
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights of molecular metal polyselenides for catalytic hydrogen generation†
Alexander
Elliott‡
 a,
James
McAllister‡
a,
Liudvika
Masaityte
a,
Mireia
Segado-Centellas
b,
De-Liang
Long
a,
Alexey Y.
Ganin
a,
Yu-Fei
Song
*c,
Carles
Bo
*b and
Haralampos N.
Miras
*a
a,
James
McAllister‡
a,
Liudvika
Masaityte
a,
Mireia
Segado-Centellas
b,
De-Liang
Long
a,
Alexey Y.
Ganin
a,
Yu-Fei
Song
*c,
Carles
Bo
*b and
Haralampos N.
Miras
*a
aSchool of Chemistry, The University of Glasgow, Glasgow G12 8QQ, UK. E-mail: charalampos.moiras@glasgow.ac.uk
bInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avgda. Països Catalans 16, 43007 Tarragona, Spain. E-mail: cbo@iciq.cat
cState Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: songyf@mail.buct.edu.cn
First published on 26th May 2022
Abstract
Molecular metal chalcogenides have attracted great attention as electrocatalysts for the hydrogen evolution reaction (HER). However, efficient utilisation of the active sites and catalytic performance modulation has been challenging. Here we explore the design of immobilized molecular molybdenum polyselenides [Mo2O2S2(Se2)(Sex)]2− that exhibit efficient hydrogen evolution at low overpotential and stability over 1000 cycles. Density functional calculations provide evidence of a unimolecular mechanism in the HER process via the exploration of viable reaction pathways. The discussed findings are of a broad interest in the development of efficient molecular electrocatalytic materials.
Development of catalysts for the hydrogen evolution reaction (HER) is one of the key challenges in the realisation of a hydrogen economy based on electrocatalytic water splitting. The best performing HER catalyst is platinum, however its high cost and low natural abundance makes the development of noble-metal free catalysts an important area of research.1 A variety of transition metal chalcogenides (MEx) (M = Co, Fe, Ni, V, Nb, Ta, Mo, W; E = S, Se, Te) have been explored2–9 in a quest for discovery of low production cost catalysts with high catalytic activity, and stability under aqueous corrosive conditions. Despite the extensive research conducted in this area, even the well explored chalcogenide, MoS29–11 significantly underperforms as electrocatalyst for HER compared to Pt. This is due to the fact that most of the activity in MoS2 comes from the edge sites which represent only a small fraction of the overall number of active sites in solid state materials like MoS2. Interestingly, molecular oxochalcogenides can incorporate by design a significant number of functional groups (which mimic very effectively the edge of solid-state chalcogenides) and have been shown to be at least as stable and effective as solid-state analogues. For example, the [MoV2(S2)6]2− cluster contains three disulphides per molybdenum,12 while the hybrid family of LMoVIO(S2)2 (where L = bidentate ligand) type catalysts exhibited an indicative cooperative electronic effect between the inorganic and organic components.13–15 When immobilised on an appropriate surface, they can effectively act as heterogeneous catalysts. Additionally, the design of the catalyst's key components (e.g. metal centres, type and number of chalcogens) can be pre-determined using a precise molecular chemistry approach. The all-inorganic molybdenum- and tungsten-based sulphide catalysts has shown overpotentials as low as −114 ± 3 mV at 10 mA cm−2.16 The presence of well-defined molecular structures rendered the computational modelling more informative and reinforced our understanding of the HER mechanism. Theoretical calculations demonstrated that the reduction occurs on the disulphide ligand, while the terminal oxo ligand stabilise the negative charge developed during the catalytic process. Interestingly, investigation of other molecular chalcoxide equivalents17–20 for the HER and exploration of the edge based catalytic sites has not previously been attempted.
In this work, we investigated the effects of stoichiometric and structural control resulting from the appropriate combination of chalcogen elements and their ratio (e.g. O2−/Se22−) within the coordination sphere of earth abundant transition metals and compare their mechanistic differences with their sulfur analogues.
