
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and preliminary evaluation of octreotate conjugates of bioactive synthetic amatoxins for targeting somatostatin receptor (sstr2) expressing cells†
Alla
Pryyma
 a,
Kaveh
Matinkhoo
a,
Yong Jia
Bu
a,
Helen
Merkens
b,
Zhengxing
Zhang
b,
Francois
Bénard
b and
David M.
Perrin
*a
a,
Kaveh
Matinkhoo
a,
Yong Jia
Bu
a,
Helen
Merkens
b,
Zhengxing
Zhang
b,
Francois
Bénard
b and
David M.
Perrin
*a
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: dperrin@chem.ubc.ca
bDepartment of Molecular Oncology, BC Cancer, Vancouver, BC V5Z 1L3, Canada
First published on 7th October 2021
Abstract
Targeted cancer therapy represents a paradigm-shifting approach that aims to deliver a toxic payload selectively to target-expressing cells thereby sparing normal tissues the off-target effects associated with traditional chemotherapeutics. Since most targeted constructs rely on standard microtubule inhibitors or DNA-reactive molecules as payloads, new toxins that inhibit other intracellular targets are needed to realize the full potential of targeted therapy. Among these new payloads, α-amanitin has gained attraction as a payload in targeted therapy. Here, we conjugate two synthetic amanitins at different sites to demonstrate their utility as payloads in peptide drug conjugates (PDCs). As an exemplary targeting agent, we chose octreotate, a well-studied somatostatin receptor (sstr2) peptide agonist for the conjugation to synthetic amatoxins via three tailor-built linkers. The linker chemistry permitted the evaluation of one non-cleavable and two cleavable self-immolative conjugates. The immolating linkers were chosen to take advantage of either the reducing potential of the intracellular environment or the high levels of lysosomal proteases in tumor cells to trigger toxin release. Cell-based assays on target-positive Ar42J cells revealed target-specific reduction in viability with up to 1000-fold enhancement in bioactivity compared to the untargeted amatoxins. Altogether, this preliminary study enabled the development of a highly modular synthetic platform for the construction of amanitin-based conjugates that can be readily extended to various targeting moieties.
Introduction
Cancer treatment with standard chemotherapeutics is limited by severe systemic toxicity owing to poor selectivity for neoplastic tissue.1 Targeting agents designed to bind specific tumor markers expressed on cancer cell surfaces can improve the therapeutic index of chemotherapeutics2,3 and harness the potential of toxins not typically used in therapy.4,5 Thus, bioconjugates comprising a targeting moiety linked to a toxin display up to 1000-fold lower IC50 values against target-expressing cells compared to the IC50 values of unconjugated cytotoxins, thus enabling deadly compounds to be used at tolerable doses.6 Generally, antibodies are employed to selectively deliver agents to malignant cells.1,6–9 To wit, nine antibody drug conjugates (ADCs) have received FDA approval (five in the past two years alone) while over 80 ADCs are undergoing clinical trials.7,10,11 This recent clinical success is accompanied by intense research on aptamer- and peptide-targeted drugs and suggests that targeted conjugates of all kinds are poised for success.12–14Whereas the majority of targeted therapeutics in clinical development exploit microtubule inhibitors (e.g. auristatins, maytansinoids, vincristine)3,15–18 or DNA-reactive cytotoxins (e.g. camptothecin, calicheamicin, doxorubicin)19–21 as standard payloads, instances of induced resistance are documented.22,23 This highlights the need for new payloads targeting alternative intracellular targets. One such payload is α-amanitin (1), a potent RNA polymerase II (pol II) inhibitor isolated from the fatal “death-cap” mushroom, Amanita phalloides (Fig. 1).24,25 Because RNA pol II is required for both cellular growth and homeostasis, α-amanitin, a potent cytotoxin, kills both actively dividing and quiescent cells, presenting a unique advantage over other payloads that act preferentially on rapidly growing cells. Additional favorable properties of amanitin include water solubility and plasticity of conjugation at multiple sites on the toxin.
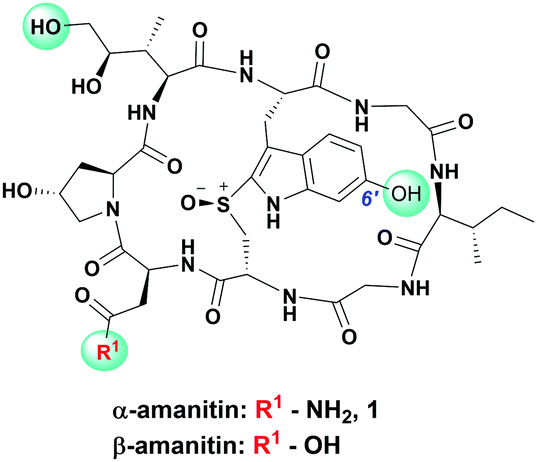 | ||
| Fig. 1 Chemical structure of α/β-amanitin with commonly used conjugation sites highlighted with green. | ||
Albeit one of the deadliest known toxins, in 1981, α-amanitin was successfully used as an ADC payload26 and subsequently validated in more potent amanitin–ADCs that exhibit negligible hepatotoxicity in vivo.27–32 By contrast, peptide drug conjugates (PDCs) of amanitin have been limited to integrin-targeting31,33 and pH-responsive “acidic” peptides.34,35 Irrespective of linker design (i.e. non-cleavable or cleavable), these particular compositions would appear to have questionable utility owing to a general lack of potency and/or minimal selectivity for target tissue.31,33 Since this work was submitted, Gallo et al. described a novel, highly efficacious class of amanitin drug constructs, that incorporates a small molecule peptidomimetic for targeting prostate specific membrane antigen and an immunoglobin Fc domain for improved pharmacokinetics.36
Peptides are attractive as they show excellent tumor specificity while overcoming known disadvantages of antibodies e.g. high manufacturing cost, low tissue penetration, aggregation, and immunogenicity.37 In light of recent synthetic breakthroughs for accessing bioactive amatoxins by us33,38–40 and others,33,38–40 we were compelled to use this practical knowledge to expand the repertoire of amanitin-based cancer therapeutics with new, synthetic amanitins.
Towards these ends, we report on the design of a modular and user-friendly approach to enable bio-orthogonal conjugation of synthetic amanitins to three different linkers that are elaborated to three potent peptide–amanitin conjugates (Fig. 2) that differ by their mode of toxin release, as defined by the linker. Octreotate (TATE–N3, 2) was chosen as it is a circulation-stable octapeptide somatostatin analog that agonizes somatostatin receptors (Kd ∼ 0.4 nM for sstr2).41,42 Targeting sstr2 has been successfully exploited for drug delivery,43–45 cancer imaging,46,47 and radiotherapy42,47 owing to the high levels of sstr2 expression on neuroendocrine tumors (e.g. carcinoids, pancreatic islet cell tumors, thyroid carcinomas, and small lung cancer to name a few). While this is the first report of an octreotate–amanitin conjugate, herein, octreotate serves as an exemplar to highlight the potential for developing peptide–amanitin conjugates in targeted applications.
 | ||
| Fig. 2 (A) Chemical structures of sstr2 targeting agent octreotate (TATE–N3, 2); (B) synthetic bioactive amatoxins 3 and 4; (C) three bioconjugates 5–7 synthesized in this study. Highlighted in blue and red are conjugation sites; (a) and (b) show two proposed pathways for self-immolative drug release of bio-reducible linker conjugate 6. | ||
Cell proliferation studies revealed that the conjugates are cytotoxic (or at least cytostatic) in the nanomolar range. This observation is further supported by kinetics of cell death, fluorescence microscopy studies, and blocking controls. To the best of our knowledge, this work is the first example of conjugating amanitin to a high-affinity peptide that targets a G-coupled protein receptor of significant interest to cancer therapy and which improves the apparent selectivity of the toxin for the intended target over the unconjugated toxin.
