorporation in schwertmannite does not seem to significantly affect its arsenate adsorption capacity, whereas As adsorption prevents Al release from schwertmannite, which could indicate that not all Al is replacing the Fe position and could also be located in Al-oxide nanodomains. Positively noteworthy is that the presence of Al increases the proportion of covalent bonds between As(V) oxyanions and the mineral surface, which has important environmental implications. Covalent bonds result in increasing stability of not only oxyanion complexes, retarding oxyanion desorption reactions, but also delaying the solid transformation into more stable phases. Hence, the precipitation of schwertmannite with high Al contents would allow a more efficient decontamination of acid water since arsenate would remain fixed for a longer period of time in AMD- and ASS-affected environments.These findings are relevant for most of the acid river water with a pH range of 2.5–3.5 buffered by Fe-hydrolysis reactions. However, in areas where acid rivers merge with alkaline water (e.g. river estuary and river confluences), schwertmannite supersaturation increases, where these newly formed precipitates are exposed to strong changes in pH, redox and salinity. These changes in field conditions could increase the incorporation of Al in schwertmannite, affect the binding between oxyanions and the mineral surface and accelerate the schwertmannite aging towards more stable phases with less adsorption capacity. It is expected that a deeper understanding of how different pH, redox and salinity conditions affect Al-incorporation in Fe phases will ensure a better prediction of the transport and retention of metals and oxyanions in these areas impacted by acid rivers and with a high environmental value.
Conflicts of interest
There are not conflicts of interest to declare.Acknowledgements
This paper is part of the PID2020-119196RB-C2 TRAMPA project funded by MCIN/AEI/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”. S. Carrero was supported by IDAEA-CSIC as a Severo Ochoa Centre of Research Excellence (Spanish Ministry of Science and Innovation, Project CEX2018-000794-S). ESRF data were acquired during the experiment EV-198 at ID22. Funding from the Labex OSUG@2020 (Investissements d'avenir–ANR10 LABX56), supporting the use of TGA-DSC analysis at ISTerre, is acknowledged. Chemical analyses were performed at the laboratories of IDAEA (CSIC) in Barcelona.References
- J. M. Bigham, U. Schwertmann, S. J. Traina, R. L. Winland and M. Wolf, Schwertmannite and the chemical modeling of iron in acid sulfate waters, Geochim. Cosmochim. Acta, 1996, 60(12), 2111–2121, DOI:10.1016/0016-7037(96)00091-9.
- J. M. Bigham and D. K. Nordstrom, Iron and aluminum hydroxysulfates from acid sulfate waters, in Sulfate Minerals: Crystallography, Geochemistry, and Environmental Significance, Walter de Gruyter GmbH, 2019, vol. 40, pp. 351–403, DOI:10.2138/rmg.2000.40.7.
- S. Carrero, R. Pérez-López, A. Fernandez-Martinez, P. Cruz-Hernández, C. Ayora and A. Poulain, The potential role of aluminium hydroxysulphates in the removal of contaminants in acid mine drainage, Chem. Geol., 2015, 417, 414–423, DOI:10.1016/j.chemgeo.2015.10.020.
- E. D. Burton, R. T. Bush, L. A. Sullivan, S. G. Johnston and R. K. Hocking, Mobility of arsenic and selected metals during re-flooding of iron- and organic-rich acid-sulfate soil, Chem. Geol., 2008, 253(1–2), 64–73, DOI:10.1016/j.chemgeo.2008.04.006.
- D. K. Nordstrom, Hydrogeochemical processes governing the origin, transport and fate of major and trace elements from mine wastes and mineralized rock to surface waters, Appl. Geochem., 2011, 26(11), 1777–1791, DOI:10.1016/j.apgeochem.2011.06.002.
- X. Wang, C. Gu, X. Feng and M. Zhu, Sulfate local coordination environment in schwertmannite, Environ. Sci. Technol., 2015, 49(17), 10440–10448, DOI:10.1021/acs.est.5b02660.
- A. Fernandez-Martinez, V. Timon, G. Romaman-Ross, G. J. Cuello, J. E. Daniels and C. Ayora, The structure of schwertmannite, a nanocrystalline iron oxyhydroxysulfate, Am. Mineral., 2010, 95(8–9), 1312–1322, DOI:10.2138/am.2010.3446.
- V. A. Schoepfer and E. D. Burton, Schwertmannite: a review of its occurrence , formation , structure , stability and interactions with oxyanions, Earth-Sci. Rev., 2021, 221, 103811, DOI:10.1016/j.earscirev.2021.103811.