The dimeric nanocluster, [Mo2O2(μ-Se)2(Se2)(Se2)]2−1, was synthesised following a facile method under mild conditions. The synthetic approach exploits the selenophilicity of molybdenum by exposing an alkaline molybdate solution to selenizing conditions. Alternatively, at elevated temperature and pressure regimes, the equilibrium shifts towards the formation of a mixture of species with the general formulae [Mo2O2(μ-Se)2(Se2)(Sex)]2− (where x = 2–4) 2. During the reaction the species [Mo2O2(μ-Se)2(Se2)(Se2)]2−, [Mo2O2(μ-Se)2(Se2)(Se3)]2− and [Mo2O2(μ-Se)2(Se2)(Se4)]2−, seem to be in an equilibrium under harsh experimental conditions and found to co-crystallise within the same unit cell. Each method yielded orange-red crystals which were characterized by single crystal and powder X-ray structural analysis, FT-IR, 77Se NMR and high-resolution electrospray ionization mass spectroscopy (ESI-MS). Subsequently, the [Mo2O2(μ-Se)2(Se2)2]2−1 species was investigated as potential electrocatalyst to produce H2 gas from acidic media where was found to exhibit excellent activity towards the hydrogen evolution reaction (HER). The oxoselenometalate compounds were synthesized under nitrogen atmosphere, following an optimised facile two-step process (see ESI†) utilizing low-cost materials, thus illustrating their potential for economically viable scalability.
The facile two-step synthetic procedure yields orange-red crystals and their single crystal X-ray diffraction (XRD) analysis shows that [(CH3)4N]2[Mo2O2(μ-Se)2(Se2)2] 1 crystallises in the orthorhombic system space group Pca21 with unit cell dimensions of a = 18.614(10), b = 11.422(6), c = 11.422(6) Å. The powder X-ray diffraction study conducted on the freshly synthesised material 1 (Fig. S5, ESI†) further confirm the purity of the synthesised material in the hexa-selenide form. Alternatively, the more challenging high temperature and pressure synthesis gives crystals containing a mixture of seleno-containing species with the formula [(CH3)4N]2[Mo2O2(μ-Se)2(Se2)2]0.25[Mo2O2(μ-Se)2(Se2)(Se3)]0.25[Mo2O2(μ-Se)2(Se2) (Se4)]0.52 which crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with unit cell dimensions of a = 10.098(4), b = 12.766(5), c = 18.713(7) Å, (α =78.432(4)°, β = 80.140(4)° and γ = 86.939(4)°). Fig. 1 displays the crystal structures of the [Mo2O2(μ-Se)2(Se2)2]2−1 and the [Mo2O2(μ-Se)2(Se2)(Se4)]2− anion found in 2 respectively. The [Mo2O2(μ-Se)2(Se2)2]2− and [Mo2O2(μ-Se)2(Se2)(Se4)]2− anions are constructed by two molybdenum centres that are mutually linked by two μ-Se2− bridges, and a MoV–MoV separation of 2.885(1) and 2.884(2) Å. In addition, the coordination sphere of each molybdenum centre is completed by a terminal oxo group of Mo
with unit cell dimensions of a = 10.098(4), b = 12.766(5), c = 18.713(7) Å, (α =78.432(4)°, β = 80.140(4)° and γ = 86.939(4)°). Fig. 1 displays the crystal structures of the [Mo2O2(μ-Se)2(Se2)2]2−1 and the [Mo2O2(μ-Se)2(Se2)(Se4)]2− anion found in 2 respectively. The [Mo2O2(μ-Se)2(Se2)2]2− and [Mo2O2(μ-Se)2(Se2)(Se4)]2− anions are constructed by two molybdenum centres that are mutually linked by two μ-Se2− bridges, and a MoV–MoV separation of 2.885(1) and 2.884(2) Å. In addition, the coordination sphere of each molybdenum centre is completed by a terminal oxo group of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distances 1.687(8) and 1.68(1) Å and two selenide bridging atoms with an average Mo–(μ-Se) bond distances of ca. 2.447 and 2.461 Å, respectively.
O bond distances 1.687(8) and 1.68(1) Å and two selenide bridging atoms with an average Mo–(μ-Se) bond distances of ca. 2.447 and 2.461 Å, respectively.
