
Adsorption and keto–enol-tautomerisation of butanal on Pd(111)†
Jessica
Wulfes
,
Ann-Katrin
Baumann
,
Tobias
Melchert
,
Carsten
Schröder
and
Swetlana
Schauermann
 *
*
Institute of Physical Chemistry, Christian-Albrechts-University Kiel, Max-Eyth-Str. 1, 24118 Kiel, Germany. E-mail: schauermann@pctc.uni-kiel.de
First published on 30th November 2022
Abstract
Microscopic-level understanding of the interaction of hydrocarbons with transition metal surfaces is an important prerequisite for rational design of new materials with improved catalytic properties. In this report, we present a mechanistic study on the keto–enol tautomerisation of butanal on Pd(111), which was theoretically predicted to play a crucial role in low-barrier hydrogenation of carbonyl compounds. These processes were addressed by a combination of reflection-absorption infrared spectroscopy, molecular beam techniques and theoretical calculations at the density functional theory level. Spectroscopic information obtained on Pd(111) suggests that butanal forms three different aldehyde species, which we indicate as A1–A3 as well as their enol counterpart E1. The electronically strongest perturbed and strongest binding species A1 is most likely related to the η2(C,O) adsorption configuration, in which both C and O atoms are involved in the bonding with the underlying metal. The species A2 weakly binds and is less electronically perturbed and can be associated with the η1(O) adsorption configuration. The third type of aldehyde species A3, which is nearly unperturbed and is found only at low temperatures, results from the formation of the butanal multilayer. Importantly, the enol form of butanal was observed on the surface, which gives rise to a new characteristic band at 1104 cm−1 related to the stretching vibration of the C–O single bond (ν(C–O)). With increasing temperature, the multi-layer related species A3 disappears from the surface above 136 K. The population of aldehyde species A1 and the enol species E1 noticeably increases with increasing temperature, while the band related to the aldehyde species A2 becomes strongly attenuated and finally completely disappears above 120 K. These observations suggest that species E1 and A1 are formed in an activated process and – in view of the strongly anti-correlated population of the species E1 and A2 – it can be concluded that enol species E1 is most likely formed from the weakly bound aldehyde species A2 (η1(O)). Finally, we discuss the possible routes to enol stabilization via intermolecular bonding and provide the possible structure of the enol-containing stabilized complex, which is compatible with all spectroscopic observations. The obtained results provide important insights into the process of keto–enol tautomerisation of simple carbonyl compounds.
Introduction
Understanding the mechanisms of heterogeneously catalyzed reactions and their reaction intermediates is a crucial prerequisite for rational based design of new heterogeneous catalysts with tailor-made catalytic properties. One of the technically highly important and fundamentally challenging classes of reactions is the hydrogenation of carbonyl compounds to alcohols, which typically requires high reaction temperatures since a significant activation barrier needs to be surmounted to insert a H atom in a stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.1–3 Recently, a new reaction pathway for this class of reactions was predicted theoretically,4–7 which relies on a two-step process: keto–enol tautomerisation of the original carbonyl compound to an unsaturated alcohol (enol) followed by H insertion into the newly formed CC double bond. Both these reaction steps were predicted to exhibit significantly lower activation barriers as compared to the direct process, opening up the prospect of low-barrier hydrogenation of carbonyl compounds via two consecutive steps.
O bond.1–3 Recently, a new reaction pathway for this class of reactions was predicted theoretically,4–7 which relies on a two-step process: keto–enol tautomerisation of the original carbonyl compound to an unsaturated alcohol (enol) followed by H insertion into the newly formed CC double bond. Both these reaction steps were predicted to exhibit significantly lower activation barriers as compared to the direct process, opening up the prospect of low-barrier hydrogenation of carbonyl compounds via two consecutive steps.
The major challenge of this potential reaction mechanism is the instability of the enol species, which are normally less thermodynamically favored than their ketone counterparts.8 These problems can be overcome by stabilization of the enol form of carbonyl compounds on surfaces via lateral interactions with the neighboring adsorbates.9–17 This interaction can be realized, e.g. via establishing hydrogen bonds between the OH-group of an enol with the O atom of the co-adsorbed species, resulting in the formation of stable adsorbates in the enol form capable of entering the second reaction step – hydrogenation of the CC bond. Recently, we provided several examples of this kind of stabilization of the enol form of a carbonyl compound via formation of oligomer complexes, in which the enol species were stabilized by hydrogen bonding in oligomers.9–16
Specifically, in our recent studies on acetophenone adsorption and hydrogenation over a Pt(111) model catalyst, we showed that a stable enol form of acetophenone can be formed.9–11,13,15 However, it requires the formation of surface ketone–enol dimers or ketone–enol–enol trimers, in which the newly formed OH group of enol is stabilized by establishing a hydrogen bond with the O atom of a carbonyl group of neighboring ketone species.9–11,13,15 The formation of dimer or trimer acetophenone species was found to be an activated process and the ketone–enol–enol trimers were shown to be stable on this surface even at relatively high (up to 350 K) temperatures. Furthermore, hydrogenation of the ketone–enol dimers was also observed on this surface at surface temperatures as low as 240 K, providing thus the first experimental observations of the enol-mediated low-barrier hydrogenation pathway.15 The formation of stabilized enol species was also hypothesized in previous studies by McBreen et al.,16,17 in which the intermolecular interactions based on hydrogen bonding were suggested to be responsible for stabilization of enols on transition metal surfaces. It should be emphasized that strong intermolecular interactions between neighboring species might also result in strong weakening of the bonds involving oxygen atoms as shown e.g. for the CO double bond in acetophenone9,10,13–15 or the C–O single bond in a carbonyl ester ethyl pyruvate.12,18 In the reported cases, a strong red shift of the vibrational frequencies of either CO or C–O bonds was observed by infrared spectroscopy when the O atoms involved in these bonds were hydrogen bonded to the –OH groups of the neighboring molecules.
Despite the recent progress in understanding these processes for relatively large molecules, such as derivatives of acetophenone9,10,13–16 and alkyl pyruvates12,17–19 offering numerous possibilities for hydrogen bonding to stabilize enol species, the chemistry of the smaller aldehydes remains less understood. Previously, the interaction of small aldehydes and ketones with transitions metal surfaces was investigated for a broad range of compounds, including acetone,20–25 acetaldehyde,26–29 propanal30–33 and others. Generally, two types of binding of a carbonyl group with the underlying metal were put forward in these studies: binding via the lone-pair of the O atom to form the “end-on” η1(O) adsorption configuration and binding via both O and C atoms, in which the “side-on” η2(C,O) configuration is formed. In these studies, acetone was the most extensively investigated adsorbates on several metal surfaces including Pd(111)and Pt(111).20–25,34–36 Based on the combination of IRAS and off-specular high-resolution electron energy loss spectroscopy (HREELS), it was possible to unambiguously differentiate between the η1(O) and η2(C,O) adsorption configurations of this molecule.20,23,24 The exceptionally large frequency shift observed for the η2(C,O) configuration (120–190 cm−1 as compared to 1710 cm−1 in unperturbed acetone) was attributed to pronounced mixing of its molecular orbitals with the d-orbitals of underlying metal atom(s).
Similar spectroscopic observations were carried out for different types of aldehydes, including formaldehyde, acetaldehyde and propanal, adsorbed on different metal surfaces and alloys.25–33,37,38 Specifically, the formation of vibrational bands in the range of 1390 and 1580 cm−1 was reported in these studies, which is close to the spectroscopic signature of the η2(C,O) configuration in acetone.20,23,24 For this broader range of aldehydes there is, however, no generally accepted conclusion on the origin of this band: while some authors attribute this vibration to the η2(C,O) configuration of non-dissociated aldehyde molecules,20,25,29,37,38 others attribute this band to the formation of enolate species.21,35 Thus, the possibility of keto–enol tautomerisation of small aldehydes and ketones on transition metal surfaces still remains a controversial issue.
To better understand the mechanistic details of the keto–enol tautomerisation of small aldehydes and the stabilization of their enol form on catalytically relevant surfaces, we performed a spectroscopic study on butanal adsorption and keto–enol tautomerisation over Pd(111) under a broad range of temperature and coverage conditions. In this study, we employ a combination of molecular beam techniques and infrared reflection absorption spectroscopy (IRAS) under ultra-high vacuum (UHV) conditions to address the mechanisms of the underlying elementary processes. Complementarily, theoretical calculations of the gas phase IR spectra of butanal at the density functional theory (DFT) level were carried out to perform a detailed assignment of the vibrational modes of butanal. Specifically, we report the formation of three types of aldehyde species, exhibiting different degrees of electronic structure perturbation by interaction with the underlying metal – the strongly bound and substantially electronically perturbed species A1, which we assign to the η2(C,O) adsorption configuration, and a weaker bound and less electronically perturbed species A2, most likely related to the configuration η1(O). Additionally, the formation of enol species E1 was observed in the whole investigated temperature range. With increasing surface temperature, the population of the species A1 and E1 substantially grows, indicating that the formation of both species occurs in an activated process, while the population of species A2 was found to strongly decrease. Based on the clear anti-correlated evolution of the aldehyde species A2 and enols E1 as a function of temperature, we conclude that the enol form of butanal results from keto–enol tautomerisation of the weakly perturbed aldehyde adsorbate A2. We propose a structural model showing how the enol form of butanal can be stabilized by intermolecular interactions with a neighboring molecule, which is consistent with our experimental observations. The obtained results provide important insights into the details of keto–enol tautomerisation of small aldehyde compounds on transition metal surfaces.
Experimental
The experiments were conducted on a Pd(111) single crystal (MaTeck GmbH, 10 × 10 mm) in a UHV chamber with a base pressure <2 × 10−10 mbar equipped with two crossing effusive molecular beams, an IRAS spectrometer (Bruker, Vertex 80v, resolution 2 cm−1, MCT detector) and a quadrupole mass spectrometer (QMS, Hiden HAL 301/3F). The full details of the experimental setup are described elsewhere.39 The sample crystal was cleaned by repeating cycles of sputtering with Ar+ ions (6 × 10−6 mbar, 1 keV, 45°), annealing at 1100 K and oxidation (1 × 10−6 mbar, 750 K). Before starting the experiment, the sample was flashed to 900 K and cooled down to the desired temperature. The order and the purity of the sample surface was checked by low energy electron diffraction (LEED) and additionally by inspecting the CO (Merck KGaA, redistilled 99.5+%) adsorption spectra in IRAS. Butanal was cleaned in several cool-pump cycles prior to each experiment. During the experiment, butanal was dosed on the surface with a flux rate of 3.70 × 1012 mol cm−2 s−1via an effusive molecular beam.DFT calculation was performed with Gaussian 09 software40 by using the B3LYP method with the aug-cc-pVQZ basis set and the empirical dispersion correction Grimme-Dispersion GD3. The butanal molecules, their vibrational modes and transition dipole moments were visualized using Gauss View 5.0.41 The calculated modes were compared to an experimentally measured reference spectrum obtained at multilayer coverages, at which the most adsorbed molecules do not directly interact with the surface. The calculated modes were scaled to the CO stretching vibration at 1715 cm−1 (ν(CO)) by a factor of 0.953 in order to optimize the fitting to the experimental data.
Results and discussion
The adsorption of butanal was investigated on a well-defined Pd(111) surface under UHV conditions in the temperature range of 100 K and 250 K employing IRAS and molecular beam techniques. In each experiment, the surface was exposed to a series of butanal pulses at a constant temperature and the IR spectra were obtained after each pulse. The resulting surface coverages amount from sub-monolayer to a monolayer coverage; furthermore, the multilayer coverages were achieved at 100 K. The multilayer spectrum obtained at 100 K was utilized as a reference for butanal molecules, which are not perturbed by the interaction with the underlying metal surface, and therefore can be employed for comparison to the theoretical calculation.