- J. Song, S.-Y. Jis, H.-T. Ren, S.-H. Wu and X. Han, Application of a high-surface-area schwertmannite in the removal of arsenate and arsenite, Int. J. Environ. Sci. Technol., 2015, 12, 1559–1568, DOI:10.1007/s13762-014-0528-9.
- E. D. Burton, R. T. Bush, S. G. Johnston, K. M. Watling, R. K. Hocking, L. A. Sullivan and G. K. Parker, Sorption of arsenic(V) and arsenic(III) to schwertmannite, Environ. Sci. Technol., 2009, 43(24), 9202–9207, DOI:10.1021/es902461x.
- S. Paikaray, J. Göttlicher and S. Peiffer, Removal of As ( III ) from acidic waters using schwertmannite : surface speciation and effect of synthesis pathway, Chem. Geol., 2011, 283(3–4), 134–142, DOI:10.1016/j.chemgeo.2010.08.011.
- J. Antelo, S. Fiol, D. Gondar, R. López and F. Arce, Comparison of arsenate, chromate and molybdate binding on schwertmannite: surface adsorption vs anion-exchange, J. Colloid Interface Sci., 2012, 386(1), 338–343, DOI:10.1016/j.jcis.2012.07.008.
- F. Maillot, G. Morin, F. Juillot, O. Bruneel, C. Casiot, G. Ona-nguema, Y. Wang, S. Lebrun, E. Aubry, G. Vlaic and G. E. Brown, Structure and reactivity of As ( III ) - and As ( V ) -rich schwertmannites and amorphous ferric arsenate sulfates acid mine drainage, France : comparison from the Carnoule with biotic and abiotic model compounds and implications for As remediation, Geochim. Cosmochim. Acta, 2013, 104, 310–329, DOI:10.1016/j.gca.2012.11.016.
- S. Carrero, A. Fernandez-Martinez, R. Pérezpérez-Lópezlópez, A. Poulain, E. Salas-Colera and J. J. Nieto, Arsenate and selenate scavenging by basaluminite: insights into the reactivity of aluminum phases in acid mine drainage, Environ. Sci. Technol., 2017, 51, 28–37, DOI:10.1021/acs.est.6b03315.
- X. Wang, H. Ying, W. Zhao, X. Feng, W. Tan, K. A. Beyer, Q. Huang, F. Liu and M. Zhu, Molecular-scale understanding of sulfate exchange from schwertmannite by chromate versus arsenate, Environ. Sci. Technol., 2021, 55(9), 5857–5867, DOI:10.1021/acs.est.0c07980.
- P. Acero, C. Ayora, C. Torrentó and J. M. Nieto, The Behavior of Trace Elements during Schwertmannite Precipitation and Subsequent Transformation into Goethite and Jarosite, Geochim. Cosmochim. Acta, 2006, 70(16), 4130–4139, DOI:10.1016/j.gca.2006.06.1367.
- J. M. Bigham and D. K. Nordstrom, Iron and Aluminum Hydroxysulfates from Acid Sulfate Waters, in Sulfate Minerals: Crystallography, Geochemistry, and Environmental Significance, Walter de Gruyter GmbH, 2019, vol. 40, pp. 351–403, DOI:10.2138/rmg.2000.40.7.
- U. Schwertmann, The influence of aluminium on iron oxides : IX . DISSOLUTION of AL-goethites in 6 M HCL, Clay Miner., 1984, 19, 9–19 CrossRef CAS.
- E. Bazilevskaya, D. D. Archibald, M. Aryanpour, J. D. Kubicki and C. E. Martı, Aluminum coprecipitates with Fe (Hydr)oxides: Does isomorphous substitution of Al3+ for Fe3+ in goethite Occur?, Geochim. Cosmochim. Acta, 2011, 75, 4667–4683, DOI:10.1016/j.gca.2011.05.041.
- A. C. Cismasu, F. M. Michel, A. P. Tcaciuc, T. Tyliszczak and G. E. Brown, Composition and structural aspects of naturally occurring ferrihydrite composition et Proprie, C. R. Geosci., 2011, 343(2–3), 210–218, DOI:10.1016/j.crte.2010.11.001.