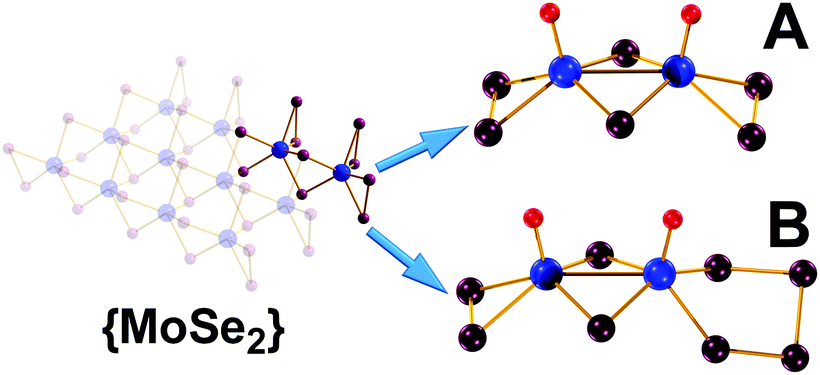 | ||
| Fig. 1 Ball-and-stick representation of: (A) [Mo2O2(μ-Se)2 (Se2)2]2− and (B) [Mo2O2(μ-Se)2(Se2)(Se4)]2−. Colour code: Mo, Blue; Se, plum; O, red; Counterions are omitted for clarity. | ||
Finally, in the case of the [Mo2O2(μ-Se)2(Se2)2]2− anion the coordination sphere of Mo is completed by two terminal diselenide (Se22−) groups with an average Se–Se bond distance of ca. 2.323 Å, while in the case of [Mo2O2(μ-Se)2(Se2)(Se4)]2− that is by a diselenide (Se22−) and a tetraselenide (Se42−) group with average Se–Se bond distance of ca. 2.333 Å. The overall charge of the compound is compensated by two tetramethyl-ammonium, (CH3)4N+, cations per dimeric cluster.
In an effort to further characterize the isolated compounds, we employed high resolution electrospray ionization mass spectrometry (ESI-MS)21–23 to determine the composition of the oxoselenometalate species in solution. The ESI-MS studies were performed in CH3CN. A series of singly charged distribution envelopes were observed, all in the region of 500–1000 m/z (Fig. S4, ESI†). More specifically, the distribution envelopes centred at 699.18 m/z value can be assigned to the intact compound [Mo2O2(μ-Se)2(Se2)2]2−. Additionally, 77Se NMR spectrum of 1 recorded in DMF. The spectrum shows two distinct peaks corresponding to the two unique environments in the dimer located at shifts of 134 and 711 ppm relative to Me2Se (Fig. S6, ESI†).
To investigate the HER activity of the molecular catalyst we immobilised the catalyst on a glassy carbon electrode (GCE) support (see ESI†).
The amount of catalyst on the electrode surface was controlled by adjusting the loading concentration and volume. The performance of the catalyst improves as a function of the catalyst loading up to the value of 3.38 μmol cm−2. Beyond this value the quality and mechanical stability of the produced electrocatalytic film degrades which leads either to similar or inferior performance due to detachment of the film from the electrode's surface. The results reported in Fig. 2 indicate that the cluster is highly active towards HER revealing a very small onset (40 mV) and low overpotential of −165 ± 3 mV (at j = 10 mA cm−2) as a function of the catalyst loading. Consequently, further increase of the applied overpotential results in a rapid rise of the cathodic current, which is indicative of HER activity, in marked contrast to the bare GCE with carbon powder.