Fig. 1 shows the spectrum of butanal in the gas phase calculated by B3LYP (aug-cc-pVQZ/GD3, Gaussian 0940) and the spectrum of the butanal multilayer measured at 100 K on pristine Pd(111) at a high butanal exposure. At this temperature, continuous growth of all vibrational bands was observed in the experimental spectra, which confirms the formation of multilayer butanal. The abscissa of the calculated spectrum is scaled by a factor 0.953 with an origin at 1715 cm−1, which is related to the stretching vibration of the prominent CO band (ν(CO)). The overall shape and the intensity distribution of the calculated IR spectrum is in excellent agreement with the measured multilayer spectrum. The assignment performed based on the comparison of these two spectra is summarized in Table 1 and, additionally, is given in parentheses close to each individual vibrational band below in the text. Note that this table also shows the vibrational bands related to the sub-monolayer and near monolayer coverages, which will be discussed later.
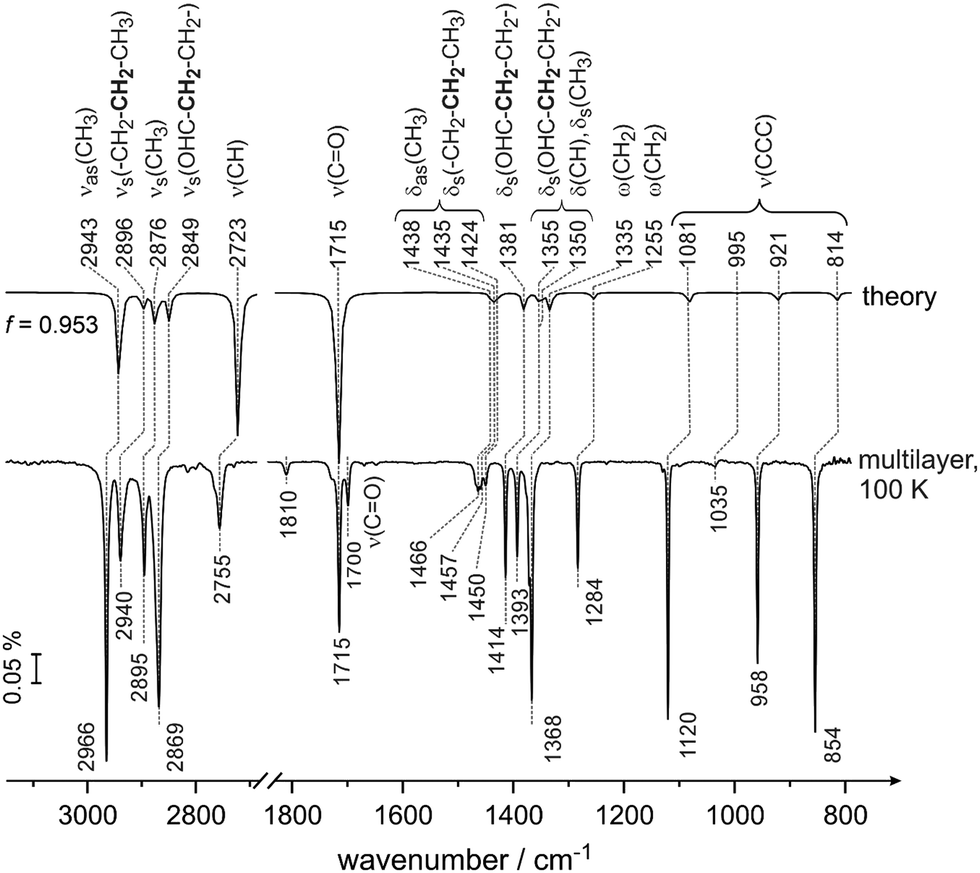 | ||
| Fig. 1 Calculated gas-phase spectrum of butanal at the DFT-B3LYP level of theory (upper spectrum) compared to the IR spectrum of butanal adsorbed on pristine Pd(111) at 100 K at multilayer coverage (bottom spectrum). The multilayer spectrum was obtained in a flux of 5.1 × 1013 mol cm−2 s−1 and a resulting exposure of 1.7 × 1016 mol cm−2. | ||
| Wavenumber/cm−1 | Assignment based on calculated spectra | |||
|---|---|---|---|---|
| Sub-mono-layer, 100 K | Near mono-layer, 100 K | Multilayer, 100 K | B3LYP/aug-cc-pvqz (f = 0.953) | |
| 2955 | 2955/2966 | 2966 | 2943 | ν as(CH3) |
| 2940 | 2940 | 2896 | ν s(–CH2–CH2–CH3) | |
| 2895 | 2876 | ν s(CH3) | ||
| 2881 | 2881 | 2869 | 2849 | ν s(OHC–CH2–CH2–) |
| 2755 | 2723 | ν(CH) | ||
| 1726 | 1715 | 1715 |
ν(CO) |
|
| 1700 | See text | |||
| 1466 | 1438 | δ as(CH3) | ||
| δ s(–CH2–CH2–CH3) | ||||
| 1457 | 1457 | 1457 | 1435 | δ as(CH3) |
| — | 1450 | 1424 | δ as(CH3) | |
| δ s(–CH2–CH2–CH3) | ||||
| 1402 | 1414 | 1414 | 1381 | δ s(OHC-CH2–CH2-) |
| 1382 | 1393 | 1355 and 1350 | δ s(CH3) | |
| δ(CH) | ||||
| δ s(OHC–CH2–CH2–) | ||||
| 1362 | 1368 | 1368 | 1335 | ω(CH2) |
| 1284 | 1255 | ω(CH2) | ||
| 1120 | 1081 | ν(CCC) | ||
| 1035 | 1035 | 1035 | 995 | ν(CCC) |
| 958 | 958 | 958 | 921 | ν(CCC) |
| 854 | 814 | ν(CCC) | ||
The vibrational region typical for C–H stretching vibrations, IR modes appear in the experimentally measured spectra at 2966 cm−1 (νas(CH3)), 2940 cm−1 (νs(–CH2–CH2–CH3)), 2895 cm−1 (νs(CH3)) and 2869 cm−1 (νs(OHC–CH2–CH2–)). Note that the bold font marks the entity that is mostly involved in the deformation vibration. The band at 2755 cm−1 (ν(CH)) is assigned to the C–H stretching mode in the aldehyde group. The prominent band at 1715 cm−1 (ν(CO)) lies in the vibrational range typical for the CO stretching mode. Additionally, a small band at 1700 cm−1 (ν(CO)) was observed in the experimental spectra but was not predicted by our calculation and will be discussed briefly below. The deformation modes of CH2 and CH3 groups appear close together at 1466 cm−1 (δas(CH3) and δs(–CH2–CH2–CH3)), 1457 cm−1 (δas(CH3)), 1450 cm−1 (δas(CH3) and δs(–CH2–CH2–CH3)). Note that the related deformation vibrational modes are complex and involve nearly all atoms. The further deformation vibrational bands comprise the modes at 1414 cm−1 (δs(OHC–CH2–CH2–)), 1393 cm−1 (δs(OHC–CH2–CH2–), δs(CH3) and δ(CH)) as well as the wagging vibrational modes at 1368 cm−1 (ω(CH2)) and 1284 cm−1 (ω(CH2)). The vibrational modes involving skeletal carbon atoms occur at 1120 cm−1 (ν(CCC)), 1035 cm−1 (ν(CCC)), 958 cm−1 (ν(CCC)) and 854 cm−1 (ν(CCC)). All skeletal modes exhibit a high intensity except of the band at 1035 cm−1 (ν(CCC)), which is almost not visible in the multilayer spectrum.
Fig. 2 shows a visualization of the selected vibrational modes discussed above. The blue arrows indicate the displacement of the individual atoms in each particular vibrational mode, while the dark orange arrow shows the resulting transition dipole moment. The arrow corresponding to the transition dipole moment is placed at an arbitrary point in space for clarity of presentation.
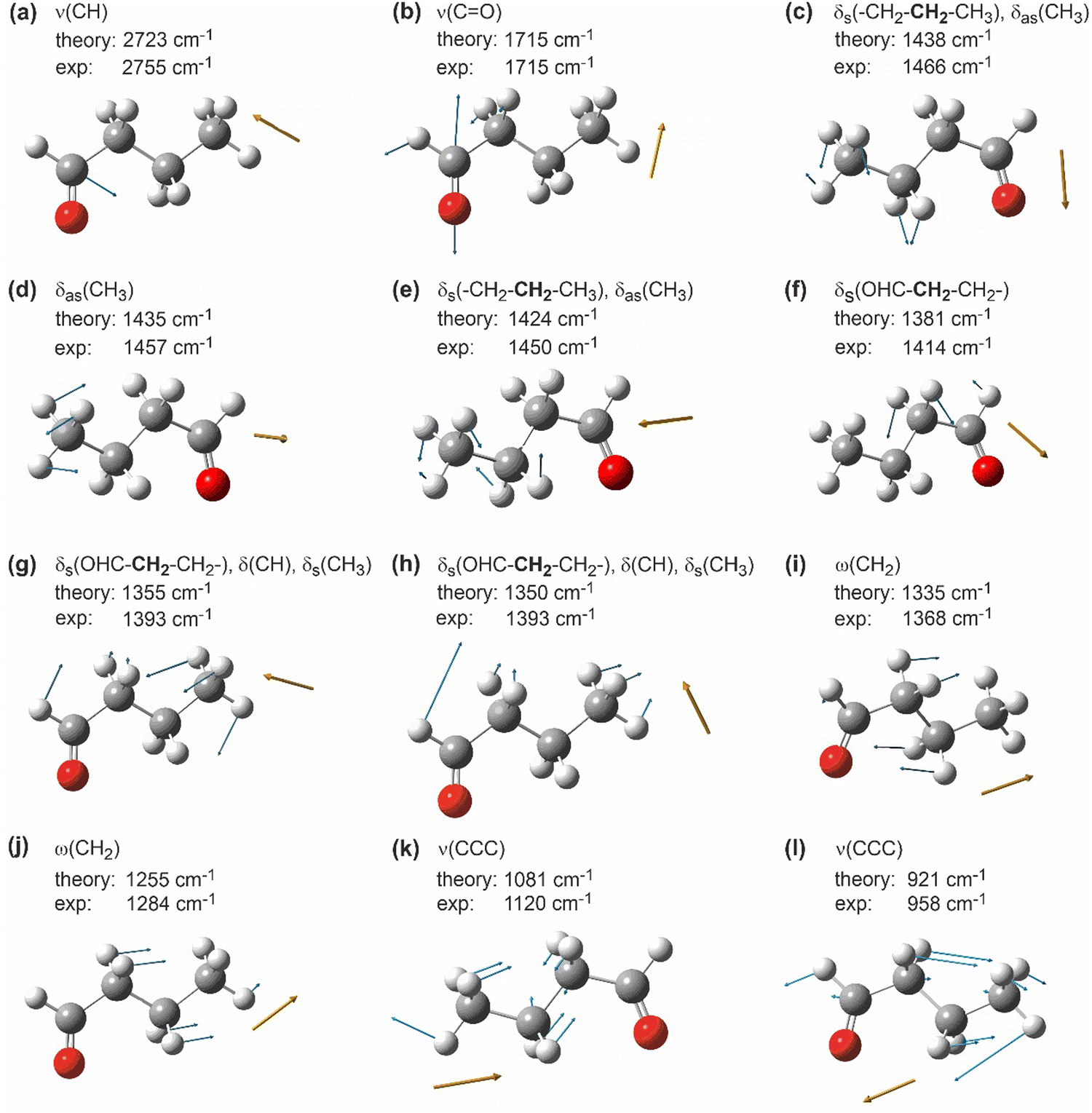 | ||
| Fig. 2 Selected calculated vibrational modes of butanal visualized by GaussView.41 White: hydrogen, grey: carbon, red: oxygen; blue arrows indicate the displacement vectors of individual atoms; dark orange arrows show the orientation of the resulting dynamic dipole moment of the corresponding mode. Note that the dark orange arrows are placed at an arbitrary point in space for a more clear representation. | ||
Previously, IR spectra of unperturbed crystalline butanal were reported in the literature,42 as well the vibrational spectra of structurally similar compounds propanal43–45 and 1-butanol.46 A detailed comparative discussion of the assignment previously performed in the literature and the assignment carried out in this study based on the theoretical calculations is provided in the ESI† (Chapter S1). In general, our assignment agrees well with the previously reported literature data for the majority of the bands.42–46 There are, however, some discrepancies mostly related to the deformation vibrations of the CHx groups and skeletal stretching vibrations (for the details see the ESI†). This observation is most likely related to the fact that the previously reported assignment for butanal42 is mainly based on the comparison of the measured IR spectra with the IR spectra of structurally similar compounds, which is not always very reliable especially for complex vibrations involving a large number of atoms. It should also be pointed out that we were able to provide a more detailed assignment of the modes associated with two different methylene groups, which are non-identical with respect to the distance from the carbonyl entity and therefore exhibit substantially different vibrational frequencies. To the best of our knowledge, this discrimination has not been reported so far.