- A. C. Cismasu, F. M. Michel, J. F. Stebbins and G. E. Brown, Properties of impurity-bearing ferrihydrite I. Effects of Al content and precipitation rate on the structure of 2-line ferrihydrite, Geochim. Cosmochim. Acta, 2012, 92, 275–291, DOI:10.1016/j.gca.2012.06.010.
- A. Adra, G. Morin, G. Ona-nguema, N. Menguy, F. Maillot, C. Casiot, O. Bruneel, S. Lebrun, F. Juillot and J. Brest, Arsenic scavenging by aluminum-substituted ferrihydrites in a circumneutral pH river impacted by acid mine drainage, Environ. Sci. Technol., 2013, 47, 12784–12792, DOI:10.1021/es4020234.
- A. Adra, G. Morin, G. Ona-nguema and J. Brest, Arsenate and arsenite adsorption onto Al-containing ferrihydrites . Implications for arsenic immobilization after neutralization of acid mine drainage, Appl. Geochem., 2016, 64, 2–9, DOI:10.1016/j.apgeochem.2015.09.015.
- J. Sánchez-España, I. Yusta, J. Gray and W. D. Burgos, Geochemistry of dissolved aluminum at low PH: extent and significance of Al-Fe(III) coprecipitation below PH 4.0, Geochim. Cosmochim. Acta, 2016, 175, 128–149, DOI:10.1016/j.gca.2015.10.035.
- D. K. Nordstrom, Mine waters: acidic to circumneutral, Elements, 2011, 7, 393–398, DOI:10.2113/gselements.7.6.393.
- M. Loan, J. M. Cowley, R. Hart and G. M. Parkinson, Evidence on the structure of synthetic schwertmannite, Am. Mineral., 2004, 89(11–12), 1735–1742, DOI:10.2138/AM-2004-11-1220.
- D. L. Parkhurst and C. A. Appelo, J. User's Guide to PhreeqC (Version 2) A Computer Program for Speciation, and Inverse Geochemical Calculations, 1999 Search PubMed.
- J. D. Allison, D. S. Brown and K. J. Novogradac, A Geochemical Assessment Model for Environmental Systems: Version 4.0 User's Manual, EPA/600/3-91/021, 1998 Search PubMed.
- J. Kieffer and D. Karkoulis, PyFAI, a Versatile Library for Azimuthal Regrouping, J. Phys.: Conf. Ser., 2013, 425(PART 20), 0–5, DOI:10.1088/1742-6596/425/20/202012.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, PDFgetX3: A rapid and highly automatable program for processing powder diffraction data into total scattering pair distribution functions, J. Appl. Crystallogr., 2013, 46(2), 560–566 CrossRef.
- C. L. Farrow, P. Juhás, J. W. Liu, D. Bryndin, E. S. Bozin, J. Bloch, T. Proffen and S. J. L. Billinge, PDFgui User Guide, 2009 Search PubMed.
- J. M. Bigham, U. Schwertmann, L. Carlson and E. Murad, A poorly crystallized oxyhydroxysulfate of iron formed by bacterial oxidation of Fe(II) in acid mine waters, Geochim. Cosmochim. Acta, 1990, 54(10), 2743–2758 CrossRef CAS.
- J. Sánchez-españa, I. Yusta and M. Diez-ercilla, Schwertmannite and hydrobasaluminite : A re-evaluation of their solubility and control on the iron and aluminium concentration in acidic pit lakes, Appl. Geochem., 2011, 26, 1752–1774, DOI:10.1016/j.apgeochem.2011.06.020.
- M. A. Caraballo, J. D. Rimstidt, F. Macías, J. M. Nieto and M. F. Hochella, Metastability, nanocrystallinity and pseudo-solid solution effects on the understanding of schwertmannite solubility, Chem. Geol., 2013, 360–361, 22–31, DOI:10.1016/j.chemgeo.2013.09.023.
- M. Sestu, G. Navarra, S. Carrero, S. M. Valvidares, G. Aquilanti and R. Pe, Whole-nanoparticle atomistic modeling of the schwertmannite structure from total scattering data research papers, J. Appl. Crystallogr., 2017, 50, 1617–1626, DOI:10.1107/S160057671701336X.
- P. Cruz-Hernández, S. Carrero, R. Pérez-López, A. Fernandez-Martinez, M. B. J. Lindsay, C. Dejoie and J. M. Nieto, Influence of As(V) on precipitation and transformation of schwertmannite in acid mine drainage-impacted waters, Eur. J. Mineral., 2019, 31(2), 237–245, DOI:10.1127/ejm/2019/0031-2821.