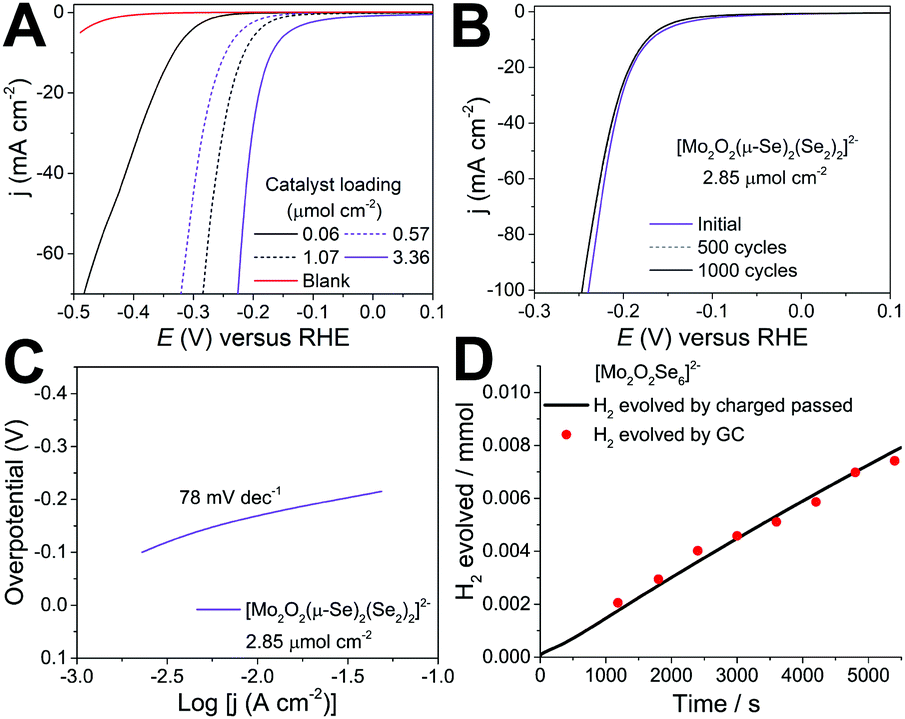 | ||
| Fig. 2 Electrochemical measurements of the molecular chalcoxide for HER electrocatalysis in 1 M H2SO4. (A) Observed polarisation curves as a function of the concentration of the catalyst; (B) Stability of the catalyst after 1000 cycles; (C) Corresponding Tafel plot and (D) Hydrogen evolved over time determined by charge passed (black line) and gas chromatography (red squares) (Me4N)2[Mo2O2(μ-Se)2(Se2)2] deposited on glassy carbon electrode. | ||
An important prerequisite of functional components used for large scale applications e.g. electrochemical water-splitting devices is the durability of the materials used. To assess this, we employed potential cycling as a simulation of the start-up/shutdown conditions found in an electrolyser to provide us with information on the catalysts’ long-term stability. Using catalyst 1 (3.38 μmol cm−2), we applied an accelerated scan rate of 100 mV s−1 between a potential range of 0.10 to −0.25 V (vs RHE) for 1000 cycles, Fig. 2b. At the end of the cycling, the catalyst showed negligible loss in cathodic current density, reaching an impressive density of j > 100 mA cm−2 below 250 mV. These results attest to the electrochemical stability of [Mo2O2(μ-Se)2(Se2)2]2−1 and its potential use in electrochemical HER devices. Finally, to confirm that the reaction taking place on the working electrode corresponds to the hydrogen evolution reaction (HER), the evolved gas was analysed using gas chromatography (Fig. S8, ESI†). As such, the faradaic efficiency of the molecular catalyst in 1 M H2SO4 was determined to be 102.3% ± 3.0 with a TOF of 0.019 s−1 at −190 mV demonstrating the evolution of hydrogen at full faradaic efficiency.
To investigate the underlying processes and reaction pathways, the electronic structure of these molecules and their reduced analogues were studied on the basis of density functional theory (DFT). The geometries of both anions were optimised starting from the single crystal X-ray diffraction data (see ESI†). The electronic structure of these anions is similar and according to orbital analysis and charge distribution, both ligands – bidentate κ1,4-Se42− and side on η2-Se22− – formally contribute −2 charges each. Mo centres present +5 formal oxidation state with a Mo–Mo bond (2.855 Å) as corroborated by the bond valence sum calculations analyses and the crystal structure. Each perselenide moiety has a doubly filled π*2π*2 group molecular orbital (MO) pair.
