
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On surface chemical reactions of free-base and titanyl porphyrins with r-TiO2(110): a unified picture
Luca
Schio
 a,
Daniel
Forrer
b,
Maurizio
Casarin
bc,
Andrea
Goldoni
d,
Celia
Rogero
e,
Andrea
Vittadini
*b and
Luca
Floreano
*a
a,
Daniel
Forrer
b,
Maurizio
Casarin
bc,
Andrea
Goldoni
d,
Celia
Rogero
e,
Andrea
Vittadini
*b and
Luca
Floreano
*a
aCNR-IOM, Laboratorio TASC, Basovizza S.S. 14, Km 163.5, I-34149 Trieste, Italy. E-mail: floreano@iom.cnr.it
bCNR-ICMATE and INSTM, via Marzolo 1, I-35131 Padova, Italy. E-mail: andrea.vittadini@unipd.it
cDipartimento di Scienze Chimiche, Università di Padova, via Marzolo 1, I-35131 Padova, Italy
dElettra-Sincrotrone Trieste S.C.p.A., Basovizza SS-14, Km 163.5, 34149 Trieste, Italy
eMaterials Physics Center MPC, Centro de Física de Materiales (CSIC-UPV/EHU) and Donostia International Physics Center (DIPC), E-20018 San Sebastian, Spain
First published on 22nd April 2022
Abstract
In this Perspective we present a comprehensive study of the multiple reaction products of metal-free porphyrins (2H-Ps) in contact with the rutile TiO2(110) surface. In the absence of peripheral functionalization with specific linkers, the porphyrin adsorption is driven by the coordination of the two pyrrolic nitrogen atoms of the macrocycle to two consecutive oxygen atoms of the protruding Obr rows via hydrogen bonding. This chemical interaction favours the iminic nitrogen uptake of hydrogen from near surface layers at room temperature, thus yielding a stable acidic porphyrin (4H-P). In addition, a mild annealing (∼100 °C) triggers the incorporation of a Ti atom in the porphyrin macrocycle (self-metalation). We recently demonstrated that such a low temperature reaction is driven by a Lewis base iminic attack, which lowers the energy barriers for the outdiffusion of Ti interstitial atoms (Tiint) [Kremer et al., Appl. Surf. Sci., 2021, 564, 150403]. In the monolayer (ML) range, the porphyrin adsorption site, corresponding to a TiO-TPP configuration, is extremely stable and tetraphenyl-porphyrins (TPPs) may even undergo conformational distortion (flattening) by partial cyclo-dehydrogenation, while remaining anchored to the O rows up to 450 °C [Lovat et al., Nanoscale, 2017, 9, 11694]. Here we show that, upon self-metalation, isolated molecules at low coverage may jump atop the rows of five-fold coordinated Ti atoms (Ti5f). This configuration is associated with the formation of a new coordination complex, Ti–O–Ti5f, as determined by comparison with the deposition of pristine titanyl-porphyrin (TiO-TPP) molecules. The newly established Ti–O–Ti5f anchoring configuration is found to be stable also beyond the TPP flattening reaction. The anchoring of TiO-TPP to the Ti5f rows is, however, susceptible to the cross-talk between phenyls of adjacent molecules, which ultimately drives the TiO-TPP temperature evolution in the ML range along the same pathway followed by 2H-TPP.
1 Introduction
Porphyrin films1 and titania surfaces2,3 are almost ubiquitously employed in photocatalysis and photovoltaic applications due to their intrinsic chemical and electronic properties, which are mutually enhanced when brought into contact. A large amount of literature dealt with the energy level alignment, exciton dynamics and charge transfer/extraction at the interface of porphyrins with titania, as they can be tailored by the choice of chelated metal element, functionalization with meso-substituents and the choice of titania facet/stoichiometry: the metal chelated to the tetra-pyrrolic macrocycle mostly determines the hierarchy and population of the occupied and unoccupied molecular orbitals;4–6 peripheral functionalization is used to promote anchoring and to steer the interfacial molecular orientation and packing, which also affect the exciton dynamics;7 and the TiO2 crystal structure and orientation determine its charge transport and reactivity properties upon electron injection.8Among the different TiO2 polymorphs and facets, the (110) surface of the rutile crystal, r-TiO2(110), is the most common and easily produced substrate and its charge transport properties closely match those of the anatase (101) surface, which is commonly regarded as the most conductive/reactive one.9 The r-TiO2(110) surface is best suited to the investigation and modelling of molecular adsorbates for a number of practical reasons: (i) it is available in large size single crystals that can be prepared in a vacuum by legacy surface science techniques, and (ii) its highly corrugated and anisotropic termination may drive the orientation of molecules. In particular, the bulk-truncated (1 × 1) surface is characterised by rows of two-fold coordinated (bridging) oxygen atoms (Obr), which protrude by ∼1.2 Å off the surface and are spaced by 6.495 Å along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction (see the drawing in Fig. 1). Such a corrugation has been found to drive by simple van der Waals (vdW) interactions the oriented and commensurate growth of several uniaxial planar polycyclic aromatic hydrocarbons (PAHs), such as anthracene,10 pentacene,11 perylene,12 PTCDA,13,14 and PTCDI.15
0] direction (see the drawing in Fig. 1). Such a corrugation has been found to drive by simple van der Waals (vdW) interactions the oriented and commensurate growth of several uniaxial planar polycyclic aromatic hydrocarbons (PAHs), such as anthracene,10 pentacene,11 perylene,12 PTCDA,13,14 and PTCDI.15
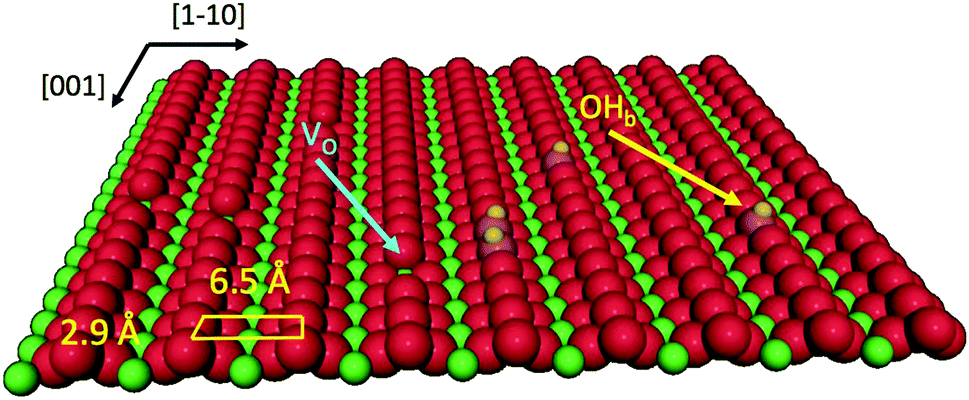 | ||
| Fig. 1 Perspective view of the TiO2(110) surface termination where oxygen and titanium atoms are represented by red and green spheres, respectively. Point defects, such as oxygen vacancies (VO) and hydroxyls (OHb), are also indicated. | ||
When porphyrins are brought into contact with the r-TiO2(110) surface, a more complex scenario of interfacial interactions develops, due to possible chemical coordination to the substrate of the macrocycle (with or without the chelated metal) and of the meso-substituents. The peripheral terminations of porphyrins are often functionalized with specific linkers to the TiO2 surface in order to tailor both the anchoring configuration and the molecular aggregation. Functionalization with carboxylic (COOH) groups of one or more phenyl rings in tetraphenyl-porphyrins (TPPs) is a common practice to establish a chemical bond between the molecule and the atoms of the five-fold coordinated titanium rows, thanks to the bidentate anchoring of carboxyl to two consecutive Ti5f atoms (spaced by 2.959 Å along the [001] direction) in a Ti5f–O–C–O–Ti5f configuration.16 At low coverage, carboxylated porphyrins lay flat on the surface independently of the number and relative position of COOH-functionalized phenyl terminations.17–20 Instead, molecules with two carboxyphenyls in the cis configuration or a single carboxyphenyl may tilt up at high coverage, forming a very dense layer anchored to the substrate only through the carboxylic linker.21–23 In addition, porphyrin functionalization with carboxylic or phosphonic acids improves the solubility of porphyrins in low hazard polar solvents (e.g. ethanol), thus facilitating the implementation of the protocols of growth on r-TiO2(110) by wet chemistry.24,25
In the absence of peripheral linkers, the vdW interactions drive a flat-lying porphyrin orientation, which brings the chelated metal atom close to the surface, where it may chemically interact with the substrate, thus dictating the adsorption site (on either Obr or Ti5f rows). Simple coordination chemistry arguments suggest that chelated metal ions with coordination vacancies will be able to form bonds with undercoordinated surface atoms.26 This is the case expected for Co-TPP (having two axial coordination sites), which has been reported to establish a covalent bond with coinage metal surfaces via surface trans-effects.27–29 Even Zn-TPP, displaying simple tetra-coordination in the gas phase, may develop strong axial coordination (single site), when the Zn atom is displaced from the porphyrin plane,30e.g. by the most commonly observed saddle-like distortion of adsorbed porphyrins. Consistently, zinc-porphyrins deposited at room temperature on r-TiO2(110) are observed by scanning tunnelling microscopy (STM) to adsorb atop the protruding Obr rows,18,31,32 where weak chemisorption (charge transfer) was inferred from the measured interface dipole.33 In contrast, Ni-TPP molecules, synthesized by sublimation of Ni on previously deposited 2H-TPP, have been reported to lie atop the Ti5f rows.34 Remarkably, these adsorption sites on TiO2 are matching predictions for MTPPs deposited on the (1 × 1) O-Fe(001) surface, where Zn-TPP and Co-TPP display a preferential adsorption site atop an oxygen atom, while Ni-TPP rather adsorbs atop a substrate Fe atom.35 To our knowledge, no STM measurements of the adsorption sites of other metal-porphyrins directly deposited on r-TiO(110) have been reported so far, apart from the specific cases of Cu-porphyrins20 and Co-pyrphyrins36 functionalized with linkers, whose bonding scheme is not primarily driven by the coordination of the chelated metal ion to the substrate.
Within this context, metal-free porphyrins offer the largest degree of flexibility to explore the chemical interactions with TiO2, as a number of protocols for the on-surface modification of 2H-Ps have been developed in the past decade, which include the possibility of incorporating different atomic species in the hydrogen saturated tetrapyrrolic pocket,37–40 as well as of changing the molecular conformation and degree of conjugation of its meso-substituents.41–44 Hence, in this Perspective we focus our attention on the interaction with the (1 × 1)-TiO2(110) surface of 2H-Ps having different meso-substituents, namely octaethyl-, tetraphenyl- and di-tert-butyltetraphenyl-porphyrins (2H-OEP, 2H-TPP and 2H-TBTPP, respectively) and their metalated homologues. The many different molecular configurations examined are comprehensively rationalized and strictly contextualized within the framework of the current literature, with the intent of providing a unified picture of the evolutionary patterns. Our former publications, as addressed in the corresponding sections, are not simply summarized, but revisited within a wider perspective and corroborated by additional experimental and theoretical data, either complementing or refining the published results. Overall, we present three new tables and fifteen new figures with unpublished data.
We first consider the interaction of pristine nitrogen atoms of the macrocycle with the substrate Obr and Ti5f atoms, as well as with dissociated hydrogen (present both at the surface and, diluted, in the bulk), which defines the porphyrin adsorption configuration, as addressed in our previous work.45 Starting from this structure, we deal with the issue of metal incorporation into the macrocycle either by metal sublimation on a TPP monolayer or by self-metalation upon sample annealing. Contrary to expectations for a strongly (covalently) bound compound substrate, self-metalation on r-TiO2(110) was formerly observed to take place at temperatures as low as ∼100 °C.46,47 We recently unveiled the atomistic mechanism of extraction and incorporation of a substrate Ti atom, which is made possible by the specific adsorption geometry of 2H-P, whose iminic nitrogen atoms lower the energy barrier for the outdiffusion of interstitial Ti atoms by a Lewis base nucleophilic attack.48 By comparison with the case of direct deposition of titanyl-porphyrin (TiO-TPP), we additionally address the migration to Ti5f rows of self-metalated TiO2br-TPP, which yields TiO-TPP species and different orientations. We will follow the conformational change of both titanyl-TPP species upon high temperature annealing. We have formerly shown that self-metalated TiO2br-TPP in the monolayer phase is flattened via partial cyclo-dehydrogenation without affecting the molecular adsorption site and orientation.49 Here we show that even the TiO-TPP@Ti5f species formed at low coverage bear a similar conformational change and preserve the pristine adsorption site. Most interestingly, TiO-TPP molecules in the ML range rather evolve along the same pathway as 2H-TPP species, due to the intermolecular interaction of the meso-substituents.
We made use of STM topography and electron diffraction to characterize the molecular adsorption geometry and phase symmetry, while synchrotron radiation photoemission spectroscopy (SXPS) provided the chemical status of the different porphyrin constituents from the binding energy (BE) of the corresponding core levels. We present complementary structural (and chemical) information on the conformational configuration of porphyrins, as obtained by means of near-edge X-ray absorption spectroscopy (NEXAFS) at the relevant carbon and nitrogen ionization thresholds and as a function of the surface orientation with respect to the (linear) polarization of the synchrotron beam. The experimental data have been interpreted with the support of hybrid-DFT calculations of the porphyrin/TiO2 interface, as modelled by using Grimme dispersion corrections in conjunction with PBE functionals. For the simulation of the photoemission core level shifts associated with the chemical reactions, we rather adopted the PBE0 functionals,50 which reproduce with better accuracy the binding energies of the different atomic species.51 Finally, we made use of the Hubbard U correction to calculate the minimum energy path of the mechanism of Tiint extraction.
2 Experimental and theoretical methods
2.1 Synchrotron X-ray spectroscopy
Both X-ray photoemission and absorption spectroscopy measurements (XPS and XAS, respectively) were performed at the ALOISA beamline of the Elettra Synchrotron facility (Trieste, Italy).52 The ultra-high-vacuum (UHV) end-station was equipped with a six-degree of freedom manipulator coaxial to the photon beam, and was moved forth and back between the preparation chamber (base pressure of 2–3 × 10−10 mbar) and the measurements chamber (10−11 mbar range). XPS measurements were performed by means of a homemade hemispherical analyzer (mean radius of 66 mm) hosted on a rotating frame which allows freely orienting the emission direction for any orientation of the surface with respect to the linear polarization of the (grazing) photon beam. The electron analyzer was equipped with a 2-dimensional delay-line detector and provided an energy resolution of 1% of the pass energy (max 25 meV) with an acceptance angle of 2° (FWHM). All XPS measurements shown hereafter were performed at a grazing angle of 4° in normal emission and with the photon magnetic field oriented transverse to the scattering plane (TM polarization, corresponding to quasi p-polarization). The O 1s and Ti 2p photoemission spectra were measured at a photon energy of 650 eV (overall energy resolution ΔE ≈ 200 meV) and calibrated to the substrate Ti 2p3/2 peak at a BE of 459.1 eV, corresponding to the Ti(IV) oxidation state. All the presented N 1s and C 1s photoemission spectra were measured with a photon energy of 500–515 eV (overall energy resolution of ΔE ≈ 160 meV) and calibrated to the Ti 3p peak at a BE of 37.6 eV. Extended valence band (VB) spectra were measured with a photon energy of 140 eV (overall energy resolution ΔE ≈ 120 meV).We performed NEXAFS measurements in terms of partial electron yield by means of a channeltron equipped with a polarizable grid in order to reject low energy secondary electrons (bias of −250 and −370 V for C and N, respectively). The harmonic of the undulator was continuously tuned to its maximum intensity while scanning the photon energy with the monochromator. The orientation of the surface with respect to the linear polarization of the photon beam was changed from transverse magnetic (TM) to transverse electric (TE, corresponding to s-polarization) by sample rotation around the beam axis, while keeping a constant grazing angle (and illuminated area) of 6°. All NEXAFS measurements were performed with the scattering plane oriented along the [001] direction. Absolute energy calibration at the C and N K-edges was obtained by simultaneous acquisition of the drain current (I0) on the last refocusing mirror (Au coated) downstream the exit slits. The I0 bore the adsorption lines due to contaminants that were independently calibrated by real time XAS measurements in the gas phase (CO and N2).52 The absorption spectra were normalized in intensity by NEXAFS measurements on the clean substrate, following the procedure described in ref. 53.
2.2 STM topography
STM measurements were performed in two different experimental chambers; however, using the same type of microscope, the Aarhus model by SPECS. In particular, measurements on the 2H-TPP and 2H-TBTPP films were performed in the UHV instrument of the Centro de Fisica de Materiales (CSIC-UPV, San Sebastian, Spain) using either a PtIr tip or a cantilever tip (Colibrí sensor, operated in STM mode), as specified in the caption of figures. The TiO-TPP films were measured at the IOM–Elettra joint laboratory for microscopy (OSMOS) in Trieste, using a W tip. All topographic images were collected by operating the STM at room temperature. In this condition, both the molecular mobility and the tip sensitivity to residual surface contaminants (strongly affecting the bare tungsten tip at negative sample bias and the Colibrí sensor tip at positive sample bias) limited the study of the intramolecular contrast as a function of the bias voltage. Independently of the tip, the rows of Ti5f atoms always appear as bright stripes. Point defects like oxygen vacancies (VO) and hydroxyls at Obr rows (OHb) are seen as bright spots in between the Ti5f rows, in agreement with a consolidated model interpretation of the STM topography.542.3 Sample preparation
The experiments were performed and repeated on a set of 5–6 different r-TiO2(110) samples (8 mm diameter and 0.5 mm thickness) purchased from MaTecK. The samples were always glued to a Mo thin spacer with Ag paste and dried in air up to 400 °C before insertion in the UHV chamber; eventually, we dried the samples under a flow of N2 in order to decrease the degree of hydrogen content in the bulk. Samples were cleaned and ordered by repeated cycles of sputtering (0.8–1 kV Ar+ ions) and annealing to 700 °C or slightly higher according to the sample ageing. The sample temperature was measured with K-type thermocouples in direct contact with the Mo spacer at ALOISA and at the OSMOS STM, thus providing an accuracy better than 20 °C. At the STM setup of CFM, the thermocouple was rather placed on the clips of the sample holder, yielding an uncertainty on the sample temperature up to 50 °C, depending on the time duration of annealing. Molecules were sublimed from homemade boron nitride crucibles and the rate was monitored using quartz microbalances (ALOISA, CFM and OSMOS). At ALOISA, a reflection high-energy electron diffraction (RHEED) system was also used for independent monitoring in real time the evolution of the pattern during molecular deposition, as well as during thermal treatment. The rate calibration was obtained a posteriori by thermal desorption of a multilayer, which defines one ML as the equivalent thickness of 2.8–3.0 Å for free-base porphyrins and 4–4.5 Å for TiO-TPP (using the same nominal molecular density of 1.26 g cm−3 for any porphyrin). This value was fully consistent with real time RHEED measurements of the ordering of 2H-TPP molecules. In order to prevent electron radiation damage, samples prepared for XPS and XAS were eventually checked by RHEED only after completion of the spectroscopy measurements. We sublimed commercially supplied molecules that are: tetraphenyl-porphine (CAS: 917-23-7) from Aldrich (purity 99.5%); tetra-(3,5-di-t-butylphenyl)-porphine (CAS: 89372-90-7) from Frontier Scientific (purity ≥99%); octaethyl-porphine (CAS: 2683-82-1) from Aldrich (purity 99.3%); and oxo-titanium(IV)-tetraphenyl-porphine (CAS: 58384-89-7) from PorphyChem (purity ≥98%). Metals were sublimed by means of a commercial electron bombardment cell (Omicron type) from Fe, Co and Ti rods (purity ≥99.98%). The metal to molecule stoichiometric ratio was determined from the effective thickness (in Ångström) assuming the atomic planar density in the preferred direction of growth, namely Fe(100) and Co/Ti(0001).2.4 Calculations and simulations
The structure and the stability of all the systems were investigated using density functional theory (DFT) in conjunction with periodically repeated slab models. To this end, we adopted the Quantum ESPRESSO package,55,56 which is based on the plane-wave pseudopotential (PP) approach. The standard computational setup included the PBE57 functional in conjunction with Grimme dispersion corrections,58,59i.e. the so-called PBE-D2 functional and Vanderbilt ultra-soft PPs.60 All the supercells were built using the theoretical PBE-D2 value of the TiO2 lattice constant.61 Coarse explorations of the potential energy surface (PES) of the TPP–surface interactions were carried out by using three-layer TiO2 slab models and “large core” PPs for Ti, including 1s–3p electrons in the core. Once the most favourable configurations were singled out, we refined the calculations on four-layer slabs and “small core” PPs, where the 3s and 3p subshells were included in the variational set. Because as many as ten TiO2 surface unit cells are required to model the densest film, only the Γ-point was used to sample the Brillouin zone. For low coverage TPP (isolated molecule), we adopted an oblique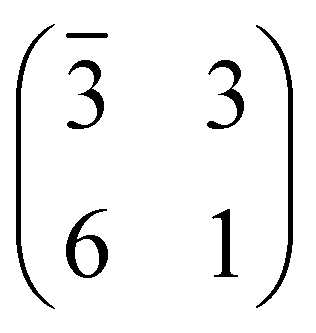 supercell, hereafter called (3 × 6)-obl, corresponding to 21 surface unit cells. Smaller supercells, as described in the text, were used to describe the specific symmetry phases, where an intermolecular correlation sets in. The above described setup has to be improved when modeling processes involving surface defects, such as the capture of Ti interstitials. In that case we increased the number of slab TiO2 layers from four to six, and we added a Hubbard U correction to the PBE functional, following the so-called rotationally-invariant DFT+U formulation.62,63
supercell, hereafter called (3 × 6)-obl, corresponding to 21 surface unit cells. Smaller supercells, as described in the text, were used to describe the specific symmetry phases, where an intermolecular correlation sets in. The above described setup has to be improved when modeling processes involving surface defects, such as the capture of Ti interstitials. In that case we increased the number of slab TiO2 layers from four to six, and we added a Hubbard U correction to the PBE functional, following the so-called rotationally-invariant DFT+U formulation.62,63
Finally, the XPS simulations of the adsorbed 2H-TPP and 4H-TPP species were carried out using clusters extracted from the optimized slab models, where the broken Ti–O bonds were saturated with a convenient number of hydrogen atoms.45 These calculations were carried out using the ADF package64 adopting the PBE0 functional,65 the zero order regular approximation (ZORA) to the relativistic Hamiltonian,66,67 and the frozen-core approximation for all the atoms but the one involved in the photoemission process.