- J. Sanchez-España, I. Yusta, J. Gray and W. D. Burgos, Geochemistry of dissolved aluminum at low PH : extent and significance of Al – Fe ( III ) coprecipitation below PH 4 . 0, Geochim. Cosmochim. Acta, 2016, 175, 128–149, DOI:10.1016/j.gca.2015.10.035.
- S. Carrero, A. Fernandez-Martinez, R. Pérez-Lopez, D. Lee, G. Aquilanti, A. Poulain, A. Lozano and J.-M. Nieto, The nanocrystalline structure of Basaluminite , an aluminum hydroxide sulfate from acid mine drainage, Am. Mineral., 2017, 102, 2381–2389 CrossRef.
- A. Lozano, A. Fernández-martínez, C. Ayora and A. Poulain, Local structure and ageing of Basaluminite at different pH values and sulphate concentrations, Chem. Geol., 2018, 496(July), 25–33, DOI:10.1016/j.chemgeo.2018.08.002.
- Y. Maseu, R. H. Loeppert and T. A. Kramer, Arsenate and arsenite adsorption and desorption behavior on coprecipitated aluminum : iron hydroxides, Environ. Sci. Technol., 2007, 41(3), 837–842 CrossRef PubMed.
- R. Harrington, D. B. Hausner, N. Bhandari, D. R. Strongin, K. W. Chapman, P. J. Chupas, D. S. Middlemiss, C. P. Grey and J. B. Parise, Investigation of surface structures by powder diffraction : a differential pair distribution function study on arsenate sorption on ferrihydrite, Inorg. Chem., 2010, 11(8), 325–330, DOI:10.1021/ic9022695.
- D. M. Sherman and S. R. Randall, Surface complexation of arsenic(V) to iron(III) (Hydr)Oxides: structural mechanism from Ab Initio molecular geometries and EXAFS Spectroscopy, Geochim. Cosmochim. Acta, 2003, 67(22), 4223–4230, DOI:10.1016/S0016-7037(03)00237-0.
- G. A. Waychunas, B. A. Rea, C. C. Fuller and J. A. Davis, Surface chemistry of ferrihydrite : Part 1 . EXAFS studies of the geometry of coprecipitated and adsorbed arsenate, Geochim. Cosmochim. Acta, 1993, 57, 2251–2269 CrossRef CAS.
- W. Li, R. Harrington, Y. Tang, J. D. Kubicki, M. Aryanpour, R. J. Reeder, J. B. Parise and B. L. Phillips, Differential pair distribution function study of the structure of arsenate adsorbed on nanocrystalline γ -alumina, Environ. Sci. Technol., 2011, 45(22), 9687–9692 CrossRef CAS PubMed.
- Y. Arai, E. J. Elzinga and D. L. Sparks, X-Ray absorption spectroscopic investigation of arsenite and arsenate adsorption at the aluminum oxide-water interface, J. Colloid Interface Sci., 2001, 235(1), 80–88 CrossRef CAS PubMed.
- J. Peña, K. D. Kwon, K. Refson and J. R. Bargar, Mechanisms of nickel sorption by a bacteriogenic birnessite, Geochim. Cosmochim. Acta, 2010, 74, 3076–3089, DOI:10.1016/j.gca.2010.02.035.
- J. Antelo, S. Fiol, D. Gondar, C. Pérez, R. López and F. Arce, Cu(II) incorporation to schwertmannite: effect on stability and reactivity under AMD conditions, Geochim. Cosmochim. Acta, 2013, 119, 149–163, DOI:10.1016/j.gca.2013.05.029.
- A. Parviainen, P. Cruz-Hernández, R. Pérez-López, J. M. Nieto and J. M. Delgado-López, Raman identification of Fe precipitates and evaluation of As fate during phase transformation in Tinto and Odiel river basins, Chem. Geol., 2015, 398, 22–31, DOI:10.1016/j.chemgeo.2015.01.022.
- M. P. Asta, C. Ayora, G. Román-Ross, J. Cama, P. Acero, A. G. Gault, J. M. Charnock and F. Bardelli, Natural attenuation of arsenic in the Tinto Santa Rosa acid stream (Iberian Pyritic Belt, SW Spain): the role of iron precipitates, Chem. Geol., 2010, 271(1–2), 1–12, DOI:10.1016/j.chemgeo.2009.12.005.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2em00029f |
| This journal is © The Royal Society of Chemistry 2022 |