Examination of the HOMO–LUMO gap provides a preliminary good estimation of the chemical potential of the species and indication of orbitals populated by the incoming electrons. 0 e− and 1 e− reduction of catalytic species are depicted in Fig. S9 (ESI†). In case of 0 e−, the energy gap is higher in [Mo2O2(μ-Se)2(Se2)2]2− than [Mo2O2(μ-Se)2(Se2)(Se4)]2− and follows the same trend as sulphur analogues of these compounds. However, the HOMO–LUMO gap and LUMO energy are smaller in selenium analogues compared to their respective sulphur anions (Fig. S9 and S10, ESI†). The first unoccupied MOs involve d metallic orbitals and a σ-antibonding Se–Se orbital. Interestingly, in case of 1 e− reduced species, the energy needed to insert the second electron is higher for Se compounds than S analogues (Fig. S9 and S10, ESI†). The half occupied orbital present Sigma Se–Se antibonding character. In order to investigate the electrochemical steps that are taking place it is important to identify the different pathways following the two-fold reduction of the molecular catalysts leading to protonation equilibria. Therefore, the stability of all possible isomeric forms was evaluated. Even though those unprotonated and short-lived species may not be detected and characterised in solution, evaluating their relevant stability is relevant as it can serve as a reference state for subsequent steps. Considering the symmetric structure of the [Mo2O2(μ-Se)2(Se2)2]2− complex, we explored a series of isomers that can be formed during the two-variable proton/electron addition leading to the global panorama of possible HER equilibria (Fig. 3). The cleavage of perselenide bond upon 1 e− and 2 e− reduction was observed. It is expected based on molecular orbital analysis, because these anions feature σ-antibonding Se–Se orbitals which might in principle be occupied by incoming electrons inducing the breaking of Se–Se bond. This behaviour was also observed in the sulphur analogues of these anions. The complete nomenclature of the obtained isomers and proton affinities are listed in the Fig. S12–S19 (ESI†). For compound 1, the most stable doubly reduced and single-protonated species is B1 since it stabilises the mitigation of charge developed in the newly formed diselenol species. Consequently, the second protonation step which derives from B1 is BB1, which exhibits a free energy value of −40.7 kcal mol−1 (see Fig. S12, ESI†). Taking into consideration the proximity of the neighbouring hydrogen atoms in this structure, that raised the possibility of the existence of a Tafel-type re-combination step. Further examination of this step for both anions revealed activation energies of 62 kcal mol−1 from double-protonated BB1 (Fig. S21 and S26, ESI†) which are too high to be operative at room temperature. To this extend, departing from BA1 whereas one selenium and one oxygen atom are protonated, Taffel reaction activation energy is more favourable with an activation energy of 37.45 kcal mol−1 (Fig. S22, ESI†). In a similar manner, we aimed to examine the Heyrovsky step departing from single- and double-protonated [H2Mo2O2(μ-Se)2(Se2)(Sex)]2− (x = 2, 4) structures. This step is thermodynamically favourable across the range of molecular systems with calculated released energies within −46.12 to −4.51 kcal mol−1 (Fig. 4 and Fig. S20, S23–S25, ESI†). The activation energy for this step is in range of +2.54 to +18.62 kcal mol−1 for both anions and all reactant complexes taken into consideration. These Ea values indicate that activation can happen at room temperature conditions. For compound 1, BB1 intermediate [H2Mo2O2(μ-Se)2(Se2)2]2−, the obtained transition state has a relative free energy value ΔG‡ = +18.62 kcal mol−1 (Fig. S20, ESI†) which is comparable to the activation energy value obtained for compound 2 when the protons interact with the side on η2-Se22− terminal group (BB1, Fig. S24, ESI,† ΔG‡ = +16.52 kcal mol−1). Interestingly, when the interaction takes place on the bidentate κ1,4-Se42− terminal site the calculated activation energy and transition state for [H2Mo2O2(μ-Se)2(Se2)(Se4)]2− (BB3) was found to be lower (ΔG‡ = +14.4 kcal mol−1 (Fig. S25, ESI†).