Results and discussion
In designing peptide–amanitin conjugates, we recognize that the majority of amanitin-based ADCs and PDCs have relied nearly exclusively on naturally-sourced α- and β-amanitin (Fig. 1). Loci for conjugation on the toxin include the δ-hydroxyl of dihydroxyisoleucine,27,48 the asparagine side chain,49 and the 6′-hydroxyl of the tryptathionine staple.28,29,31,50,51 In some cases, these have been linked either to reducible or proteolyzable linkers, or to linkers assumed to be non-cleavable for which the metabolic fate is usually unknown.A key advance in the development of any cytotoxic payload is access to synthetic toxins and analogs thereof. In 2018, we disclosed the first total synthesis of this venerated toxin,38 which was followed by two others.39,40 Whereas a priori these routes now provide synthetic access, challenges remain regarding efficiency and scalability, particularly in terms of accessing a 6′-hydroxytryptathionine staple and the (R)-sulfoxide (although both the thioether and sulfone are equally cytotoxic). To expand the potential for new amanitin derivatives, we recently reported the synthesis of an unnatural 5′-hydroxy-6′-deoxy-amanitin (3) that is equipotent to α-amanitin (along with the corresponding (R)-sulfoxide, and the sulfone).52 With this more synthetically accessible toxin in hand, here we report on conjugates that would exploit the 5′-hydroxy-6′-deoxy-amanitin 3 (Fig. 2B). We further expand this study on octreotate conjugates to include conjugation to N-propargyl-dideoxy-amanitin 4 (Fig. 2B),33 an alternative, equally synthetically accessible derivative that lacks the 6′-hydroxyindole yet shows near-native toxicity (IC50 on CHO cells 1–2 μM compared to 0.3–0.7 μM for α-amanitin) and is equipped with an alkyne conjugation handle for copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC).
In considering a non-cleavable linker, we note that PEGylation of various toxins has been shown to enhance pharmacokinetic properties53 and aid in overcoming hydrophobicity.54 Whereas PEG linkers were used in the context of RGD–amanitin,31 because they are assumed to be non-cleavable, they would likely degrade slowly inside cells, which is consistent with decreased overall potency. Nevertheless, several antecedent studies showed that non-cleavable linkers to amanitin still provide highly cytotoxic conjugates (see Discussion).
More promising therefore are linkers that liberate the toxin from the targeting vector once inside cells. A review of the vast literature on circulation-stable linkers reveals two well-established classes of cleavable linkers that are advancing to clinical studies. These include: (i) disulfide-linked, first introduced in 2006,55–57 which decompose upon reductive cleavage in the intracellular reducing environment via cyclization either to a thiocarbonate58–60 or an episulfide with explusion of CO261,62 and concomitant release of the active drug (Fig. 2C);55,57,63,64 and (ii) protease cleavable (e.g. Cathepsin B) linkers, which are degraded by lysosomal proteases that are overexpressed in cancer cells.65,66
The use of N-propargyl-amanitin 4 enables production of both cleavable and non-cleavable constructs. Hence, we designed a PEG-based non-cleavable conjugate 5 and a disulfide-containing bio-reducible, self-immolative conjugate 6 (Fig. 2C). In the context of grafting to the tryptathionine staple, we noted antecedent reports of amanitin–RGD peptide conjugates linked to the 6′-hydroxyl group in amanitin.31 Following our recent report on the more synthetically accessible 5′-hydroxytryptathionine-stapled amanitin,52 we took advantage of the nucleophilicity of the 5′-hydroxyl group for grafting linkers to the 5′-hydroxytrypthathionine as well. Towards this end, we appreciated the recent success of valine–citrulline (Val–Cit) Cathepsin B cleavable linkers in FDA approved ADCs.65,67 A favorable feature of Val–Cit linkers is the presence of a self-immolative p-aminobenzyloxycarbonyl (PABC) spacer which decomposes upon deacylation, prompting a spontaneous release of the toxin in its free unmodified form (Fig. 2C). Hence, we designed bioconjugate 7 employing a Val–Cit–PABC linker conjugated to 5′-hydroxy-6′-deoxy-amanitin 3 (Fig. 2C).
Considerations for designing modular linkers for bioorthogonal toxin conjugation
In pursuit of linkers for amatoxins 3 and 4, we sought a degree of modularity for biorthogonal conjugation while providing optionality for state-of-the-art immolation. The linkers designed for conjugation to amatoxin 4 were equipped with two orthogonally protected conjugation handles that take advantage of CuAAC conjugation, namely an azide and a TES-protected alkyne, that follow elegant reports by Aucagne68,69 and others70 (Scheme 1). For conjugation, we favored the CuAAC reaction owing to its fast kinetics, bio-orthogonality, and compatibility with a broad range of solvents.71–74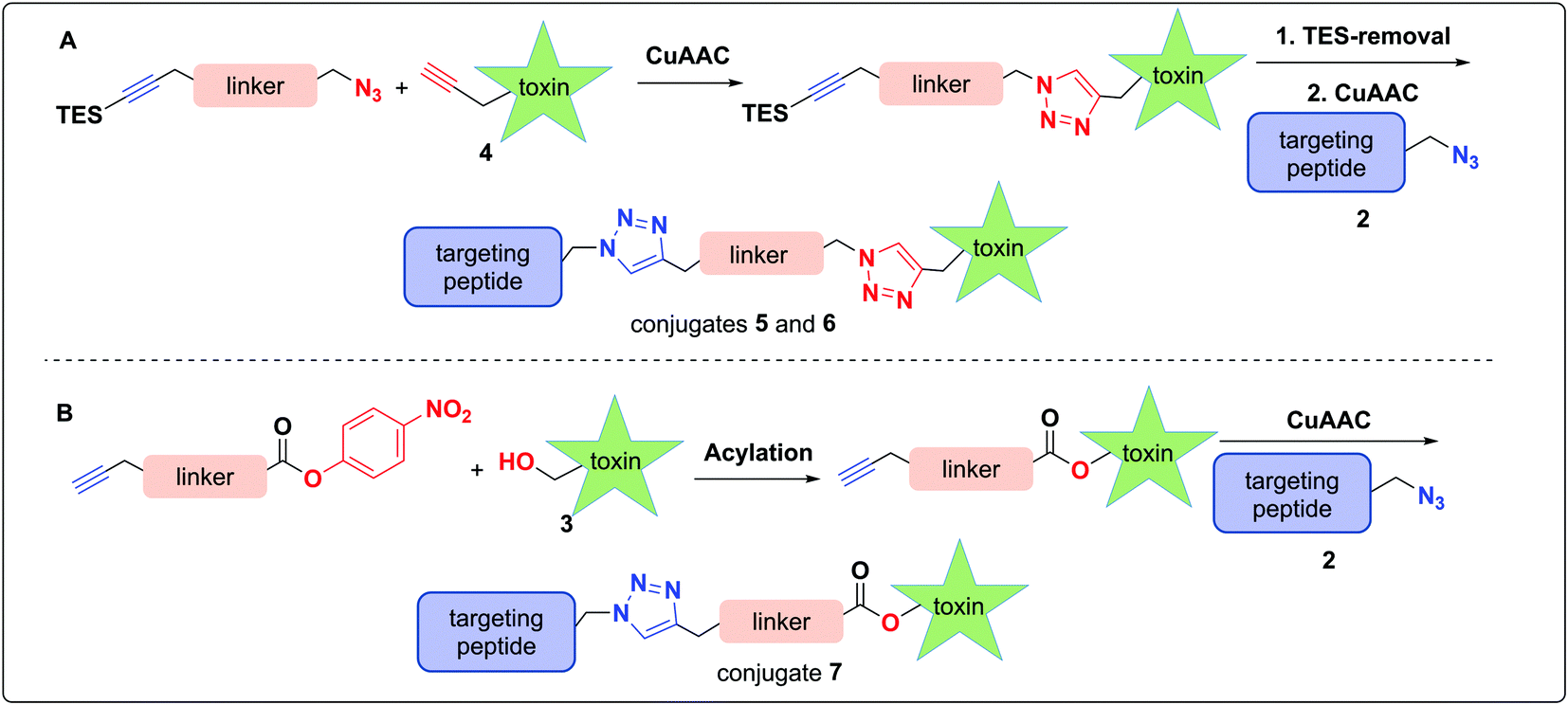 | ||
| Scheme 1 Synthetic approach towards a modular assembly of conjugates 5, 6, and 7 employing linkers with suitable orthogonal protecting groups; (A) PEG-linked and disulfide-containing bio-reducible conjugate 5 and 6 of amatoxin 4; (B) Cathepsin B cleavable self-immolative conjugate of amatoxin 3. | ||
To assemble the conjugates, 4 was first reacted with the azide of the linker via CuAAC (Scheme 1A). Removal of the TES group was then carried out to reveal the alkyne for use in the second CuAAC reaction with the azide of targeting agent 2, yielding two peptide–amanitins 5 and 6 (Scheme 1A). In a similar vein, we created a second class of alkyne-based linker with an activated p-nitrophenyl carbonate for selective acylation of the 5′-hydroxytryptathionine of 3 and CuAAC conjugation to azide containing peptide 2 to provide conjugate 7 (Scheme 1B).
Linker synthesis
The non-cleavable PEG-linker 10 was constructed by reacting the freshly prepared TES-protected propargyl amine 8 with a commercially available azido–PEG8–NHS ester 9 (Fig. 3A) in 73% yield. The bio-reducible linker 16 was designed with a disulfide bridge in proximity to the carbamate ester (Fig. 3B). We opted for a carbamate linkage instead of a carbonate one, due to the known sensitivity of carbonates to hydrolysis,55,64 which could potentially result in handling difficulties and pre-mature release of the toxin as well as decomposition during purification, as we observed (data not shown). Synthesis of 16 was carried out as shown in Fig. 3B. | ||
| Fig. 3 Synthesis of bifunctional linkers 10, 16 and 18: (A) PEG-based linker containing TES protected alkyne and azide handles; (B) linker 16 containing a bio-reducible disulfide, a TES protected alkyne; (C) Cathepsin B cleavable linker 18 functionalized with an alkyne and a p-nitrophenyl carbonate ester; py – pyridyl. | ||
To assemble the carbamate ester, azidoethylamine 11 was first reacted with p-nitrophenyl chloroformate to give 12 followed by reaction with the alcohol of the mixed disulfide-bridged 2-(pyridine-2-yl-disulfanyl) ethanol 13 to produce 14. The mixed disulfide-bridged 14 was exchanged with mercaptopropionic acid in water/DMF at pH 6 under argon to give 15 in 82% yield (Fig. 3B). Finally, the carboxylic acid of 15 was coupled to the TES-protected propargylamine 8 to afford the final linker 16 (Fig. 3B).
A Cathepsin B cleavable linker 18 was assembled by a combination of solid- and solution-phase synthesis as described previously (Fig. 3C).75,76 Briefly, 2-chlorotrityl chloride resin was elaborated with Cit–Val–pentynoate under Oxyma-buffered solid-phase conditions.75 Resin cleavage followed by direct coupling to p-aminobenzyl alcohol produced 17, which was activated with bis-p-nitrophenyl carbonate to yield target linker 18 (Fig. 3B).
Assembly of TATE–amanitin bioconjugates 5–7
This convergent synthesis enabled bioconjugates to be efficiently and rapidly constructed from propargyl–amanitin 4 in three steps (Scheme 2A) to give 5 and 6 in good yields. First, the azides of linkers 10 and 16 were coupled to 4via CuAAC to yield 19 and 20, respectively (Scheme 2A). We found that the addition of tris(benzyltriazolylmethyl)amine (TBTA) as a ligand for Cu(II) greatly increased the reaction rate and allowed for the reaction to proceed to completion in one hour and at a low concentration of amatoxin 4 (0.5 mM). The alkyne functionality was then unmasked via facile and clean TES removal with excess KHF2 in dimethyl sulfoxide (DMSO) at 60 °C. Excess KHF2 was readily removed by a simple C18 Sep-Pak work-up. The free alkyne was then coupled to TATE–N32 to afford the final conjugates TATE–PEG–Ama 5 and TATE–SS–Ama 6 (Scheme 2A).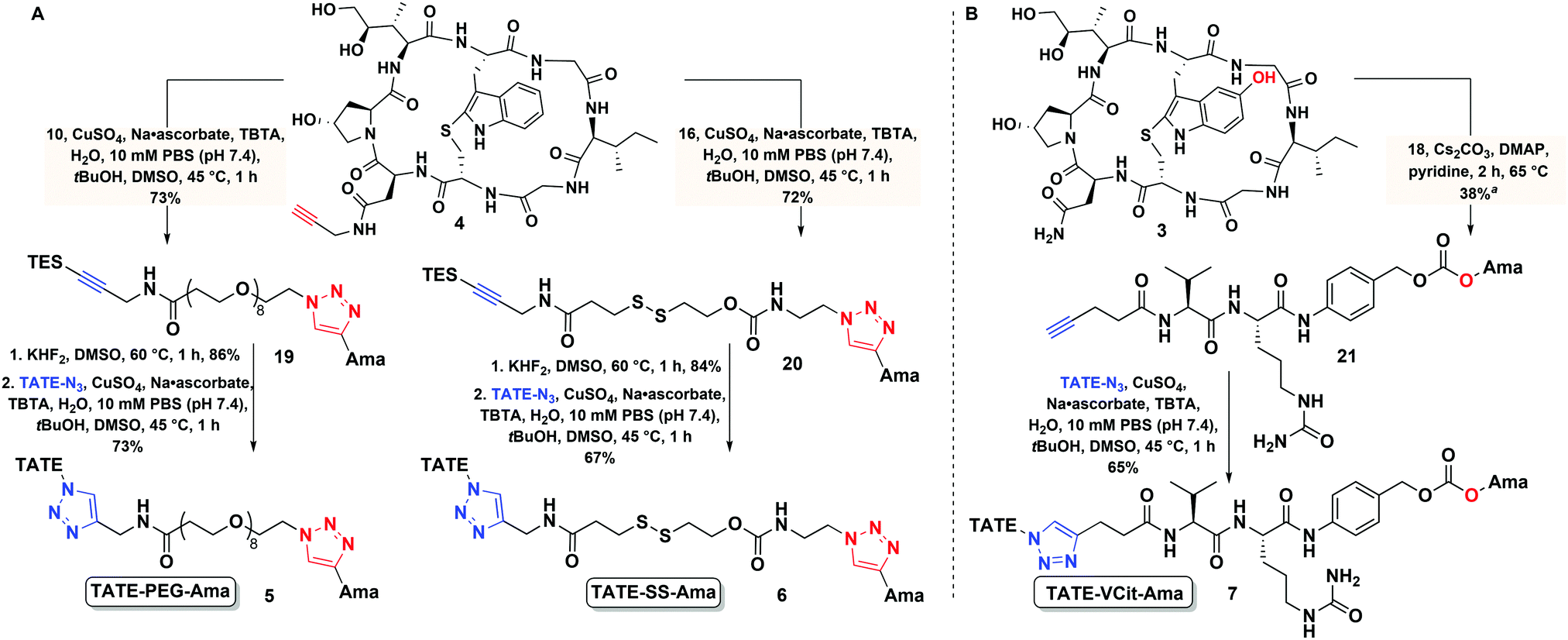 | ||
| Scheme 2 (A) Final steps of the synthesis of non-cleavable and disulfide containing bioconjugates 5 an 6 through CuAAC reaction; (B) final steps of the synthesis of Cathepsin B cleavable bioconjugate 7; astarting material (3) was partially recovered. | ||
Conjugate 7 was efficiently assembled in two steps from amanitin 3 and linker 18 (Scheme 2B). The 5′-hydroxyl of amanitin 3 was reacted with Val–Cit–PABC–PNP linker 18 in the presence of DMAP and pyridine as a solvent to afford compound 21. This transformation was accompanied by a change to the UV-Vis absorption spectrum resulting from the acylation of the 5′-hydroxytryptathionine chromophore. CuAAC reaction of the alkyne 21 with TATE–N32 yielded final conjugate 7 (Scheme 2B).
Biological evaluation
With bioconjugates 5–7 in hand, we evaluated their cytotoxicity by assaying cell viability on an sstr2-positive rat pancreatic cancer Ar42J cells, using free amatoxins as controls. The MTS cell viability assay showed that all three PDCs were effective at reducing the viability of Ar42J cells in the low nM range, providing calculated EC50 values of 4.2, and 2.3 nM for conjugates 6 and 7, respectively (Fig. 4); these displayed up to 1000-fold enhancement in apparent cytotoxicity compared to free amatoxins 3 and 4, both of which gave calculated EC50 values of ∼2 μM. This points to successful targeting and intracellular accumulation of toxin inside cells through an sstr2-mediated uptake inside the cells.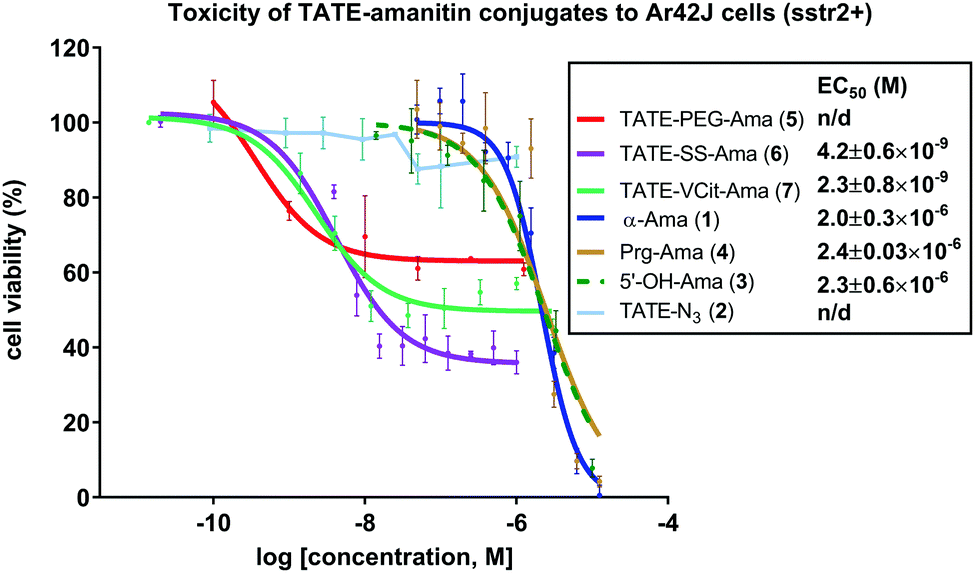 | ||
| Fig. 4 Cytotoxicity of amanitin-based conjugates 5, 6 and 7 to Ar42J cells expressing sstr2 in cell proliferation MTS assay; cells were incubated with TATE–PEG–Ama 5, TATE–SS–Ama 6, TATE–VCit–Ama 7, TATE–N32 and amatoxins 1, 3 and 4 for 72 h at 37 °C (n = 3), EC50 (effective concentration giving half-maximal response), data represented as mean value ± S.D., n/d – not determined. | ||
Further evidence supporting target-mediated uptake is the micromolar toxicity of bioconjugates to the sstr2-negative CHO cell line that is otherwise sensitive to non-targeted amanitin (see Fig. S1, ESI†). In comparison to α-amanitin and synthetic amatoxins 3 and 4, bioconjugates 5–7 are 7- to 14-fold less toxic to CHO cells (control cell line), likely owing to impaired uptake of octreotate–amanitin conjugates that must enter via diffusion mediated processes or other non-specific import mechanisms that are currently unknown. In addition, a blocking study was run in the presence of free TATE–N32, which led to the expected reduction in apparent toxicity (see Fig. S2, ESI†).
When comparing the two cleavable bioconjugates 6 and 7, it is important to consider the identity of the active toxin liberated inside the cell following the targeted delivery. Based on considerable precedent with these types of linkers, the self-immolation of bioconjugate 7 is expected to release the unmodified toxin 3, whereas, degradation of bioconjugate 6 liberates amanitin 22 with a pendant ammonium-ethyl (Fig. 5) triazole, which might be expected to reduce toxicity, particularly if the ammonium cation hinders lysosomal escape.
 | ||
| Fig. 5 Structure of the toxin liberated inside of the targeted cells as a result of the disulfide reduction and self-immolation of bioconjugate TATE–SS–Ama 6. | ||
Indeed, following a deliberate degradation of disulfide-containing bioconjugate to obtain 22, we found 22 to be about 10-fold less toxic to CHO cells compared to 4 (EC50 ∼ 5 μM, see Fig. S1, ESI†). The exact reason for apparent diminished toxicity is difficult to establish at this juncture: 22 may have a diminished affinity for RNA pol II or it may penetrate cells relatively inefficiently due to the presence of the charged ammonium-ethyl residue, or both.
Interestingly, even though conjugates 6 and 7 immolate via two different mechanisms, both elicited a similar response (Fig. 4). Reduction in cell viability in the case of 6 and 7 will be governed by complex effects: (i) the affinity of the targeting agent for its receptor, (ii) the extent of toxin internalization, (iii) the inherent IC50 of the liberated toxin once internalized, and (iv) the effective intracellular concentration of toxin. While the confluence of these effects and the various kinetic steps that govern them makes it difficult to assess which of the above (i–iv) is limiting, these observations show that a combination of target-specific binding, active uptake, internalization, and immolation contribute to a sufficiently high concentration of toxin that results in substantial reduction in viability.
As anticipated, octreotate–amanitin conjugates linked by the immolating linkers showed greater overall toxicity than the non-cleavable PEG-linked conjugate 5, interestingly however, 5 still showed activity. Nevertheless, the data obtained using non-cleavable PEG-linked construct 5 were not sufficiently robust to allow us to reliably calculate an EC50 value. This result adds to a growing body of critical knowledge on reports of amanitin-conjugates comprising linkers prepared by amide-bond formation with β-amanitin that are presumed to be non-cleavable. To wit, amanitin-conjugates linked via an amide bond to albumin,49 epidermal growth factor,77 as well as to both RGD and PEG-rhodamine33 have all shown potent activity, despite the use of non-cleavable linkers linked to the Asp/Asn. In none of the above reports, however, did the authors establish that the amanitin toxin was liberated. In contrast, however, non-cleavable linkers grafted to the 6′-hydroxytryptathionine and linked to RGD were found to be essentially non-toxic.31
Notably, only partial killing of Ar42J cells was observed during the MTS assays for all three PDCs. This phenomenon is often observed in in vitro cell viability studies on cancer cells and has spurred numerous studies.78–81 It is well-known that cellular heterogeneity and cell-to-cell variability are present to some degree in any cell population and the overall behaviour of the population gleaned by the means of cell-population-averaged techniques does not represent an individual cell.79 Stochastic fluctuations inherent to biological systems can cause changes in gene and protein expression levels.80,82,83 Consequently, the levels of mRNA, proteins, metabolites, and other cellular components may result in the emergence of phenotypically distinct subpopulation and genetically identical cells to respond differently to sudden stress and become more or less sensitive to a drug, which may prove either cytotoxic or simply cytostatic.80,82,83 Because amanitin is known to induce cell death, we contend that the concentration-dependent depletion of sstr2-expressing cells in the presence of 5–7 is likely explained by cytotoxicity and not simply cytostasis.
In an effort to understand the partial reduction in cell viability, we applied FACS scanning to assess the sstr2 levels as function of time as well as in response to Ama–SS–TATE 6. To do this, we synthesized a TATE–PEG2–fluorescein (S7) as a fluorescent reporter ligand. Ar42J cells were plated for varying lengths of time (48, 72, 96, and 120 hours). In our hands, in the absence of 6, Ar42J cells appeared to undergo a time-dependent loss of sstr2 resulting in a subpopulation that shows significantly lower sstr2 expression and approaches approximately 50% of the overall population (Fig. 6). When the same cells were incubated with 6 (30 nM) for the same length of time, we observed a depletion in the sstr2-expressing population. Similar results were obtained when cells were treated with 6 at 60 nM (see ESI†). We should caution that this analysis proved technically difficult and additional attempts were confounded by excessive clumping of these cells in culture while FACS scans showed even more dramatic reduction in apparent sstr2 expression (data not shown).
 | ||
| Fig. 6 FACS scan of Ar42J cells that were untreated (gray) show a time-dependent increase in an sstr2-negative population from 48–96 h. Cells treated with 6 (30 nM) show a similar effect (black), however, the sstr2-positive population is depleted in comparison to the sstr2-negative population. Similar results were also observed when cells were treated with 6 at 60 nM (see ESI†). | ||
Although an explanation for this striking and altogether unexpected reduction in the sstr2 expression levels over time is not readily intuited, this observation suggests that the apparent loss of Ar42J viability in the presence of 6, as measured by MTS assays, is due to a loss of target expression in a sub-population of cells and otherwise cytotoxic action of 6 on those cells that expressed the target. Notwithstanding, this result also suggests that 6 is highly potent against those cells that are sstr2-positive since by 96 h, there appears to be a dramatic reduction in the number of sstr2-positive cells.
Fluorescence studies – receptor binding of TATE-linked conjugates
To further substantiate uptake and internalization of TATE-linker conjugates, albeit indirectly, we designed a fluorescent surrogate 23 where TATE–N32 was conjugated to rhodamine via the same linker chemistries used for the synthesis of 6 (Fig. 7a). Given the structural similarity between TATE–SS–rhodamine 23 and TATE–SS–Ama 6, we expected that 23 would also bind selectively to Ar42J cells.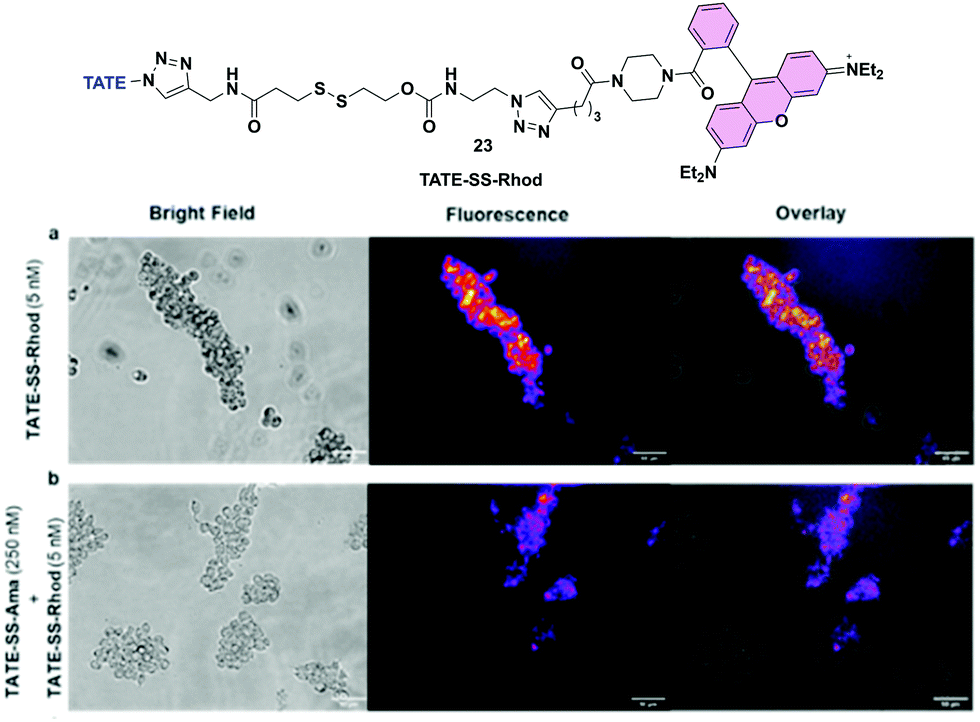 | ||
| Fig. 7 Fluorescence imaging of Ar42J cancer cells (sstr2-positive) using TATE–SS–Rhod 23; (a) cells were incubated with 5 nM TATE–SS–Rhod 23 for 30 min – control; (b) blocking – cells were pre-treated with 250 nM TATE–SS–Ama 6 (30 min) followed by incubation with 5 nM TATE–SS–Rhod 23 (30 min). | ||
The synthesis was effected by coupling rhodamine–piperazine–hexynamide equipped with disulfide linker 16 to octreotate 2 (see Scheme S1, ESI†). We used rhodamine instead of fluorescein to red-shift the fluorescent emission. Using fluorescence microscopy, we then evaluated cell-specific binding and uptake by sstr2-positive cells. We first assessed the binding capability of TATE–SS–Rhod 23 to Ar42J cells; in the presence of 5 nM 23, strong fluorescence was observed. Hence, Ar42J cells were treated with a 5 nM concentration of 23 in the presence of a 50-fold excess of 6 to block the binding of 23 (Fig. 7a and b). Using fluorescence microscopy, we observed that indeed TATE–SS–Ama 6 successfully blocked the binding of 23 (Fig. 7b). This observation further validates the targeted enrichment of toxin inside the cells.
Cell death time-course studies
Measuring cell viability as a function of time revealed a relatively rapid loss of cell viability with disulfide-linked conjugate 6 (60 nM) after only 24 hours of incubation with no further reduction thereafter (Fig. 8). In contrast, the same cells when treated with a much higher concentration (10 μM) of free α-amanitin 1 remained fully viable after 24 hours (Fig. 8) and loss of cell viability was only observed after 48 hours. It is well-known that in the absence of organic anion transport proteins normally found on hepatocytes, α-amanitin enters cells by passive diffusion through the plasma membrane.84 With toxicity directly correlated to plasma membrane permeability along with increased lipophilicity,85 the comparatively larger conjugates are less active than the synthetic amanitins used for conjugation. The relatively repaid kinetics of cell death observed here along with the fact that cell death occurs at nearly 1000-fold lower concentrations suggests an increase in the rate of accumulation of amanitin in the cells treated with the bioconjugate, suggesting a receptor-mediated mechanism of cell entry. We note that these data were generated by MTS assays which may result in discrepancies compared to the data obtained by FACS scanning, which suggested that cell death may take a slightly longer time. While we do not have an immediate explanation for this discrepancy, we suggest that qualitatively these data are consistent with the notion of sstr2-targeting by novel octreotate–amanitin conjugates, the synthesis of which has been the focus of this work.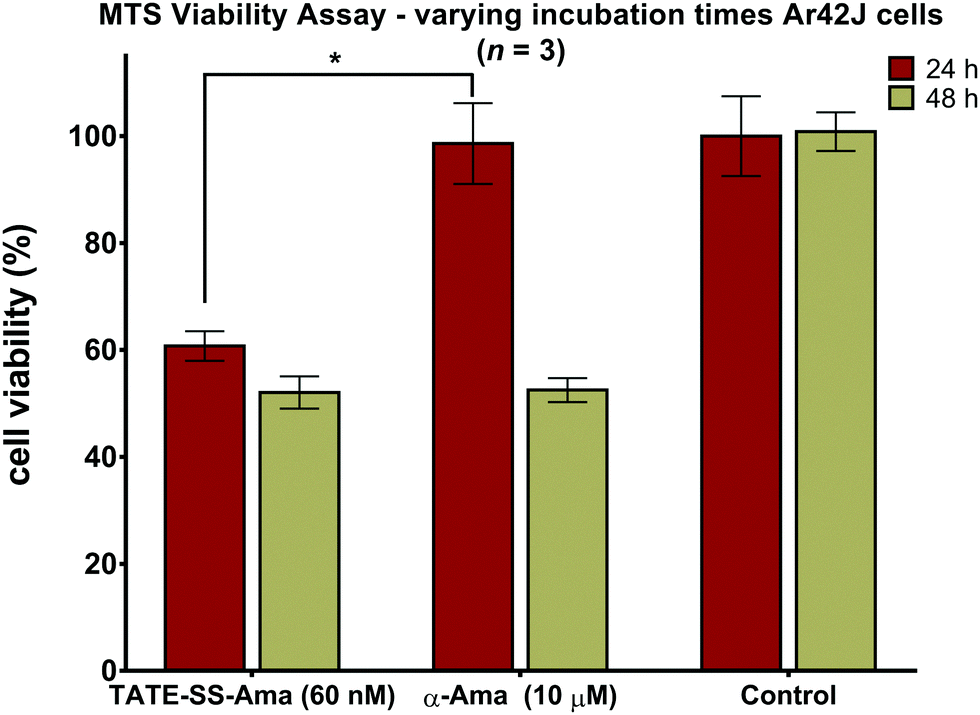 | ||
| Fig. 8 Cell death kinetics of bioconjugate TATE–SS–Ama 6 and α-amanitin 1 evaluated on Ar42J cells following 24 and 48 hours of incubation (MTS, n = 3), data reported as mean ± S.D.; *P < 0.01; control experiments are cells grown under the same conditions and treated with 0.5% DMSO (vehicle) culture media. | ||
Conclusions
Here we report on the development of three novel somatostatin receptor-targeting PDCs of two different amatoxins and octreotate peptide. Whereas previous reports on amanitin based bioconjugates largely relied on naturally sourced α- and β-amanitin, this work highlights the use of synthetically accessible amatoxins 3 and 4. Moreover, this work elaborates a modular synthesis and conjugation chemistry of three linkers specifically designed to react with these analogs. The use of 5′-hydroxy-6′-deoxy-amanitin 3 as a payload for targeted therapy is demonstrated for the first time along with successful conjugation chemistry compatible with this analog.Two cleavable conjugates, taking advantage of the reducing intracellular environment and elevated levels of Cathepsin B protease in cancerous cells, and a non-cleavable PEG-linked bioconjugate, were evaluated on somatostatin-expressing Ar42J cell line. We believe the modularity of the synthetic platform described in this work can be immediately applicable to other targeting agents equipped to undergo CuAAC reaction.
All three constructs represent the first examples of peptide–amanitin conjugates that elicit a cellular response consistent with target-specific toxin delivery in the nM-range. Nevertheless, cell viability inhibition was incomplete even though bioconjugates reduced the number of viable Ar42J cells in a concentration dependent fashion. To probe this, FACS scanning demonstrated that Ar42J cells in culture media underwent time-dependent divergence to give a second, distinct population with a significantly lower target expression. This may account for the observed response in the cell viability assay, which showed approximately 50% maximum cell lethality.
When the same conjugates were applied to cells that did not express the target (i.e. CHO cells), the conjugates were far less active than even amanitin itself, an observation that is consistent with lower passive uptake of a conjugate comprising two peptides. Use of a fluorescent octreotate conjugate demonstrated that binding of this fluorescent probe could be blocked with TATE–SS–Ama, which is consistent with receptor binding. Similarly, when TATE–N3 was added, toxicity was significantly blocked.
Although we did not conclusively establish that receptor mediated endocytosis is responsible for the activity of these conjugates on sstr2-expressing cells, such is likely to be operative to account for reduction in cell viability elicited at low-to-mid nanomolar concentrations. In addition, as the MTS assay only quantifies cell viability, we cannot distinguish cytoxicity from cytostasis. Notwithstanding, because amanitin is a known cytotoxin, we submit that the reduction in cellular viability is due to cell death.
Notably, salient points of this work include: (i) use of fully synthetic and unnatural amanitin analogs to expand access to this class of toxin, (ii) modular design of cleavable and non-cleavable linkers for concise directional conjugation, and (iii) the first report of amanitin conjugation to octreotate which is of considerable interest in cancer targeting and imaging. Finally, in view of the urgent need for anti-cancer therapeutics that inhibit non-standard intracellular targets (i.e. RNA pol II), the findings presented here are of mechanistic and therapeutic significance. It is anticipated that this work would expand the repertoire of octreotate-based therapeutics while providing empowering synthetic routes to consider in the design of numerous linkers of varied composition including the use of alternative immolative functionalties, which could be conjugated to octreotate, octreotide, or to other PDCs based on amanitin or other synthetic amatoxins. This work may also inform the design of antibody drug conjugates based on similar linkers.