Having assigned the vibrational modes in the multilayer of butanal, we will address in the following the interaction of butanal with Pd(111) at 100 K. Fig. 3 shows the IR spectra obtained after exposure to increasing amounts of butanal. Butanal was dosed at a flux of 3.7 × 1012 mol cm−2 s−1via the effusive molecular beam in a sequence of pulses; after a certain exposure indicated in Fig. 3, the molecular beam was interrupted and an IR spectrum was recorded (Fig. 3, spectra 1–5). Afterwards, the molecular beam was left continuously running, while the IR spectrum was simultaneously recorded (Fig. 3, spectrum 6).
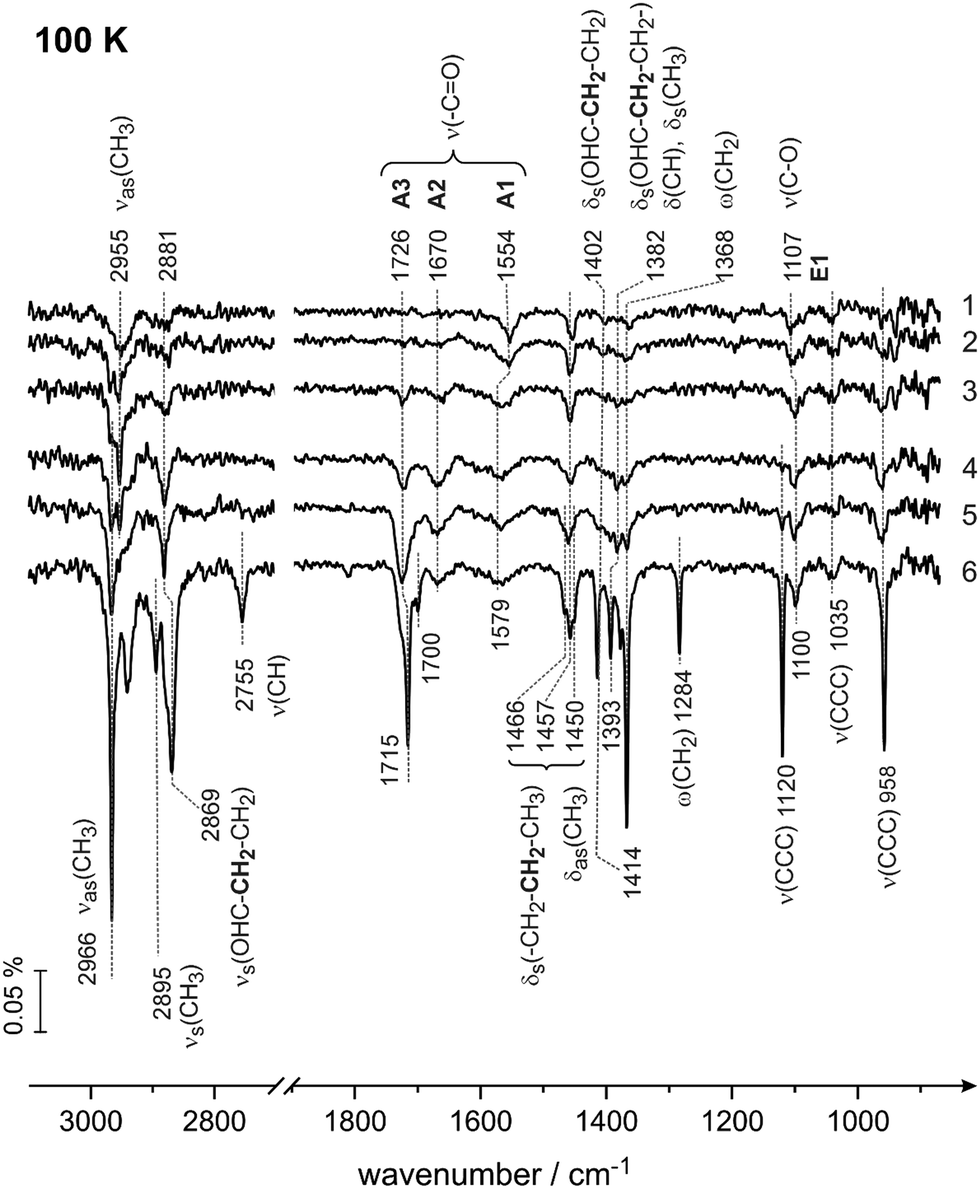 | ||
| Fig. 3 IR spectra of butanal adsorbed on Pd(111) at 100 K at different butanal exposures. Butanal was dosed with a flux of 3.7 × 1012 mol cm−2 s−1 resulting in an exposure of (1) 1.9 × 1014, (2) 2.6 × 1014, (3) 4.1 × 1014, (4) 5.6 × 1014, and (5) 7.8 × 1014 mol cm−2. Spectrum (6) was obtained during continuous butanal dosing via the molecular beam resulting in an overall exposure of 22.0 × 1014 mol cm−2. | ||
The IR spectra obtained in this coverage-resolved way allow us to directly follow the evolution and the shifts of each vibrational band from the sub-monolayer to the multilayer regime and in this way to unambiguously assign the bands evolving at sub-monolayer coverages. This procedure is especially important for strongly interacting systems, such as hydrocarbons adsorbed on transition metal surfaces, since some bands in the sub-monolayer regime can shift with respect to their unperturbed state so substantially that their unambiguous assignment would be impossible.
In the lowest coverage range (Fig. 3, spectra 1 and 2) six vibrational modes appear at 2955 cm−1 (νas(CH3)), 2881 (νs(OHC–CH2–CH2–)), 1554 cm−1 (ν(CO)), 1457 cm−1 (δas(CH3)), 1107 cm−1 (ν(C–O)) and at 1035 cm−1 (ν(CCC)). Note that neither the band at 1554 cm−1 nor the band at 1107 cm−1 are present in the multilayer spectrum of butanal, so they must originate from butanal interaction with Pd(111). Also note that the vibrational range 1050–1250 cm−1 is typical for the stretching vibration of a single C–O bond in the alcohol and alkoxy-groups,47,48 and for this reason the newly evolving band at 1107 cm−1 can be assigned to the stretching vibration ν(C–O) of the C–O single bond. The origin and the reason for the assignment of the vibrational band at 1554 cm−1 to the stretching vibration ν(CO) of the strongly electronically perturbed CO double bond will be discussed in detail below (see also the literature overview given in the introduction).
With increasing exposure (Fig. 3, spectrum 3), the broad band at 2955 cm−1 splits into two more distinct bands at 2966 (νas(CH3)) and 2955 cm−1 (νas(CH3)); the band at 1554 cm−1 (ν(CO)) broadens and shifts to 1579 cm−1 (ν(CO)), also the C–O band shifts by 7 cm−1 to 1100 cm−1 (ν(C–O)). Additionally, the deformation and wagging modes between 1402 cm−1 (δs(OHC–CH2–CH2–)) and 1368 cm−1 (ω(CH2)) appear as well as the skeletal stretching mode at 958 cm−1 (ν(CCC)). The bands in the range of CHx deformations −1466 cm−1 (δas(CH3), δs(CH2–CH2–CH3)) and 1450 cm−1 (δas(CH3), δs(CH2–CH2–CH3)) – slightly shift and become more pronounced.
Further increasing the exposure results in the appearance of two new bands at 1726 cm−1 and at 1670 cm−1 (spectrum 4, Fig. 3), which are located in the typical region of CO stretching vibrations (ν(CO)), while all other bands merely grow in intensity. The intensity of the band at 1726 cm−1 (ν(CO)) further increases in spectra 5 and 6, while the band at 1670 cm−1 (ν(CO)) saturates already in spectrum 4. Note that the band at 1726 cm−1 (ν(CO)) finally shifts to the value 1715 cm−1 (spectrum 6, Fig. 3), which exactly corresponds to the value of the CO stretching vibration observed in the multilayer (see Fig. 1). The origin and the reason for the assignment of the vibrational band at 1670 cm−1 to the stretching vibration ν(CO) of the CO double bond will be discussed in detail below (see also the literature overview given in the introduction). Additionally, the bands at 2895 cm−1 (νa(CH3)), 2755 cm−1ν(CH), and 1120 cm−1ν(CCC) appear in the very last spectrum 6 related to the formation of multilayer butanal.
Thus two major observations can be made based on the coverage dependence of the IR spectra of butanal at 100 K. First, three new vibrational bands appear at the frequencies 1554–1579 cm−1 (ν(CO)), 1670 cm−1 (ν(CO)) and 1107–1100 (ν(C–O)), which are not present in the multilayer spectrum of butanal. The appearance of these bands points to a strong perturbation of the electronic structure of the original butanal molecules by the interaction with the underlying Pd metal (the bands at 1554–1579 cm−1 and 1670 cm−1, both ν(CO)), or even to its chemical transformation to form a C–O single bond (the band at 1007–1100 cm−1, ν(C–O)). Second, the evolution of all other bands shows a very strong coverage dependence, i.e. some of the bands, which are very pronounced in the multilayer butanal, are missing at sub-monolayer coverages. This latter observation is a consequence of the metal surface selection rule (MSSR),49 which predicts that only those vibrational modes are visible in the IR spectra obtained on metals, whose projection of the dynamic dipole moment on the surface normal is different from zero. In contrast, if the dynamic dipole moment is oriented parallel to the surface plane, the intensity of the corresponding vibration should be zero.49 Based on the MSSR, it is principally possible to infer the adsorption configuration of adsorbed butanal from the distribution of the vibrational bands, which are seen at the lowest adsorbate coverages. However, in view of the high structural flexibility of butanal and possible strong deformation of the skeletal structure upon interaction with the metal, the adsorption configuration based on MSSR would be highly speculative. For this reason, we will not perform this type of analysis. The only important observation that we would like to emphasize here is that even at the lowest butanal coverages, the vibrational modes related to the CO and C–O bonds are visible in the spectra suggesting that these bonds are not perfectly parallel to the surface, i.e. they are either tilted or oriented perpendicular with respect to the surface plane.
The three vibrational bands (1554–1579, 1670 and 1726–1715 cm−1) observed in the frequency range characteristic for the CO double bond suggest that three different butanal species are formed on the surface, which we will further refer to as A1, A2 and A3 species, respectively. The newly formed species appearing at 1107–1100 cm−1 (ν(C–O)) will be indicated as E1 species. Additionally, based on the coverage dependence of the IR spectra, the exposure needed to form a monolayer of butanal adsorbates can be estimated. The species A3 exhibiting the vibrational frequency of 1726–1715 cm−1, which is very close to the value of nearly unperturbed molecules, appear most likely when a multilayer starts to form. Since this species appears pronouncedly in spectra 3 and 4 (Fig. 3), it can be concluded that at exposure above 4.1 × 1014 mol cm−2 the multilayer starts to evolve. Below this value, butanal is most likely adsorbed as a monolayer. Taking into account the density of surface Pd atoms 1.5 × 1015 atoms cm−2 and assuming that the sticking coefficient of butanal is equal to unity at 100 K, the saturation coverage of butanal can be roughly estimated as 0.3 butanal molecules per one surface Pd atom. It should also be noted that all species, which can be related to the butanal adsorbates strongly interacting with the underlying metal – the species A1 (1554–1579 cm−1), A2 (1670 cm−1) and E1 (1007–1000 cm−1) – saturate between spectra 3 and 4. This observation additionally supports the estimate of the butanal exposure, which is required to form a full monolayer (>4.1 × 1014 mol cm−2) as achieved above. Indeed, when the coverage exceeds a full monolayer, the concentration of the strongly interacting species A1, A2 and E1 cannot increase further, which should result in saturation of the corresponding vibrational bands, which is indeed observed experimentally.
Previously, the interaction of aldehydes and ketones with transition metal surfaces was investigated for a broad range of acetone,20–25 acetaldehyde,26–29 and propanal.30–33 Generally, two types of binding of a carbonyl group to the underlying metal were put forward in these studies: binding via the O atom to form the “end-on“ η1(O) adsorption configuration and binding via both O and C atoms, in which the “side-on“ η2(C,O) configuration is formed. In these studies, acetone20–25 was the most extensively investigated adsorbate on various transition metal surfaces and alloys. In the liquid form, acetone exhibits a CO stretching vibration at 1710 cm−1 (ν(CO)),50 which is related to the unperturbed carbonyl group. The vibrational bands in the range 1630–1695 cm−1 (ν(CO)) observed on different transition metal surfaces were typically attributed to the η1(O) species, which was proposed to be bonded to the metal through the loan pair electrons of O.20–23,36 The unambiguous spectroscopic identification of the η2(C,O) adsorption configuration is more difficult, since the orientation of the dynamic dipole moment of the CO vibration in this configuration (with both O and C binding to the surface) might be not substantially tilted with respect to the metal surface and therefore difficult to detect due to the MSSR.49 Typically, vibrational frequencies in the range 1520–1585 cm−1 were attributed to the η2(C,O) configuration in these studies on acetone.20,23,24 Specifically, Ibach et al.20 investigated acetone adsorption on Pt(111) by a combination of two types of vibrational spectroscopies: IRAS and off-specular high-resolution electron energy loss spectroscopy (HREELS). In this study, the formation of vibrational peaks in the range 1520–1585 cm−1 (IRAS) and 1550 cm−1 (HREELS) was observed. These vibrational bands were assigned to a “side-on” η2(C,O) configuration of acetone,20 in which the original carbonyl group forms a fragment Pt⋯C⋯O⋯Pt, either in π or di–σ configuration. This assignment was further supported by the studies on the decomposition of isopropanol,51 leading to the formation of the η2(C,O) entity, which exhibits a characteristic vibration in the same frequency range (1584 cm−1). The large frequency shift (120–190 cm−1) as compared to the value 1710 cm−1 in unperturbed acetone was attributed to pronounced mixing of its molecular orbitals with the d-orbitals of underlying metal atom(s).
An even larger vibrational range related to the η2(C,O) configuration (1390–1580 cm−1) was discussed for a broader range of aldehydes, e.g. including also propanal and acetaldehyde.25–33,37,38 There is, however, no generally accepted conclusion on the origin of the vibrational bands in this frequency range: while some authors attribute this band to a partly dissociated form of aldehyde resulting from aldehyde-H abstraction at elevated temperatures,37,52 in the other studies this band is attributed either to η1(O) or to η2(C,O) configuration of non-dissociated aldehyde molecules.20,25,29,37,38 Also the formation of enolate species was discussed to be responsible for the appearance of the vibrational bands in this range.21,35 Many of these assignments remain somewhat speculative though, since an unambiguous conclusion on the presence of the nearly flat lying η2(C,O) species by vibrational spectroscopy can be achieved only by a method allowing the MSSR to be overcome,49e.g. by employing off-specular HREELS or X-ray absorption near edge structure (NEXAFS) spectroscopy, which were applied only for a very limited number of investigated systems.
Summarizing, two major adsorption models exist currently in the literature, in which the vibrational band appearing in the range close to 1670 cm−1 is usually assigned to the η1(O) configuration of the carbonyl group, while the vibrations observed in the range 1520–1585 cm−1 are usually attributed to the more electronically perturbed η2(C,O) configuration. In the first case, the carbonyl group interacts with the metal surface via the lone pair of the O atom, while in the latter adsorption configuration both C and O atoms are binding to the surface, either via π- or di–σ bonds. Specifically for the adsorption of aldehydes, some authors speculate that formation of the η2(C,O) configuration might be accompanied by the abstraction of aldehyde hydrogen.37,52
Based on the above discussion of the literature reports, the species A2 (vibrational band at 1670 cm−1, (ν(CO)) can be most likely attributed to the η1(O) adsorption configuration of butanal bonded to the surface via the O atom. The species A1 with the characteristic vibrational band at 1554–1579 cm−1 results most likely from the η2(C,O) adsorption configuration of butanal. This latter conclusion is particularly strongly supported by the assignment of Ibach et al.20,51 performed in the study for acetone adsorption, in which a highly reliable combination of IRAS and HREELS was applied (see the discussion above). Note that the orientation of the dynamic dipole moment on this configuration should not be perfectly parallel to the surface plane, otherwise this band would not be seen in our IR spectra due to MSSR.49 It is difficult to unambiguously determine based on our experimental data, whether the formation of the A1 species is accompanied by subtraction of aldehyde H as was previously speculated in some previous reports.52 Based on the very low temperature applied in our experiment, we tend to rather exclude decomposition of butanal and will refer to this species as η2(C,O) in the following. Indeed, in the previously reported studies on aldehyde decomposition on Pd(111),37 a temperature as high as 170 K was required for aldehyde H abstraction, which is significantly higher than the 100 K applied in our sets of experiments. It should be kept in mind though that this is not a finally resolved question.
Both species A1 and A2 clearly belong to sub-monolayer species since they appear at the earliest stages of butanal exposure and saturate with an increasing amount of adsorbed butanal. First, species A1 (η2(C,O) configuration) appears on the surface indicating that this is the strongest binding species. This observation is in line with the previously reported studies on acetone, in which the formation of a strongly binding η2(C,O) configuration was already observed at the lowest acetone exposures.20 Only after species A1 nearly reached their saturation coverage, did species A2 (η1(O) configuration) appear (Fig. 3 spectrum 3), which was accompanied by the blue shift of the vibrational band related to the species A1 from 1554 to 1579 cm−1. This shift might be potentially related to two possible effects: (i) direct short-range intermolecular interaction between the species A1 and A2, e.g. via the H bonding; (ii) long range interaction via dipole coupling typically resulting in blue frequency shifts.49
The other prominent vibration that clearly belongs to the newly formed surface species is related to the band at 1107 cm−1, which shifts with increasing butanal coverage to 1100 cm−1 (Fig. 3). The vibrational range is typical for a single C–O bond in alcohols, alkoxy-groups and enols.9,12,18,47,48,53–57 There are two principle routes to forming a C–O bond starting from butanal–keto–enol tautomerisation to the enol form of butanal and (partial) hydrogenation either to a butoxy group or to butanol. In view of the absence of hydrogen and very low surface temperature (100 K), the hydrogenation pathway seems to be rather unlikely. For this reason, we assign the band appearing in the range 1107–1100 cm−1 to the stretching vibration of the C–O single bond in enol form of butanal (ν(C–O)) resulting from keto–enol tautomerisation. It should be highlighted that direct comparison of the experimentally measured vibrational bands on surfaces with the vibrational data obtained for enols in the gas or in the liquid phase is hardly possible. The enols are typically less thermodynamically stable than ketones,8 therefore there are nearly no spectroscopic experimental data on the alone-standing enol species. However, there are some cases, in which the vibrational frequencies of the C–O single bonds in enols were experimentally determined for enol complexes, in which the –C–OH group of the enol species was stabilized by hydrogen bonding with a neighbouring CO group. Thus, in the previously reported studies on keto–enol tautomerization in diketones and ketoesters, the formation of chelated ring-like ketone/enol tautomers was observed.58–63 In these chelating ring-like structures, one carbonyl group transforms to the C–O(H) bond to form enol, while the O atom of the second carbonyl group stabilizes the newly formed OH group of the enol by hydrogen bonding resulting in the –CO⋯H–O– entity. The frequency range found for the C–O(H) bond of the enol part agrees well with the vibrational band appearing at 1107–1100 cm−1 in our study. Additionally, in our recent studies on keto–enol tautomerisation of acetophenone over Pt(111), the formation of dimer and trimer complexes containing enol species was spectroscopically proven.9,10 In these ketone–enol dimers9 and enol-ketone–enol trimers,9,10 a vibrational band in a similar frequency range was observed at temperatures above 140 K and 280 K, correspondingly. Note that the new vibrational band at 1107–1100 cm−1 (ν(C–O)) does not coincide with the vibrational band at 1120 cm−1 related to the skeletal vibration ν(CCC) in butanal, which appears only at high coverages in the multilayer (see Fig. 3, spectrum 6 and Table 1). This can be even more clearly deduced from spectrum 5 in Fig. 3, showing that the band at 1120 cm−1 (ν(CCC)) starts to evolve and later continuously grows in intensity with increasing coverage, while the band at 1107–1100 cm−1 (ν(C–O)) forms at sub-monolayer coverages and saturates well before the transition to the multilayer.
Summarizing, three types of surface species are formed upon adsorption of butanal on Pd(111) at 100 K, which we attribute to two different ketone species – strongly perturbed A1 (band at 1554–1579 cm−1, ν(CO), most likely η2(C,O) configuration), less strongly perturbed A2 species (band at 1670 cm−1, ν(CO), η1(O) configuration), and one type of enol species E1 (band at 1107–1100 cm−1, ν(C–O)). It should also be noted that enol species adsorbed on metal surfaces are typically stabilized by intermolecular interaction with neighbouring adsorbates,9,10,17e.g. via interaction with the carbonyl group of co-adsorbed ketones as evidenced by the formation of different oligomers observed by scanning tunnelling microscopy (STM). In analogy with these previous observations, we can hypothesize that also the enol species E1 formed in our case is most likely stabilized by intermolecular interactions with the neighbouring carbonyls or other enol molecules. The exact proposed models for this interaction will be discussed below. In view of the possible strong changes of the butanal structure upon interaction with the underlying metal, we omit an extensive discussion of the possible orientation of the individual functional groups with respect to the surface plane based purely on MSSR. It can only be safely concluded that all bands visible in IRAS, most importantly the CO and C–O bonds, are tilted with respect to the surface plane.
As the next step, the chemical transformations of butanal-related species were investigated in the temperature range 120–250 K. Specifically, the possible keto–enol tautomerisation of ketone species to their enol counterpart was examined in the broad coverage range. For this reason, we will focus in the following mostly on the frequency ranges characteristic for ν(CO) and ν(C–O) vibrations (1850 cm−1 to 1000 cm−1).
Fig. 4 shows the IRAS spectra obtained for different exposures of butanal at 120 (Fig. 4a) and 136 K (Fig. 4b), correspondingly. Butanal was dosed at a flux of 3.7 × 1012 mol cm−2 s−1via the effusive molecular beam applying an identical sequence of pulses as described for Fig. 3. After a certain exposure was reached, the molecular beam was interrupted and an IR spectrum was recorded (Fig. 4, spectra 1a–5a and 1b–5b). Afterwards, the beam was left continuously running, while the IR spectrum was simultaneously recorded (Fig. 4, spectra 6a and 6b).