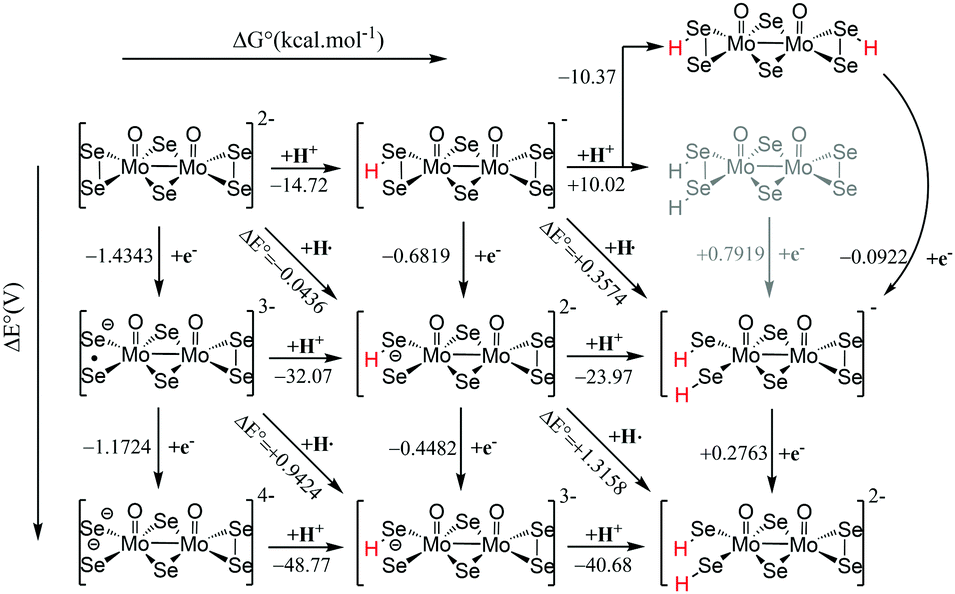 | ||
| Fig. 3 Thermodynamic routes for compound [Mo2O2(μ-Se)2(Se2)2]2−1 with two variables: electron addition (vertical) and proton addition (horizontal). | ||
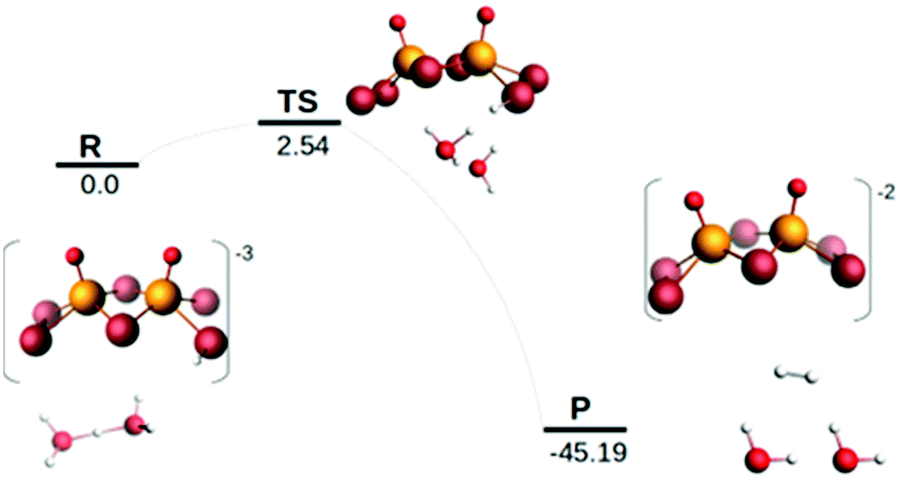 | ||
| Fig. 4 Activation energy and transition state for B1 H[Mo2O2(μ-Se)2 (Se2)2]3−. | ||
It is worth noting that single-protonated forms have significantly lower activation energies than the double-protonated ones. The activation barriers for compounds 1 (B1) and 2 are only 2.54 (Fig. 4) and 5.18 kcal mol−1 (Fig. S23, ESI†), respectively. Thus, the model of the transition state for single protonated forms is the most probable candidate. For the non-reduced, single- and double-reduced species, their double-protonated forms are the most thermodynamically stable. Considering electrochemical routes, 2e− addition to double-protonated non-reduced species is non-favourable. In contrast, reduction reactions from single protonated and non-protonated forms are more feasible (Fig. 3). Thus, our results clearly point towards the Heyrovsky mechanism based on thermodynamic favourability and the high activation energies observed for the alternative Taffel step.
In conclusion, a more accessible and high-yield synthesis for the selenium analogue to the well-known [Mo2O2S2(S2)2]2− structure makes it possible to explore the electronic and chemical properties of the molecule, which have previously remained unexplored. The performance of the molecular catalyst for the hydrogen evolution reaction was evaluated while we propose viable mechanistic pathways in relation to the interaction between catalysts and substrate while we identified intriguing differences with their sulfur analogues. The initial 1 e− reduction is easier compared to the sulphur containing analogue however the second reduction is more energetically demanding which is reflected to the observed slightly elevated Tafel slope. This indicates the significant electronic changes of the selenium containing structure relative to the sulphur. Notably, these results represent an attractive strategy for preparing next generation HER electrocatalysts. Understanding the electronic underpinnings of trends in activity as a function of the metal or chalcogen incorporated in the molecular species, can help inform the design of new catalysts and exploitation of cooperative and structural effects. Moreover, the development of facile low-cost synthesis of a molecular catalyst is highly desirable for the easier scale up in large scale applications.