Author contributions
The manuscript was written by AP and DMP and synthetic work was executed by AP, KM, and YJB with the exception of the synthesis of TATE–N3 that was effected by ZZ. FACS scanning was performed by HM in the laboratory of FB who consulted on the use of cell lines and evaluated the FACS data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Canadian Institutes for Health Research #220656 and the Canadian Cancer Society Research Initiative Grant #703374 and thank Dr Maria Ezhova for help with NMR acquisition, Dr Elena Polishchuk, and Ms Jessie Chen for help with cytotoxicity assays.Notes and references
- R. V. J. Chari, Acc. Chem. Res., 2008, 41, 98–107 CrossRef CAS PubMed.
- T. Ghose and S. P. Nigam, Cancer, 1972, 29, 1398–1400 CrossRef CAS.
- B. C. Laguzza, C. L. Nichols, S. L. Briggs, G. J. Cullinan, D. A. Johnson, J. J. Starling, A. L. Baker, T. F. Bumol and J. R. F. Corvalan, J. Med. Chem., 1989, 32, 548–555 CrossRef CAS PubMed.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef CAS.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219 CrossRef CAS.
- L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5–13 CrossRef CAS PubMed.
- F. Pretto and R. E. FitzGerald, Regul. Toxicol. Pharm., 2021, 122, 5 CrossRef.
- P. L. Salomon, E. E. Reid, K. E. Archer, L. Harris, E. K. Maloney, A. J. Wilhelm, M. L. Miller, R. V. J. Chari, T. A. Keating and R. Singh, Mol. Pharmaceutics, 2019, 16, 4817–4825 CrossRef CAS.
- R. He, B. Finan, J. P. Mayer and R. D. DiMarchi, Molecules, 2019, 24, 1855 CrossRef CAS.
- M. Roveri, M. Bernasconi, J. C. Leroux and P. Luciani, J. Mat. Chem. B, 2017, 5, 4348–4364 RSC.
- Y. Gilad, M. Firer and G. Gellerman, Biomedicines, 2016, 4, 24 CrossRef.
- A. Maderna and C. A. Leverett, Mol. Pharmaceutics, 2015, 12, 1798–1812 CrossRef CAS PubMed.
- R. V. Chari, B. A. Martell, J. L. Gross, S. B. Cook, S. A. Shah, W. A. Blättler, S. J. McKenzie and V. S. Goldmacher, Cancer Res., 1992, 52, 127–131 CAS.
- G. D. L. Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons, E. Mai, W. A. Blättler, J. M. Lambert, R. V. Chari and R. J. Lutz, Cancer Res., 2008, 68, 9280–9290 CrossRef.
- M. Barok, H. Joensuu and J. Isola, Breast Cancer Res., 2014, 16, 1–12 CrossRef.
- P. J. Burke, P. D. Senter, D. W. Meyer, J. B. Miyamoto, M. Anderson, B. E. Toki, G. Manikumar, M. C. Wani, D. J. Kroll and S. C. Jeffrey, Bioconjugate Chem., 2009, 20, 1242–1250 CrossRef CAS.
- P. Trail, D. Willner, S. Lasch, A. Henderson, S. Hofstead, A. Casazza, R. Firestone, I. Hellstrom and K. Hellstrom, Science, 1993, 261, 212–215 CrossRef CAS PubMed.
- A. D. Ricart, Clin. Cancer Res., 2011, 17, 6417–6427 CrossRef CAS.
- S. García-Alonso, A. Ocaña and A. Pandiella, Cancer Res., 2018, 78, 2159–2165 CrossRef.
- D. M. Collins, B. Bossenmaier, G. Kollmorgen and G. Niederfellner, Cancer, 2019, 11, 394 CrossRef CAS.
- T. J. Lindell, F. Weinberg, P. W. Morris, R. G. Roeder and W. J. Rutter, Science, 1970, 170, 447–449 CrossRef CAS PubMed.
- T. Wieland and H. Faulstich, Experientia, 1991, 47, 1186–1193 CrossRef CAS.
- M. T. B. Davis and J. F. Preston, Science, 1981, 213, 1385–1388 CrossRef CAS PubMed.
- G. Moldenhauer, A. V. Salnikov, S. Luttgau, I. Herr, J. Anderl and H. Faulstich, J. Natl. Cancer Inst., 2012, 104, 622–634 CrossRef CAS.
- S. Park, S. Y. Kim, J. Cho, D. Jung, D. Seo, J. Lee, S. Lee, S. Yun, H. Lee, O. Park, B. Seo, S. Kim, M. Seol, S. H. Woo and T. K. Park, Bioconjugate Chem., 2019, 30, 1969–1978 CrossRef CAS PubMed.
- S. Park, S. Y. Kim, J. Cho, D. Jung, D. Seo, J. Lee, S. Lee, S. Yun, H. Lee, O. Park, B. Seo, S. H. Woo and T. K. Park, Bioconjugate Chem., 2019, 30, 1957–1968 CrossRef CAS PubMed.
- T. Hechler, M. Kulke, C. Mueller, A. Pahl and J. Anderl, Cancer Res., 2014, 74, 1 Search PubMed.
- L. Bodero, P. L. Rivas, B. Korsak, T. Hechler, A. Pahl, C. Muller, D. Arosio, L. Pignataro, C. Gennari and U. Piarulli, Beilstein J. Org. Chem., 2018, 14, 407–415 CrossRef CAS PubMed.
- V. Figueroa-Vazquez, J. Ko, C. Breunig, A. Baumann, N. Giesen, A. Palfi, C. Muller, C. Lutz, T. Hechler, M. Kulke, C. Muller-Tidow, A. Kramer, H. Goldschmidt, A. Pahl and M. S. Raab, Mol. Cancer Ther., 2021, 20, 367–378 CrossRef CAS PubMed.
- L. Zhao, J. P. May, A. Blanc, D. J. Dietrich, A. Loonchanta, K. Matinkhoo, A. Pryyma and D. M. Perrin, ChemBioChem, 2015, 16, 1420–1425 CrossRef CAS.
- A. Moshnikova, V. Moshnikova, O. A. Andreev and Y. K. Reshetnyak, Biochemistry, 2013, 52, 1171–1178 CrossRef CAS.
- D. Weerakkody, A. Moshnikova, N. S. El-Sayed, R.-C. Adochite, G. Slaybaugh, J. Golijanin, R. K. Tiwari, O. A. Andreev, K. Parang and Y. K. Reshetnyak, Sci. Rep., 2016, 6, 1–13 CrossRef.
- F. Gallo, B. Korsak, C. Muller, T. Hechler, D. Yanakieva, O. Avrutina, H. Kolmar and A. Pahl, J. Med. Chem., 2021, 64, 4117–4129 CrossRef CAS PubMed.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS.
- K. Matinkhoo, A. Pryyma, M. Todorovic, B. O. Patrick and D. M. Perrin, J. Am. Chem. Soc., 2018, 140, 6513–6517 CrossRef CAS PubMed.
- M. A. J. Siegert, C. H. Knittel and R. D. Sussmuth, Angew. Chem., Int. Ed., 2020, 59, 5500–5504 CrossRef CAS.
- C. Lutz, W. Simon, S. Werner-Simon, A. Pahl and C. Muller, Angew. Chem., Int. Ed., 2020, 59, 11390–11393 CrossRef CAS.
- Q. Wang, K. Graham, T. Schauer, T. Fietz, A. Mohammed, X. X. Liu, J. Hoffend, U. Haberkorn, M. Eisenhut and W. Mier, Nucl. Med. Biol., 2004, 31, 21–30 CrossRef CAS.
- P. A. Whetstone, H. Akizawa and C. F. Meares, Bioconjugate Chem., 2004, 15, 647–657 CrossRef CAS.
- B. Redko, E. Ragozin, B. Andreii, T. Helena, A. Amnon, S. Z. Talia, O.-H. Mor, K. Genady and G. Gary, Pept. Sci., 2015, 104, 743–752 CrossRef CAS.
- J. B. Engel, A. V. Schallly, J. Dietl, L. Rieger and A. Honig, Mol. Pharmaceutics, 2007, 4, 652–658 CrossRef CAS.
- L. C. Sun, L. V. Mackey, J. Luo, J. A. Fuselier and D. H. Coy, Clin. Med. Oncol., 2008, 2, 491–499 CrossRef PubMed.
- Y. Menda and D. Kahn, Semin. Nucl. Med., 2002, 32, 92–96 CrossRef PubMed.
- M. de Jong, W. A. Breeman, D. J. Kwekkeboom, R. Valkema and E. P. Krenning, Acc. Chem. Res., 2009, 42, 873–880 CrossRef CAS PubMed.
- A. Pahl, C. Lutz and T. Hechler, Drug Discovery Today Technol., 2018, 30, 85–89 CrossRef.
- G. Barbantibrodano and L. Fiume, Nature, 1973, 243, 281–283 CrossRef CAS.
- K. W. Swiderska, A. Szlachcic, L. Opalinski, M. Zakrzewska and J. Otlewski, Int. J. Mol. Sci., 2018, 19, 16 Search PubMed.
- M. A. Krzyscik, L. Opalinski and J. Otlewski, Mol. Pharmaceutics, 2019, 16, 3588–3599 CrossRef CAS.
- A. Pryyma, K. Matinkhoo, A. A. Wong and D. M. Perrin, Chem. Sci., 2020, 11(43), 11927–11935 RSC.
- P. B. Lawrence and J. L. Price, Curr. Opin. Chem. Biol., 2016, 34, 88–94 CrossRef CAS PubMed.
- A. Maruani, M. E. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- L. R. Jones, E. A. Goun, R. Shinde, J. B. Rothbard, C. H. Contag and P. A. Wender, J. Am. Chem. Soc., 2006, 128, 6526–6527 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- C. Muniesa, V. Vicente, M. Quesada, S. Saez-Atienzar, J. R. Blesa, I. Abasolo, Y. Fernandez and P. Botella, RSC Adv., 2013, 3, 15121–15131 RSC.
- P. A. Wender, E. A. Goun, L. R. Jones, T. H. Pillow, J. B. Rothbard, R. Shinde and C. H. Contag, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10340–10345 CrossRef CAS.
- W. Zhang, J. J. Song, B. Z. Zhang, L. W. Liu, K. R. Wang and R. Wang, Bioconjugate Chem., 2011, 22, 1410–1415 CrossRef CAS PubMed.
- M. Sengee, J. J. Eksteen, S. L. Nergard, T. Vasskog and L. K. Sydnes, Bioconjugate Chem., 2019, 30, 1489–1499 CrossRef CAS.
- I. R. Vlahov, F. You, H. K. R. Santhapuram, Y. Wang, J. F. Vaughn, S. J. Hahn, P. J. Kleindl, M. J. Fan and C. P. Leamon, Bioorg. Med. Chem. Lett., 2011, 21, 1202–1205 CrossRef CAS.
- D. L. Zhang, T. H. Pillow, Y. Ma, J. dela Cruz-Chuh, K. R. Kozak, J. D. Sadowsky, G. D. L. Philips, J. Guo, M. Darwish, P. Fan, J. T. Chen, C. R. He, T. Wang, H. Yao, Z. J. Xu, J. H. Chen, J. Wai, Z. H. Pei, C. Hop, S. C. Khojasteh and P. S. Dragovich, ACS Med. Chem. Lett., 2016, 7, 988–993 CrossRef CAS PubMed.
- A. K. Jain, M. G. Gund, D. C. Desai, N. Borhade, S. P. Senthilkumar, M. Dhiman, N. K. Mangu, S. V. Mali, N. P. Dubash, S. Halder and A. Satyam, Bioorg. Chem., 2013, 49, 40–48 CrossRef CAS PubMed.
- S. A. Kularatne, C. Venkatesh, H. K. R. Santhapuram, K. Wang, B. Vaitilingam, W. A. Henne and P. S. Low, J. Med. Chem., 2010, 53, 7767–7777 CrossRef CAS PubMed.
- J. Lu, F. Jiang, A. P. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 22 Search PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- S. O. Doronina, B. A. Mendelsohn, T. D. Bovee, C. G. Cerveny, S. C. Alley, D. L. Meyer, E. Oflazoglu, B. E. Toki, R. J. Sanderson and R. F. Zabinski, Bioconjugate Chem., 2006, 17, 114–124 CrossRef CAS PubMed.
- I. E. Valverde, A. F. Delmas and V. Aucagne, Tetrahedron, 2009, 65, 7597–7602 CrossRef CAS.
- V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505–4507 CrossRef CAS.
- O. D. Montagnat, G. Lessene and A. B. Hughes, Tetrahedron Lett., 2006, 47, 6971–6974 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- A. Pryyma, S. Gunasekera, J. Lewin and D. M. Perrin, Bioconjugate Chem., 2020, 31, 2685–2690 CrossRef CAS.
- B. Q. Wei, J. Gunzner-Toste, H. Yao, T. Wang, J. Wang, Z. J. Xu, J. H. Chen, J. Wai, J. Nonomiya, S. P. Tsai, J. Chuh, K. R. Kozak, Y. C. Liu, S. F. Yu, J. Lau, G. M. Li, G. D. Phillips, D. Leipold, A. Kamath, D. A. Su, K. Y. Xu, C. Eigenbrot, S. Steinbacher, R. Ohri, H. Raab, L. R. Staben, G. L. Zhao, J. A. Flygare, T. H. Pillow, V. Verma, L. A. Masterson, P. W. Howard and B. Safina, J. Med. Chem., 2018, 61, 989–1000 CrossRef CAS PubMed.
- U. Bermbach and H. Faulstich, Biochemistry, 1990, 29, 6839–6845 CrossRef CAS.
- V. Almendro, A. Marusyk and K. Polyak, in Annual Review of Pathology: Mechanisms of Disease, Vol 8, ed. A. K. Abbas, S. J. Galli and P. M. Howley, Annual Reviews, Palo Alto, 2013, vol. 8, pp. 277–302 Search PubMed.
- S. J. Altschuler and L. F. Wu, Cell, 2010, 141, 559–563 CrossRef CAS PubMed.
- M. K. Jolly, P. Kulkarni, K. Weninger, J. Orban and H. Levine, Front. Oncol., 2018, 8, 50 CrossRef.
- A. Zimmer, S. Amar-Farkash, T. Danon and U. Alon, BMC Syst. Biol., 2017, 11, 33 CrossRef PubMed.
- M. Kaern, T. C. Elston, W. J. Blake and J. J. Collins, Nat. Rev. Genetics, 2005, 6, 451–464 CrossRef CAS PubMed.
- Z. Inde and S. J. Dixon, Crit. Rev. Biochem. Mol. Biol., 2018, 53, 99–114 CrossRef CAS PubMed.
- K. Letschert, H. Faulstich, D. Keller and D. Keppler, Toxicol. Sci, 2006, 91, 140–149 CrossRef CAS.
- H. Faulstich, H. Trischmann, T. Wieland and E. Wulf, Biochemistry, 1981, 20, 6498–6504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00036e |
| This journal is © The Royal Society of Chemistry 2022 |