3 Free-base meso-substituted porphyrins at room temperature
Contrary to their synthetic counterparts (i.e. free-base phthalocyanines), the tetrapyrrolic macrocycle of meso-substituted porphyrins is quite flexible, and is subject to large conformational changes upon deposition on surfaces, as a result of chemical interactions with the substrate of the pyrrolic units and relaxation of the peripheral terminations. On most of the metal surfaces, STM imaging reveals an uniaxial inflexion of 2H-TPP, which is commonly attributed to a saddle-like distortion of the macrocycle, where the pyrrolic nitrogen (NH) and iminic one (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) point outward the macrocycle plane, but with a different degree of misorientation.68,69 Such a structural distortion might eventually break the macrocycle aromaticity, which further enhances the STM topographic contrast by electronic effects.70,71 The nitrogen ligands eventually become equivalent when coordinated to a metal atom, which recovers the aromaticity of the macrocycle, and reduces its uniaxial inflexion, and it is normally expected to increase the STM intensity at the centre of the macrocycle, due to the increase of the charge density.39,40
) point outward the macrocycle plane, but with a different degree of misorientation.68,69 Such a structural distortion might eventually break the macrocycle aromaticity, which further enhances the STM topographic contrast by electronic effects.70,71 The nitrogen ligands eventually become equivalent when coordinated to a metal atom, which recovers the aromaticity of the macrocycle, and reduces its uniaxial inflexion, and it is normally expected to increase the STM intensity at the centre of the macrocycle, due to the increase of the charge density.39,40
3.1 Adsorption site and geometry
Upon deposition of 2H-TPP at room temperature (RT), the molecular features display a unique adsorption configuration, atop the Obr rows (appearing as dark rows in the left panel of Fig. 2), displaying a pronounced inflection along the short axis, which is in turn oriented transverse to the substrate rows.34,45 This saddle-shaped appearance with two well distinct lobes is practically independent of the tip bias and level of spatial resolution, which makes discriminating TPP molecules from other contaminants and/or molecular fragments easy. The fuzzy appearance of some of the molecules is due to the fast mobility along the substrate rows, [001] direction, at RT. As will be shown in the next sections, at higher coverage locally ordered domains are observed, which yield a long range ordered and commensurate 2H-TPP phase at saturation of the first molecular layer that defines the monolayer coverage (ML).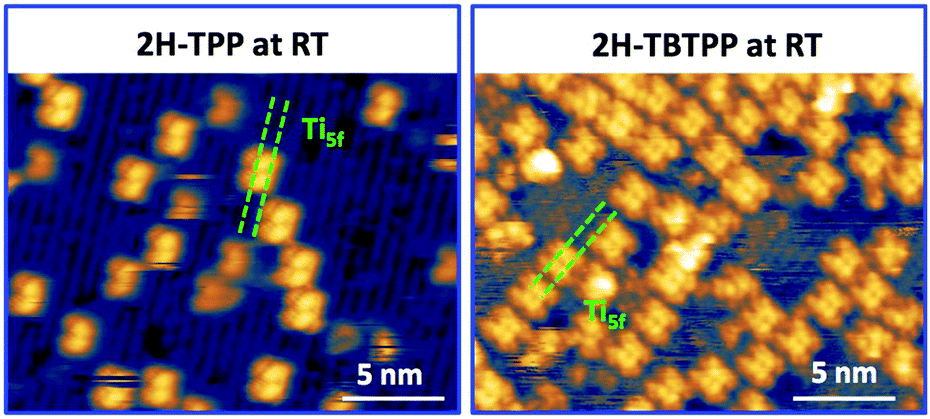 | ||
| Fig. 2 Left: STM image (Colibrí tip) of 2H-TPP deposited at RT (60 pA, −1.5 V); the Ti5f rows along the [001] direction appear as bright rows at both positive and negative biases; saddle-shaped 2H-TPP molecules adsorb exclusively onto the dark Obr rows. Right: STM image (PtIr tip) of 2H-TBTPP deposited at RT (90 pA, +1.3 eV); 2H-TBTPP molecules also adsorb onto Obr rows and display the same orientation and saddle-shape as 2H-TPP. As discussed in the text, 2H-P molecules cannot be discriminated from their acidic counterpart 4H-P. | ||
The same molecular configuration is found by STM for RT deposition of 2H-TBTPP; that is, ubiquitous adsorption atop the Obr rows and a pronounced saddle-shaped distortion contrast (see the right panel of Fig. 2).48 From a topographic point of view, TBTPP molecules only differ from TPP ones by displaying a slightly larger footprint due to their more extended peripheral terminations. In fact, we never observed the onset of ordered domains of 2H-TBTPP at any substrate temperature. These pieces of topographic evidence indicate that the adsorption of 2H-Ps is only driven by the interaction of the macrocycle (namely the pyrrolic and iminic nitrogen atoms) with the Obr rows, while the different meso-substituents merely govern the intermolecular organization in terms of spacing, molecular mobility and order, without affecting the adsorption site nor the molecular orientation.
The C2v symmetry appearance of the adsorbed species simplifies the theoretical modelling of the adsorption configuration. We restrained calculations to the case of TTP species, which can be fairly modeled as isolated molecules using supercells including 21 primitive surface cells. We considered two azimuthal orientations α = 0° and 45°, defined as the in-plane angle between the [001] direction (Obr and Ti5f rows) and the vertical plane bisecting the pyrrole rings. As expected, the orientation at α = 45° is unfavoured for any site on either Obr or Ti5f rows. Adsorption with the orientation α = 0° is largely favoured on Obr rows rather than on Ti5f rows. In particular, the bridge site between two Obr atoms is favoured by ∼0.2 eV with respect to an on-top site. This configuration corresponds to the effective formation of two hydrogen bonds of each pyrrolic nitrogen with the oxygen atom beneath, N–H⋯Obr.45
3.2 Substrate hydrogen capture by iminic nitrogen
The change in bonding configuration of the nitrogen atoms in the tetrapyrrolic pocket can be effectively monitored by measuring the core level shift (CLS) of the corresponding N 1s photoemission spectrum. The two pairs of iminic and pyrrolic nitrogen atoms give rise to two well separated peaks (∼2 eV) in the gas phase N 1s spectrum. These two peaks display practically the same intensity and marginally differ because of photoelectron diffraction effects (namely, backward scattering from the substrate atoms underneath) upon adsorption. Surprisingly, free-base porphyrins at the interface with the r-TiO2(110) surface display a largely dominant peak at ∼400.2–400.5 eV, corresponding to the binding energy of the pyrrolic N–H component, while the iminic nitrogen contribution (at ∼398–398.5 eV) is absent or very small (with the intensity varying from sample to sample or with sample history). We remark that an excess of the pyrrolic contribution has been reported also for carboxylated 2H-TPP (as prepared in a vacuum22 and in solution21,25) in a flat lying configuration (either because of the number of COOH terminations or because of the coverage). Representative N 1s photoemission spectra of 2H-TBTPP, 2H-TPP and 2H-OEP are shown in Fig. 3 for submonolayer depositions (0.5–0.8 ML) together with reference multilayer films. As will be shown hereafter, the bare interaction of the iminic nitrogen with the substrate atoms cannot yield such a large (∼2 eV) core level shift, due to either initial (chemical) or final (screening) state effects, rather the excess pyrrolic contribution must be attributed to the effective hydrogenation of the iminic nitrogen at the interface with TiO2 by a mechanism of hydrogen uptake. It is worth mentioning that porphyrin protonation in acidic solution forms mono- and diacidic 4H-P1+/2+ species,72,73 whereas the present doubly hydrogenated porphyrins are essentially neutral.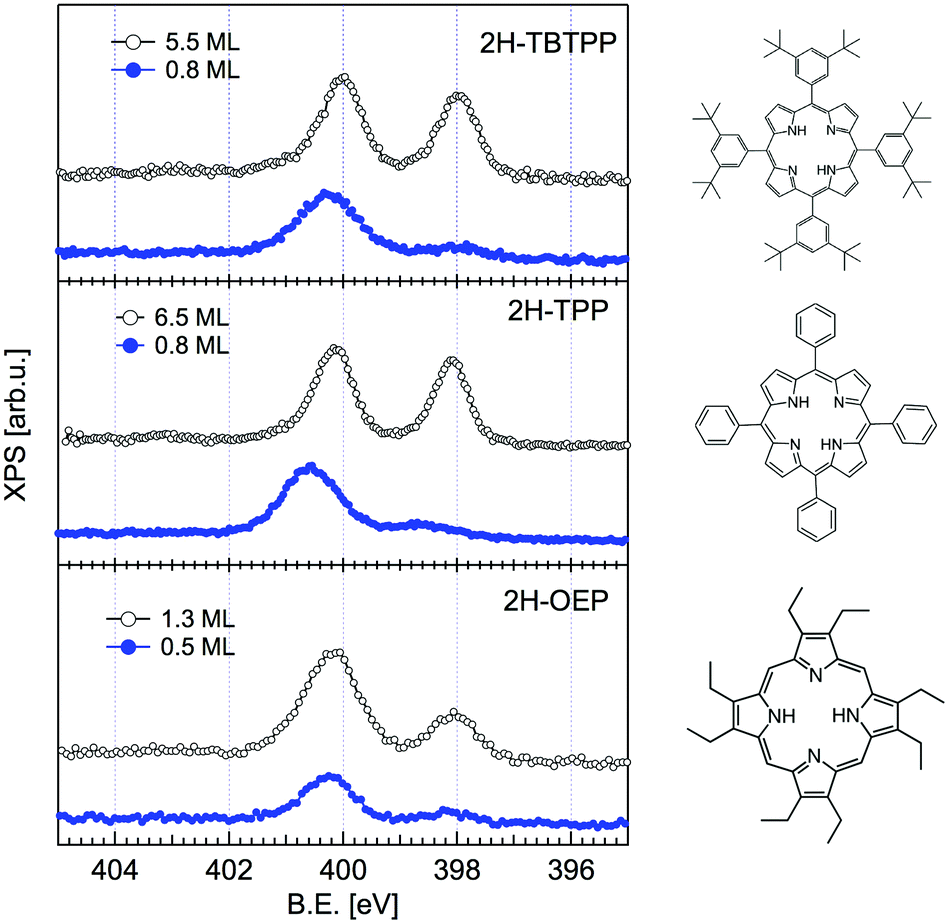 | ||
| Fig. 3 Comparison between the monolayer (filled blue markers) and multilayer (open black markers) photoemission spectra of the N 1s peak for three different porphyrins. All films were grown on the substrate at room temperature (RT). Adapted from ref. 45 with permission from the PCCP Owner Societies, Copyright 2015. | ||
A wealth of literature dealt with the presence and origin of dissociated hydrogen at the r-TiO2(110) surface, as anchored at the Obr atoms in the form of hydroxyls (OHb). While molecular hydrogen H2 does not interact with titania surfaces in an UHV environment,2 a small concentration of hydroxyls (up to 4–5%) is often left on the surface as the product of dissociation of H2O from the residual gas pressure at surface oxygen vacancies (VO).74 Such a small hydroxyl concentration cannot, however, account for the full hydrogenation of a TPP monolayer (corresponding to a density of 1 mol/10 unit cells), which would require an OHb coverage of 20%. In fact, the concentration of hydroxyls can be monitored in photoemission by measuring the OH 3σ peak at a binding energy of ∼11 eV.75 As shown in Fig. 4, by depositing the same amount of TPP molecules on a surface devoid of hydroxyls, one can effectively see a larger contribution from the iminic component, as compared to deposition on a surface with 4–5% hydroxyls, but the pyrrolic component remains, however, 4–5 times larger. Most of the hydrogen must consequently be captured either from the residual gas pressure or from subsurface layers. As a matter of fact, the bulk of TiO2 is regarded as a natural sink for dissociated hydrogen at interstitial sites. Hydrogen dilution is typically achieved by sample annealing with a relatively low vapour pressure of water (few tens of mbar at 500 °C) in the presence of oxygen76 or with hydrogen.77 In this regard, the as received samples already contain a certain amount of hydrogen, which can be possibly increased by the sample mounting operation, where single crystal wafers are glued with Ag paste and dried by heating in air prior to UHV insertion.
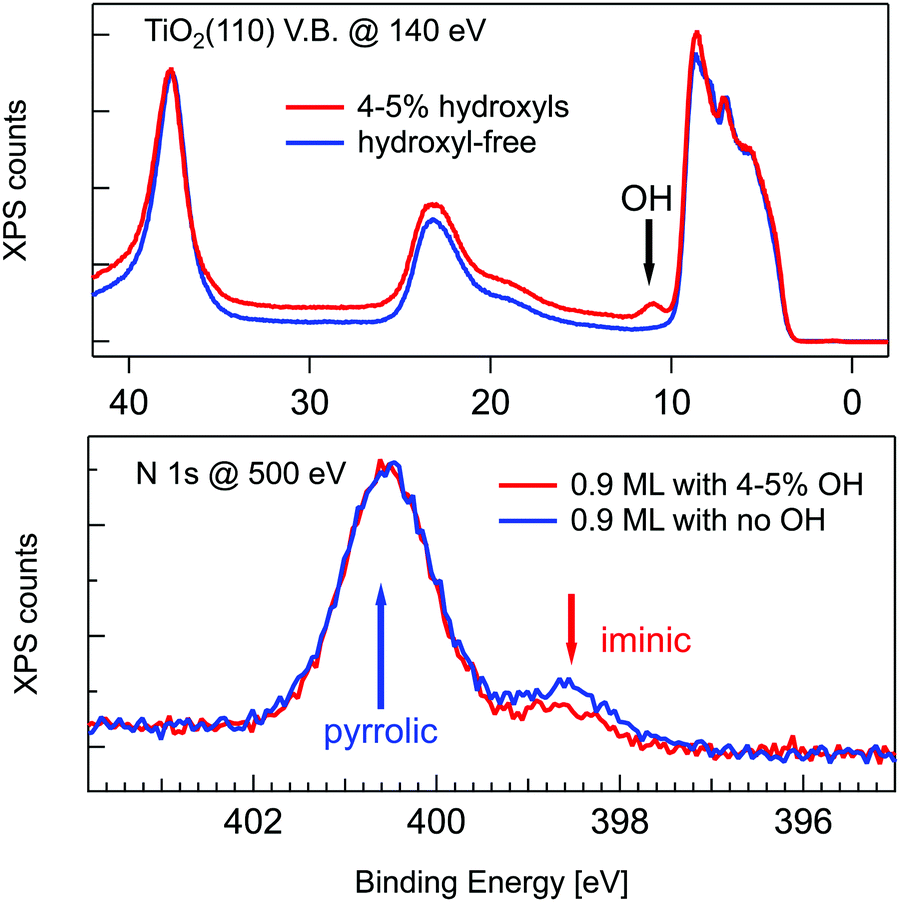 | ||
| Fig. 4 Upper panel: Valence band photoemission from a slightly hydroxylated (red line) and a hydroxyl-free surface (blue line) before deposition; the OH 3σ peak is marked with an arrow. Lower panel: Photoemission spectra of the N 1s peak for 0.9 ML of 2H-TPP deposited at RT on the same surfaces of the upper panel. Reproduced from ref. 47 with permission from the American Chemical Society, Copyright 2017. | ||
To get a better insight into the mechanism of porphyrin hydrogenation, we performed a few GGA-DFT calculations for the 2H-TPP molecule. The double hydrogenation in the gas phase gas2H-TPP + gasH2 → gas4H-TPP was slightly exothermic at 0 K (ΔG = −0.1 eV), but became endothermic at 298 K (ΔG = +0.3 eV).45 In fact, 2H-TPP is the stable species at room temperature in the gas phase, while 4H-TPP formation and stabilization has previously been reported only in acidic solution.78,79 The energetics are dramatically different for the adsorbed species, where we considered two dissociated hydrogen atoms adsorbed at well separated Obr sites (see ref. 45 for details). In this case, the adsorbed 4H-TPP species is largely favoured by an energy gain (enthalpy only) ΔE = −1.40 eV. For comparison, the energy barrier for interlayer diffusion of dissociated hydrogen across the (110) plane is about 1 eV, while the energy balance of the process is in the 0.1 eV range with the sign (+/−) depending on the details of the theoretical analysis.74,80 Significant diffusion from the bulk towards the surface was experimentally observed to take place already at 100 °C in H-implanted TiO2(110) crystals.81 Considering that the energy barrier for H outdiffusion decreases by sample reduction80 (or equivalently by bulk metal acceptor doping82), the porphyrin uptake of hydrogen at RT from the topmost and nearby subsurface layers is reasonable and quantitatively consistent.
3.3 Equilibrium configuration of 2H/4H-TPP at the Obr bridge
We can now focus on the equilibrium configuration of the adsorbed 4H-TPP molecules. From the previous discussion, we can assume that all TTP and TBTTP molecules imaged by STM in Fig. 2 rather correspond to their di-hydrogenated counterparts (4H-TPP and 4H-TBTPP) because of the overall low coverage. We calculated the adsorption energies for the same configurations, as formerly described in Section 3.1. The different adsorption sites for 4H-TPP yield the same energy hierarchy of the 2H-TPP species, only displaying an average increase of the adsorption energy by ∼2 eV.45 In detail, the adsorption in a 45° rotated configuration is always unfavored with respect to the α = 0° orientation, for any adsorption site and with minor differences among Ti5f and Obr rows, either on-top or on-bridge. For the α = 0° orientation, the Obr rows are favoured by 1 eV with respect to the Ti5f ones. The bridge site between two Obr atoms (4H-TPPEads = 5.31 eV) is favoured with respect to the on-top site, by an energy difference of 0.3 eV, slightly larger than the 0.2 eV value found for 2H-TPP. In the structural model shown in Fig. 5, one can appreciate the upward tilt of the hydrogenated iminic atoms (left panel), whereas each pristine pyrrolic nitrogen is strongly bent towards the substrate (28° and 22° for 4H-TPP and 2H-TPP, respectively), where it forms a hydrogen bond to the Obr atom underneath (right panel).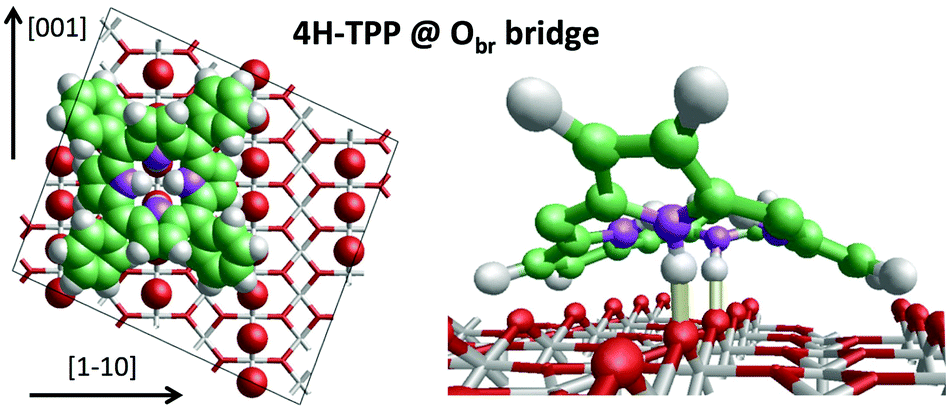 | ||
| Fig. 5 Left: Top view of the 4H-TPP equilibrium configuration; the protruding Obr atoms (in red) have been enlarged; nitrogen, carbon and hydrogen atoms are painted in purple, green and white, respectively. Right: Perspective view of the 4HTPP molecule, where the two hydrogen bonds N–H⋯Obr are highlighted with yellow sticks. Adapted from ref. 45 with permission from the PCCP Owner Societies, Copyright 2015. | ||
Such a large distortion of the tetra-pyrrolic macrocycle is confirmed by the NEXAFS measurements at the N K-edge ionization threshold (see Fig. 6), where the π-symmetry resonances of the pyrrolic (NH) and iminic (N–) components are well separated by 2–2.2 eV. When measured in TE and TM polarization (s- and close to p-, respectively) at 1 ML coverage, the residual iminic component at 397.5–397.8 eV clearly displays a large NEXAFS dichroism for any meso-substituted porphyrin;45 that is, the resonance is maximum in TM polarization and almost vanishes in TE polarization, indicating a close parallelism to the surface of the non-hydrogenated iminic π* orbital. Conversely, the pyrrolic resonance at ∼400 eV displays a relatively large intensity even in TE polarization. For the TPP case, the intensity ratio of the pyrrolic resonance in opposite polarization indicates an average tilt off the surface by ∼30° of the corresponding π* orbital, in perfect agreement with DFT calculations.†
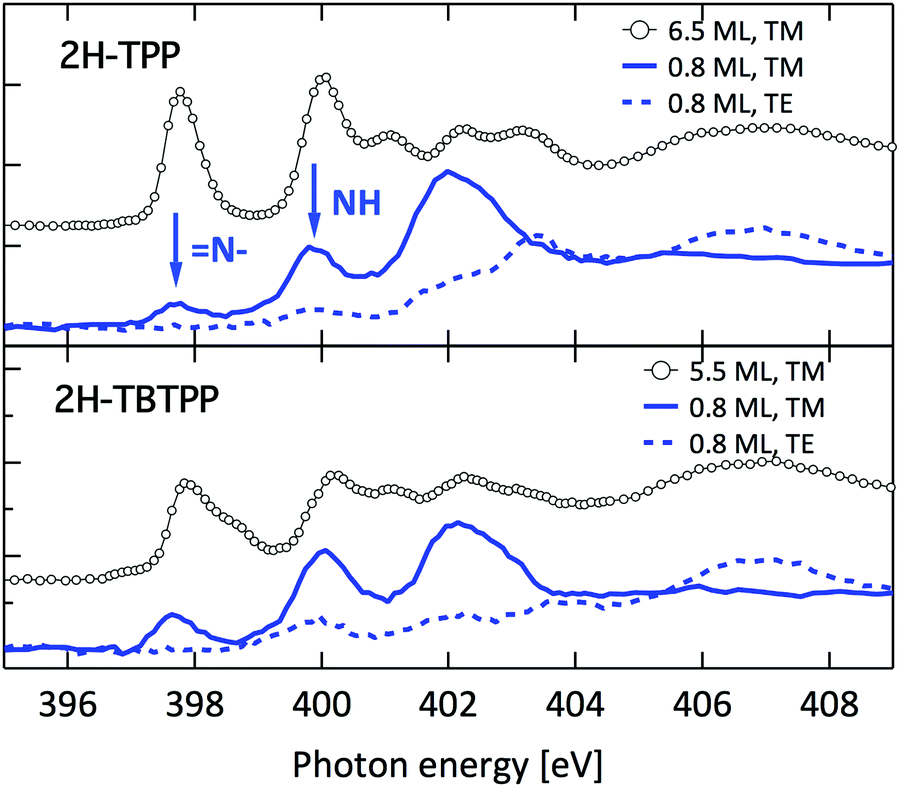 | ||
| Fig. 6 Nitrogen K-edge NEXAFS spectra of the 2H-TPP and 2H-TBTPP films of Fig. 3 (top and bottom panels, respectively). NEXAFS measurements in TM and TE polarization (full and dashed lines, respectively) are shown for the 0.8 ML films, whereas only spectra in TM polarization are shown for multilayers (open markers). | ||
Once the preferred configuration was defined, we simulated the STM images with the best intramolecular contrast. Representative STM images of the individual porphyrins at positive and negative sample biases are shown in Fig. 7 together with DFT simulations. The observed intramolecular contrast is reproduced to a good approximation. Most importantly, the simulations for the pristine and hydrogenated TPP yield closely similar modulations of the electron density with minor changes that cannot be practically detected by our STM; that is, 2H-TPP and 4H-TPP molecules cannot be discriminated in RT topographic images.
 | ||
| Fig. 7 Comparison of STM images (Colibrí tip) on a low coverage RT deposition of 2H-TPP with the calculated DOS cut at the energy corresponding to the experimentally used voltage. Simulations are shown for the equilibrium configuration of 4H-, 2H- and Ti-TPP species anchored to the Obr rows on a bridge site. Positive (negative) sample bias corresponds to empty (filled) states/orbitals. | ||
Finally, we made use of PBE0 functionals to calculate the N 1s core level shifts of the adsorbed 2H-TPP and 4H-TPP species, as bonded to the Obr atoms underneath via the pristine pyrrolic hydrogen. Calculations for a free standing 2H-TPP molecule resulted in good agreement with measurements on a multilayer film: the calculated BE shift of the iminic peak with respect to the pyrrolic one was 5% smaller than the measured one (−2.06 eV). The agreement was equally good for the adsorbed 2H-TPP species: the calculated BE shift was found to be 5% smaller than the measured one (−1.95 eV). The hydrogenated iminic peak of 4H-TPP was calculated to be shifted towards larger binding energy by +0.3 eV with respect to the pyrrolic one. Such a small CLS cannot be discriminated in our photoemission spectra; however, it is consistent with the measured broadening by 0.4 eV of the adsorbed 4H-TPP pyrrolic peak with respect to that of 2H-TPP in the multilayer.
4 Metal incorporation
For most of the practical purposes, metallo-porphyrins (MPs) are more relevant than their free-base homologues because the metal centre allows one to tune the alignment of the occupied and unoccupied molecular orbitals of the isolated molecule, as well as to modify the metal oxidation state and spin at the interface with metals and semiconductors. The choice of central metal depends on the target application; for example, for the fabrication of dye-sensitized solar cells, Zn-porphyrins are better suited for coupling with TiO2 than Cu-porphyrins.83 On the other hand, Cu-TPP at the interface with TiO2 displays degradation yields of atrazine and luminol larger than those of Zn-TPP.84,85 Titanyl–porphyrin complexes also display catalytic properties both in solution86 and in supported layers.87 When the magnetic properties are targeted, the metals of choice are rather iron, cobalt, and manganese,88 but the coupling with non-magnetic substrates may be exploited to tailor the spin state of other chelated metals, e.g. Cu and Ni.89,90 In the past ten years, several routes have been developed for the on-surface incorporation of different metal atoms in the tetrapyrrolic pocket of an adsorbed molecular layer, spanning from the vapour phase deposition91,92 to self-metalation38,93 and trans-metalation.94,954.1 Metalation by deposition
Due to the large extension of their (polarizable) heteroaromatic macrocycle, porphyrins develop significant interactions of vdW type with substrates, which favours a flat-lying orientation in most cases. This geometry, making the tetrapyrrole pocket directly accessible, favours the direct incorporation of metals deposited from vacuum. In spite of the double hydrogen bond to the Obr atoms and possible hydrogenation of the iminic nitrogen atoms, Gottfried and coworkers achieved the metalation of 2H-TPP on the r-TiO2(110) surface by postgrowth deposition of nickel.34 This coordination reaction can be monitored by measuring the N 1s photoemission, where the two pyrrolic and iminic components are replaced by a single one corresponding to the metal chelation by the four nitrogen atoms. The binding energy of the metalated nitrogen component changes slightly with the nature of the chelated metal, but is always found to be close to that of the iminic one at 0.3–0.5 eV higher BE.Representative spectra of the N 1s photoemission are shown in Fig. 8 before and after RT deposition of iron, cobalt and titanium metal atoms on top of a (sub-)monolayer of 2H-TPP. While the initial chemical state of the molecular layer mostly corresponds to the hydrogenated species, 4H-TPP, a new N 1s component, associated with the metalated species, grows at the expense of the pyrrolic one upon RT deposition of Fe, Co and Ti. As expected for this protocol of metalation, the increase of the metalated N component is not linear with the amount of metal deposit. In fact, whereas metal atoms hitting the macrocycle pocket are immediately incorporated in it and replace the hydrogen atoms, those falling aside may diffuse to form metal clusters and must overcome an extra barrier to access the macrocycle.39 Even in the presence of a large excess of metal atoms, as compared to the molecular coverage, the metalation yield of 4H-TPP remains well below 80%, in full agreement with previous findings in the case of nickel. The reversed order of deposition (first a small amount of metal, then a ML of 2H-TPP molecules), followed by annealing to ∼250 °C was formerly reported to provide an equivalent (insufficient) yield on both the (1 × 1) and reconstructed (2 × 1) surfaces.34,96 However, this protocol must be discarded on TiO2 because it rather leads to incorporation of Ti atoms from the substrate, as will be demonstrated in Section 6. Even metalation in solution, which is very effective for free-base porphyrins in solution and vacuum-deposited on gold,97 does not exceed a 75% yield for a wetting layer of carboxylated TPP on TiO2(110).21,25
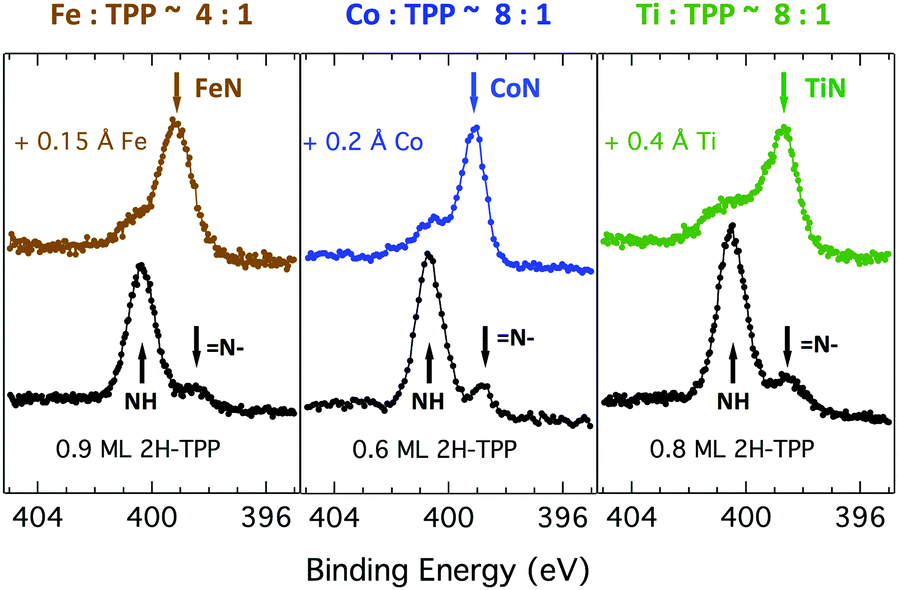 | ||
| Fig. 8 N 1s photoemission spectra for a single layer film of 2H-TPP before (black markers) and after deposition of Fe, Co and Ti (brown, blue and green markers, respectively) from left to right. The peaks of the pyrrolic (NH), iminic (N–) and metalated N components are marked with arrows. | ||
In principle, metalation by deposition offers the advantage of leaving unaffected the 2H-TPP molecular ordering eventually established by self assembly.92,98 This is not necessarily the case for the r-TiO2(110) surface, where the adsorption site appears to be driven by a direct chemical interaction of the macrocycle with the surface, even for the simple case of 2H-/4H-HTPP. The incorporation of Ni atoms was shown to rotate by 45° and shift the TPP molecules on the Ti5f rows, indeed.34 Even the fraction of metals not incorporated in the macrocycle may undergo major chemical interactions with the substrate, hence changing the chemical and electronic structures of the surface. In fact, 2H-TPP molecules do not affect the shape of either the Ti 2p peak or the valence band, but postgrowth deposition of 0.4 Å Ti yields both a large increase of the Ti 2p tail (associated with Ti3+/2+ atoms87) and that of the corresponding Defect State (DS) at BE ≈ 0.9 eV (associated with the partial filling of the Ti 3d levels,99 which normally lie just above the Fermi edge), as shown in the upper left panel of Fig. 9. Such an amount of titanium, albeit not sufficient to yield complete metalation, largely exceeds (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) the TPP monolayer density. The Ti atoms in excess do not simply condense into metallic clusters, rather they react quickly with the substrate oxygen, as witnessed by the change in the O 2p band shape (3–8 eV) and BE shift of the O 2s peak. This reaction leads to the formation of undercoordinated TiOx clusters on the surface, as well as charge redistribution among Ti lattice atoms,100 which strongly affect the substrate chemical and transport properties. For comparison, none of the aforementioned changes is observed upon direct deposition of a monolayer of TiO-TPP (see lower panels of Fig. 9), a porphyrin complex where the metal atom displays the same oxidation state Ti4+ of the substrate. The Ti 2p spectra before and after TiO-TPP deposition display the same shape, indeed; the VB oxygen spectral features also remain unchanged, apart from intensity attenuation, and the highest occupied molecular orbital (HOMO) is clearly observed at 2 eV, just aside the DS peak of the pristine surface.
:1) the TPP monolayer density. The Ti atoms in excess do not simply condense into metallic clusters, rather they react quickly with the substrate oxygen, as witnessed by the change in the O 2p band shape (3–8 eV) and BE shift of the O 2s peak. This reaction leads to the formation of undercoordinated TiOx clusters on the surface, as well as charge redistribution among Ti lattice atoms,100 which strongly affect the substrate chemical and transport properties. For comparison, none of the aforementioned changes is observed upon direct deposition of a monolayer of TiO-TPP (see lower panels of Fig. 9), a porphyrin complex where the metal atom displays the same oxidation state Ti4+ of the substrate. The Ti 2p spectra before and after TiO-TPP deposition display the same shape, indeed; the VB oxygen spectral features also remain unchanged, apart from intensity attenuation, and the highest occupied molecular orbital (HOMO) is clearly observed at 2 eV, just aside the DS peak of the pristine surface.
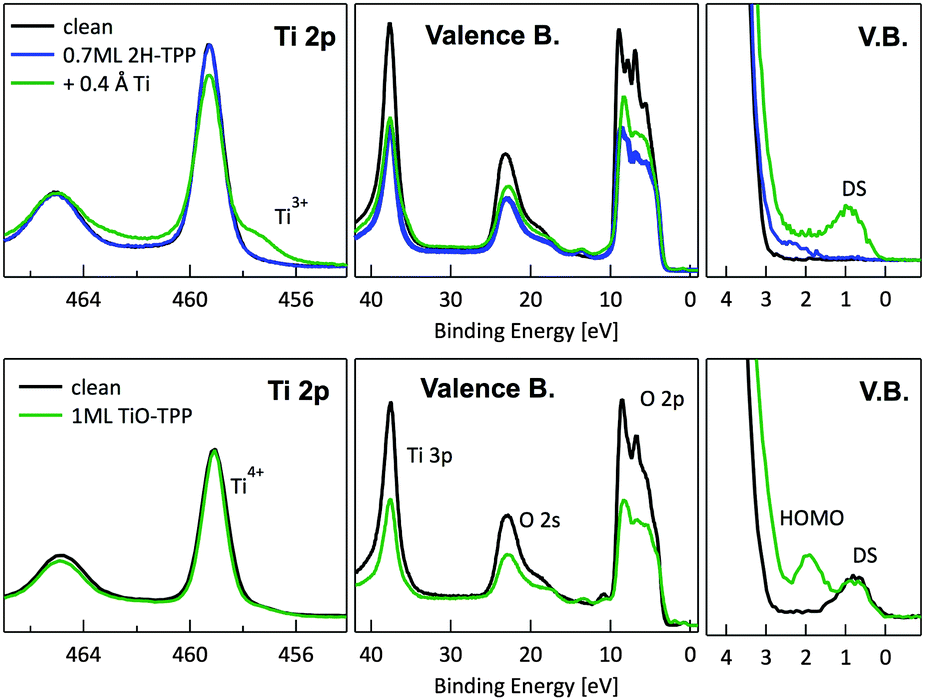 | ||
| Fig. 9 Upper panels: photoemission spectra of the Ti 2p, valence band and VB zoom-in close to the Fermi level (DS) for a layer of 2H-TPP metalated by RT deposition of 0.4 Å of Ti atoms. Lower panels: same photoemission spectra after RT deposition of 1 ML of TiO-TPP. Each molecular layer is compared with the corresponding pristine r-TiO2(110) surface. The Ti 2p spectra of the clean surface are renormalized to the intensity after TPP deposition; the Ti 2p spectrum after Ti deposition is shown without further renormalization to highlight the Ti 2p tail increase. The VB spectra are simply normalized to the flux in order to highlight the change in intensity of the DS peak. | ||
4.2 Self-metalation
As a matter of fact, the direct extraction and incorporation of surface metal atoms (self-metalation) by adsorbed free-base tetrapyrrolic macrocycles is the only reaction yielding a homogeneously 100% pure layer of metal-porphyrins. Upon thermal treatment, porphyrins can extract atoms from almost any metal surface.38,101,102 Since the metal incorporation implies the release of the two pristine hydrogen atoms (either to the surface or desorbed upon recombination), the presence of oxygen atoms may favour the reaction by synthesis and release of a molecule of water. In fact, the bare formation of a molecule of water from one molecule of hydrogen and a half-molecule of oxygen in the gas phase provides an energy gain (enthalpy) of 2.5 eV.103 For example, TPP self-metalation on clean Cu(111) displays an activation energy of ∼1.5 eV,104 which corresponds to the release of pyrrolic hydrogen towards the surface, where it may either recombine and desorb as H2 or coordinate to the peripheral phenyl terminations.105 However, self-metalation on a previously oxidized O–Cu(111) surface opens the path to the synthesis and release of H2O with an energy cost of ∼0.6–0.7 eV,106 which may lower the onset of 2H-TPP self-metalation on copper by ∼200 K.107 Equivalent results have been demonstrated by thermal treatment of 2H-TPP on clean and oxidized cobalt surfaces and nanoparticles,108–110 as well as on ZnO nanorods.111 For MgO, the TPP hydrogen release to the substrate and the consequent formation of hydroxyls has been proposed to compensate for the creation of a Mg vacancy upon metal sequestration.112,113 More recently, TPP self-metalation on MgO has been directly correlated with a net charge transfer from the substrate to the molecule, whose footprint is the partial filling of the LUMO that falls below the conduction edge, hence appearing in the gap of the VB photoemission spectra.114With such a paraphernalia of metal extraction/incorporation mechanisms, it does not come unexpected that meso-substituted porphyrins display a self-metalation reaction on r-TiO2(110) too. Seminal studies for the TPP/TiO2 interface have put in evidence a dependence on the molecular coverage (the lower the coverage, the lower the reaction temperature)47 and on the degree of TPP hydrogenation (preferential self-metalation of neutral 2H-TPP with respect to hydrogenated 4H-TPP).46 More recently, TPP self-metalation has been reported also for TPP on the anatase TiO2 polymorph115 and for carboxylic functionalized porphyrins.21,23 From the analysis of the N 1s photoemission spectra shown in Fig. 10, one observes that different meso-substituted porphyrins at the ML coverage on r-TiO2(110) display exactly the same dependence on the annealing temperature, thus indicating a common reaction mechanism.48 In particular, the minority of neutral 2H-P molecules is fully metalated already at 100 °C, as witnessed by the disappearance of the iminic N– component at 398 eV and the appearance of the metalated TiN one at 0.3–0.5 eV higher BE. Further annealing to 200 °C is, however, necessary to achieve 100% self-metalation at the ML coverage. We never observed a correlation between the self-metalation reaction and the degree of sample reduction, as monitored from the intensity of the Ti 2p low BE tail and/or the DS in the gap.
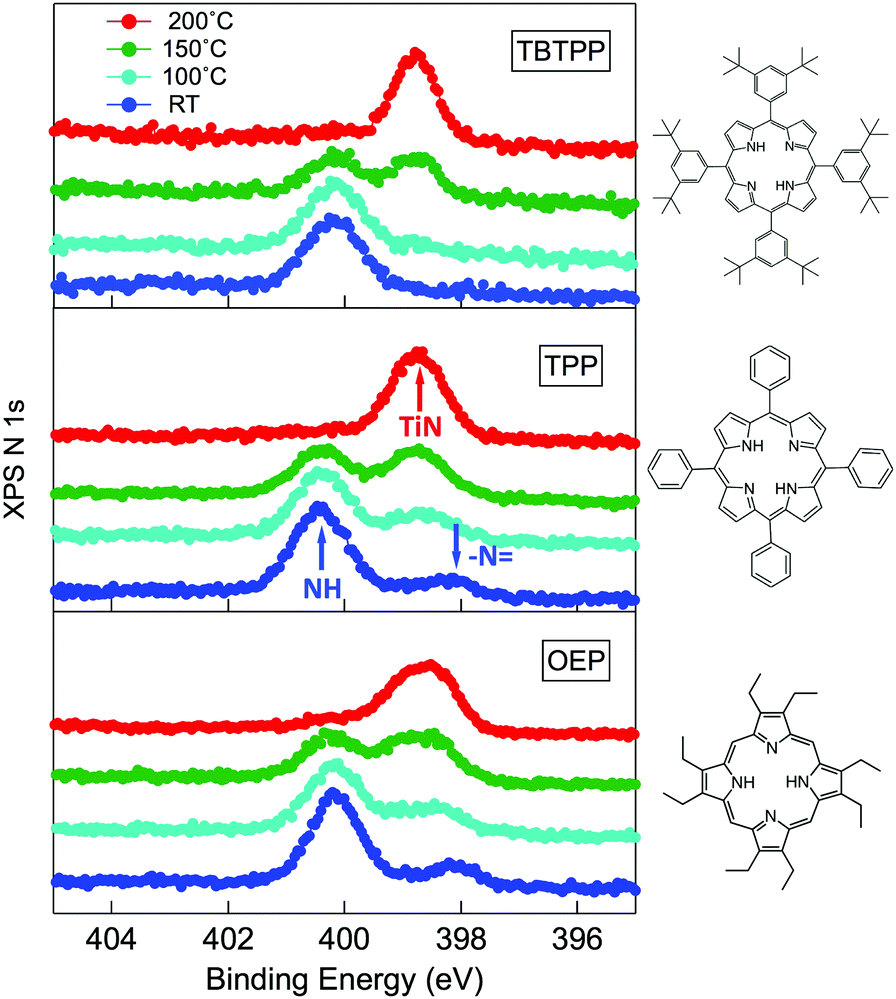 | ||
| Fig. 10 From top to bottom: the N 1s spectra of 2H-TBTPP (0.8 ML), 2H-TPP (1.1 ML) and 2H-OEP (1.1 ML) as deposited at RT and after annealing; the annealing temperatures are indicated in the top panel; arrows in the central panel (TPP) mark the binding energies of pyrrolic (NH), iminic (N–) and metalated (TiN) nitrogen. Reproduced from ref. 48 with permission from Elsevier, Copyright 2021. | ||
A close look at the Ti 2p and VB photoemission spectra, however, reveals that the mechanism of self-metalation on TiO2 is different from those mentioned above for metals and metal oxides. Although large amounts of dissociated hydrogen can be accommodated at the Obr rows,116 the release to the surface of dissociated hydrogen (like for MgO) must be discarded. In fact, the VB photoemission spectra in Fig. 11 show that neither the OH 3σ peak at BE ≈ 11 eV (vanishing upon RT porphyrin deposition45) reappears upon self-metalation, nor any (filled) LUMO state appears in the gap (in agreement with the close similarity of the ML and multilayer NEXAFS resonances, see Fig. 6). The synthesis and release of water molecules is also inconsistent with both the VB and Ti 2p spectra, because the extraction of an oxygen atom would leave an excess of charge on the nearby subsurface lattice Ti atoms,99 which manifest themselves as an increase of the DS at BE ≈ 0.9 eV (Ti 3d character) accompanied by an increase of the Ti 2p tail in the 457–458 eV range (from the Ti3+ atoms). Actually, the VB spectra only show an attenuation of the pristine DS peak (when present) due to the TPP overlayer, and the Ti 2p lineshape remains unchanged before and after self-metalation. In particular, the latter experimental evidence indicates that the incorporated Ti atom must have the same oxidation state (IV) of the metal atoms in the substrate lattice; that is, the metalated TPP molecule must be coordinated to the substrate oxygen atoms.
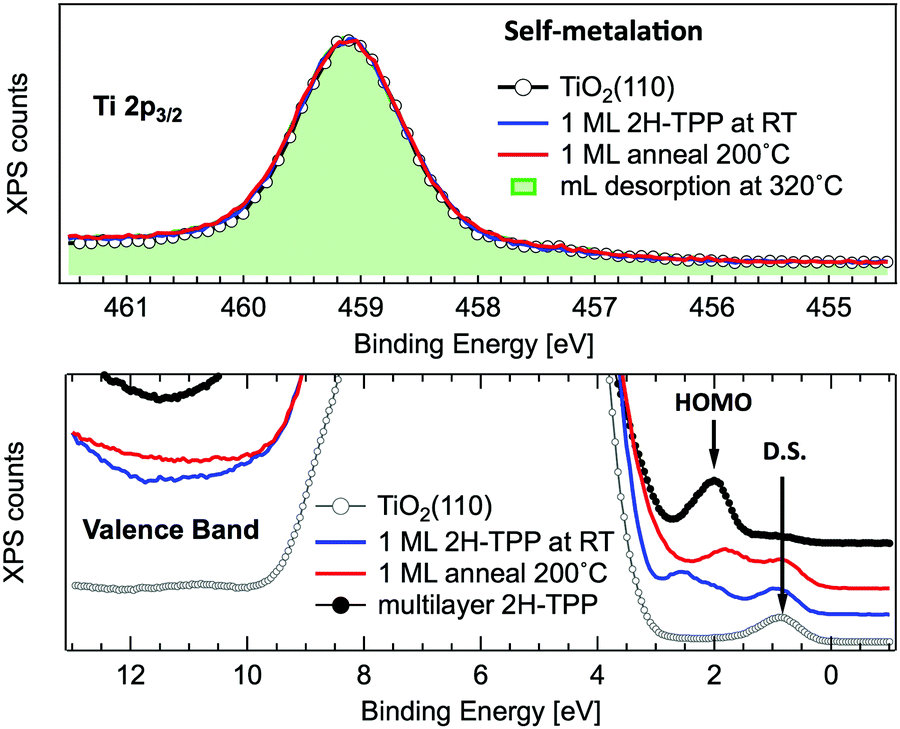 | ||
| Fig. 11 Upper panel: Normalized photoemission spectra of the Ti 2p3/2 core level for a clean r-TiO2(110) surface (open markers), after RT deposition of 1 ML of 2H-TPP (blue line) and after annealing to 200 °C (red line). For comparison, the spectrum measured on a monolayer obtained by desorption of a multilayer at 320 °C is also shown (green shaded area). Lower panel: Photoemission spectra of the valence band measured on the same substrate and films of the upper panel. The arrows mark the substrate DS peak and the 2H-TPP bulk HOMO state. Adapted from ref. 47 with permission from the American Chemical Society, Copyright 2017. | ||
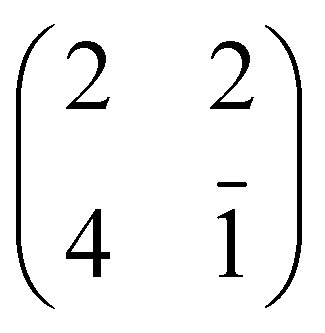 phase symmetry pattern, hereafter called (2 × 4)-obl, close to the completion of the first layer.47 This diffraction pattern, characterized by 5-fold fractional spots along the [11] direction, is preserved up to 300 °C, well beyond the self-metalation temperature, only displaying a sharpening without any change in relative intensity of the spots. We may conclude that the pristine molecules (both 2H-TPP and 4H-TPP ones) preserve the same intermolecular long range ordering and adsorption site after incorporation of the Ti atoms from the substrate. This indication is confirmed by the STM images shown in Fig. 12, where an intermolecular spatial correlation among free-base molecules (both 2H- and 4H-TPP) sets in at a coverage of ∼0.5 ML, displaying the formation of domains with local (2 × 4)-obl ordering. At this intermediate coverage, we cannot exclude the formation of local domains with the oblique
phase symmetry pattern, hereafter called (2 × 4)-obl, close to the completion of the first layer.47 This diffraction pattern, characterized by 5-fold fractional spots along the [11] direction, is preserved up to 300 °C, well beyond the self-metalation temperature, only displaying a sharpening without any change in relative intensity of the spots. We may conclude that the pristine molecules (both 2H-TPP and 4H-TPP ones) preserve the same intermolecular long range ordering and adsorption site after incorporation of the Ti atoms from the substrate. This indication is confirmed by the STM images shown in Fig. 12, where an intermolecular spatial correlation among free-base molecules (both 2H- and 4H-TPP) sets in at a coverage of ∼0.5 ML, displaying the formation of domains with local (2 × 4)-obl ordering. At this intermediate coverage, we cannot exclude the formation of local domains with the oblique 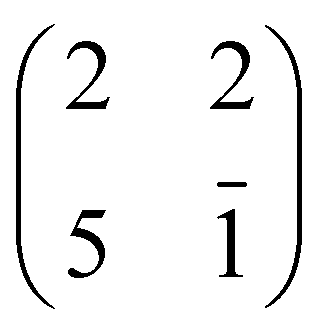 symmetry phase, hereafter called (2 × 5)-obl, as reported for Zn-TPP.32 The latter phase has a molecular density of 0.43 mol nm−2, which is 20% smaller than that of the (2 × 4)-obl (0.52 mol nm−2), a difference that cannot be easily discriminated at this coverage, because the locally ordered domains are formed by less than a dozen of molecules.
symmetry phase, hereafter called (2 × 5)-obl, as reported for Zn-TPP.32 The latter phase has a molecular density of 0.43 mol nm−2, which is 20% smaller than that of the (2 × 4)-obl (0.52 mol nm−2), a difference that cannot be easily discriminated at this coverage, because the locally ordered domains are formed by less than a dozen of molecules.
 | ||
| Fig. 12 Left: STM images (Colibrí tip) of a ∼0.5 ML film of 2H-TPP deposited at RT, where small domains with local (2 × 4)-obl order start to form; top and bottom panels correspond to the same film measured at positive (+1.3V, 40 pA) and negative (−1.3 V, 140 pA) biases, respectively. Right: Top views of the (2 × 4)-obl and (2 × 5)-obl phases are shown in the top and bottom panels, respectively. | ||
In any case, very extended and long range ordered domains are observed upon annealing a fully saturated layer beyond the self-metalation reaction, still preserving the (2 × 4)-obl symmetry, as well as the characteristic saddle-shaped molecular appearance, as shown in Fig. 13. In comparison with the isolated molecules, the STM intramolecular contrast of TTPs in the periodic (2 × 4)-obl lattice shows a deviation from the mirror symmetry along the major axis, due to the small displacement in the opposite direction of the two main lobes with respect to the Obr rows.
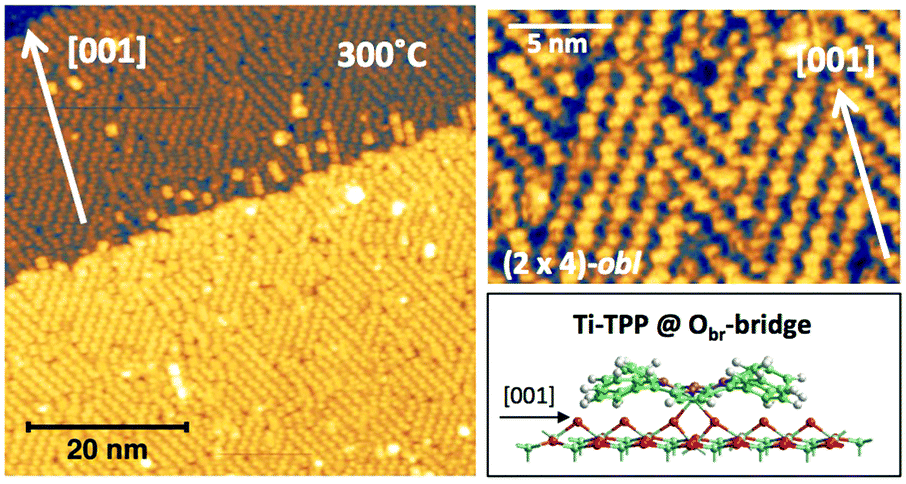 | ||
| Fig. 13 Left: Large scale image of 1 ML obtained upon thermal desorption at 300 °C of a bi-layer of 2H-TPP deposited at RT (+2.5V, 100 pA); all terraces are covered with an extended (2 × 4)-obl phase, entirely composed of metalated TPP molecules. Right: A smaller scale image of the same film puts in evidence the saddle-shape of the metalated molecules, and a small asymmetry with respect to the major axis due to the small azimuthal rotation of the molecules in the ordered phase, as compared to the isolated ones. Inset panel: A side-view of the equilibrium configuration of an isolated TiO2br-TPP molecules puts in evidence the bonding of molecular Ti with the two Obr atoms underneath. | ||
From a theoretical point of view, the incorporation of a Ti atom in the macrocycle upon dehydrogenation of the pyrrolic units does not change the equilibrium configuration of the molecule with respect to that found for the free-base molecule in Sections 3.1 and 3.2. The most stable configuration for an isolated molecule on a stoichiometric surface is simply achieved by replacing the two NH⋯Obr hydrogen bonds with two NTi⋯Obr metal bonds, as schematically shown in the drawing of Fig. 13. The molecule, hereafter designated TiO2br-TPP, remains in the bridge site on the Obr rows displaying only minor relaxations of the molecular backbone. The saddle-shaped inflection decreases by few degrees (from 29° of 4H-TPP and 25° of 2H-TPP to 22°), leaving practically unchanged the intra-molecular contrast in our STM images and in the DFT simulation of the electron density contour, as can be appreciated in Fig. 7 in the case of isolated 2H-/4H-/TiO2br-TPP molecules.
In the high density monolayer phase, the scenario remains practically unchanged for the 2H-/4H-TPP molecules, apart from a small azimuthal rotation by 8° of the molecular axis, which is responsible for the aforementioned asymmetry with respect to the [001] direction, and is due to the interpenetration of phenyls between adjacent molecules (see the model in the upper panel of Fig. 12). This structure corresponds to the equilibrium configuration also for TiO2br-TPP. However, for a better understanding of the stability of the self-metalated species, we performed additional calculations for a molecule adsorbed atop a single Obr atom (as predicted for Zn-TPP18,31,32), which corresponds to the formation of a double bond with an oxygen atom, like in the commercially available TiO-TPP compound.
For an isolated TiO2br-TPP molecule, corresponding to a (3 × 6)-obl supercell, we found the bridge site on the Obr rows to be favoured by 70 meV with respect to the on-top site, where the tendency of the Ti atom to form a double bond with a single oxygen atom is overcome by the optimization of the interaction of the substrate with the network of phenyl H-bonds. The energy balance is reversed in the (2 × 4)-obl phase, where the intermolecular phenyl interaction weakens that with the substrate, now favouring by exactly 70 meV the adsorption on-top rather than on-bridge. However, since self metalation takes place starting from the already formed (2 × 4)-obl phase, a collective displacement from the bridge adsorption site seems unlikely in such a very high density layer (ρ = 0.52 nm−2), and we may conclude that a pure TiO2br-TPP phase is, however, formed at either coverage. In fact, a small decrease of the intermolecular spacing, like in a hypothetic (2 × 5)-obl phase (ρ = 0.43 nm−2), is sufficient to recover the pristine phenyl interaction with the substrate (as well as the α = 0° azimuthal orientation) and to favour the bridge site by 100 meV. The molecular adsorption energies (Eads) for the different configurations are summarized in Table 1.‡
| Configuration | Isolated molecule | (2 × 4)-obl |
|---|---|---|
| E ads/eV | E ads/eV | |
| 2H-TPP@Obr – bridge | −2.92 | −3.03 |
| 4H-TPP@Obr – bridge | −4.92 | −4.66 |
| Ti-TPP@Obr – bridge | −1.02 | −0.66 |
| Ti-TPP@Obr – top | −0.95 | −0.73 |
As remarked before, none of the previously reported chemical pathways of self-metalation on oxide/oxidized surfaces (either water desorption or surface release of dissociated hydrogen) fits the experimental evidence on the r-TiO2(110) surface. In fact, TiO2 owes its special catalytic and transport properties to the possibility of injecting electrons in its Ti 3d empty bands, which then falls below the Fermi level originating the Defect State that makes the surface conductive and chemically reactive. Point defects, such as oxygen vacancies and Ti interstitials, are the most common origin of excess electrons populating the Ti 3d band, because they are inevitably formed during the mechanical, thermal and chemical processing of samples. Both VO and Tiint defects are experimentally found to generate the same distribution of charge at the r-TiO2(110) surface,118 which is mainly localized at a few subsurface lattice Ti sites, independently of the mechanism of charge injection.99,119 Consistently, the DS is theoretically described as a polaron, and the corresponding transport properties are independent of the nature of the defects.120–123 On the other hand, the chemical reactions with surface VO and subsurface Tiint would follow different pathways and yield different products.
In the literature, one may find many studies of the direct interactions of VO with gas and adsorbates because they are clearly visible by STM on r-TiO2(110), where they are more energetically favoured at the surface than in the bulk.124 Studying the participation of Tiint in surface chemical reactions is more elusive because their energy at subsurface sites is ∼0.6 eV more favourable than that at surface (adatom) sites.125 Furthermore, the lowest energy barrier for Tiint outdiffusion on the clean surface has been calculated as 1.40 eV,125 which is eventually lowered to 1.2 eV in the presence of adsorbed O2.126 Apart from the case of surface regrowth (or growth from beneath), when r-TiO2(110) is exposed to oxygen at high temperature,127–129 molecular reactions with Tiint defects are rather triggered by the corresponding charge excess (however localized at subsurface lattice Ti sites), and do not require their molecular coordination.130,131 In fact, the charge transfer from the DS to the unoccupied molecular orbitals of an aromatic acceptor may take place even in the absence of a linker or any atomically localized channel.132 Only very recently, the direct participation of Tiint atoms in the Ullmann coupling reaction of polymer precursors has been proposed.133 In fact, we found by DFT+U calculations that the outdiffusion of Tiint is also the mechanism driving the self-metalation reaction of porphyrins, where the key configuration for the metalation process is the adsorption of free-base porphyrins by chemical coordination of the two pristine pyrrolic nitrogen atoms to the corresponding Obr atoms underneath, independently of their meso-substituents.48
The reaction process may be split in three consecutive steps:
(1) outdiffusion, where a near-surface interstitial Tiint atom converts into a Ti adatom.
(2) sequestration, where the adatom is captured by the porphyrin core, yielding a Ti@2H–TPP(ads) complex.
(3) H recombination, where the pyrrolic hydrogen atoms desorb in the gas phase as H2.
Within this framework, we focused onto the fundamental steps (1) and (2), and we neglected the step of diffusion of bulk interstitials to a near surface site, since it has been already shown that the migration from the second subsurface layer (energetically more stable than the topmost subsurface layer by ∼0.1 eV) to a subsurface interstitial site must overcome a barrier of ∼0.84 eV.125 Because of the long range order of the TPP moiety in the ML phase, we considered a periodic (2 × 4)-obl supercell, with the 2H-/4H-TPP molecule in the corresponding (high coverage) equilibrium configuration; we also increased the slab thickness from 4 to 6 layers, which is necessary to properly describe interstitials. As compared to former DFT+U calculations, we used a Hubbard correction U = 2.3 eV, which is better suited to deal with redox reactions at the TiO2 surface134,135 than larger values that better reproduce the gap states.120 With reference to the drawings of Fig. 14, we considered different sites for the extra Ti atom, namely (i) a subsurface interstitial site F, (ii) an adatom site A, located aside two Obr atoms and coordinated to a three-fold oxygen atom O3f underneath, (iii) an adatom site B, located aside a single Obr atom and coordinated to two O3f atoms underneath, and (iv) a bridge site P on the Obr rows, where the Ti atom is already coordinated to the molecule, still holding the two pyrrolic H atoms.
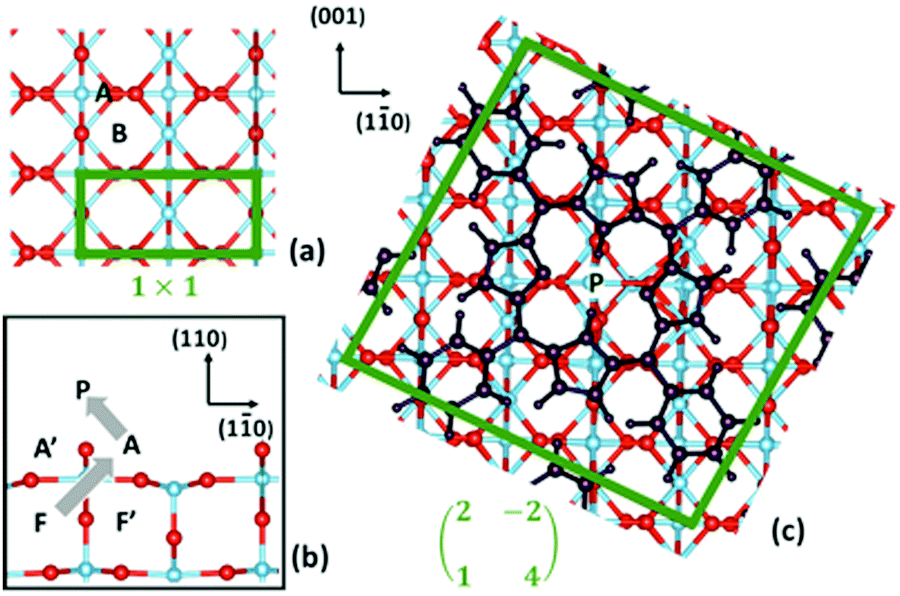 | ||
| Fig. 14 (a) Top and (b) side views of the TiO2(110) surface; and (c) top view of the oblique supercell used in the present work, where the adsorbed 2H-TPP film is also shown. Relevant sites for Ti are indicated. Ti atoms are blue, O atoms are red, and all the 2H-TPP atoms are black. The borders of the (1 × 1) unit cell and of the oblique supercell are marked with green lines. The grey arrows in (b) show the approximate path of Ti during the self-metalation reaction. Reproduced from ref. 48 with permission from Elsevier, Copyright 2021. | ||
As shown in Table 2, we found that the F site on the clean surface is more stable than the A site by 0.61 eV and the B site by 1.25 eV, which are in good agreement with the corresponding 0.57 eV and 0.91 eV values reported in ref. 125, when considering our larger supercell, our different functional PBE-D2 (better suited to deal with molecular adsorption) and different U correction. The energy balance between F and A sites is completely reversed upon adsorption of 2H-TPP: the F site is less stable than the A site by 0.49 eV that, in turn, is less stable than the P site by 0.74 eV (the B site is no longer an equilibrium configuration). Thus a robust thermodynamic driving force now exists for the outdiffusion and sequestration steps, corresponding to an overall energy gain of 1.23 eV for the F → P transition. In contrast, the bare extraction of a Ti lattice atom to the adatom site A in the absence of an extra Tiint at the nearby F site is unfavoured by 1.3 eV and the P site is no longer an equilibrium configuration.
| Site A | Site B | Site P | |
|---|---|---|---|
| TiO2(110) | 0.61 | 1.25 | — |
| 2H-TPP@Obr – bridge | −0.49 | No min | −1.23 |
| 4H-TPP@Obr – bridge | 0.23 | — | 1.33 |
We then studied the transition states from site F to site P (outdiffusion and sequestration) for the neutral molecule 2H-TPP by means of the climbing-image nudged elastic band (CI-NEB) algorithm.136 The structural configurations of the most relevant intermediate states are shown in the panels of Fig. 15, together with the corresponding minimum energy path (MEP) curve that highlights the relevant energy barriers and local minima. The mechanism involves the displacement of two titanium atoms, namely the subsurface lattice atom Ti(I), just beneath the Obr bridge coordinated to the pyrrolic NH, and an interstitial Ti(II) atom in the F site beneath the A′ site (see the model in Fig. 14). In the first step, Ti(I) is displaced by its lattice 6-fold coordinated site to a site with planar 3-fold coordination (TS1 configuration). Next, the interstitial Ti(II) atom moves towards the vacant lattice sites and Ti(I) further coordinates to the iminic nitrogen atom (coordination 4) in a pyramidal geometry (T configuration). The F → T transition is exothermic (energy gain of 0.25 eV) with an energy barrier of only 0.65 eV. Next, in a few barrierless stages, Ti(I) completes the outdiffusion step, reaching the adatom site A, while Ti(II) definitely fills the vacant lattice site. The final segment A → P is also strongly exothermic with a very small barrier of 0.21 eV and corresponds to the porphyrin capture of atom Ti(I), now in a pseudo-octahedral configuration. Thus, the interaction of iminic nitrogen with a 6-coordinated subsurface lattice atom drives its outdiffusion in the presence of a nearby Tiint and reduces the energy barrier to 0.65 eV, much less than the value calculated for the clean surface (1.40 eV) and also lower than the barrier for Tiint interlayer diffusion in the bulk (0.84 eV), which makes self-metalation at temperatures as low as 100 °C possible.
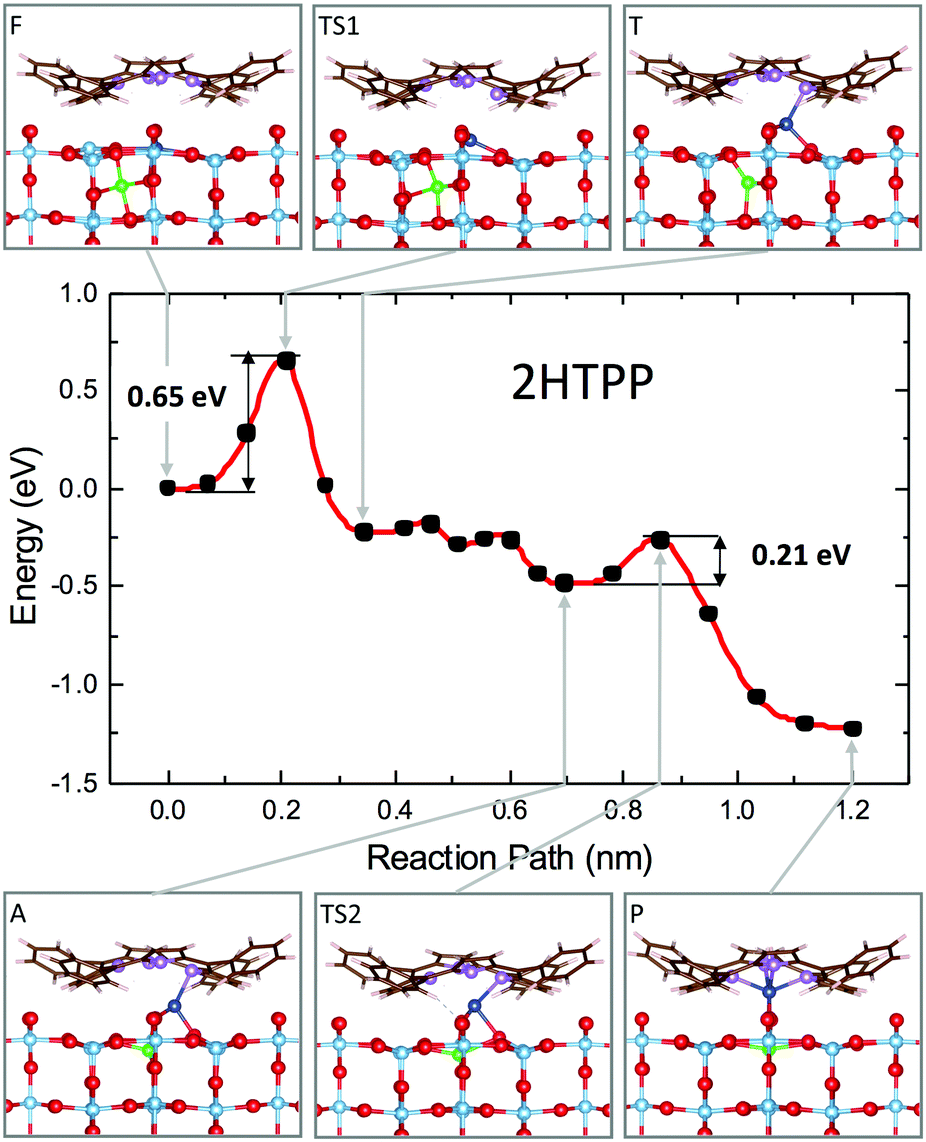 | ||
| Fig. 15 Minimum energy path for the self-metalation reaction of 2H-TPP adsorbed at TiO2(110). Sketches of relevant configurations, viewed along [001], are shown in the grey boxes and are connected to the appropriate MEP point by arrows. O atoms are red, C atoms are brown, N atoms are purple, and H atoms are white. Ti atoms are cyan, and those involved in the diffusion process are blue (Ti-I) and green (Ti-II). Note the slight azimuthal rotation of the adsorbate due to the dense film packing. The red curve is a spline interpolation, while black dots are the CI-NEB computed points. Reproduced from ref. 48 with permission from Elsevier, Copyright 2021. | ||
Overall, the outdiffusion of Tiint atoms is driven by the gradient of the chemical potential generated by the porphyrin self-metalation. Topmost interstitials are first sequestrated at mild temperature (barrier of 0.65 eV), then Tiint atoms from deeper layers outdiffuse at slightly higher temperature (barrier of 0.84 eV). This might explain why the self-metalation reaction is observed to be completed at lower temperature at low coverage (0.2 ML),47 where molecules are not aggregated and may diffuse over the surface to reach the most reactive site, in correspondence of a Tiint at F sites. Within this thermodynamical framework, no change in the concentration of interstitials in the subsurface layers is expected after self-metalation. In fact, both the Ti 2p and VB photoemission spectra (only probing 3–4 layers due to the short escape depth of 5–10 Å) indicate that the pristine concentration of Ti3+ ions (Ti 2p tail at ∼457 eV), as well as that of the excess of charge (DS peak), does not change after the reaction. In this regard, we cannot exclude the manifestation of a small transient charge unbalance (1–2%) just across the self-metalation reaction, as reported during real time monitoring of oligomer polymerization by Ullmann coupling.133
The energy landscape changes completely when considering the adsorption of the doubly hydrogenated molecule 4H-TPP. As shown in Table 2, the adatom site A is less stable than the interstitial one F; that is, the outdiffusion step is now endothermic. Most importantly, the adatom capture at the P site is strongly unfavoured (1.33 eV) because all of the porphyrin N atoms are saturated by hydrogen and the Ti atom cannot establish further coordination links. We also calculated the MEP for the outdiffusion and sequestration steps, where the intermediate configuration T (adatom with 3-fold coordination) is still a local minimum, but with an energy cost of 0.8 eV and a barrier of 1.15 eV.48 In fact, this barrier for outdiffusion is smaller than that on the clean surface thanks to the pyrrolic bonding to the Obr atoms, which loosen the crystalline bonds. With a small barrier (0.15 eV) the adatom can reach the A site, which is 0.6 eV more stable than the T configuration, but still unfavoured (endothermic) by 0.2 eV with respect to the starting configuration. In fact, the adatom is slightly displaced away from the A site because now the extra energy of stabilization is not provided by the fourth coordination link to iminic N, but by the (weaker) interaction with the π orbital of the pyrrolic ring.
From such a disadvantageous configuration, the migration to the very unstable P site is prevented by an energy barrier of 1.55 eV, which makes the overall energy barrier for the A → P transition as high as 1.75 eV. This means that the direct self-metalation of 4H-TPP cannot take place and the molecule must first release its two extra hydrogen atoms. The hydrogen release may take place before starting the outdiffusion step (F stage) or when the adatom has been already formed (stage A), but an extra cost for hydrogen release must, however, be taken into account. This is consistent with the N 1s photoemission spectra of Fig. 10, where pristine 2H-P molecules (marked by the residual iminic peak at ∼398 eV) are readily metalated at 100 °C, while 100% metalation of the majority 4H-P molecules (marked by the pyrrolic peak at BE ≥400 eV) takes place only at higher temperature. In fact, the temperature of self-metalation for 4H-TPP was formerly reported to take place at 100 K higher temperature than 2H-TPP,46 which might account for the extra cost of hydrogen release.
On a more general ground, this mechanism of Ti extraction is consistent also with the spectroscopic evidence reported for the self-metalation of carboxylated porphyrins. In fact, the onset of self-metalation for TPP with a single carboxylated phenyl was also observed to take place at 150–200 °C at low coverage, where molecules are lying flat on TiO2(110). Instead, annealing up to 400 °C was necessary to achieve self-metalation at 1 ML,137 where densely packed molecules are rather standing up.21 Such a high temperature was associated with an intermediate step of partial desorption and/or decomposition of the TPP carboxylic linker, in order to allow for a tilt down of the molecules to allow for self-metalation. However, Ti interstitials may freely outdiffuse to the surface at such high temperature (≥300 °C),127–129 opening the way to alternative mechanisms of Ti incorporation.
5 Cyclo-dehydrogenation at 400–450 °C
As mentioned before, a common practice to obtain a well ordered monolayer of poly- and hetero-conjugated molecules on surfaces is the thermal desorption of a multilayer film, which is made possible by the strong interaction with surfaces developed by aromatic compounds due to charge transfer and/or vdW type attraction, with the latter being typically stronger than the intermolecular one. This protocol is effective on inert surfaces such as Au(111), but the film annealing on a more reactive substrate might trigger unwanted reactions that change the chemical and electronic properties of the hybrid interface. Beyond the self-metalation reaction, the interaction with reactive substrates often yields conformational changes due to cyclo-dehydrogenation of the peripheral terminations. The flattening of TTPP molecules at Ts ≈ 300 °C has been reported on both Ag41,44 and Cu42,43,138 surfaces, where the phenyls establish new C–C aryl bonds with the pyrrolic peripheral carbons (each bond yielding the release of one H2 molecule), thus increasing the degree of conjugation of the molecular backbone. Further dehydrogenation takes place at higher temperature,42 leading to partial desorption, disordering and dendritic polymerization beyond C.43,44 Actually, we found that self-metalated TPP molecules remain anchored to the r-TiO2(110) surface up to an exceptionally high temperature of 450 °C, forming a fully wetting commensurate phase.495.1 Molecular flattening and ordering
The thermal evolution of 2H-TPP in the monolayer range is best followed by real time surface diffraction (RHEED), as shown in ref. 49: the (2 × 4)-obl symmetry pattern starts fading at ∼350 °C, leaving place to a new (2 × 6)-rect symmetry phase, which is best observed after annealing to 400–450 °C. The change in phase symmetry is clearly appreciated in the STM images shown in the top panel of Fig. 16. From a comparison of adjacent domains, we may notice that molecules in the (2 × 6)-rect phase are still characterized by a nodal plane along the short axis, which suggests an overall integrity of the porphyrin macrocycle. The molecules in the new phase display a slightly elongated footprint and a perfect symmetry with respect to the long molecular axis, whereas those in the (2 × 4)-obl phase display a small asymmetry, due to the aforementioned in-plane azimuthal rotation (8°) and the interpenetration of phenyls between adjacent molecules, as seen in Section 4.2.1. At the highest annealing temperature, where some disorder sets in, we observed a displacement between adjacent molecules by one substrate lattice unit along the direction (the atomic rows), still yielding locally commensurate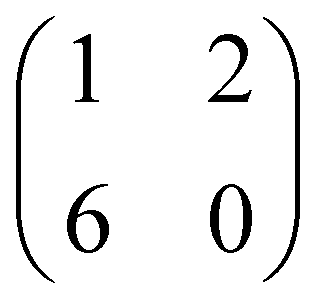 domains, hereafter called (2 × 6)-obl, with the same molecular density. In the bottom panel of Fig. 16, one may notice a significant molecular de-wetting on the (2 × 1) reconstructed regions of the surface, which can be used to assign the adsorption site on the adjacent (2 × 6)-rect molecular domains by comparison with high resolution images measured on the clean substrate.49 Also for this high temperature phase, molecules in the monolayer phase preferentially adsorb on Obr rows.
domains, hereafter called (2 × 6)-obl, with the same molecular density. In the bottom panel of Fig. 16, one may notice a significant molecular de-wetting on the (2 × 1) reconstructed regions of the surface, which can be used to assign the adsorption site on the adjacent (2 × 6)-rect molecular domains by comparison with high resolution images measured on the clean substrate.49 Also for this high temperature phase, molecules in the monolayer phase preferentially adsorb on Obr rows.
 | ||
| Fig. 16 Top: STM image (Colibrí tip) showing the coexistence of (2 × 4)-obl and (2 × 6)-rect phases, as obtained by thermal desorption at 350–400 °C of a bi-layer film grown at RT (+2.5 V, 60 pA); molecules in the new phase display two symmetric lobes, much brighter than those in the (2 × 4)-obl phase. Bottom: The same film after annealing to 450–500°, as imaged close to a (2 × 1) reconstructed region, which shows a significant dewetting (+1.5 V, 80 pA). | ||
Photoemission spectroscopy reported in ref. 49 indicated that the chemical state of the molecule remains almost unchanged: apart from a small shift to a lower BE (−0.2 eV) of the C 1s peak, the N 1s BE and peak shape remain unchanged, as well as the Ti 2p and its low BE tail; the DS is simply attenuated by the molecular overlayer and the HOMO at 2 eV simply broadens. The only relevant change concerns 20% decreases of the N 1s and C 1s peak intensities, perfectly matching the expected change in molecular density from the (2 × 4)-obl phase (ρ = 0.52 nm−2) to the (2 × 6)-rect one (ρ = 0.43 nm−2).49
As previously discussed in Section 3.3, NEXAFS at the N and C K-edges is better suited than XPS to check for possible change in molecular orientation/configuration from the intensity ratio of the π*-symmetry resonances measured in TE and TM polarization (s and close to p, respectively). In Fig. 17, we show the spectra of a 2H-TPP monolayer film measured after deposition at RT, and after annealing to 300° and 450 °C, which are representative of the different molecular species.§ The nitrogen K-edge resonances at 397.55 ± 0.02 eV and 399.80 ± 0.02 eV (corresponding to the residual iminic contribution and the majority pyrrolic one at RT) are replaced with a single one at ∼398.3 eV upon annealing to 300°, corresponding to the equivalent chelation of the four N atoms to the incorporated Ti atom. In this chemical state, corresponding to the (2 × 4)-obl phase, the NEXAFS spectra display an almost perfect dichroism, indicating that the macrocycle is closely planar and parallel to the surface (average tilt of ∼10°, as determined from the LUMO intensity ratio). Further annealing to 450 °C does not change the energy position of the N resonances and we only recorded an increase of the intensity in TE polarization, indicating either an increase of the average tilt of the macrocycle from 10° to 15° or a distortion/rehybridization of the macrocycle.
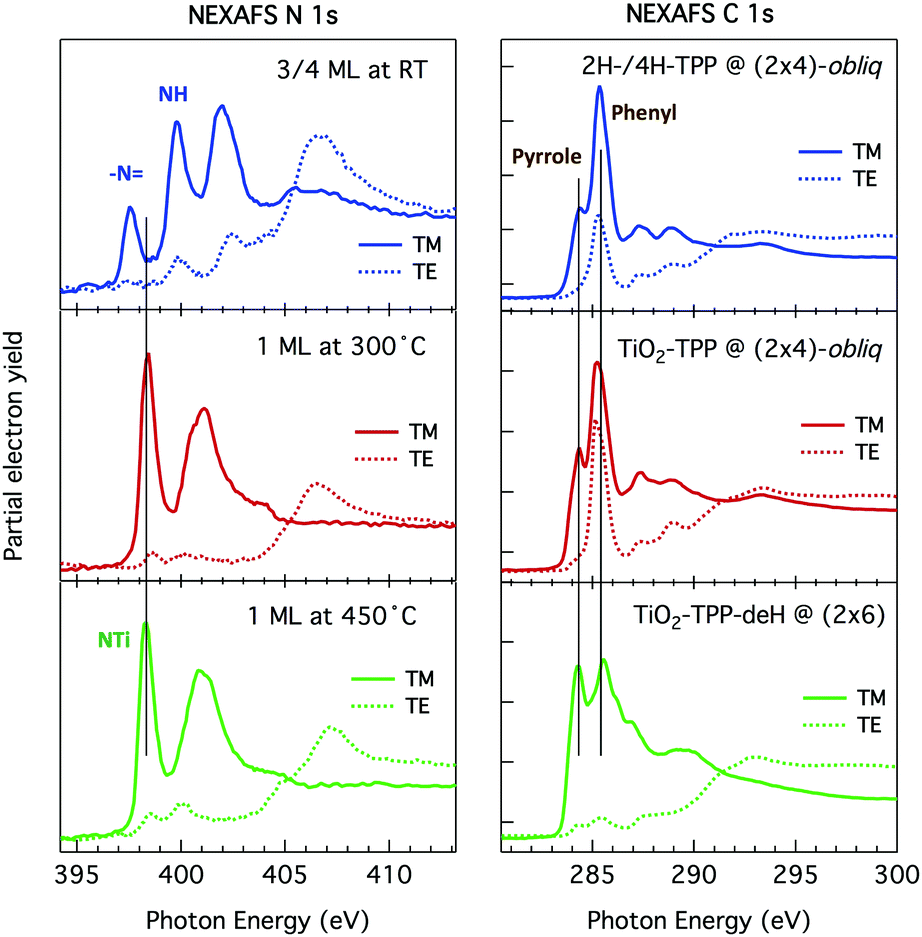 | ||
| Fig. 17 NEXAFS spectra measured in partial electron yield at the N and C K-edges, left and right panels, respectively, at RT, 300 °C and 450°, corresponding to different molecular species and symmetry phases; spectra measured in TM and TE polarization are represented as full and dotted lines, respectively. The surface was azimuthally oriented to keep the scattering plane along the [001] direction. In this geometry, the TE spectra are measured with the electric field transverse to the rows, where the molecules display a slightly larger intensity of π-symmetry resonances than along the rows, hence yielding the maximum tilt angle/molecular distortion in any chemical state. Adapted from ref. 49 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
Significant variations of the dichroism were rather observed at the C K-edge, which is characterized by two main π-symmetry resonances: a narrow shoulder at ∼284 eV, whose molecular orbital is mostly localized on the carbon atoms of the tetra-pyrrolic macrocycle, and a sharp dominant resonance at ∼285 eV, corresponding to the contribution from the phenyl meso-substituents. The pyrrolic resonance yields a large dichroism, which slightly decreases with increasing temperature, consistently with the trend observed at the N K-edge. The phenyl resonance at RT displays a large intensity also in TE polarization, corresponding to an average tilt angle of 30°, in good agreement with the theoretical value (27° for 4H-TPP). Upon metal incorporation at 300 °C, the dichroism of the phenyl resonance further decreases, corresponding to an increase of the tilt to ∼40°. Such a tilt up of the phenyls might be associated with a stiffening of the molecular backbone due to the larger delocalization of the macrocycle upon metal inclusion, as formerly reported for Co incorporation on the Ag(111) surface.139 Upon annealing to 450 °C, we observed a major change in the resonance associated with phenyls, which almost vanishes in TE polarization, thus indicating an average tilt off the surface by ∼10°. At this temperature, also the shape of the NEXAFS in TM polarization changes: the pyrrole and phenyl resonances can be easily recognized at the same energy, but display a different relative intensity. The highest energy resonances display both a change in shape (broadening) and a shift closer to the LUMO. Overall, these two lines of evidence indicate a flattening of the molecule, accompanied by an increase of the degree of conjugation of the carbon backbone, in full agreement with a model of partial cyclo-dehydrogenation and formation of new C–C aryl bonds (flat TPP-deH).
5.2 Equilibrium configuration of cyclo-dehydrogenated TPP:TiO2br-TPP-deH
For the theoretical modelling of the adsorption configuration, we considered both the (2 × 6)-rect and-obl phases. Among the different conformations that may be assumed by TTPs upon flattening,43,44 we restricted the calculations to the rectangular one, which solely may yield the perfect in-plane symmetry with respect to both the short and long molecular axes, as observed by STM. Although the molecular images indicate a straight alignment (α = 0°) along the [001] direction, we also considered ordered phases with different azimuthal orientations. Preliminary calculations for an isolated flat molecule, i.e. on a (3 × 6)-obl supercell, indicate a large preference for the orientation α = 0°, even when the molecule is adsorbed atop a single Obr atom, with any alternative azimuthal orientations being more expensive by ≥1 eV (see Section 6.2 for details). These results tell us that the straight alignment of the molecules along the [001] direction is strongly favoured by the formation of weak hydrogen bonds among the peripheral C–H terminations and the adjacent Obr rows.The network of peripheral CH⋯Obr bonds produces a bending of the molecule around its long axis that, for the equilibrium configuration in the (2 × 6)-rect phase, is predicted to reach ∼158°, as can be appreciated in Fig. 18. The corresponding molecular conformation can be described as a hogback roof tile, whose bending is stronger when the molecule is adsorbed on a Obr bridge site rather than atop a single Obr atom, because the bridge site yields a better matching, hence bonding, of the peripheral CH terminations with the protruding Obr atoms. In any case, the Obr bridge site is favoured by 0.1–0.15 eV for both the (2 × 6)-rect and-obliq phases, which now yield the same adsorption energy (−3.5 eV).49
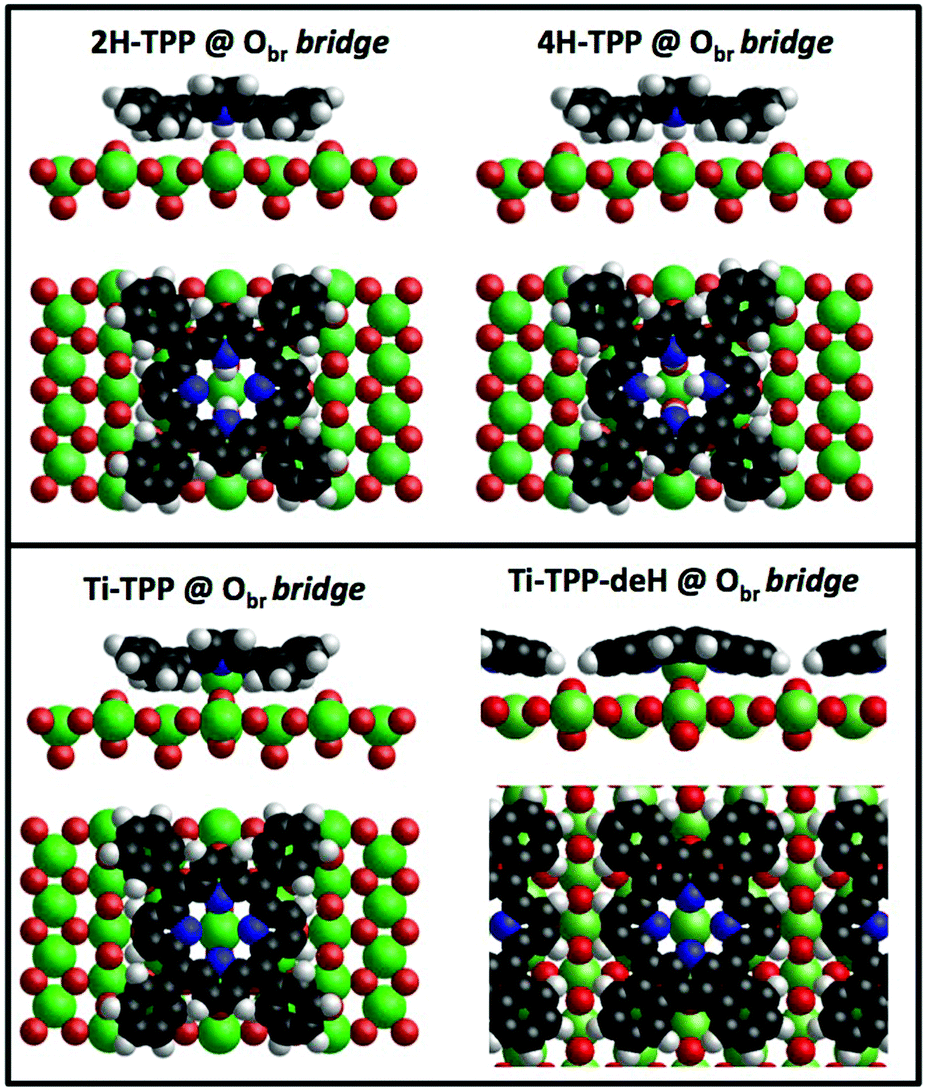 | ||
| Fig. 18 Top: Side and top views of the equilibrium configurations for isolated 2H- and 4H-TPP molecules (left and right, respectively) on the r-TiO2(110) surface. Bottom: Equilibrium configurations of an isolated TiO2br-TPP molecule (left) and a flat TiO2br-TPP-deH species in its (2 × 6)-rect phase (right). | ||
Looking at the comparison of the DOS cuts in Fig. 19, the Obr bridge and on-top sites are closely similar and dominated by an inflection along the short axis at any voltage bias (with a slightly larger density at the center of the molecule at the on-top site). This topographic feature, appearing as a nodal plane transverse to the molecule in low resolution (large area) STM scans, stems from the large intensity of the pyrroles located on the Obr rows; that is, it has an electronic origin, even if the intensity contour does not look much different from that of the 2H-/4H-/TiO2br-TPP molecules, where the electronic effect is accompanied by a significant saddle-like structural distortion. The DOS cuts of the on-top and bridge sites also differ by the apparent extension of the molecular footprint, where the on-top site yields a larger spread of the peripheral density at either positive or negative bias, due to the smaller bending of the molecular backbone (structural effect). By comparison with high resolution STM images shown in Fig. 19, the DOS cut for the bridge site yields an overall better agreement with the intramolecular contrast observed both in the periodic (2 × 6) structure and in flattened molecules embedded in the (2 × 4)-obl phase at intermediate annealing temperature.
 | ||
| Fig. 19 (a) High resolution STM image (Colibrí tip) measured after annealing to 350 °C a ML of 2H-TPP deposited at RT (−1.5 V, 63 pA); new species with two very bright and symmetric lobes (type B) appear in the middle of a (2 × 4)-obl domain formed by dim, saddle-shaped and asymmetric molecules (type A); (b) STM close view of the TiO2br-TPP-deH molecules in the (2 × 6) phase, as obtained after annealing to 450 °C (Colibrí tip, +1.8 V, 80 pA); and (c) calculated DOS cuts at +2.0 V for TPP molecules in the (2 × 6)-rect phase, as adsorbed at the Obr bridge and on-top site (left and right panels, respectively). | ||
Free-base TPP deposited on the r-TiO2(110) surface thus bears multiple chemical and conformational reactions (shown side by side in Fig. 18) as functions of temperature without ever changing the adsorption site:
• At RT, 2H- and 4H-TPP molecules self assemble into a (2 × 4)-obl ML phase, where they are directly anchored on an Obr bridge site by two hydrogen bonds N–H⋯Obr.
• Upon self-metalation (100–200°), the hydrogen bonds are simply replaced by Ti bonding to two Obr atoms underneath, where the TiO2br-TPP molecules of the (2 × 4)-obl phase remain in the on-bridge site because of the steric constraint due to the intercalation of the tilted phenyl rings among adjacent molecules.
• At 400–450 °C, the monolayer phase converts into a (2 × 6) phase by partial desorption (20%) and partial cyclo-dehydrogenation, where the Ti coordination to two Obr atoms is now favoured by peripheral hydrogen bonding to the adjacent Obr rows, thus yielding a new porphyrin conformation, hereafter called TiO2br-TPP-deH.
The exceptionally high thermal stability of self-metalated TPP on TiO2(110) can be rationalized in terms of the different degree of interaction played by the substrate with the molecular macrocycle and its meso-substituents. TiO2 is strongly reactive for the tetra-pyrrolic macrocycle (with and without the central Ti atom) favouring strong molecular bonding, while it behaves like a dielectric for the phenyl meso-substituents favouring their decoupling from the surface. As the temperature increases, the molecule remains pinned through the TiO2br coordination, while the phenyls are dynamically flapping, which favours their overlap with the peripheral carbon of the pyrroles. In particular, the rectangular shape might be favoured by the release of hydrogen to the Obr rows underneath, where H atoms immediately recombine and desorb at such a high temperature.
6 Formation of TiO-TPP
As shown before, the bare incorporation of a Ti atom by self-metalation does not change either the phase symmetry or the adsorption site of a TPP monolayer film. Nonetheless, upon self-metalation at very low coverage (0.1–0.2 ML), we also observed the formation of some molecular features located on the Ti5f rows, with a pronounced square shape and rotated by 45°, as can be appreciated in Fig. 20. It is worth mentioning here the close resemblance of such porphyrin species with those reported after metalation by deposition of nickel on a 2H-TPP film (both at RT and after annealing to 250 °C),34 which confirms the metalated nature of the new TPP species.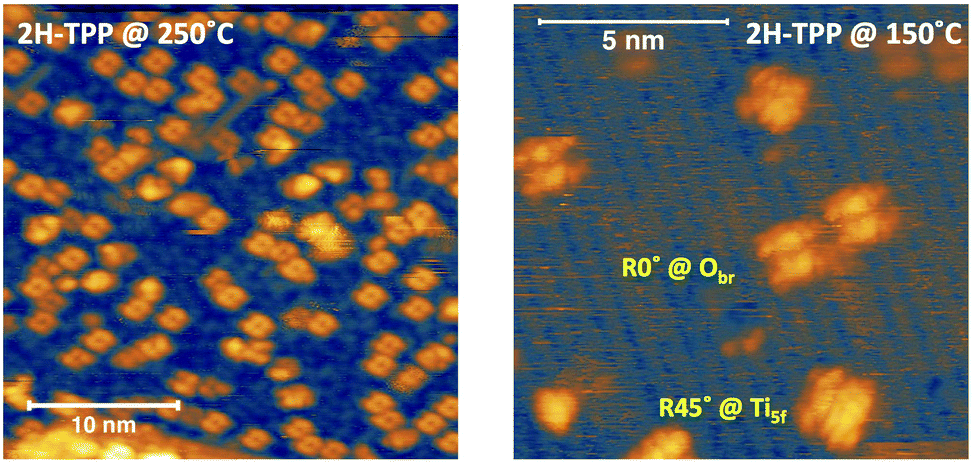 | ||
| Fig. 20 Left: STM image (PtIr tip) of ∼0.2 ML 2H-TPP molecules after self-metalation at 250 °C (+1.3 V, 96 pA). Right: A close-up view of isolated TPP molecules after self-metalation at 150 °C (+1.5 V, 40 pA). | ||
The origin of these metalated R45° porphyrins can be better understood by comparison with the direct deposition of TiO-TPP, which is available as a commercial product. When deposited, TiO-TPP may assume different adsorption configurations depending on the landing orientation: with the O atom pointing either towards (TiOdown-TPP) or off (TiOup-TPP) the surface. In the former case, TiOdown-TPP can only adsorb in between the Obr rows. In Fig. 21a, one can see a (local) majority of square molecules that are adsorbed on the Ti5f rows and rotated by 45°, practically identical to the minority species observed upon self-metalation of 2H-TPP at low coverage (right panel of Fig. 20). In addition, one can see a few saddle-shaped molecules on the Obr rows, which we can associate with TiOup-TPP molecules having lost their pristine oxygen after landing on the Obr rows, hence corresponding to TiO2br-TPP species. We observed also a minority of intermediate TiO-TPP configurations at RT; in particular a few molecules in between the Obr and Ti rows display a characteristic square R0° four-leaf intermolecular contrast (Fig. 21b). In any case, this configuration and other minority RT configurations seem to be out of equilibrium and/or metastable, since they are entirely converted into the two aforementioned square-R45° and saddle-shaped species upon mild heating. In fact, most of the pristine TiOdown-TPP square-R45° species are also converted into saddle-shaped ones, as shown in Fig. 21c and d. In brief, a low coverage film (≤0.2 ML) of 2H-/4H-TPP is converted into a majority of saddle-shaped TiO2br-TPP and a minority of square-R45° TiO-TPP species upon annealing to ∼200 °C. Remarkably, as deposited TiO-TPP molecules at coverage ≥0.4 ML are also converted into saddle-shaped TiO2br-TPP upon annealing to ∼200 °C, including most of the pristine TiOdown-TPP species in the square-R45° configuration.
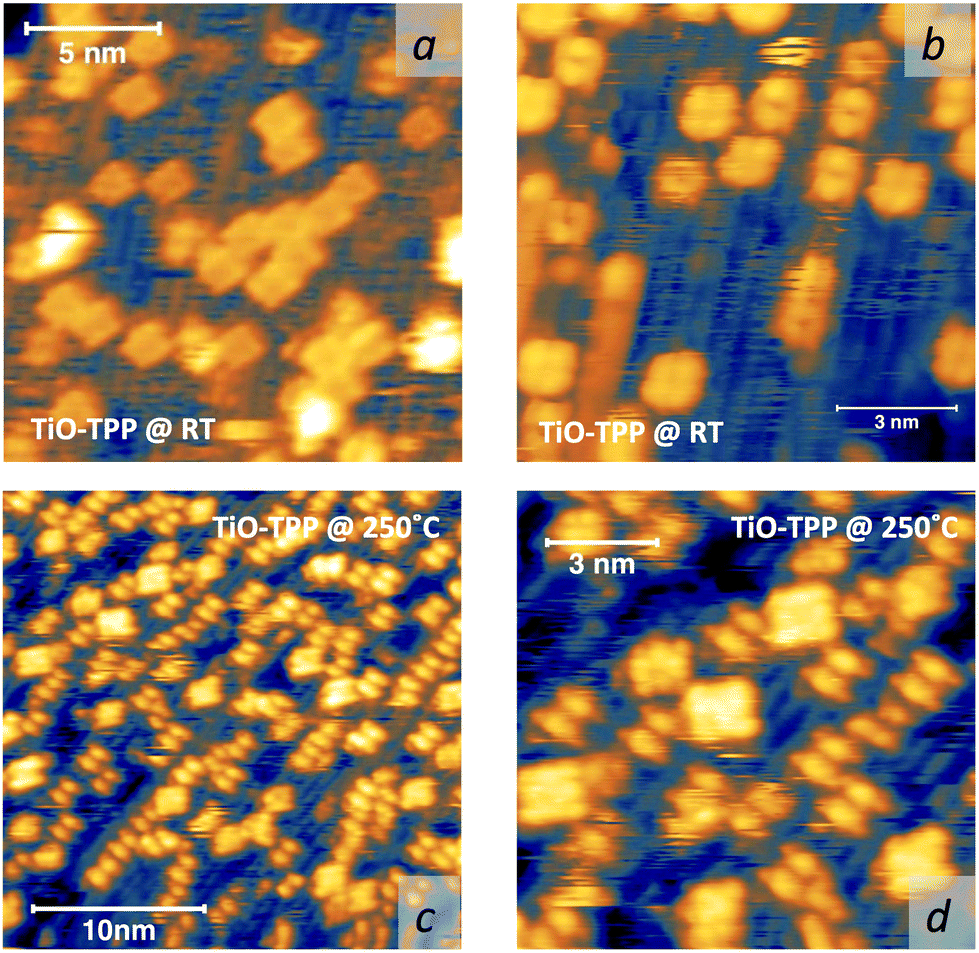 | ||
| Fig. 21 Top: STM images (W tip) of a ∼0.4 ML film of TiO-TPP deposited at RT: (a) the region is covered with a majority of square molecules adsorbed on the Ti5f rows (bright rows) and rotated by 45°; a few saddle-shaped molecules with an overall dimmer intensity are observed on the Obr rows (+1.4 V, 150 pA); and (b) in this region, most of the molecules are adsorbed on the Obr rows with a saddle-shaped conformation, and a few molecules in between the Obr and Ti5f rows rather display a four-leaf motif (+1.7 V, 190 pA). Bottom: The same film after annealing to 250 °C (+1.2 V, 170 pA): (c) most of the molecules are now observed on the Obr rows with a saddle-shape; and (d) close-up view of a few residual square-R45° molecules on Ti5f rows, surrounded with saddle-shaped molecules on the Obr rows. | ||
6.1 Comparison with deposition of TiO-TPP
In order to disentangle the possible mechanisms driving the observed molecular changes, we performed detailed calculations to compare the energetics of the multiple absorption configurations that may be assumed by TiO-TPP. We considered three molecular conformations, i.e. the aforementioned TiOdown- and TiOup-TPP orientations of the oxygen atom with respect to the surface, as well as the bare Ti-TPP case, where the molecule is assumed to have lost its oxygen atom. In front of the large number of possible molecular orientations, we considered only three azimuthal orientations, namely α = 0°, 45°, and 90° (following the same notation defined in Section 3.3). Finally, we tested the equilibrium geometry at multiple adsorption sites, i.e. Obr, Ti5f and O3f rows, both on-top and on-bridge, depending on compatibility.We start the discussion from the TiOdown-TPP conformation, whose main equilibrium structures are presented in the drawings of Fig. 22 for molecules atop a Ti5f atom, and the corresponding adsorption energies (Eads) are summarized in Table 3. As expected from experimental evidence, the most favoured orientation is with the molecule rotated by 45°, which gives the reference adsorption geometry for TiO-TPP on the r-TiO2(110) surface. In this configuration, the saddle-like distortion is less pronounced thanks to the low steric hindrance between the phenyls and the substrate, whose interaction ultimately determines the optimal adsorption geometry. Among the other equilibrium configurations, the closest in energy are those obtained at α = 90°. In particular those where the molecular Ti atom is leaning towards the adjacent Obr atom (R90°-asym) or coordinated to it (forming a TiO2–TPP complex) might correspond to the aforementioned minority four-leaf species, observed to lie in between the Obr and Ti5f rows. Refinement calculations performed on the most relevant configurations using the Hubbard correction (eV) and small core pseudopotentials (sc-PBE+U) did not change the energy hierarchy and landscape, rather strengthening the stability of the R45°-Ti5f equilibrium configuration.
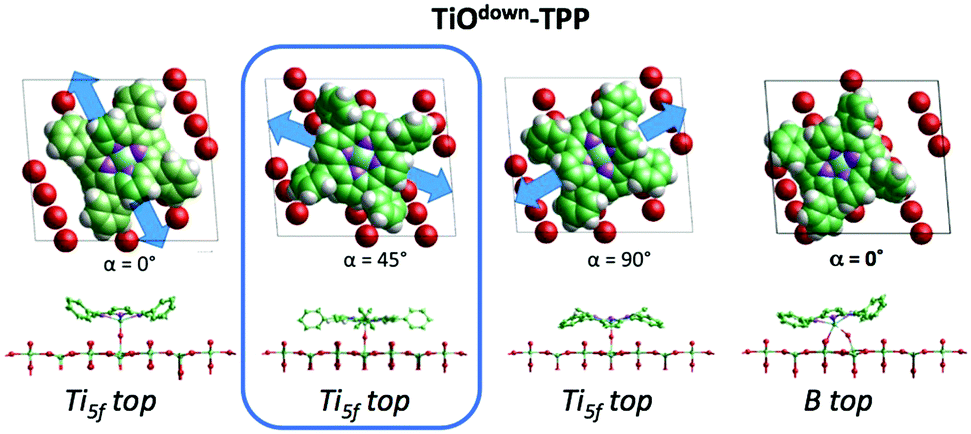 | ||
| Fig. 22 Side and top views of the equilibrium configurations for different azimuthal orientations of TiOdown-TPP adsorbed atop a Ti5f atom, as well as above the B adatom site, after coordination to an adjacent Obr atom. | ||
| Site | α | O coordination | lc-PBE (eV) | sc-PBE+U (eV) |
|---|---|---|---|---|
| Ti5f top | 45° | Odown | 0.00 | 0.00 |
| B top | 0° | Odown + Obr | 0.29 | 0.45 |
| Ti5f top | 90° | Odown | 0.34 | 0.43 |
| Ti5f top | 0° | Odown | 0.77 | 0.61 |
| Ti5f top | 90°-asym | Odown | 0.32 | 0.35 |
| Obr top | 0° | Oup | 1.19 | — |
| Ti5f top | 45° | Oup | 1.85 | — |
| Ti5f top | 90° | Oup | 2.12 | — |
| Ti5f top | 0° | Oup | 2.81 | — |
We then considered molecules landed with the oxygen atom pointing off the surface. In this conformation, adsorption on the Ti5f rows is always unfavoured by 2 eV or more with respect to the reference configuration TiOdown-TPP-R45° for any azimuthal orientation, as shown in Table 3. As expected, adsorption on the Obr rows provides the lowest energy equilibrium geometry for the α = 0° orientation, however with an absorption energy of 1.19 eV, much larger than that of TiOdown-TPP in any configuration. Such a high adsorption energy suggests that TiOup-TPP molecules rather lose their pristine oxygen upon landing, eventually coordinating to the Obr rows.
We then computed the equilibrium adsorption energies for de-oxygenated Ti-TPP species. The most stable configuration is obviously on the Obr rows in the bridge site with α = 0°, which corresponds to the TiO2br-TPP molecule, as obtained upon self-metalation of 2H-/4H-TPP. Most interestingly, we found only one equilibrium configuration on the Ti5f rows, corresponding to on-top adsorption with the α = 45° orientation. Any alternative geometry on Ti5f rows (on either the bridge or top site) did not converge to a local minimum and the molecule inevitably moved aside towards an O3f atom. The Ti-TPP coordination to an O3f atom determines its partial extraction from the lattice and a consequent bounce back of the molecule towards the Ti5f rows. In any case, adsorption of Ti-TPP on either Ti5f or O3f is largely unfavoured by 2–3 eV, as summarized in Table 4. Also in this case, refinement by including small core functionals and Hubbard correction did not change the scenario, apart from allowing for an equilibrium configuration of Ti-TPP rotated by 90° on top of Ti5f.
| Site | α (°) | O coordination | lc-PBE (eV) | sc-PBE+U (eV) |
|---|---|---|---|---|
| Obr bridge | 0 | 2 × Obr | 0.00 | 0.00 |
| Ti5f top | 45 | None | 3.69 | 2.80 |
| Ti5f top | 90 | None | No min | 3.06 |
| Ti5f–O3f | 45 | O3f | 2.79 | 2.51 |
| Ti5f–O3f | 90 | O3f | 2.62 | 2.45 |
With these results, we may attempt to rationalize the shift to the Ti5f rows of 2H-TPP upon self-metalation at low coverage. First of all, the bare detachment of a TiO–TPP complex from the Obr rows seems unlikely because it would leave behind a VO that, even if we are dealing with a low coverage (0.1–0.2 ML) film, would be detected as an increase of the intensity of the DS peak in the gap. Hence, we conjecture a direct interaction of TiO2br-TPP with diffusing oxygen adatoms Oa on the Ti5f rows. In this case, GGA calculations indicate a barrier-less capture of Oa by the molecule, which would assume the B-R0° configuration (Eads ∼ 0.3 - 0.4 eV), as depicted in the right panel of Fig. 22, finally transforming into the most stable configuration corresponding to the R45° orientation on top of a Ti5f site (i.e. the equilibrium configuration of TiOdown-TPP).
Oxygen adatoms on the Ti5f rows (as formed by O2 dissociation at a VO140) are quite stable at RT;141 however, the oxygen residual gas pressure in UHV systems is normally negligible, even as outgassed by heating filaments during sample annealing. Conversely, water in the UHV residual gas pressure is known to hydroxylate TiO2(110) within a few hours at RT,142 as well as to adsorb at Ti5f rows either in the molecular form or in the pseudo-dissociated form.143,144 In the latter case, one hydrogen is donated to an adjacent Obr to form a hydroxyl OHb and a terminal hydroxyl OHt remains on the Ti5f rows.75 While paired OHb–OHt are short lived, unpaired terminal hydroxyls, however generated (e.g. by ionization of the residual water gas pressure using vacuum gauges), are long lived at RT.141
We can reasonably assume that any OHt (and possibly H2O too) close to a TiO2br-TPP molecule would easily release its hydrogen atom and coordinate to the molecular Ti atom, thus detaching the molecule from the Obr rows and rapidly evolving towards the equilibrium TiOdown-TPP-R45° configuration. As will be shown in the next section, the amount of these porphyrin species may increase strongly (becoming the majority population), when the sample annealing is performed on a day long 2H-/4H-TPP low coverage film, which is consistent with the hypothesis that these species are formed by interaction with surface contaminants (mostly water) on defected surfaces.
The reversed conformational change of a pristine film of TiO-TPP into a majority of TiO2br-TPP species upon annealing to 250 °C cannot be straightforwardly explained in terms of the adsorption energy of isolated species. The bare capture of TiO-TPP by existing oxygen vacancies (and the corresponding molecular displacement) may be excluded as well, because we did not observe any decrease of the pristine DS peak in the gap after annealing. Rather the DS peak and Ti 2p tail slightly increase, suggesting a small release of Ti atoms and the corresponding formation of TiOx species. In any case, the r-TiO2(110) surface could not bear a VO concentration large enough to accommodate a full wetting layer of TiO-TPP.
In the monolayer range, TiO-TPP molecules neither aggregate into ordered domains nor assume a homogeneous adsorption configuration. Square molecules rotated by 45° and 90°, as well as saddle-shaped ones, are randomly distributed on the surface at RT, as shown in Fig. 23a. Upon annealing to 300 °C, all the molecules are, however, converted into saddle-shaped ones that, even if not yielding long range order, clearly show the formation of local domains mimicking the (2 × 4)-obl phase of the 2H-/4H-/TiO2br-TPP species (see Fig. 23b). Not surprisingly, further annealing to 450 °C converts the film into the (2 × 6) symmetry phases formed by flattened and dehydrogenated TiO2br-TPP-deH species, displaying their characteristic rectangular shape oriented along the [001] direction (Fig. 23c).
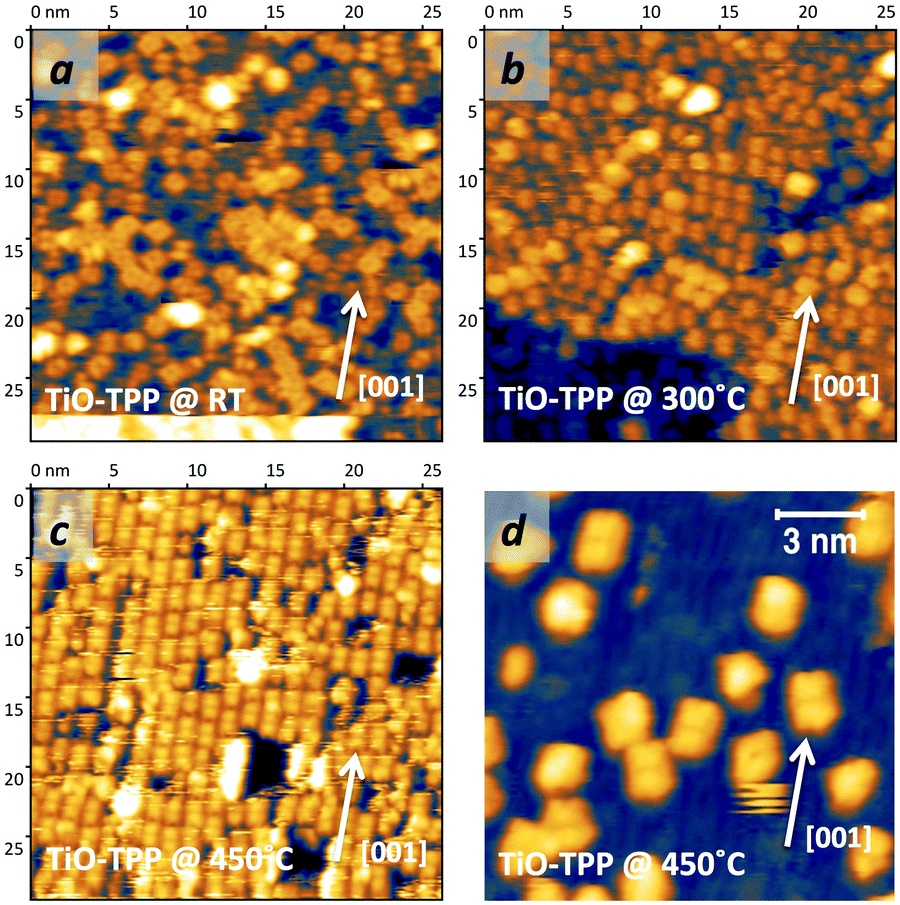 | ||
| Fig. 23 STM images (W tip) of (a) 1 ML of TiO-TPP as deposited at RT, (b) after annealing to 300 °C and (c) 450 °C; and (d) 0.2 ML of TiO-TPP after annealing to 450 °C. | ||
By comparison with the case of the self-metalated species in Fig. 20, the molecular coverage appears to be the critical parameter discriminating between the two, apparently opposite, behaviours. We may thus conjecture that the oxygen release and migration of TiOdown-TPP to the Obr rows is driven by a concerted relaxation of the phenyl-substituents between molecules entering into contact. In this regard, we recall that the (2 × 4)-obl phase is the most dense one for TPP species, thanks to the (vdW attractive) interpenetration of phenyls between adjacent molecules, which is further stabilized by annealing to 300 °C, as witnessed by the sharpening of the corresponding diffraction pattern.47 In contrast, isolated TiOdown-TPP molecules would anyway preserve their anchoring site, and eventually follow a different thermal evolution with respect to the saddle-shaped ones upon further heating. The annealing to 450 °C of a very low coverage film of TiO-TPP, as shown in Fig. 23d, seems to confirm this hypothesis, displaying the coexistence of rectangular shaped TiO2br-TPP-deH species on Obr rows together with other species on the Ti5f rows displaying other molecular orientations (mostly rotation by 30°) or conformations (including residual square-R45° species).
6.2 Flattening of TiO-TPP atop Ti5f atoms
Clearly, also the isolated 2H-/4H-TPP species that move to the Ti5f rows, due to interaction with terminal hydroxyls, are expected to follow such a distinct evolution path. As mentioned before, when the surface has a large concentration of defects/contaminants, we observed a significant increase of the number of 2H-/4H-TPP molecules that migrate on the Ti5f rows upon annealing a low coverage film. These conditions are easily met by sample exposure for a few hours to the residual gas pressure even in an UHV environment.142 In Fig. 24a, we show a representative STM image obtained by annealing to 400–450 °C the low coverage 2H-TPP film of Fig. 2, after overnight exposure to the background pressure (∼2 × 10−10 mbar). The panels in Fig. 24b put in evidence the large concentration of residual defects on the Obr rows. Under these tip conditions, the defects appearing as bright and dim spots in between the thin bright stripes of Ti5f rows are mostly OHb and VO, respectively. The majority of (metalated) molecules are characterized by a rectangular shape, either parallel to the [001] direction or rotated by ∼60°. The non-rotated species are located on the Obr rows and display the same intramolecular contrast of the molecules participating in the (2 × 6) phases (also see Fig. 19); that is, they can be identified with isolated TiO2br-TPP-deH molecules. The azimuthally rotated species are located on the Ti5f rows, and their rectangular shape indicates that they also underwent a flattening structural transition due to partial cyclo-dehydrogenation.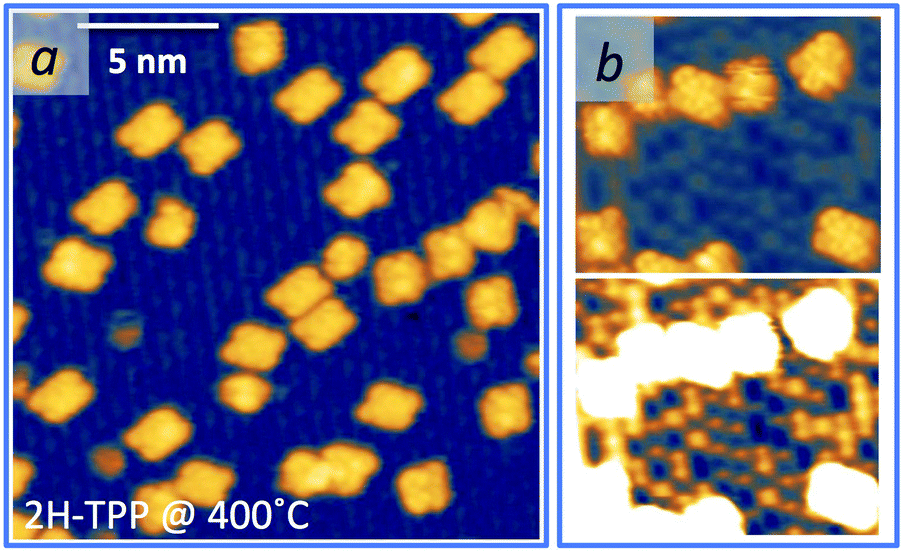 | ||
| Fig. 24 STM images (Colibrí tip) of a low coverage 2H-TPP film (the same as Fig. 2), as annealed to 400–450 °C after 24 h from deposition. (a) Rectangular molecules rotated by ∼60° are observed on the Ti5f rows (the bright ones), whereas those sitting on the Obr rows preserve the original orientation R0° of their long axis along the [001] direction (−1.5 V, 160 pA); and (b) two representations of the same area with different contrast levels are shown to highlight the large concentration of defects on the uncovered surface under these preparation conditions (+0.6 V, 40 pA). | ||
Even if the flattened conformation of molecules on Ti5f rows was evident from STM, we performed a few calculations for an isolated flat molecule with alternative azimuthal orientations of 30°, 60° and 90°. In this case, we only considered relaxation of the molecular atoms, while keeping frozen the corresponding three-layer (2 × 6)-obl supporting slab. As mentioned in Section 5.2, when the molecule is located on the Obr rows, its Ti atom is directly coordinated to two Obr atoms underneath, and the molecular long axis is consistently predicted to be strictly parallel to the [001] direction. Any alternative azimuthal orientation was largely unfavoured by more than 1 eV, as summarized in Table 5.
| Molecule | Site | ΔEads (eV) | |
|---|---|---|---|
| TiO2br-TPP-deH | Obr bridge | 0° | 0.00 |
| 30° | 1.19 | ||
| 60° | 0.98 | ||
| 90° | 1.55 | ||
| TiOdown-TPP-deH | Ti5f top | 0° | 1.01 |
| 30° | 0.00 | ||
| 60° | 0.34 | ||
| 90° | 0.08 |
The energy landscape is reversed when we consider the adsorption on the Ti5f rows, corresponding to a TiOdown-TPP-deH configuration. In this case, the orientation parallel to the substrate atomic rows is the only one to be largely unfavoured, while the adsorption energies of the other orientations are found to be within 0.3 eV (see Table 5). The most stable configuration is predicted for the R30° orientation, where two phenyls at the opposite corners of the flat molecule lean down towards one Obr atom of adjacent rows, as shown in the drawings of Fig. 25. This orientation is in good agreement with measurements on TiO-TPP low coverage films (Fig. 23d), as well as for 2H-TPP, when annealed immediately after RT deposition. Under these experimental conditions, the large majority of the molecules are anyway adsorbed on Obr rows in the TiO2br-TPP-deH R0° configuration. The preferred R60° orientation observed on hydroxylated surfaces might be due to either interaction with the numerous defects (e.g. VOs) or the limitation of our simplified structural model. In any case, a simple DOS cut calculated for the R30° orientation reproduces also the fine details of the intramolecular contrast observed by STM on a molecule with the R60° orientation, as can be appreciated in Fig. 25.
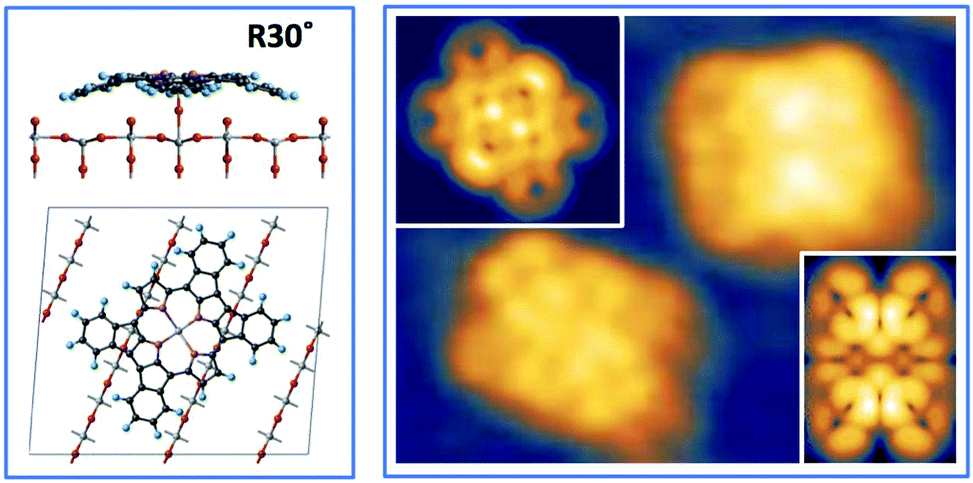 | ||
| Fig. 25 Left: Side and top views of the equilibrium configuration for the most favoured R30° orientation of the TiOdown-TPP-deH species atop a Ti5f atom. Right: Close-up STM image of the two main types of flat TPP-deH molecules, R60° and R0° on Ti5f and Obr rows, respectively (+0.6 V, 40 pA); the corresponding simulations (DOS cuts) are shown in the inset. | ||
The analysis of large scale images shows that other minority configurations are present (e.g. residual square R45° molecules on Ti5f rows, as well as some elongated and asymmetric ones located in between the Obr and Ti5f rows); however, almost all of the molecular species can be associated with a flat rectangular conformation. In this regard, at the end of Section 5.2 we suggested that this conformation would be favoured for the TiO2br-TPP-deH-R0° species by the spatial proximity of the phenyl H atoms to the Obr rows underneath. The experimental evidence for the TiOdown-TPP-deH molecules rotated by 30°/60° points to a more general rule, where the substrate anisotropy, however, dictates the merging direction of phenyls with the pyrrole units.
7 Summary and conclusions
We have shown that 2H-TPP and TiO-TPP molecules display a complex, but overlapping, phase diagram on the r-TiO2(110) surface, with each phase yielding different molecular configurations that may additionally interact with typical contaminants, like bulk diluted hydrogen, terminal hydroxyls OHt and H2O. We may thus recapitulate the main reactions as follows:(1) On a stoichiometric sample, 2H-TPP molecules anchor to the Obr rows in a bridge site through the hydrogen bonding of the pristine pyrrolic groups to the oxygen atoms underneath; the iminic nitrogen favours the outdiffusion of Ti interstitials, hence pyrrole dehydrogenation and self-metalation starting at ∼100 °C; further cyclo-dehydrogenation takes place at 350–400 °C, leading to a flat molecular conformation; the molecules do not change their adsorption site and orientation throughout the reactions, rather they form a commensurate (2 × 4)-obl phase at RT, which is preserved upon self-metalation, and transforms into a (2 × 6) phase upon flattening; the reaction path is:
| 2H-TPP → TiO2br-TPP → TiO2br-TPP-deH-R0° |
(2) 2H-TTP readily reacts with dissociated hydrogen at RT, yielding an acidic 4H-TPP molecule with the same adsorption site and symmetry phase; the self-metalation of 4H-TPP takes place at higher temperature (>150 °C) because the saturated iminic groups must first release their hydrogen atoms in order to extract and coordinate a substrate Ti atom; the self-metalated species then follow the reaction path (i):
| 2H-TPP → 4H-TPP → TiO2br-TPP → TiO2br-TPP-deH-R0° |
(3) Isolated TiO2br-TPP molecules (≤0.2 ML) may jump to adjacent Ti5f rows by capture of a diffusing oxygen atom, as provided by physisorbed H2O or terminal hydroxyls OHt; in the new configuration, the chelated Ti atom is doubly coordinated to an oxygen atom atop a Ti5f site and azimuthally rotated by 45°; this molecular species also undergoes flattening by partial cyclo-dehydrogenation at 350–400 °C, remaining atop the Ti5f atom, but rotated by 30° (or 60°); the reaction path following the capture of adsorbed oxygen by self-metalated molecules is:
| TiO2br-TPP → TiOdown-TPP-R45°→ TiOdown-TPP-deH-R30° |
(4) TiO-TPP molecules directly deposited on the surface mostly adsorb on Ti5f and Obr rows assuming the TiOdown-TPP-R45° and TiO2br-TPP conformations, respectively, while a minor fraction assumes other configurations; isolated molecules simply follow the corresponding reaction path, (iii) or (i), as outlined before; at higher density (≥0.2 ML), the interactions of phenyls between molecules entering into contact converts all the molecular species into the TiO2br-TPP conformation at 200–250 °C, which then follows the reaction path (i), including the formation of the (2 × 4)-obl and (2 × 6) phases:
| TiO-TPP → TiO2br-TPP → TiO2br-TPP-deH-R0°. |
These findings have some general implications and also shed light on the surface chemistry of TiO2, as well as on the processes involving molecules adsorbed thereon, as pointed out below.
The capture of hydrogen by iminic nitrogen takes place at RT up to a coverage of ∼1 ML and independently of the specific meso-substituents, provided molecules to adsorb in a flat configuration. These lines of evidence indicate that the major source of hydrogen is the diluted (and dissociated) one in the subsurface layers. The mechanism of H outdiffusion is expected to be equivalent to that of Ti extraction and incorporation. In the latter case the iminic nucleophilic attack lowers the energy barrier for Tiint outdiffusion by ∼0.8 eV and reverses the stability of the outdiffused Ti atom from an energy cost of ∼0.6 eV (endothermic)125 to an energy gain of ≥1.2 eV (exothermic). On a clean surface, the equilibrium energies for H above and below the surface are closely similar (±0.1 eV) and the barrier for H diffusion across the (110) plane is in the range of 0.8–1 eV.74,80 As a consequence, a decrease of the H outdiffusion barrier to hundred meV due to the iminic attack is quite reasonable in front of the large energy gain (1.4 eV) of hydrogen coordination to the macrocycle rather than on Obr atoms.
More generally, the mechanism of Ti sequestration outlined in Section 4.2.2 proves the direct involvement of Tiint in chemical reactions at the surface, rather than the simple exploitation of its surface induced excess of charge.126 This makes possible the physical participation of substrate Ti atoms in the synthesis/modification of molecular adsorbates, as first proposed for the Ullmann coupling reaction of polymer precursors.133
Focusing on the specific issue of porphyrin metalation, the low temperature onset of self-metalation practically precludes the route of hetero-metalation by in vacuum metal sublimation on a free-base porphyrin film. In fact, the latter protocol intrinsically provides an insufficient metalation yield at RT, which cannot be improved by post-sublimation annealing because it would result in a mixed composition layer of M-TPPs. As an alternative, free-base porphyrin metalation in solution,21 as well as metal-porphyrin transmetalation,97 provided much higher yields at RT. The in-vacuum chemical vapour deposition of metals from small gas precursors is also a promising route, but hitherto has not been explored for the metalation of free-base tetra-pyrrolic molecules on TiO2.
Along with the issue of unwanted side effects by film annealing on TiO2, one should also consider the possible metal exchange between metal-porphyrins and the r-TiO2(110) surface in front of the high reactivity of Ti interstitials. This issue was clearly outlined for pyrphyrin and porphyrin molecules on metal substrates, where the replacement of the central metal atom with a different one from pre-deposited metal clusters was found to follow the expected rank of metal oxidation activity (e.g. Fe chelation was more stable than Ni chelation, which in turn was more stable than Cu chelation).94,95 However, counterintuitive cases of metal exchange with substrate atoms were reported for other metal-organic tetrapyrroles on Cu(111), e.g. Ni-TPP,145 Pb-TPP,146 and Co-phthalocyanine.147 The situation is further complicated on r-TiO2(110) due to the mechanism of Ti extraction that cannot be directly generalized to metal-porphyrins, because the adsorption site of M-Ps depends on the specific chelated metal. Trans-metalation for M-Ps adsorbing on the Ti5f rows, like Ni-TPP,34 should be inhibited at least up to a temperature of 300–350 °C, where Tiint atoms may freely outdiffuse. Metal-porphyrins rather adsorbing on the Obr rows, like Zn-TPP,18,19,32 are also not expected to undergo trans-metalation at mild temperature because their iminic terminations already chelate a metal atom. In that case, the coordination to Obr atoms of the pristine porphyrin metal might, however, loosen the tetra-pyrrolic ligand network and favour the release of the metal atom by annealing. Also in view of the unexpected change in configuration observed for TiO-TPP in the ML range, the thermal stability of different metal-porphyrins on r-TiO2(110) is a relevant issue that needs to be settled for the correct functional modelling of any application oriented porphyrin/TiO2 interface.
An important aspect foreshadowing an alternative route to the design of porphyrin/TiO2 architectures for photovoltaic and photocatalytic applications is the extremely high temperature stability (450–500 °C) of the self-metalated molecules, as well as TiO-TPP ones. For practical purposes, porphyrins are normally functionalized with peripheral linkers in order to achieve a tight molecular anchoring to TiO2 substrates. On one hand, this facilitates deposition by wet chemistry methods; on the other hand, single/double linker porphyrins yield larger molecular densities but in the unfavourable standing-up orientation, while multiple linker porphyrins, although improving the charge injection efficiency by the lying down orientation, limit the density of active centres (by steric arguments).83 Conversely, the high temperature flattening of titanyl-porphyrin may be regarded as a perfect grafting of the r-TiO2(110) surface with compact and long range ordered aromatic tiles.
The evidence that the anchoring of titanyl-porphyrins (however formed) is robust against thermal treatment, even on the Ti5f rows, suggests that a similar reaction path and thermodynamic behaviour may be obtained on other crystalline orientations and polymorphs, as partly confirmed by the observation of TPP self-metalation on anatase TiO2 in the same temperature range of the rutile.115 The direct TPP anchoring to the substrate through the Ti⋯Obr coordination is reasonably expected to quench the molecular reactivity, hence limiting its direct participation in photo-conversion and/or catalysis. However, it does not hamper either the aromaticity of the molecule or the efficiency of charge transfer into the substrate. The (2 × 6) phases made of TiO2br-TPP-deH molecules are thus best suited to accommodate further layers of porphyrins oriented parallel to the surface and functionalized with meso-substituents maximizing the molecular density.
Author contributions
L. S.: investigation, data curation, writing – review & editing. D. F.: formal analysis, software, writing – review & editing. M. C.: formal analysis, supervision, writing – review & editing. A. G.: investigation, resources, supervision, writing – review & editing. C. R.: investigation, resources, supervision, writing – review & editing. A. V.: conceptualization, formal analysis, methodology, resources, supervision, writing – review & editing. L. F.: conceptualization, investigation, methodology, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. F. and C. R. are grateful to Dr Giacomo Lovat, Dr Mikel Abadia and Dr Marcos Dominguez-Rivera for support in data acquisition. A. V. is grateful to Markus K. Kremer for computational support. C. R. acknowledges the financial support through program MCIN/AEI/10.13039/501100011033 of Spain under grants PID2019-107338RB-C63 and PID2020-114252GB-I00. We acknowledge the CINECA award under the ISCRA initiative (code: HP10B7T83G) and the C3P computer facility at the University of Padova for the availability of high performance computing resources and support.References
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2002, 117, 205–219 CrossRef CAS.
- E. V. Shah, V. Kumar, B. K. Sharma, K. Rajput, V. P. Chaudhary and D. R. Roy, J. Mol. Model., 2018, 24, 239 CrossRef CAS PubMed.
- R. Büchner, M. Fondell, R. Haverkamp, A. Pietzsch, V. Vaz da Cruz and A. Föhlisch, Phys. Chem. Chem. Phys., 2021, 23, 24765–24772 RSC.
- S. Verma and H. N. Ghosh, J. Phys. Chem. Lett., 2012, 3, 1877–1884 CrossRef CAS PubMed.
- W.-J. Yin, B. Wen, C. Zhou, A. Selloni and L.-M. Liu, Surf. Sci. Rep., 2018, 73, 58–82 CrossRef CAS.
- A. G. Thomas, W. R. Flavell, A. K. Mallick, A. R. Kumarasinghe, D. Tsoutsou, N. Khan, C. Chatwin, S. Rayner, G. C. Smith, R. L. Stockbauer, S. Warren, T. K. Johal, S. Patel, D. Holland, A. Taleb and F. Wiame, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035105 CrossRef.
- D. V. Potapenko, N. J. Choi and R. M. Osgood, J. Phys. Chem. C, 2010, 114, 19419–19424 CrossRef CAS.
- V. Lanzilotto, C. Sanchez-Sanchez, G. Bavdek, D. Cvetko, M. F. Lopez, J. A. Martin-Gago and L. Floreano, J. Phys. Chem. C, 2011, 115, 4664–4672 CrossRef CAS.
- G. Otero-Irurueta, J. I. Martínez, G. Lovat, V. Lanzilotto, J. Méndez, M. F. López, L. Floreano and J. A. Martín-Gago, J. Phys. Chem. C, 2015, 119, 7809–7816 CrossRef CAS.
- S. Godlewski, A. Tekiel, W. Piskorz, F. Zasada, J. S. Prauzner-Bechcicki, Z. Sojka and M. Szymonski, ACS Nano, 2012, 6, 8536–8545 CrossRef CAS PubMed.
- S. Rangan, C. Ruggieri, R. Bartynski, J. I. Martínez, F. Flores and J. Ortega, J. Phys. Chem. B, 2018, 122, 534–542 CrossRef PubMed.
- V. Lanzilotto, G. Lovat, G. Otero, L. Sanchez, M. F. López, J. Méndez, J. A. Martín-Gago, G. Bavdek and L. Floreano, J. Phys. Chem. C, 2013, 117, 12639–12647 CrossRef CAS.
- C. Lun Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328–2353 RSC.
- R. Jöhr, A. Hinaut, R. Pawlak, A. Sadeghi, S. Saha, S. Goedecker, B. Such, M. Szymonski, E. Meyer and T. Glatzel, J. Chem. Phys., 2015, 143, 094202 CrossRef PubMed.
- Ł. Zajac, P. Olszowski, S. Godlewski, L. Bodek, B. Such, R. Jöhr, R. Pawlak, A. Hinaut, T. Glatzel, E. Meyer and M. Szymoński, Appl. Surf. Sci., 2016, 379, 277–281 CrossRef.
- R. Jöhr, A. Hinaut, R. Pawlak, Ł. Zajac, P. Olszowski, B. Such, T. Glatzel, J. Zhang, M. Muntwiler, J. J. Bergkamp, L. M. Mateo, S. Decurtins, S. X. Liu and E. Meyer, J. Chem. Phys., 2017, 146, 184704 CrossRef.
- R. Pawlak, A. Sadeghi, R. Jöhr, A. Hinaut, T. Meier, S. Kawai, Ł. Zajac, P. Olszowski, S. Godlewski, B. Such, T. Glatzel, S. Goedecker, M. Szymoński and E. Meyer, J. Phys. Chem. C, 2017, 121, 3607–3614 CrossRef CAS.
- D. Wechsler, C. C. Fernández, H. P. Steinrück, O. Lytken and F. J. Williams, J. Phys. Chem. C, 2018, 122, 4480–4487 CrossRef CAS.
- C. C. Fernández, D. Wechsler, T. C. Rocha, H. P. Steinrück, O. Lytken and F. J. Williams, Surf. Sci., 2019, 689, 121462 CrossRef.
- D. Wechsler, P. Vensaus, N. Tsud, H. P. Steinrück, O. Lytken and F. J. Williams, J. Phys. Chem. C, 2021, 125, 6708–6715 CrossRef CAS.
- C. C. Fernández, D. Wechsler, T. C. Rocha, H. P. Steinrück, O. Lytken and F. J. Williams, J. Phys. Chem. C, 2019, 123, 10974–10980 CrossRef.
- D. Wechsler, C. C. Fernández, J. Köbl, L. M. Augustin, C. Stumm, N. Jux, H. P. Steinrück, F. J. Williams and O. Lytken, Molecules, 2021, 26, 2871 CrossRef CAS PubMed.
- A. Orbelli Biroli, A. Calloni, A. Bossi, M. S. Jagadeesh, G. Albani, L. Duó, F. Ciccacci, A. Goldoni, A. Verdini, L. Schio, L. Floreano and G. Bussetti, Adv. Funct. Mater., 2021, 31, 2011008 CrossRef CAS.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H.-P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- Y. H. Chang, H. Kim, S.-J. Kahng and Y.-H. Kim, Dalton Trans., 2016, 45, 16673–16681 RSC.
- S. Carlotto, I. Cojocariu, V. Feyer, L. Floreano and M. Casarin, Nanomaterials, 2022, 12, 218 CrossRef CAS PubMed.
- S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880–3889 RSC.
- M. Lackinger, M. S. Janson and W. Ho, J. Chem. Phys., 2012, 137, 234707 CrossRef PubMed.
- S. Rangan, C. Ruggieri, R. Bartynski, J. I. Martínez, F. Flores and J. Ortega, J. Phys. Chem. C, 2016, 120, 4430–4437 CrossRef CAS PubMed.
- J. I. Martínez, F. Flores, J. Ortega, S. Rangan, C. M. Ruggieri and R. A. Bartynski, Phys. Chem. Chem. Phys., 2017, 19, 24412–24420 RSC.
- C. Wang, Q. Fan, S. Hu, H. Ju, X. Feng, Y. Han, H. Pan, J. Zhu and J. M. Gottfried, Chem. Commun., 2014, 50, 8291–8294 RSC.
- G. Fratesi, S. Achilli, A. Ugolotti, A. Lodesani, A. Picone, A. Brambilla, L. Floreano, A. Calloni and G. Bussetti, Appl. Surf. Sci., 2020, 530, 147085 CrossRef CAS.
- Y. Gurdal, S. Luber, J. Hutter and M. Iannuzzi, Phys. Chem. Chem. Phys., 2015, 17, 22846–22854 RSC.
- T. E. Shubina, H. Marbach, K. Flechtner, A. Kretschmann, N. Jux, F. Buchner, H.-P. Steinrück, T. Clark and J. M. Gottfried, J. Am. Chem. Soc., 2007, 129, 9476–9483 CrossRef CAS PubMed.
- R. González-Moreno, C. Sánchez-Sánchez, M. Trelka, R. Otero, A. Cossaro, A. Verdini, L. Floreano, M. Ruiz-Bermejo, A. García-Lekue, J. Á. Martín-Gago and C. Rogero, J. Phys. Chem. C, 2011, 115, 6849–6854 CrossRef.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS PubMed.
- K. Diller, A. C. Papageorgiou, F. Klappenberger, F. Allegretti, J. V. Barth and W. Auwärter, Chem. Soc. Rev., 2016, 45, 1629–1656 RSC.
- G. Di Santo, C. Castellarin-Cudia, M. Fanetti, B. Taleatu, P. Borghetti, L. Sangaletti, L. Floreano, E. Magnano, F. Bondino and A. Goldoni, J. Phys. Chem. C, 2011, 115, 4155–4162 CrossRef CAS.
- M. Röckert, M. Franke, Q. Tariq, S. Ditze, M. Stark, P. Uffinger, D. Wechsler, U. Singh, J. Xiao, H. Marbach, H. P. Steinrück and O. Lytken, Chem. – Eur. J., 2014, 20, 8948–8953 CrossRef.
- A. Wiengarten, J. A. Lloyd, K. Seufert, J. Reichert, W. Auwärter, R. Han, D. A. Duncan, F. Allegretti, S. Fischer, S. C. Oh, O. Saglam, L. Jiang, S. Vijayaraghavan, D. Écija, A. C. Papageorgiou and J. V. Barth, Chem. – Eur. J., 2015, 21, 12285–12290 CrossRef CAS PubMed.
- C. Ruggieri, S. Rangan, R. A. Bartynski and E. Galoppini, J. Phys. Chem. C, 2016, 120, 7575–7585 CrossRef CAS.
- G. Lovat, D. Forrer, M. Abadia, M. Dominguez, M. Casarin, C. Rogero, A. Vittadini and L. Floreano, Phys. Chem. Chem. Phys., 2015, 17, 30119–30124 RSC.
- J. Köbl, T. Wang, C. Wang, M. Drost, F. Tu, Q. Xu, H. Ju, D. Wechsler, M. Franke, H. Pan, H. Marbach, H. P. Steinrück, J. Zhu and O. Lytken, ChemistrySelect, 2016, 1, 6103–6105 CrossRef.
- G. Lovat, D. Forrer, M. Abadia, M. Dominguez, M. Casarin, C. Rogero, A. Vittadini and L. Floreano, J. Phys. Chem. C, 2017, 121, 13738–13746 CrossRef CAS.
- M. K. Kremer, D. Forrer, C. Rogero, L. Floreano and A. Vittadini, Appl. Surf. Sci., 2021, 564, 150403 CrossRef CAS.
- G. Lovat, D. Forrer, M. Abadia, M. Dominguez, M. Casarin, C. Rogero, A. Vittadini and L. Floreano, Nanoscale, 2017, 9, 11694–11704 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- G. Fratesi, V. Lanzilotto, S. Stranges, M. Alagia, G. P. Brivio and L. Floreano, Phys. Chem. Chem. Phys., 2014, 16, 14834–14844 RSC.
- L. Floreano, G. Naletto, D. Cvetko, R. Gotter, M. Malvezzi, L. Marassi, A. Morgante, A. Santaniello, A. Verdini, F. Tommasini and G. Tondello, Rev. Sci. Instrum., 1999, 70, 3855–3864 CrossRef CAS.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- T. Minato, Y. Sainoo, Y. Kim, H. S. Kato, K.-I. Aika, M. Kawai, J. Zhao, H. Petek, T. Huang, W. He, B. Wang, Z. Wang, Y. Zhao, J. Yang and J. G. Hou, J. Chem. Phys., 2009, 130, 124502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-laRoza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934–939 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- D. Forrer and A. Vittadini, Chem. Phys. Lett., 2011, 516, 72–75 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed.
- B. Himmetoglu, A. Floris, S. de Gironcoli and M. Cococcioni, Int. J. Quantum Chem., 2014, 114, 14–49 CrossRef CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J.-A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E.-v Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- S. P. Jarvis, S. Taylor, J. D. Baran, D. Thompson, A. Saywell, B. Mangham, N. R. Champness, J. A. Larsson and P. Moriarty, J. Phys. Chem. C, 2015, 119, 27982–27994 CrossRef CAS.
- P. T.-P. Ryan, P. L. Lalaguna, F. Haag, M. M. Braim, P. Ding, D. J. Payne, J. V. Barth, T.-L. Lee, D. P. Woodruff, F. Allegretti and D. A. Duncan, Chem. Commun., 2020, 56, 3681–3684 RSC.
- M. Lepper, J. Köbl, T. Schmitt, M. Gurrath, A. de Siervo, M. A. Schneider, H.-P. Steinrück, B. Meyer, H. Marbach and W. Hieringer, Chem. Commun., 2017, 53, 8207–8210 RSC.
- J. C. Moreno-López, D. J. Mowbray, A. Pérez Paz, R. C. de Campos Ferreira, A. Ceccatto dos Santos, P. Ayala and A. de Siervo, Chem. Mater., 2019, 31, 3009–3017 CrossRef.
- H. Nagatani and H. Watarai, Anal. Chem., 1996, 68, 1250–1253 CrossRef CAS PubMed.
- D.-M. Chen, X. Liu, T.-J. He and F.-C. Liu, Chem. Phys., 2003, 289, 397–407 CrossRef CAS.
- Y. Du, N. G. Petrik, N. A. Deskins, Z. Wang, M. A. Henderson, G. A. Kimmel and I. Lyubinetsky, Phys. Chem. Chem. Phys., 2012, 14, 3066–3074 RSC.
- L. E. Walle, A. Borg, P. Uvdal and A. Sandell, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 1–5 CrossRef.
- O. W. Johnson, W. D. Ohlsen and P. I. Kingsbury, Phys. Rev., 1968, 175, 1102–1109 CrossRef CAS.
- L. B. Mo, Y. Wang, Y. Bai, Q. Y. Xiang, Q. Li, W. Q. Yao, J. O. Wang, K. Ibrahim, H. H. Wang, C. H. Wan and J. L. Cao, Sci. Rep., 2015, 5, 17634 CrossRef CAS PubMed.
- F. Hibbert and K. P.-P. Hunte, J. Chem. Soc., Perkin Trans. 2, 1977, 1624–1628 RSC.
- H. Yamashige, S. Matsuo, T. Kurisaki, R. C. Perera and H. Wakita, Anal. Sci., 2005, 21, 635–639 CrossRef CAS PubMed.
- B. Wei and M. Calatayud, Top. Catal., 2022, 65, 270–280 CrossRef CAS.
- M. I. Nandasiri, V. Shutthanandan, S. Manandhar, A. M. Schwarz, L. Oxenford, J. V. Kennedy, S. Thevuthasan and M. A. Henderson, J. Phys. Chem. Lett., 2015, 6, 4627–4632 CrossRef CAS PubMed.
- O. W. Johnson, J. DeFord and J. W. Shaner, J. Appl. Phys., 1973, 44, 3008–3012 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- G. Granados-Oliveros, E. A. Páez-Mozo, F. M. Ortega, C. Ferronato and J.-M. Chovelon, Appl. Catal., B, 2009, 89, 448–454 CrossRef CAS.
- G. Granados-Oliveros, E. A. Páez-Mozo, F. Martínez Ortega, M. Piccinato, F. N. Silva, C. L.-B. Guedes, E. Di Mauro, M. F.-D. Costa and A. T. Ota, J. Mol. Catal. A: Chem., 2011, 339, 79–85 CrossRef CAS.
- E. Mansour, P. Maillard, P. Krausz, S. Gaspard and C. Giannotti, J. Mol. Catal., 1987, 41, 361–366 CrossRef CAS.
- D. A. Duncan, P. S. Deimel, A. Wiengarten, R. Han, R. G. Acres, W. Auwärter, P. Feulner, A. C. Papageorgiou, F. Allegretti and J. V. Barth, Chem. Commun., 2015, 51, 9483–9486 RSC.
- C. Wäckerlin, K. Tarafder, D. Siewert, J. Girovsky, T. Hählen, C. Iacovita, A. Kleibert, F. Nolting, T. A. Jung, P. M. Oppeneer and N. Ballav, Chem. Sci., 2012, 3, 3154–3160 RSC.
- C. Krull, R. Robles, A. Mugarza and P. Gambardella, Nat. Mater., 2013, 12, 337–343 CrossRef CAS PubMed.
- H. M. Sturmeit, I. Cojocariu, A. Windischbacher, P. Puschnig, C. Piamonteze, M. Jugovac, A. Sala, C. Africh, G. Comelli, A. Cossaro, A. Verdini, L. Floreano, M. Stredansky, E. Vesselli, C. Hohner, M. Kettner, J. Libuda, C. M. Schneider, G. Zamborlini, M. Cinchetti and V. Feyer, Small, 2021, 17, 2104779 CrossRef CAS PubMed.
- J. M. Gottfried, K. Flechtner, A. Kretschmann, T. Lukasczyk and H.-P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644–5645 CrossRef CAS PubMed.
- W. Auwärter, A. Weber-Bargioni, S. Brink, A. Riemann, A. Schiffrin, M. Ruben and J. V. Barth, Chem. Phys. Chem., 2007, 8, 250–254 CrossRef PubMed.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- A. Rieger, S. Schnidrig, B. Probst, K. H. Ernst and C. Wäckerlin, J. Phys. Chem. Lett., 2017, 8, 6193–6198 CrossRef CAS PubMed.
- D. Hötger, P. Abufager, C. Morchutt, P. Alexa, D. Grumelli, J. Dreiser, S. Stepanow, P. Gambardella, H. F. Busnengo, M. Etzkorn, R. Gutzler and K. Kern, Nanoscale, 2018, 10, 21116–21122 RSC.
- C. Wang, Q. Fan, Y. Han, J. I. Martínez, J. A. Martín-Gago, W. Wang, H. Ju, J. M. Gottfried and J. Zhu, Nanoscale, 2016, 8, 1123–1132 RSC.
- M. Franke, F. Marchini, N. Jux, H. P. Steinrück, O. Lytken and F. J. Williams, Chem. – Eur. J., 2016, 22, 8520–8524 CrossRef CAS PubMed.
- F. Buchner, V. Schwald, K. Comanici, H.-P. Steinrück and H. Marbach, ChemPhysChem, 2007, 8, 241–243 CrossRef CAS PubMed.
- P. Krüger, S. Bourgeois, B. Domenichini, H. Magnan, D. Chandesris, P. Le Févre, A. M. Flank, J. Jupille, L. Floreano, A. Cossaro, A. Verdini and A. Morgante, Phys. Rev. Lett., 2008, 100, 055501 CrossRef PubMed.
- M. Nolan, S. D. Elliott, J. S. Mulley, R. A. Bennett, M. Basham and P. Mulheran, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235424 CrossRef.
- A. Goldoni, C. A. Pignedoli, G. Di Santo, C. Castellarin-Cudia, E. Magnano, F. Bondino, A. Verdini and D. Passerone, ACS Nano, 2012, 6, 10800–10807 CrossRef CAS PubMed.
- J. Mielke, F. Hanke, M. V. Peters, S. Hecht, M. Persson and L. Grill, J. Am. Chem. Soc., 2015, 137, 1844–1849 CrossRef CAS PubMed.
- NIST Chemistry WebBook, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg (MD), 2022 Search PubMed.
- S. Ditze, M. Stark, M. Drost, F. Buchner, H.-P. Steinrück and H. Marbach, Angew. Chem., Int. Ed., 2012, 51, 10898–10901 CrossRef CAS PubMed.
- M. Röckert, M. Franke, Q. Tariq, D. Lungerich, N. Jux, M. Stark, A. Kaftan, S. Ditze, H. Marbach, M. Laurin, J. Libuda, H. P. Steinrück and O. Lytken, J. Phys. Chem. C, 2014, 118, 26729–26736 CrossRef.
- A. Verdini, P. Shinde, G. L. Montanari, S. T. Suran-Brunelli, M. Caputo, G. Di Santo, C. A. Pignedoli, L. Floreano, D. Passerone and A. Goldoni, Chem. – Eur. J., 2016, 22, 14672–14677 CrossRef CAS PubMed.
- J. Nowakowski, C. Wäckerlin, J. Girovsky, D. Siewert, T. A. Jung and N. Ballav, Chem. Commun., 2013, 49, 2347–2349 RSC.
- F. Kollhoff, J. Schneider, G. Li, S. Barkaoui, W. Shen, T. Berger, O. Diwald and J. Libuda, Phys. Chem. Chem. Phys., 2018, 20, 24858–24868 RSC.
- D. Wechsler, C. C. Fernández, Q. Tariq, N. Tsud, K. C. Prince, F. J. Williams, H. Steinrück and O. Lytken, Chem. – Eur. J., 2019, 25, 13197–13201 CrossRef CAS PubMed.
- C. Wang, R. Wang, J. Hauns and T. Fauster, J. Phys. Chem. C, 2020, 124, 14167–14175 CrossRef CAS.
- R. González-Moreno, P. L. Cook, I. Zegkinoglou, X. Liu, P. S. Johnson, W. Yang, R. E. Ruther, R. J. Hamers, R. Tena-Zaera, F. J. Himpsel, J. E. Ortega and C. Rogero, J. Phys. Chem. C, 2011, 115, 18195–18201 CrossRef.
- J. Schneider, F. Kollhoff, J. Bernardi, A. Kaftan, J. Libuda, T. Berger, M. Laurin and O. Diwald, ACS Appl. Mater. Interfaces, 2015, 7, 22962–22969 CrossRef CAS PubMed.
- J. Schneider, M. Franke, M. Gurrath, M. Röckert, T. Berger, J. Bernardi, B. Meyer, H.-P. Steinrück, O. Lytken and O. Diwald, Chem. – Eur. J., 2016, 22, 1744–1749 CrossRef CAS PubMed.
- L. Egger, M. Hollerer, C. S. Kern, H. Herrmann, P. Hurdax, A. Haags, X. Yang, A. Gottwald, M. Richter, S. Soubatch, F. S. Tautz, G. Koller, P. Puschnig, M. G. Ramsey and M. Sterrer, Angew. Chem., Int. Ed., 2021, 60, 5078–5082 CrossRef CAS PubMed.
- J. Schneider, T. Berger and O. Diwald, ACS Appl. Mater. Interfaces, 2018, 10, 16836–16842 CrossRef CAS PubMed.
- X. L. Yin, M. Calatayud, H. Qiu, Y. Wang, A. Birkner, C. Minot and C. Wöll, ChemPhysChem, 2008, 9, 253–256 CrossRef CAS PubMed.
- C. B. Honsberg and S. G. Bowden, Photovoltaics Education Website, 2019, https://www.pveducation.org.
- J. Li, R. Lazzari, S. Chenot and J. Jupille, Phys. Rev. B, 2018, 97, 041403 CrossRef CAS.
- P. Krüger, J. Jupille, S. Bourgeois, B. Domenichini, A. Verdini, L. Floreano and A. Morgante, Phys. Rev. Lett., 2012, 108, 126803 CrossRef PubMed.
- N. A. Deskins, R. Rousseau and M. Dupuis, J. Phys. Chem. C, 2009, 113, 14583–14586 CrossRef CAS.
- P. M. Kowalski, M. F. Camellone, N. N. Nair, B. Meyer and D. Marx, Phys. Rev. Lett., 2010, 105, 146405 CrossRef PubMed.
- K. Morita, T. Shibuya and K. Yasuoka, J. Phys. Chem. C, 2017, 121, 1602–1607 CrossRef CAS.
- S. Chrétien and H. Metiu, J. Phys. Chem. C, 2011, 115, 4696–4705 CrossRef.
- H. Li, Y. Guo and J. Robertson, J. Phys. Chem. C, 2015, 119, 18160–18166 CrossRef CAS.
- P. A. Mulheran, M. Nolan, C. S. Browne, M. Basham, E. Sanville and R. A. Bennett, Phys. Chem. Chem. Phys., 2010, 12, 9763–9771 RSC.
- S. Wendt, P. T. Sprunger, E. Lira, G. K.-H. Madsen, Z. Li, J. O. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Laegsgaard, B. Hammer and F. Besenbacher, Science, 2008, 320, 1755–1759 CrossRef CAS PubMed.
- P. Stone, R. A. Bennett and M. Bowker, New J. Phys., 1999, 1, 008 CrossRef.
- R. D. Smith, R. A. Bennett and M. Bowker, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 035409 CrossRef.
- M. Bowker and R. A. Bennett, J. Phys.: Condens. Matter, 2009, 21, 474224 CrossRef PubMed.
- L. Benz, J. Haubrich, R. G. Quiller, S. C. Jensen and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 15026–15031 CrossRef CAS PubMed.
- L. Benz, J. Haubrich, S. C. Jensen and C. M. Friend, ACS Nano, 2011, 5, 834–843 CrossRef CAS PubMed.
- V. Lanzilotto, G. Lovat, G. Fratesi, G. Bavdek, G. P. Brivio and L. Floreano, J. Phys. Chem. Lett., 2015, 6, 308–313 CrossRef CAS PubMed.
- M. Abadia, G. Vasseur, M. Kolmer, L. Zajac, A. Verdini, J. E. Ortega, L. Floreano, C. Rogero and J. Brede, J. Phys. Chem. C, 2020, 124, 16918–16925 CrossRef CAS.
- Z. Hu and H. Metiu, J. Phys. Chem. C, 2011, 115, 5841–5845 CrossRef CAS.
- S. Lutfalla, V. Shapovalov and A. T. Bell, J. Chem. Theory Comput., 2011, 7, 2218–2223 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- D. Wechsler, P. Vensaus, N. Tsud, H.-P. Steinrück, O. Lytken and F. J. Williams, J. Phys. Chem. C, 2021, 125, 6708–6715 CrossRef CAS.
- J. Xiao, S. Ditze, M. Chen, F. Buchner, M. Stark, M. Drost, H. P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2012, 116, 12275–12282 CrossRef CAS.
- G. Di Santo, C. Sfiligoj, C. Castellarin-Cudia, A. Verdini, A. Cossaro, A. Morgante, L. Floreano and A. Goldoni, Chem. – Eur. J., 2012, 18, 12619–12623 CrossRef CAS PubMed.
- Y. Du, Z. Dohnálek and I. Lyubinetsky, J. Phys. Chem. C, 2008, 112, 2649–2653 CrossRef CAS.
- Y. Du, N. A. Deskins, Z. Zhang, Z. Dohnálek, M. Dupuis and I. Lyubinetsky, Phys. Rev. Lett., 2009, 102, 096102 CrossRef CAS PubMed.
- Y. Du, N. A. Deskins, Z. Zhang, Z. Dohnálek, M. Dupuis and I. Lyubinetsky, J. Phys. Chem. C, 2009, 113, 666–671 CrossRef CAS.
- Z.-T. Wang, Y.-G. Wang, R. Mu, Y. Yoon, A. Dahal, G. K. Schenter, V.-A. Glezakou, R. Rousseau, I. Lyubinetsky and Z. Dohnálek, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1801–1805 CrossRef CAS PubMed.
- U. Diebold, J. Chem. Phys., 2017, 147, 040901 CrossRef PubMed.
- C. M. Doyle, J. P. Cunniffe, S. A. Krasnikov, A. B. Preobrajenski, Z. Li, N. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2014, 50, 3447–3449 RSC.
- J. Herritsch, S. R. Kachel, Q. Fan, M. Hutter, L. J. Heuplick, F. Münster and J. M. Gottfried, Nanoscale, 2021, 13, 13241–13248 RSC.
- K. Shen, B. Narsu, G. Ji, H. Sun, J. Hu, Z. Liang, X. Gao, H. Li, Z. Li, B. Song, Z. Jiang, H. Huang, J. W. Wells and F. Song, RSC Adv., 2017, 7, 13827–13835 RSC.
Footnotes |
| † This tilt value must be regarded as a qualitative indication of the out of plane distortion of the macrocycle, because the orbital rehybridization, due to iminic hydrogenation and pyrrolic bonding to oxygen, likely breaks the π-symmetry, thus hampering the NEXAFS dichroism analysis. |
| ‡ Present data are calculated with small core pseudopotentials, whereas values reported in Fig. 4 of ref. 45 are those obtained using large core pseudopotentials. |
| § Note that the N K-edge spectra in ref. 49 are affected by an erroneous graphical offset of 2.8 eV towards higher energy. |
| This journal is © the Owner Societies 2022 |