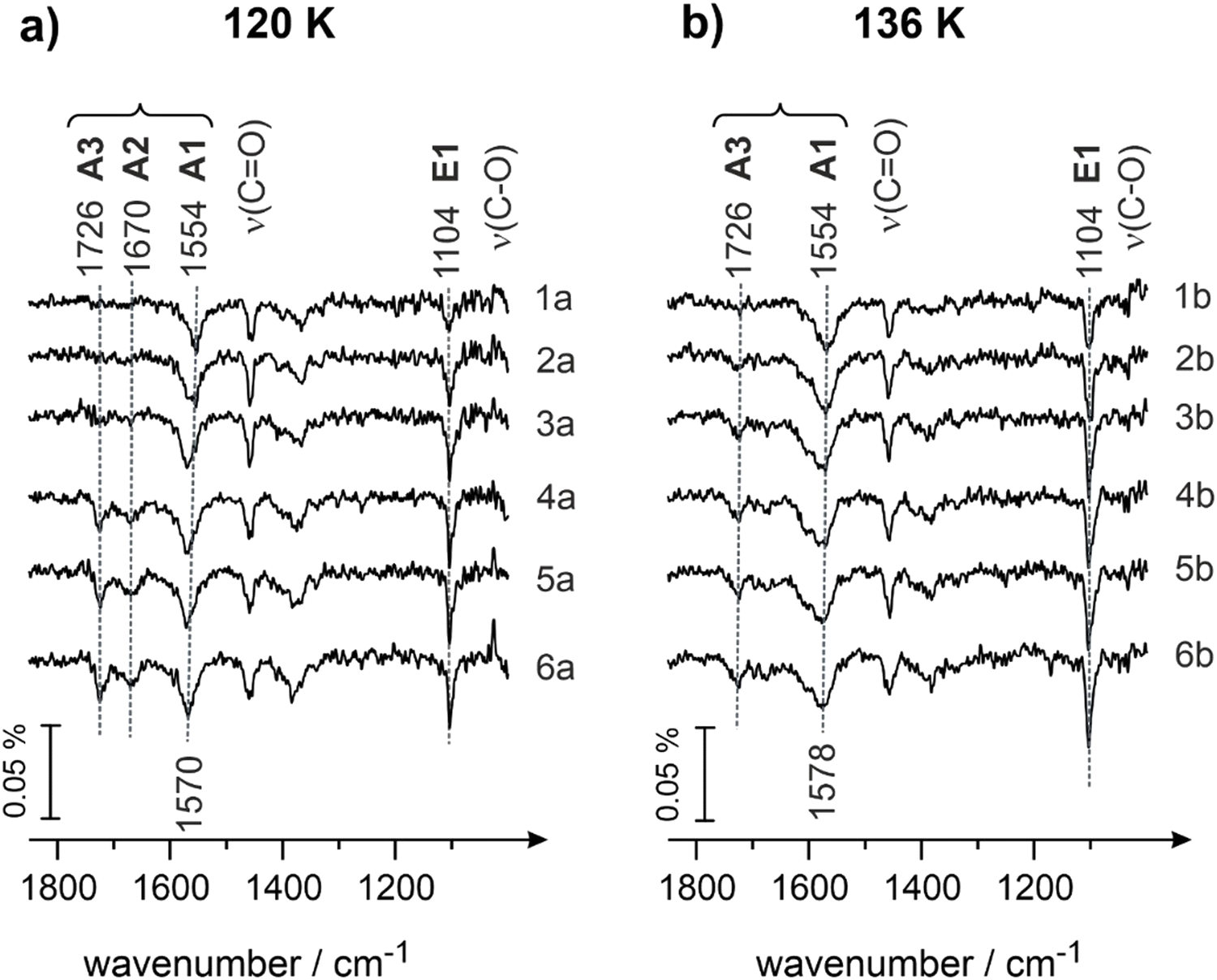 | ||
| Fig. 4 IR spectra of butanal adsorbed on Pd(111) at (a) 120 K and (b) 136 K at different butanal exposures. Butanal was dosed with a flux of 3.7 × 1012 mol cm−2 s−1 resulting in exposures of (1a and 1b) 1.9 × 1014, (2a and 2b) 2.6 × 1014, (3a and 3b) 4.1 × 1014, (4a and 4b) 5.6 × 1014, and (5a and 5b) 7.8 × 1014 mol cm−2. Spectra (6a and 6b) were obtained during continuous butanal dosing via the molecular beam resulting in an overall exposure of 22.0 × 1014 mol cm−2. | ||
It can be clearly seen that the evolution of both ketone (A1, A2 and A3) and enol (E1) species at 120 K (Fig. 4a) with increasing coverage closely resembles that behavior observed at 100 K. At the lowest butanal exposure, the ketone species A1 (1554 cm−1, ν(CO)) and the enol species E1 (1104 cm−1, ν(C–O)) coexist and the corresponding bands grow in intensity (Fig. 4, spectra 1–3). With further increasing the coverage, the ketone species A2 (1670 cm−1, ν(CO), spectra 4–5) evolves, while the intensity of the bands related to A1 and E1 species seem to be already saturated at these exposures. At coverages exceeding one monolayer, the band at 1726 cm−1 appears at a very small intensity (spectra 4–6), however, this band does not continuously grow with growing exposure. This latter observation suggests that multilayer butanal cannot be formed at 120 K anymore; the small amounts of A3 species, which are related to the nearly unperturbed form of butanal, are formed most likely in the second layer, which can be populated only at very high butanal exposures at this temperature. Despite the similarity in the evolution of the different surface species with the data displayed in Fig. 3 for 100 K, there is one striking discrepancy that can be clearly seen at 120 K: the band at 1104 cm−1 (ν(C–O)) has a significantly higher intensity than at 100 K. This observation suggests that the surface concentration of enol species E1 grows with increasing temperature.
The same trend continues also at the surface temperature of 136 K (Fig. 4b): the intensity of the vibrational band at 1104 cm−1 (ν(C–O), species E1) becomes even larger as compared to the surface temperature of 120 K (Fig. 4a). Simultaneously, another important observation can be made at 136 K: the intensity of the band at 1570 cm−1 (species A1, ν(CO)) remains nearly the same as at 120 K, however the band at 1670 cm−1 (species A2, ν(CO)) does not appear at all. There is still a very low-intensity band at the position 1726 cm−1, which is related most likely to small amounts of unperturbed butanal adsorbed in the second layer.
In view of the observed pronounced temperature dependence of enol (E1) formation, it can be quite safely concluded that in this temperature range (100–136 K) keto–enol tautomerisation is an activated process on Pd(111), which requires surmounting an activation barrier. This conclusion is in excellent agreement with our previous spectroscopic and microscopic observations on keto–enol tautomerization of acetophenone on Pt(111).9,10 In these previous studies, acetophenone was observed to form enol species at temperatures exceeding 140 K. In the present study, the onset of keto–enol tautomerisation is somewhat lower – already at 100 K the small concentration of enol can be detected by IRAS, which is most likely related to the lower barrier for the hydrogen transfer process in butanal adsorbed on Pd(111).
Another important observation is related to the fact that the increase of the surface concentration of enols E1 is strongly anti-correlated with the amount of the surface species A2 (band 1670 cm−1, ν(CO)): while the band related to the enol species E1 substantially grows in intensity with increasing temperature, the IR band related to the species A2 becomes strongly attenuated and finally completely disappears. On the other hand, the abundance of the ketone species A1 (band at 1554 cm−1, ν(CO)) seems to remain very similar in both data sets shown in Fig. 4. The combination of these observations suggests that species A1, which are most likely related to the strongly bonded η2(C,O) configuration of butanal, do not directly interconvert to enols, while species A2, associated with the weaker bonded η1(O) configuration of butanal, undergoes keto–enol tautomerisation and forms enol species. Note that this conclusion is valid if only two aldehyde species A1 and A2, which can be detected experimentally, are present on the surface.
This hypothesis is further supported by the experimental data shown in Fig. 5a and b obtained at the surface temperatures 160 and 200 K, correspondingly. Here, two major vibrational bands are present for each temperature in the entire investigated range of exposures: (i) the band related to the enol species E1 (1104 cm−1, ν(C–O), for both temperatures) and (ii) the band associated with the strongly bound A1 species (1558 cm−1, ν(CO)). In neither of these spectra is the band related to the weakly bound ketone species A2 (1670 cm−1) present. The intensities of the bands related to A1 are similar for 160 and 200 K and comparable to the intensities of the same species measured at 120 and 136 K (see Fig. 4a and b). On the other hand, the intensity of the band related to enol species E1 (1104 cm−1) grows in the range 120–136 K and then remains nearly unchanged at the further temperature increase from 136 to 200 K. This overall trend is again clearly anti-correlated to the abundance of the species A2: they decrease in intensity within the temperature range 120–136 K and remain absent in the range 136 to 200 K. This observation additionally supports our hypothesis that species A1 aren’t most likely interconverted to species E1, while species A2 is a direct precursor for E1.
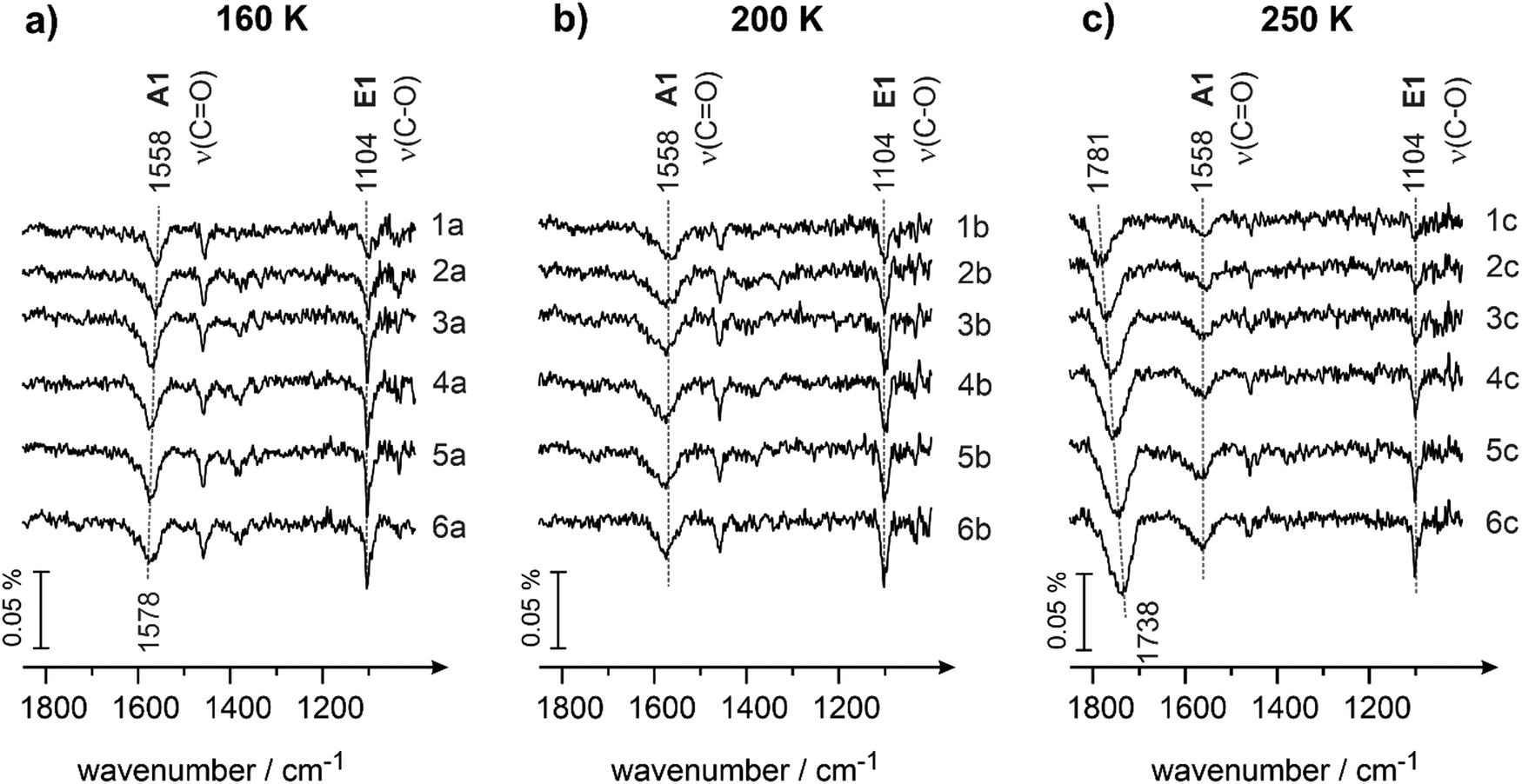 | ||
| Fig. 5 IR spectra of butanal adsorbed on Pd(111) at (a) 160 K, (b) 200 K and (c) 250 K at different butanal exposures. Butanal was dosed with a flux of 3.7 × 1012 mol cm−2 s−1 resulting in an exposure of (1a, 1b and 1c) 1.9 × 1014, (2a, 2b and 2c) 2.6 × 1014, (3a, 3b and 3c) 4.1 × 1014, (4a, 4b and 4c) 5.6 × 1014, and (5a, 5b and 5c) 7.8 × 1014 mol cm−2. Spectra (6a, 6b and 6c) were obtained during continuous butanal dosing via the molecular beam resulting in an overall exposure of 22.0 × 1014 mol cm−2. | ||
At 250 K (Fig. 5c), both species A1 and E1 are present on the surface, however at somewhat lower intensities as compared to the temperature range 120–200 K. On the other hand, a new prominent band at 1781 cm−1 appears, which shifts to 1738 cm−1 with increasing butanal exposure. The appearance of this band can be most likely explained by the onset of partial decomposition of butanal to form –HxCO fragments on the surface, which is in line with the previous reports on decomposition of aldehydes.37 Indeed, the band at 1781 cm−1 lies in the typical range of carbonyl compounds,49 however, it is significantly blue shifted as compared to unperturbed butanal molecules (1715 cm−1 in the butanal multilayer, see Fig. 1). On the other hand, the decomposition product cannot be an alone-standing CO molecule, as the vibrational frequency of adsorbed CO on Pd(111) appears typically well above 1800 cm−1.64 For this reason, we attribute the newly evolving band at 1781 cm−1 to a partial decomposition product of butanal, involving the carbonyl group and the rest of the original molecule. The diminishing intensity of the bands at 1558 cm−1 (species A1) and 1104 cm−1 (species E1) might be related to the decomposition of butanal: a fraction of all butanal derivatives can partly decompose starting from 250 K, which results in the decreasing overall concentration of A1 and E1 species. On the other hand, some fraction of butanal can also desorb with increasing temperature and contribute in this way to the decrease of the overall concentration of butanal species.
Fig. 6 shows the comparison of IR spectra obtained on Pd(111) at different surface temperatures following identical butanal exposure (7.8 × 1014 mol cm−2). Note that in view of the different sticking coefficients at varying surface temperature, identical butanal exposure does not necessarily result in identical surface coverage. It can be clearly seen that at 100 K, this butanal exposure results in the formation of all three aldehyde species – the species A1 and A2 adsorbed directly on the metal surface, as well as the unperturbed species A3, which are most likely adsorbed in the upper lying layers. The enol species E1 is also formed at 100 K, however, the intensity of the related band remains quite low. With increasing temperature, the unperturbed species A3 completely disappear from the surface, indicating that no multilayer of butanal can be formed above 136 K. Simultaneously, the intensities of the two surface species A1 and E1 noticeably grow, while the intensity of the band related to the species A2 strongly decreases. Above 160 K, only the aldehyde surface species A1 (strongly bound η2(C,O) configuration) and the enol species E1 are present on the surface at relatively high concentrations. Based on the well pronounced anti-correlated abundance of the species A2 (weakly bound η1(O) configuration) and the enol species E1, we conclude that the enols species are rather formed from the precursor A2 species, while species A1 does not undergo keto–enol tautomerisation to any noticeable extent.
 | ||
| Fig. 6 Comparison of the IR spectra of butanal adsorbed on Pd(111) obtained at different surface temperatures indicated next to the spectra (100–250 K). The exposure of butanal in all spectra amounts to 7.8 × 1014 mol cm−2; the butanal was dosed at a flux of 3.7 × 1012 mol cm−2 s−1. | ||
Fig. 7 shows the integrated intensities of the IR bands related to the species A1–A3 and E1 plotted as a function of surface temperature. It should be highlighted that in a general case, the IR intensity might not be directly proportional to the concentration of the particular surface species. The nonlinearity can arise from a number of effects, such as the changing inclination angles of the related bands, dipole–dipole coupling etc. For this reason, possible non-linearity of the integrated IR bands shown in Fig. 7 should be kept in mind. However, one conclusion can be quite safely drawn based in these data: species A2 (weakly bound η1(O) species) completely disappears at increasing temperature, while the intensity of the enol species E1 simultaneously grows. Indeed, the tilted adsorption geometry of the η1(O) species implies that if species A2 is present on the surface, they would be most likely visible in the IR spectra based on MSSR. It is very unlikely that the orientation of the CO bond in the “end-on” η1(O) species can change with increasing temperature so much, that the CO bond would become perfectly parallel to the surface and therefore invisible in IRAS. For this reason, we explain the vanishing of the band at 1670 cm−1 not by the strong change of the orientation geometry of the CO bond, but by the vanishing of these species, e.g. due to their keto–enol tautomerization to enols.
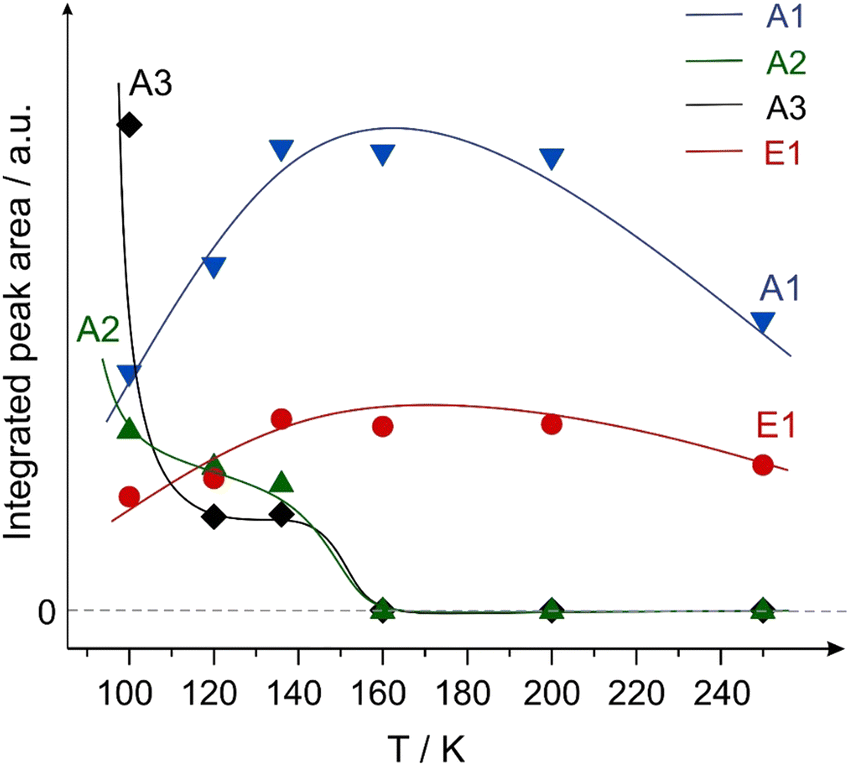 | ||
| Fig. 7 Integrated peak areas of the vibrational bands of the aldehyde species A1–A3 (ν(CO)) and the enol species E1 (ν(C–O)) plotted as a function of surface temperature. The areas were integrated within the rectangles shown in Fig. 6. | ||
Summarizing the observed temperature dependence shown in Fig. 4–7, it can be concluded that the enol species, evolving on the surface at elevated temperatures in an activated process, are rather formed from the weakly bound η1(O) aldehyde precursor (A2 species), while the strongly bound η2(C,O) aldehyde A1 species do not undergo keto–enol tautomerisation and remain on the surface in the original form up to at least 250 K.
It should also be noted that the enol species are first observed at 100 K and their surface concentration noticeably increases in the temperature range 120–200 K as shown in Fig. 6 and 7. To explain these experimental results, two types of co-existing surface species should be most likely taken into consideration: the first type of highly active sites should be capable of catalyzing the enol formation already at 100 K, while the second type of surface sites requires higher temperatures for keto–enol tautomerisation and is responsible for vanishing of species A2 (concomitant to the increase of species E1) in the temperature range 120–200 K. The most likely candidate for the highly active sites might be surface defects, while the less active sites can be related to the regular surface sites.
In the following, we will discuss possible adsorption configurations of enol species, which are displayed in Fig. 8. Normally, enol species are thermodynamically less stable than their ketone counterparts. There is a number of experimental studies showing that the adsorbed species in enol form must be stabilized.9–11,13,16,17 Known examples are related e.g. to enol stabilization in dicarbonyl compounds via intramolecular interactions resulting in building chelate-like ring complexes, in which one of the carbonyl groups tautomerizes to enol and the OH group of the enol entity makes a hydrogen bond with the O atom of the second carbonyl group.58–63 On metal surfaces, different types of enol complexes were put forward to explain stabilization of the enol form of acetophenone: (i) hydrogen bonding between the OH group of one enol adsorbate and the O atom of a carbonyl group of the neighboring enol species;9–11,13 and (ii) hydrogen bonding between two OH groups of neighboring enol species;16,17 in this latter configuration an O atom of one OH group is bonded to the H atom of the other OH groups. In all experimental studies reported so far, no indications were found that the enol species can be present on the surface without any stabilization via intermolecular interactions. In line with these previous experimental observations, we can hypothesize that enol species spectroscopically observed in this study, should also be stabilized by some sort of intermolecular interaction with the neighboring molecules. Fig. 8 shows the possible adsorption configurations that might account for this bonding. The most common assumption on the configuration of the enol complex is indicated as the structure D1, in which the H atom of the OH group is hydrogen bonded to the carbonyl group of the neighboring butanal molecule. This structure is, however, not very likely in our opinion, as it was previously shown that the H bonding between a carbonyl group and the acidic H of the enol OH group results in a red frequency shift of about 40 cm−1.9 In our case, this kind of structure should lead to the appearance of new bands in the range of carbonyl vibrations, which are by appr. 40 cm−1 lower than the frequencies of the surface-adsorbed species A1 or A2, which is not observed experimentally. For this reason, we rather exclude the model, in which the O atom involved in the carbonyl group directly participates in the interaction with the OH group of the enol. In contrast, it appears to be more likely, that hydrogen bonding is realized via the O atom involved in the OH group of enol species. Two possible scenarios for this interaction are shown in Fig. 8 as structure D2 and D3. In the case of the D2-model, the O atom of the OH group establishes H bonding with the aldehyde H of the neighboring butanal species, while in the case of the D3-model, the O atom of the OH group is hydrogen bonded to the H atom of the other OH group involved in the enol species. We have chosen these two structures since in both cases O establishes the strongest hydrogen bond with two highly acidic H atoms: either aldehyde-H or H involved in the OH group. According to previous reports, the strength of hydrogen bonding directly depends on the acidity of the participating hydrogen: the stronger the acidity of the hydrogen atom, the stronger the hydrogen bond.65,66 It cannot be excluded based on our experimental results that an O atom can establish hydrogen bonding also with some H atoms of the CHx groups of butanal. However, the strength of this bond would be lower than in the D2 and D3 models displayed in Fig. 8 and therefore less likely.
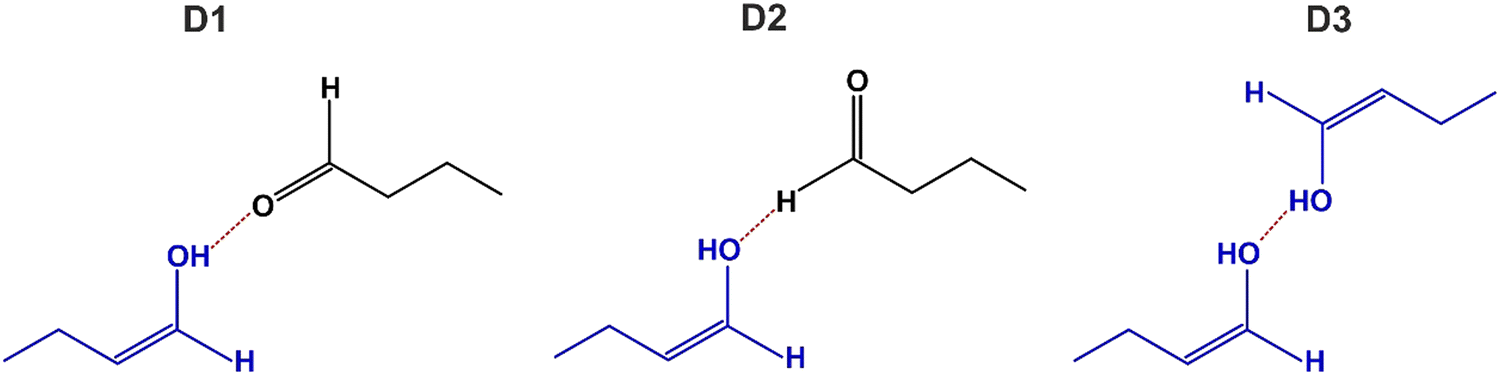 | ||
| Fig. 8 Schematic representation of the possible configurations of enol-containing complexes, in which the normally unstable enol species are stabilized by establishing hydrogen bonding with the neighboring adsorbates (see the text for more details). | ||
It should also be noted that the D3 structure contains two non-identical C–O(H) bonds: in one C–O(H) group, the O atom is directly involved in hydrogen bonding with the neighboring H atom, while in the other C–O(H) group the O atom does not participate in hydrogen bonding. These two non-equivalent C–O(H) groups might result in two different vibrations of the related C–O bond, which is not observed experimentally. Taking into account this consideration, we rather believe that the structure D2 is the most likely one as it contains only one C–O(H) entity, which should result in a single IR band in the range of C–O vibration, which exactly corresponds to our experimental observations. The hydrogen bonding in this model is most likely realized through the bonding to aldehyde H of the species A1. We would like to point out that the presented model D2 fits our experimental observations in the best way. However, we cannot exclude that other enol-containing structures can also co-exist on the surface. For the additional discussion on the possible adsorption configuration of the complex D2 see Chapter S2 in the ESI.†
Conclusions
Adsorption and keto–enol tautomerisation of butanal on a well-defined Pd(111) surface was investigated by a combination of IRAS and molecular beam techniques in a broad range of temperature and coverage conditions. Complementarily, theoretical calculations of the IR spectra of butanal were performed at the DFT level, which were employed for the accurate assignment of individual vibrational bands.Specifically, butanal adsorption was first investigated at 100 K, at which butanal forms a multilayer that can serve as a reference for an unperturbed molecule. The IR spectra obtained for multilayer butanal coverages were compared to the theoretically computed spectra and a detailed assignment of evolving vibrational bands was performed. Besides some well-known previously reported bands, it was possible to more accurately assign the stretching and deformation vibrations of different CHx groups and differentiate between the vibrations involving terminal CH3 groups and two non-identical CH2 groups.
Upon adsorption on pristine Pd(111), butanal was found to form three different aldehyde species, which are indicated as A1–A3 as well as the enol counterpart E1. The strongest perturbed species A1 exhibits a vibrational band at the frequency 1554 cm−1 (red-shifted by about 160 cm−1 as compared to the unperturbed state) and can be most likely related to the η2(C,O) adsorption configuration, in which both C and O atoms are involved in bonding with the underlying metal. Species A2 appear at higher butanal exposures, indicating that their binding energy on Pd(111) is lower than that of species A1. The associated vibrational frequency amounts to 1670 cm−1, suggesting that the degree of electronic perturbation of this configuration is significantly lower than in species A1. This adsorbate adopts most likely the weaker binding η1(O) configuration, in which the interaction with the metal is realized via the lone pair of the O atom involved in the carbonyl group. The assignment of species A1 and A2 to η2(C,O) and η1(O) configurations, respectively, is in perfect agreement with previously published reports on structurally similar carbonyl compounds. The third type of aldehyde species A3 evolves at the highest butanal exposures at vibrational frequencies of 1726–1715 cm−1 and results from the formation of a multilayer. The last type of adsorbates formed on the surface is the enol form of butanal, which gives rise to the evolution of a new vibrational band at 1104 cm−1, the typical frequency of the stretching vibration of the C–O single bond (ν(C–O)).
With increasing temperature, species A3 quickly disappear from the surface above 136 K, indicating that multilayer butanal cannot be formed above this temperature. The intensities of the vibrational bands related to the aldehyde species A1 (configuration η2(C,O)) and the enol species E1 noticeably increase, while the band related to the aldehyde species A2 (configuration η1(O)) becomes strongly attenuated and finally completely disappears above 120 K. Two important conclusions can be quite safely drawn from this observation: (i) the formation of enol species E1 and aldehyde species A1 is an activated process and (ii) the enol species E1 is most likely formed from the weakly bound aldehyde species A2 and not from the strongly bound species A1. This latter conclusion is based on a strongly anti-correlated abundance of the species E1 and A2.
At temperatures as high as 250 K, an onset of butanal decomposition was observed. Spectroscopically, a new band at 1745 cm−1 was detected, which is most likely related to a butanal fragment containing a carbonyl group (HxCO).
Finally, we discuss the possible route to enol stabilization on the surface, which is most likely realized via intermolecular interactions with neighboring adsorbates. We put forward the model D2, which is compatible with all spectroscopic observations and is based on the hydrogen bonding between the OH group of enol and aldehyde H of neighboring aldehyde species. It should be kept in mind that other enol complexes might also be thermodynamically stable on the surface under the given conditions.
The obtained results provide important insights into the process of keto–enol tautomerisation of simple carbonyl compounds. The related effects are expected to play a role in the low-temperature hydrogenation pathway of carbonyl compounds involving consecutive processes of keto–enol tautomerisation followed by low-barrier hydrogenation of the newly formed olefinic bond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the German Science Foundations (DFG, Grant SCHA 1477/6-1 and DFG, INST 257/543-1 FUGG) is gratefully acknowledged.References
- F. Shi, H. Wang and X. Dai, Carbonyl Compounds: Reactants, Catalysts and Products, John Wiley & Sons, Incorporated: ProQuest Ebook Central, 2021 Search PubMed.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- F. Meemken and A. Baiker, Recent Progress in Heterogeneous Asymmetric Hydrogenation of CO and CC Bonds on Supported Noble Metal Catalysts, Chem. Rev., 2017, 117(17), 11522–11569 CrossRef CAS.
- A. M. H. Rasmussen, M. N. Groves and B. Hammer, Remote Activation of Chemical Bonds in Heterogeneous Catalysis, ACS Catal., 2014, 4(4), 1182–1188 CrossRef CAS.
- B. Yang, D. Wang, X.-Q. Gong and P. Hu, Acrolein hydrogenation on Pt(211) and Au(211) surfaces: a density functional theory study, Phys. Chem. Chem. Phys., 2011, 13(47), 21146–21152 RSC.
- J. E. De Vrieze, J. W. Thybaut and M. Saeys, Role of Keto–Enol Tautomerization in the Copper-Catalyzed Hydrogenation of Ketones, ACS Catal., 2019, 9(5), 3831–3839 CrossRef CAS.
- B. Bandyopadhyay, P. Pandey, P. Banerjee, A. K. Samanta and T. Chakraborty, CH⋯O Interaction Lowers Hydrogen Transfer Barrier to Keto–Enol Tautomerization of β-Cyclohexanedione: Combined Infrared Spectroscopic and Electronic Structure Calculation Study, J. Phys. Chem. A, 2012, 116(15), 3836–3845 CrossRef CAS PubMed.
- Z. Rappoport, The Chemistry of Enols (Chemistry of Functional Groups Series), John Wiley & Sons Ltd Chichester, 1990 Search PubMed.
- S. Attia, M.-C. Schmidt, C. Schröder, P. Pessier and S. Schauermann, Surface-Driven Keto–Enol Tautomerization: Atomistic Insights into Enol Formation and Stabilization Mechanisms, Angew. Chem., Int. Ed., 2018, 57(51), 16659–16664 CrossRef CAS PubMed.
- S. Attia, M. C. Schmidt, C. Schröder and S. Schauermann, Formation and Stabilization Mechanisms of Enols on Pt through Multiple Hydrogen Bonding, ACS Catal., 2019, 9(8), 6882–6889 CrossRef CAS.
- M. C. Schmidt, S. Attia, C. Schröder, A.-K. Baumann and S. Schauermann, Formation and Real-Space Distribution of Acetophenone Dimers on H-containing Pt(111), J. Phys. Chem. C, 2021, 125(35), 19311–19324 CrossRef.
- C. Schröder, M. C. Schmidt, C. Witt, S. Attia, J. Weber, A.-K. Baumann, B. Hartke and S. Schauermann, Tuning the Strength of Molecular Bonds in Oxygenates via Surface-Assisted Intermolecular Interactions: Atomistic Insights, J. Phys. Chem. C, 2020, 124(51), 28159–28168 CrossRef.
- M. C. Schmidt, S. Attia, C. Schröder, A.-K. Baumann, P. Pessier and S. Schauermann, Temperature-Dependent Formation of Acetophenone Oligomers Accompanied by Keto–Enol Tautomerism: Real Space Distribution, J. Phys. Chem. C, 2020, 124(26), 14262–14271 CrossRef CAS.
- S. Attia and S. Schauermann, Coverage-Dependent Adsorption Geometry of Acetophenone on Pt(111), J. Phys. Chem. C, 2020, 124(1), 557–566 CrossRef CAS.
- S. Attia, M. C. Schmidt, C. Schröder, J. Weber, A.-K. Baumann and S. Schauermann, Keto–Enol Tautomerization as a First Step in Hydrogenation of Carbonyl Compounds, J. Phys. Chem. C, 2019, 123(48), 29271–29277 CrossRef CAS.
- V. Demers-Carpentier, M.-A. Laliberté, S. Lavoie, G. Mahieu and P. H. McBreen, Two-Dimensional Self-Assembly and Catalytic Function: Conversion of Chiral Alcohols into Self-Assembled Enols on Pt(111), J. Phys. Chem. C, 2010, 114(16), 7291–7298 CrossRef CAS.
- S. Lavoie, M.-A. Laliberté, G. Mahieu, V. Demers-Carpentier and P. McBreen, Keto–Enol Driven Assembly of Methyl Pyruvate on Pt(111), J. Am. Chem. Soc., 2007, 129(38), 11668–11669 CrossRef CAS PubMed.
- C. Witt, M.-C. Schmidt, C. Schröder, S. Schauermann and B. Hartke, Disordered Two-Dimensional Self-Organization of Ethyl Pyruvate Molecules on the Pt(111) Surface, J. Phys. Chem. C, 2021, 125(47), 26167–26179 CrossRef CAS.
- M. Castonguay, J. R. Roy, A. Rochefort and P. H. McBreen, Orientation and Conformation of Methyl Pyruvate on Ni(111), J. Am. Chem. Soc., 2000, 122(3), 518–524 CrossRef CAS.
- M. A. Vannice, W. Erley and H. Ibach, A RAIRS and HREELS study of acetone on Pt(111), Surf. Sci., 1991, 254(1), 1–11 CrossRef CAS.
- W.-S. Sim, T.-C. Li, P.-X. Yang and B.-S. Yeo, Isolation and Identification of Surface-Bound Acetone Enolate on Ni(111), J. Am. Chem. Soc., 2002, 124(18), 4970–4971 CrossRef CAS.
- J. L. Davis and M. A. Barteau, The influence of temperature and surface composition upon the coordination of acetone to the Pd(111) surface, Surf. Sci., 1989, 208(3), 383–403 CrossRef CAS.
- N. R. Avery, W. H. Weinberg, A. B. Anton and B. H. Toby, End-On and Side-On Bonding of Ketones to Surfaces: Acetone on the Ru(001) and Pt(111) Surfaces, Phys. Rev. Lett., 1983, 51(8), 682–685 CrossRef CAS.
- N. R. Avery, EELS identification of the adsorbed species from acetone adsorption on Pt(111), Surf. Sci., 1983, 125(3), 771–786 CrossRef CAS.
- A. B. Anton, N. R. Avery, B. H. Toby and W. H. Weinberg, Adsorption of acetone both on the clean ruthenium(001) surface and on the ruthenium(001) surface modified chemically by the presence of an ordered oxygen adatom overlayer, J. Am. Chem. Soc., 1986, 108(4), 684–694 CrossRef CAS.
- J.-R. Roy, M.-A. Laliberté, S. Lavoie, M. Castonguay and P. H. McBreen, Adsorption states of acetaldehyde and butane-2,3-dione on Ni(111), Surf. Sci., 2005, 578(1), 43–56 CrossRef CAS.
- G. Wu, D. Stacchiola, M. Collins and W. T. Tysoe, The Adsorption and Reaction of Acetaldehyde On Clean Ag(111), Surf. Rev. Lett., 2000, 07(03), 271–275 CrossRef CAS.
- C. L. A. Lamont, W. Stenzel, H. Conrad and A. M. Bradshaw, The oxidation of acetaldehyde on Cu{111}: a high resolution electron energy loss spectroscopy study, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 287–296 CrossRef CAS.
- R. W. McCabe, C. L. DiMaggio and R. J. Madix, Adsorption and reactions of acetaldehyde on platinum(S)-[6(111).times. (100)], J. Phys. Chem., 1985, 89(5), 854–861 CrossRef CAS.
- K.-H. Dostert, C. P. O'Brien, F. Mirabella, F. Ivars-Barcelo and S. Schauermann, Adsorption of acrolein, propanal, and allyl alcohol on Pd(111): a combined infrared reflection-absorption spectroscopy and temperature programmed desorption study, Phys. Chem. Chem. Phys., 2016, 18(20), 13960–13973 RSC.
- N. F. Brown and M. A. Barteau, Reactions of 1-propanol and propionaldehyde on rhodium(111), Langmuir, 1992, 8(3), 862–869 CrossRef CAS.
- N. F. Brown and M. A. Barteau, Reactions of unsaturated oxygenates on rhodium(111) as probes of multiple coordination of adsorbates, J. Am. Chem. Soc., 1992, 114(11), 4258–4265 CrossRef CAS.
- D. A. Esan and M. Trenary, Surface chemistry of propanal, 2-propenol, and 1-propanol on Ru(001), Phys. Chem. Chem. Phys., 2017, 19(17), 10870–10877 RSC.
- N. R. Avery, Adsorption of Hexafluoroacetone on Pt(111) - Is Bonded End-on (η-1) or Side-on (η-2), Langmuir, 1985, 1(1), 162–166 CrossRef CAS.
- E. L. Jeffery, R. K. Mann, G. J. Hutchings, S. H. Taylor and D. J. Willock, A density functional theory study of the adsorption of acetone to the (111) surface of Pt: Implications for hydrogenation catalysis, Catal. Tod., 2005, 105(1), 85–92 CrossRef CAS.
- C. Houtman and M. A. Barteau, Adsorbed states of acetone and their reactions on rhodium(111) and rhodium(111)-(2.times.2)oxygen surfaces, J. Phys. Chem., 1991, 95(9), 3755–3764 CrossRef CAS.
- J. L. Davis and M. A. Barteau, Polymerization and decarbonylation reactions of aldehydes on the Pd(111) surface, J. Am. Chem. Soc., 1989, 111(5), 1782–1792 CrossRef CAS.
- L. E. Murillo and J. G. Chen, Adsorption and reaction of propanal, 2-propenol and 1-propanol on Ni/Pt(111) bimetallic surfaces, Surf. Sci., 2008, 602(14), 2412–2420 CrossRef CAS.
- S. Attia, E. J. Spadafora, J. Hartmann, H.-J. Freund and S. Schauermann, Molecular beam/infrared reflection-absorption spectroscopy apparatus for probing heterogeneously catalyzed reactions on functionalized and nanostructured model surfaces, Rev. Sci. Inst., 2019, 90(5), 053903 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- R. D. Dennington, T. A. Keith and J. M. Millam, GaussView Version 5.0.8, Gaussian, Inc., Wallingford, 2008 Search PubMed.
- G. Sbrana and V. Schettino, Vibrational Spectra and Isomerism in Propyl- and Butyraldehydes, J. Mol. Spect., 1970, 33, 100–108 CrossRef CAS.
- K.-H. Dostert, C. P. O'Brien, F. Mirabella, F. Ivars-Barceló and S. Schauermann, Adsorption of acrolein, propanal, and allyl alcohol on Pd(111): a combined infrared reflection-adsorbtion spectroscopy and temperature programmed desorption study, Phys. Chem. Chem. Phys., 2016, 18, 13960 RSC.
- G. A. D. Guirgis, B. R. Gounev and T. K. Durig, J.R., Conformational stability and vibrational assignment of propanal, Spectrochim. Acta, Part A, 1998, 54, 123–143 CrossRef.
- E. F. Worden, The infrared spetra of propionaldehyde and propionaldehyde-d1*, Spectrochim. Acta, 1962, 18, 1121–1126 CrossRef CAS.
- K. Ohno, H. Yoshida, H. Watanabe, T. Fujita and H. Matsuura, Conformational Study of 1-Butanol by the Combined Use of Vibrational Spectroscopy and ab Initio Molecular Orbital Calculations, J. Phys. Chem., 1994, 98(28), 6924–6930 CrossRef CAS.
- W. J. Mitchell, J. Xie, T. A. Jachimowski and W. H. Weinberg, Carbon Monoxide Hydrogenation on the Ru(001) Surface at Low Temperature Using Gas-Phase Atomic Hydrogen: Spectroscopic Evidence for the Carbonyl Insertion Mechanism on a Transition Metal Surface, J. Am. Chem. Soc., 1995, 117(9), 2606–2617 CrossRef CAS.
- M. K. Weldon and C. M. Friend, Probing Surface Reaction Mechanisms Using Chemical and Vibrational Methods:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Alkyl Oxidation and Reactivity of Alcohols on Transitions Metal Surfaces, Chem. Rev., 1996, 96(4), 1391–1412 CrossRef CAS PubMed.
Alkyl Oxidation and Reactivity of Alcohols on Transitions Metal Surfaces, Chem. Rev., 1996, 96(4), 1391–1412 CrossRef CAS PubMed. - F. M. Hoffmann, Infrared reflection-absorption spectroscopy of adsorbed molecules, Surf. Sci. Rep., 1983, 3(2–3), 107–192 CrossRef CAS.
- G. Dellepiane and J. Overend, Vibrational spectra and assignment of acetone, ααα acetone-d3 and acetone-d6, Spectrochim. Acta, 1966, 22(4), 593–614 CrossRef CAS.
- M. A. Vannice, W. Erley and H. Ibach, A RAIRS and HREELS study of isopropyl alcohol on Pt(111), Surf. Sci., 1991, 254(1), 12–20 CrossRef CAS.
- E. Jeroro and J. M. Vohs, Exploring the Role of Zn in PdZn Reforming Catalysts: Adsorption and Reaction of Ethanol and Acetaldehyde on Two-dimensional PdZn Alloys, J. Phys. Chem. C, 2009, 113(4), 1486–1494 CrossRef CAS.
- C. Schröder, A.-K. Baumann, M. C. Schmidt, J. Smyczek, P. A. Haugg, O.-C. Graap and S. Schauermann, Mechanisms Acting in Ligand-Directed Heterogeneous Catalysis: Electronic, Geometric, and Enol-Induced Effects, J. Phys. Chem. C, 2022, 126(10), 4907–4920 CrossRef.
- C. Schröder, M. C. Schmidt, P. A. Haugg, A.-K. Baumann, J. Smyczek and S. Schauermann, Understanding Ligand-Directed Heterogeneous Catalysis: When the Dynamically Changing Nature of the Ligand Layer Controls the Hydrogenation Selectivity, Angew. Chem., Int. Ed., 2021, 60(30), 16349–16354 CrossRef PubMed.
- S. Schauermann, Partial Hydrogenation of Unsaturated Carbonyl Compounds: Toward Ligand-Directed Heterogeneous Catalysis, J. Phys. Chem. Lett., 2018, 9(18), 5555–5566 CrossRef CAS PubMed.
- C. P. O'Brien, K.-H. Dostert, S. Schauermann and H.-J. Freund, Selective Hydrogenation of Acrolein Over Pd Model Catalysts: Temperature and Particle-Size Effects, Chem. – Eur. J., 2016, 22(44), 15856–15863 CrossRef PubMed.
- K.-H. Dostert, C. P. O’Brien, F. Ivars-Barceló, S. Schauermann and H.-J. Freund, Spectators Control Selectivity in Surface Chemistry: Acrolein Partial Hydrogenation Over Pd, J. Am. Chem. Soc., 2015, 137(42), 13496–13502 CrossRef CAS PubMed.
- V. Humblot, C. J. A. Bingham, D. Le Roux, E. Mateo Marti, A. McNutt, T. S. Nunney, M. Ortega Lorenzo, A. J. Roberts, J. Williams, M. Surman and R. Raval, Synchrotron far-infrared RAIRS studies of complex molecules on Cu(110), Surf. Sci., 2003, 537(1), 253–264 CrossRef CAS.
- M. Garvey, Y. Bai, J. A. Boscoboinik, L. Burkholder, T. E. Sorensen and W. T. Tysoe, Identifying Molecular Species on Surfaces by Scanning Tunneling Microscopy: Methyl Pyruvate on Pd(111), J. Phys. Chem. C, 2013, 117(9), 4505–4514 CrossRef CAS.
- T. E. Jones and C. J. Baddeley, Investigating the Mechanism of Chiral Surface Reactions: The Interaction of Methylacetoacetate with (S)-Glutamic Acid Modified Ni{111}, Langmuir, 2006, 22(1), 148–152 CrossRef CAS PubMed.
- T. E. Jones and C. J. Baddeley, Influence of Modification Conditions on the Interaction of Methylacetoacetate with (R,R)-Tartaric Acid-Modified Ni{111}, J. Phys. Chem. C, 2007, 111(47), 17558–17563 CrossRef CAS.
- D. B. Skliar, C. Gelmi, T. Ogunnaike and B. G. Willis, Interaction of 2,2,6,6-tetramethyl-3,5-heptanedione with the Si(100)-2 × 1 surface: Scanning tunneling microscopy and density functional theory study, Surf. Sci., 2007, 601(14), 2887–2895 CrossRef CAS.
- J. Ontaneda, R. E. J. Nicklin, A. Cornish, A. Roldan, R. Grau-Crespo and G. Held, Adsorption of Methyl Acetoacetate at Ni{111}: Experiment and Theory, J. Phys. Chem. C, 2016, 120(48), 27490–27499 CrossRef CAS.
- S. Bertarione, D. Scarano, A. Zecchina, V. Johánek, J. Hoffmann, S. Schauermann, M. M. Frank, J. Libuda, G. Rupprechter and H.-J. Freund, Surface Reactivity of Pd Nanoparticles Supported on Polycrystalline Substrates As Compared to Thin Film Model Catalysts: Infrared Study of CO Adsorption, J. Phys. Chem. B, 2004, 108(11), 3603–3613 CrossRef CAS.
- S. D. Fried, S. Bagchi and S. G. Boxer, Measuring Electrostatic Fields in Both Hydrogen-Bonding and Non-Hydrogen-Bonding Environments Using Carbonyl Vibrational Probes, J. Am. Chem. Soc., 2013, 135(30), 11181–11192 CrossRef CAS PubMed.
- M. Nolasco Mariela and P. J. A. Ribeiro-Claro Paulo, C−H⋯O Hydrogen Bonds in Cyclohexenone Reveal the Spectroscopic Behavior of C−H and C−H Donors, ChemPhysChem, 2005, 6, 496–502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04398j |
| This journal is © the Owner Societies 2022 |