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. C. McGlynn, T. Dankwort, L. Kienle, N. A.-G. Bandeira, J. P. Fraser, E. K. Gibson, I. Cascallana-Matías, K. Kamarás, M. D. Symes, H. N. Miras and A. Y. Ganin, Nat. Commun., 2019, 10, 4916 CrossRef PubMed.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, B. Martín-García, M. Prato, L. Pasquale, J.-K. Panda, P. Marvan, Z. Sofer and F. Bonaccorso, ACS Catal., 2020, 10, 3313–3325 CrossRef CAS PubMed.
- J. C. McGlynn, M. Friskey and A. Y. Ganin, Sustainable Energy Fuels, 2020, 4, 4473–4477 RSC.
- J. Yang, A. R. Mohmad, Y. Wang, R. Fullon, X. Song, F. Zhao, I. Bozkurt, M. Augustin, E. J.-G. Santos, H. S. Shin, W. Zhang, D. Voiry, H. Y. Jeong and M. Chhowalla, Nat. Mater., 2019, 18, 1309–1314 CrossRef CAS PubMed.
- H. Kwon, D. Bae, H. Jun, B. Ji, D. Won, J.-H. Lee, Y.-W. Son, H. Yang and S. Cho, Appl. Sci., 2020, 10, 3087 CrossRef CAS.
- J. Shi, Y. Huan, X. Zhao, P. Yang, M. Hong, C. Xie, S. Pennycook and Y. Zhang, ACS Nano, 2021, 15, 1858–1868 CrossRef CAS PubMed.
- A. S. Goloveshkin, N. D. Lenenko, A. V. Naumkin, A. Y. Pereyaslavtsev, A. V. Grigorieva, A. V. Shapovalov, V. N. Talanova, A. V. Polezhaev, V. I. Zaikovskii, V. V. Novikov, A. A. Korlyukov and A. S. Golub, ChemNanoMat, 2021, 7, 447–456 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed.
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 15181–15185 CrossRef CAS PubMed.
- B. R. Garrett, S. M. Polen, K. A. Click, M. He, Z. Huang, C. M. Hadad and Y. Wu, Inorg. Chem., 2016, 55, 3960–3966 CrossRef CAS PubMed.
- B. R. Garrett, K. A. Click, C. B. Durr, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2016, 138, 13726–13731 CrossRef CAS PubMed.
- B. R. Garrett, S. M. Polen, M. Pimplikar, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2017, 139, 4342–4345 CrossRef CAS PubMed.
- J. McAllister, N. A.-G. Bandeira, J. C. McGlynn, A. Y. Ganin, Y.-F. Song, C. Bo and H. N. Miras, Nat. Commun., 2019, 10, 370 CrossRef CAS PubMed.
- J. H. Liao, L. Hill and M. G. Kanatzidis, Inorg. Chem., 1993, 32, 4650–4652 CrossRef CAS.
- C. nin Chau, R. W.-M. Wardle and J. A. Ibers, Inorg. Chem., 1987, 26, 2740–2741 CrossRef.
- B. W. Eichhorn, D. R. Gardner, A. Nichols-Ziebarth, K. J. Ahmed and S. G. Bott, Inorg. Chem., 1993, 32, 5412–5414 CrossRef CAS.
- G.-C. Guo and T. C.-W. Mak, Inorg. Chem., 1998, 37, 6538–6540 CrossRef CAS PubMed.
- H. N. Miras, E. F. Wilson and L. Cronin, Chem. Commun., 2009, 1297–1311 RSC.
- H. N. Miras, M. Sorus, J. Hawkett, D. O. Sells, E. J.-L. McInnes and L. Cronin, J. Am. Chem. Soc., 2012, 134, 6980–6983 CrossRef CAS PubMed.
- J. Yan, D.-L. Long, H. N. Miras and L. Cronin, Inorg. Chem., 2010, 49, 1819–1825 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental method, materials characterization, electrochemical data and calculation method. CCDC 2155514 (1) and 2155450 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01226j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |