
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePurely organic light-harvesting phosphorescence energy transfer by β-cyclodextrin pseudorotaxane for mitochondria targeted imaging†
Fang-Fang
Shen
 ,
Yong
Chen
,
Xianyin
Dai
,
Hao-Yang
Zhang
,
Bing
Zhang
,
Yaohua
Liu
and
Yu
Liu
*
,
Yong
Chen
,
Xianyin
Dai
,
Hao-Yang
Zhang
,
Bing
Zhang
,
Yaohua
Liu
and
Yu
Liu
*
College of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn
First published on 9th December 2020
Abstract
A new type of purely organic light-harvesting phosphorescence energy transfer (PET) supramolecular assembly is constructed from 4-(4-bromophenyl)-pyridine modified β-cyclodextrin (CD-PY) as a donor, cucurbit[8]uril (CB[8]) as a mediator, rhodamine B (RhB) as an acceptor, and adamantane modified hyaluronic acid (HA-ADA) as a cancer cell targeting agent. Interestingly, the complexation of free CD-PY, which has no RTP emission in aqueous solution, with CB[8] results in the formation of CD-PY@CB[8] pseudorotaxane with an RTP emission at 510 nm. Then the addition of RhB leads to an efficient light-harvesting PET process with highly efficient energy transfer and an ultrahigh antenna effect (36.42) between CD-PY@CB[8] pseudorotaxane and RhB. Importantly, CD-PY@CB[8]@RhB assembles with HA-ADA into nanoparticles with further enhanced delayed emission at 590 nm. The nanoparticles could be successfully used for mitochondria targeted imaging in A549 cancer cells. This aqueous-state PET based on a supramolecular assembly strategy has potential application in delayed fluorescence cell imaging.
Introduction
Purely organic emitting materials with room-temperature phosphorescence (RTP) have shown great potential in bioimaging,1–3 anti-counterfeiting materials,4,5 organic light-emitting diodes,6–8 and so on.9–12 Accelerating intersystem crossing (ISC) between singlet and triplet excited states and suppressing non-radiative transitions and intermolecular collisions have been proven to contribute to efficient RTP. Several strategies have been reported to obtain efficient RTP based on the above two methods, such as introducing heavy halogen atoms13,14 or carbonyl groups,15–17 crystallization,18–23 polymerization,24–30 and so forth.31–36 Although many breakthroughs have been made in the research of purely organic phosphorescent materials in recent years, purely organic phosphorescence energy transfer (PET) in the aqueous state especially light-harvesting PET systems is very rarely reported, and most PET systems reported so far are organometallic or solid materials.37–40 For example, Yan and co-workers put forward a strategy to achieve a high-efficiency PET system by incorporating both the donor (isophthalic acid) and the acceptor (eosin Y) into 2D Zn–Al-LDH nanosheets.41 George reported phosphorescence energy transfer in purely organic donor–acceptor (coronene tetracarboxylate salt – sulpharhodamine 101) as a strategy to realize “afterglow fluorescence” in the solid state.40In recent years, supramolecular complexation and assembly strategies have been used extensively to construct RTP systems.42–45 As the main supramolecular macrocyclic molecules, cyclodextrins (CDs)46 and cucurbiturils (CBs)47 with hydrophobic cavities can interact with guest molecules through supramolecular interactions, such as electrostatic interactions, hydrogen bonding, hydrophobic effects and so forth.48–51 Moreover, the advantages of using macrocyclic molecules to achieve RTP are as follows: (1) stable host–guest complexation can provide a rigid environment for guest molecules, inhibiting non-radiative transitions caused by molecular rotation; (2) hydrophobic cavities can protect phosphorescence from quenching by water or oxygen from the outside.
More recently, some interesting purely organic RTP systems in aqueous solution have also been reported.53 Wu and Chen reported self-assembled nanoparticles with a red RTP from organic difluoroboron β-diketonate compounds in aqueous solution.54 Ma and co-workers reported that triazinyl-bridged 4-(4-bromophenyl)-pyridine and CB[8] could form a peculiar 2:2 quaternary structure with a yellow RTP emission via “assembling-induced emission”. Additionally, tunable photoluminescence by adding different amounts of CB[8] can be achieved in the solution state.55 Inspired by these interesting RTP phenomena based on self-assembly or supra-molecular strategies in solution and examples of highly efficient solid-state PET mentioned above, we wish to report the first supramolecular assembly with a purely organic light-harvesting PET system in solution.
In our research, phosphor group 4-(4-bromophenyl)-pyridine modified β-cyclodextrin (CD-PY) has no phosphorescence in aqueous solution and can form pseudorotaxane with CB[8] accompanied by the emergence of green phosphorescence at 510 nm (Scheme 1). To our delight, with the further addition of a small amount of RhB, the obtained CD-PY@CB[8]@RhB complexes showed a new delayed fluorescence emission (590 nm) based on a light-harvesting PET system through host–guest interactions. As a result, the PET system demonstrated high energy transfer efficiency (84%) and exhibited an ultrahigh antenna effect (36.42) when the ratio of acceptor/donor was 1/50. More interestingly, CD-PY@CB[8]@RhB complexes continued to assemble with adamantane modified hyaluronic acid (HA-ADA), which was used as a cancer cell targeting agent, into nanoparticles, resulting in the enhancement of the delayed emission intensity of RhB by suppressing non-radiative decay and shielding quenchers. In addition, the nanoparticles could aggregate in the mitochondria of A549 cancer cells.
 | ||
| Scheme 1 Construction of the supramolecular assembly for a purely organic light-harvesting PET system and related molecules. | ||
The phosphor unit 4-(4-bromophenyl)-pyridine modified β-CD (CD-PY) was synthesized according to Tian's method56 (Fig. S1–S3†). As reported in previous literature, cucurbit[n]urils (CB[n]s) could complex with some positively charged phosphorescent guest molecules and enhance their phosphorescence intensity in the solid state or induce visible phosphorescence in the liquid state.42,43,55 In order to investigate the effect of host–guest interactions on the optical properties of the CD-PY molecule in aqueous solution, we selected two macrocyclic host molecules, i.e. CB[7] and CB[8]. The host–guest binding mode of CB[7,8] and CD-PY was investigated by 1H NMR experiments. In 1H NMR titration spectra (Fig. 1), with the gradual addition of CB[8] into the aqueous solution of CD-PY, peaks related to protons Ha, Hb, Hc, and Hd of CD-PY all underwent upfield shifts, indicating the complexation of the 4-(4-bromophenyl)-pyridine (PY) unit within the cavity of CB[8]. When the amount of CB[8] increased to >0.5 eq., proton chemical shifts were no longer changed. The final chemical shifts of Hd, Hc, Ha, and Hb were 0.02 ppm, 0.46 ppm, 0.98 ppm, and 1.25 ppm respectively. Moreover, through analysis of the 2D ROSEY spectrum of CD-PY in the presence of 0.5 equiv. of CB[8] (Fig. S5†), a strong ROE cross-peak between Hb and Hc was easily observed, suggesting that the two protons must come from two head–tail-stacking PY units. These results provided evidence that two CD-PY molecules were encapsulated into the cavity of CB[8] with a head-to-tail orientation to form a 2:1 complex. In addition, the results of the 1H NMR titration experiment demonstrated that the stoichiometric ratio of CD-PY![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CB[7] was 1:1 (Fig. S6†).
:CB[7] was 1:1 (Fig. S6†).
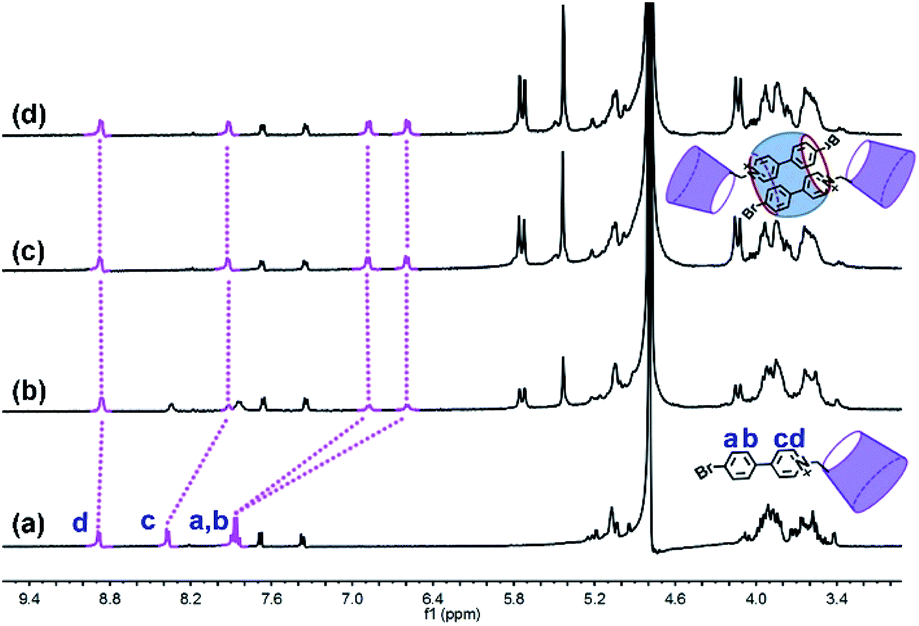 | ||
| Fig. 1 1H NMR titration spectra (400 MHz, D2O, 298 K) of CD-PY (1.0 mM) in the presence of (a) 0.00, (b) 0.25, (c) 0.50, and (d) 0.70 equiv. of CB[8]. | ||
Subsequently, the optical properties of the host–guest complexes were characterized with UV/vis absorption and PL emission experiments in aqueous solution. The absorption spectra of CD-PY displayed a strong absorption peak at around 311 nm, when CB[7] or CB[8] was added to the aqueous solution of CD-PY; the absorption peak at 311 nm gradually decreased with a clear red shift (Fig. S7 and S8†), accompanied by the formation of two isosbestic points at 252 nm and 322 nm for CB[7], and 256 nm and 326 nm for CB[8], respectively. The above results indicated that CB[7] or CB[8] and CD-PY formed host–guest complexes. It's known that free phosphorescent molecule 4-(4-bromophenyl)-N-methylpyridinium can emit phosphorescence in the crystalline state but not in aqueous solution.42 From photoluminescence emission spectra, free guest molecule CD-PY revealed just an emission peak centered at around 384 nm (Fig. 2a). In the gated spectra (Fig. 2b, delayed 0.1 ms), the peak at 384 nm disappeared, and only the emission peak at 510 nm was observed, revealing the short-lived emission at 384 nm and long-lived emission at 510 nm, respectively. We speculated that the emission peak that gradually became intense at 510 nm may be attributed to phosphorescence emission. When N2 was introduced into the aqueous solution of CD-PY@CB[8], the emission peak at 510 nm was further enhanced. However, the emission peak at 384 nm was insensitive to O2, and no change occurred. Meanwhile, the time-resolved decay curves of free CD-PY (384 nm) and pseudorotaxane CD-PY@CB[8] (384 nm and 510 nm) were measured under ambient conditions. In Fig. S9,† the lifetime measured at 384 nm was on a picosecond scale (204 ps for CD-PY and 235 ps for CD-PY@CB[8]), revealing that it was a typical fluorescence emission. In Fig. 2d, the lifetime at 510 nm showed a microsecond scale (504 μs). These above results confirmed our previous speculation about phosphorescence identification of 510 nm.
 | ||
| Fig. 2 The prompt photoluminescence (a and c) (inset of (c): the enlarged luminescence peak at 510 nm) and gated (0.1 ms) spectra (b) of CD-PY with or without CB[7,8] and N2 in aqueous solution. (d) Decay curve of CD-PY@CB[8] (1:0.5) aqueous solution at 510 nm ([CD-PY] = 0.5 mM,44,54,60 Ex = 340 nm, 298 K). | ||
In contrast, after adding 1.0 eq. CB[7] into CD-PY aqueous solution, the emission peak of the CD-PY molecule at 384 nm did not disappear but increased instead, and no new peak appeared near 510 nm (Fig. 2c). Even if we injected N2 into the CD-PY@CB[7] (1:1) aqueous solution, phosphorescence was hardly observed (Fig. 2c). Under the gated mode, the peak around 510 nm was still very weak (Fig. 2b). Besides, the lifetime of CD-PY@CB[7] (1:1) aqueous solution at 510 nm was 25.8 μs (Fig. S10 and Table S1†), which was much smaller than that of pseudorotaxane CD-PY@CB[8], suggesting CB[8] was more capable of suppressing the non-radiative relaxation of CD-PY molecules than CB[7]. In view of these results, the generation of effective RTP in aqueous solution could be attributed to the fact that CB[8] not only prevents the quenching of triplet oxygen in aqueous solution, but also effectively inhibits the molecular vibration of CD-PY molecules. Furthermore, isothermal titration calorimetry (ITC) experiments were carried out to study the binding constants (KS) between CD-PY and CB[7,8]. As shown in Fig. S12,† the titration data were well fitted by means of the “two successive binding sites” model of computer simulation. The value of KS was calculated to be (2.06 ± 0.69) × 1013 M−2. In addition, the UV/vis titration data also gave a bonding constant as high as 1013 M−2 (Fig. S8†), implying an extremely strong bonding ability between CB[8] and the PY unit. On the other hand, the titration data of CD-PY@CB[7] were well-fitted using the “one set of binding sites” of computer simulation, giving a KS value of (8.08 ± 0.62) × 106 M−1, which was consistent with the UV/vis titration data (Fig. S7 and S11†). These results indicated that a more stable 1:2 inclusion complex formed between CB[8] and CD-PY (a higher association constant) than CD-PY@CB[7], in which two CD-PY units with strong π–π stacking interactions are present inside the cavity of CB[8], resulting in stronger phosphorescence emission than CD-PY@CB[7].
The interesting RTP properties of pseudorotaxane CD-PY@CB[8] in aqueous solution prompted us to fabricate an efficient phosphorescence energy transfer (PET) system in the solution state, and the RhB dye was selected as the acceptor in this PET system. As shown in Fig. 3a, the absorption spectrum of β-CD@RhB could overlap with the phosphorescence emission of pseudorotaxane CD-PY@CB[8] to a certain degree, indicating that a phosphorescence energy transfer (PET) process could take place between them. The complexes of β-CD@RhB and CD-PY@CB[8]@RhB were investigated using 1H NMR experiments. Through the analysis of chemical shifts of 1H NMR signals and the ROE cross-peaks of the 2D ROSEY spectrum (Fig. S13–S15†), we found that β-CD and RhB could form a 1:1 stable inclusion complex, where one of the diaminoethyl sides of RhB entered the β-CD cavity via the secondary hydroxyl rim. This is consistent with the results of previously reported literature.57 Furthermore, the molecular mechanics calculation results showed that the distance between RhB and the PY unit in CD-PY@CB[8]@RhB was 17.2 Å (Fig. S16†), providing theoretical support for the realization of the PET process.
 | ||
| Fig. 3 (a) Normalized absorption spectrum of β-CD@RhB and the gated emission spectrum of CD-PY@CB[8] (1:0.5). (b) Gated spectra (0.1 ms) of CD-PY@CB[8] (1:0.5) in aqueous solution with different concentrations of RhB. Inset: (1) Photographs of CD-PY@CB[8] (1:0.5), and (2) CD-PY@CB[8]@RhB (1:0.5:0.02) under UV light (365 nm). (c) Gated spectra (0.1 ms) of CD-PY@CB[8]@RhB (1:0.5:0.02) from 298 to 200 K. (d) Decay curve of CD-PY@CB[8]@RhB (1:0.5:0.02) at 590 nm at 200 K. (e) A possible mechanism diagram of the phosphorescence energy transfer (PET) process for the CD-PY@CB[8]@RhB system (Abs. stands for absorption, ISC for intersystem crossing, Fluo for fluorescence, Phos for phosphorescence and DF for delayed fluorescence). ([CD-PY] = 0.5 mM, Ex = 340 nm, Ex. slit = 10, Em. slit = 10, 298 K). | ||
As expected, in the gated spectra (Fig. 3b, delayed 0.1 ms), with the gradual addition of RhB to pseudorotaxane CD-PY@CB[8], the phosphorescence intensity at 510 nm decreased, accompanied by a blue shift from 510 nm to 486 nm, while a new emission peak at 580 nm emerged with a slight bathochromic shift of 10 nm when excited at 340 nm. These results could be ascribed to the simultaneous excitation of free and included RhB in different ratios upon excitation at 340 nm with the process of host–guest interactions. The colour of the solution changed from green to yellow during irradiation with 365 nm UV light. Hence, it implies that an efficient energy transfer happened from the triplet state of CD-PY@CB[8] to the RhB molecules. Furthermore, the normalized gated emission of CD-PY@CB[8]@RhB at 590 nm is identical to the fluorescence emission of pure CD-PY@RhB, which clearly indicates a typical delayed-fluorescence (DF) character of RhB emission (Fig. S18†). A temperature-dependent experiment was further conducted to study the DF. When lowering the temperature from room temperature to 200 K, the DF intensity enhanced notably with a lifetime of 780 μs (Fig. 3c and d) indicating that the delayed emission at 590 nm was not thermally activated delayed fluorescence (TADF). Interestingly, we found that the emission intensity of RhB at 200 K is about 100-fold higher than that at 298 K, while the emission intensity of CD-PY at 200 K is only 10-fold higher than that at 298 K (Fig. 3c). The stronger RTP and DF emission and longer lifetimes under low-temperature conditions may be ascribed to the cryogenic temperature providing a more fixed microenvironment to suppress the molecular movement. Additionally, such a significant difference in the increase of RTP and DF emission intensity at the two wavelengths may be because the carboxyphenyl ring of RhB which was not covered by the β-CD cavity can rotate in aqueous solution compared to the PY unit that was tightly immobilized by CB[8], and the molecular rotation was restrained in the low-temperature environment, so DF emission of RhB increased more than RTP intensity. As we blow N2 into the solution with a donor–acceptor ratio of 50:1, we can see that the emission intensity 500 nm and 590 nm both further enhanced, suggesting that the DF emission of the acceptor comes from the RTP emission of the donor (Fig. S17†).
However, when the ratio of acceptor/donor is higher than 1/136, the increase of the amount of RhB weakened the DF intensity at 590 nm. The reason for this phenomenon may be due to the aggregation-caused quenching (ACQ) effect. To further confirm the ACQ effect of DF emission, the gated spectra of CD-PY@RhB supramolecular complexes in the solid state were recorded to determine the DF emission lifetime (Fig. S19 and S20†). Compared with CD-PY, the DF emission decay of RhB in CD-PY@RhB showed a decrease in the lifetime from 229 to 102 μs when the ratio of acceptor/donor reached 1:100 and 3:100, respectively (Fig. S20 and Table S1†), which also indicates the ACQ effect of DF emission. Moreover, it should be mentioned that the phosphorescence lifetime of 510 nm was drastically reduced from 504 μs to 156 μs upon addition of RhB, which further validated the occurrence of the PET process via triplet-to-singlet Förster resonance energy transfer (TS-FRET) in water (Fig. 2d, 3e, S24a and Table S1†). Singlet–singlet FRET is negligible in the present system due to the smaller spectral overlap, and the fluorescence lifetime decay profiles of the donor monitored at 383 nm hardly changed clearly indicating that TS-FRET is predominant (Fig. S9 and S21†). As can be seen in Fig. S26 and S27,† the energy transfer efficiency and the antenna effect value were calculated to be 84% and 36.42 when the ratio of acceptor/donor reached 1/50. To the best of our knowledge, the antenna effect value is relatively high compared to that of the recently reported artificial light-harvesting triplet energy transfer system.39 As a control, there was no appreciable DF emission of CD-PY@RhB under direct excitation at 340 nm (Fig. S22†), revealing that CB[8] plays an important role in the light-harvesting PET process to result in DF emission of RhB in the solution.
Additionally, taking advantage of the strong binding ability between β-CD and adamantane,51,52 HA-ADA was introduced into this PET system in aqueous solution to interact with the β-CD unit. Learning from previous reports,58,59 hyaluronic acid (HA) that possesses excellent aqueous solubility, biocompatibility, and biodegradability can specifically recognize HA receptors (CD44 and RHAMM receptors) over expressed on the tumor cell surface in cancer metastasis. The optical changes after adding HA-ADA were investigated. As depicted in Fig. 4a and S23,† when adding HA-ADA to the solution with a donor–acceptor ratio of 50:1, DF intensity at 590 nm increased 2.2-fold, while the intensity at 500 nm remained nearly unchanged. Meanwhile, the lifetimes monitored at 490 nm and 590 nm were 162 μs and 289 μs, respectively (Fig. 4b, S24b and Table S1†). A reasonable explanation for the enhancement of DF intensity at 590 nm is that the hydrogen bond (H-bonds) formed by the –COOH groups of the HA unit and the –OH groups of the CD unit locked the RhB molecules which have been enclosed by β-CD, further suppressing the non-radiative relaxation and shielding quenchers of water effectively. The RTP emission at 500 nm was not significantly enhanced because when HA-ADA was added to the complexes, no similar effect acted on the PY units that were well fixed by CB[8] (Fig. 4a and S23†). In control experiments, there was no appreciable DF emission for the CD-PY@RhB@HA-ADA and RhB@HA-ADA system under the same conditions (Fig. 4a), which confirms again that CB[8] is indispensable for the DF emission of RhB.
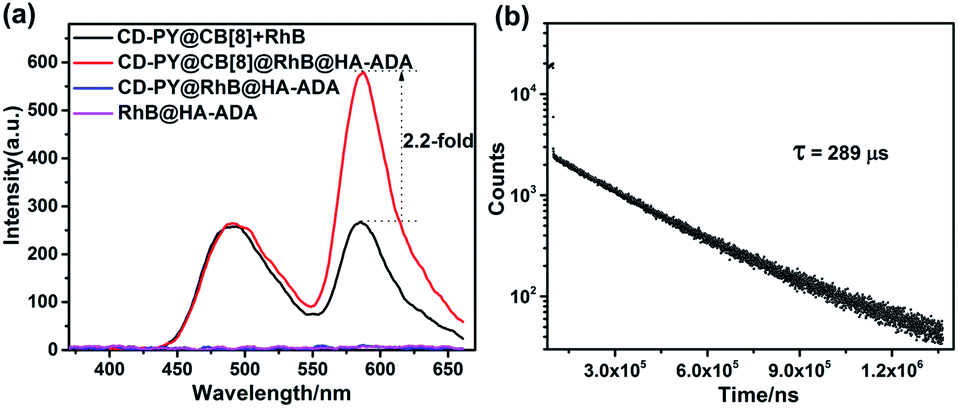 | ||
| Fig. 4 (a) Gated spectra of CD-PY@CB[8]@RhB (1:0.5:0.02), CD-PY@CB[8]@RhB@HA-ADA (1:0.5:0.02:0.31), CD-PY@RhB@HA-ADA (1:0.02:0.31), and RhB@HA-ADA (0.02:0.31). (b) Decay curves of CD-PY@CB[8]@RhB@HA-ADA (1:0.5:0.02:0.31) at 580 nm ([CD-PY] = 0.5 mM, Ex = 340 nm, Ex. slit = 10, Em. slit = 20, 298 K). | ||
In addition, transmission electron microscopy (TEM) and zeta potential experiments were employed to investigate the morphological details and surface charge of CD-PY@CB[8]@RhB@HA-ADA. The TEM images (Fig. 5b) showed that the assembly existed as spherical nanoparticles with an average diameter of ca. 120 nm. In addition, the zeta potential of the CD-PY@CB[8]@RhB@HA-ADA assembly was measured to be ca. −4.55 mV (Fig. 5c). This negatively charged surface with biocompatibility could extend the circulation time of the assembly in the body.
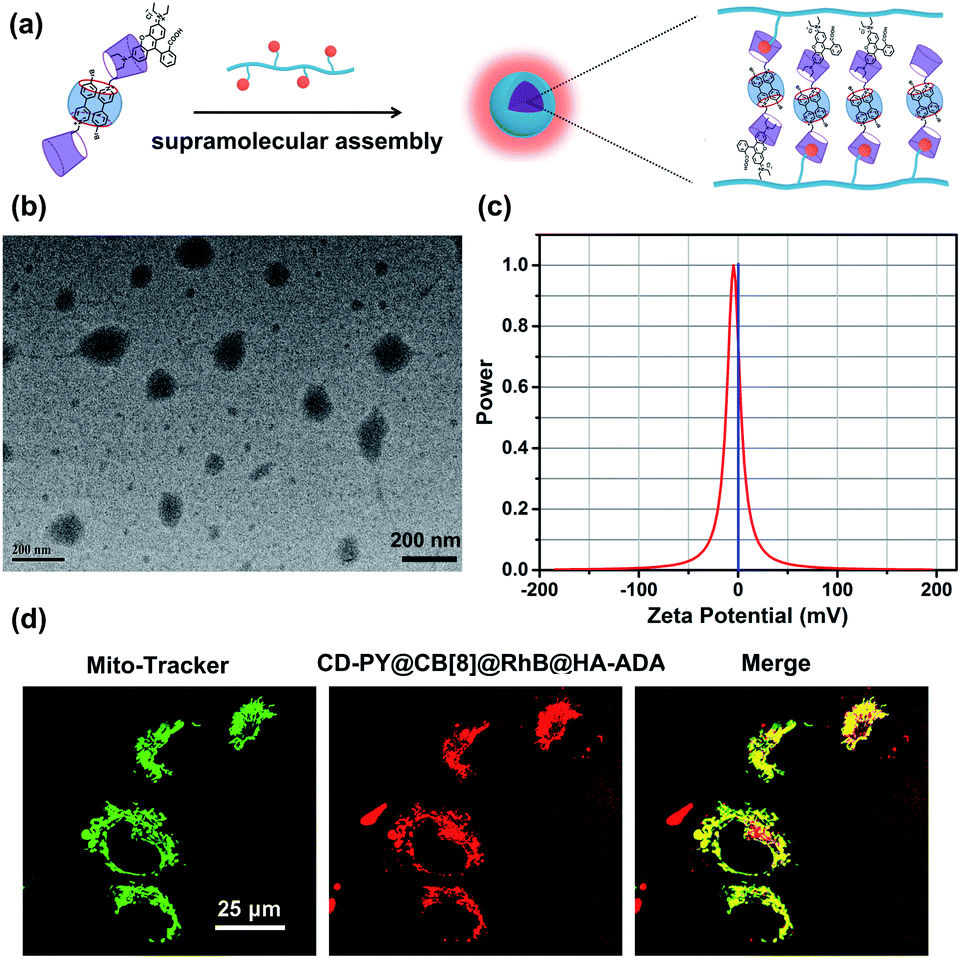 | ||
| Fig. 5 (a) Schematic illustration of the formation of supramolecular nanoparticles. (b) TEM image and (c) zeta potential of CD-PY@CB[8]@RhB@HA-ADA. (d) Laser confocal images of A549 cells co-stained with (1) Mito-Tracker Green, and (2) CD-PY@CB[8]@RhB@HA-ADA (1:0.5:0.02:0.31) ([CD-PY] = 0.005 mM); (3) merged image of (1) and (2). | ||
Benefiting from this solution-state purely organic light-harvesting PET assembly with targeting capability, the cell imaging experiment was carried out to explore the potential of the assembly for the targeted bio-imaging application. Then, human lung adenocarcinoma cells (A549 cells) were treated with CD-PY@CB[8]@RhB@HA-ADA assemblies for 12 h. Confocal laser scanning microscopy was used to examine the intracellular distribution of the CD-PY@CB[8]@RhB@HA-ADA assemblies. A549 cells were co-stained with commercially available mitochondria staining dye Mito-Tracker Green and CD-PY@CB[8]@RhB@HA-ADA assemblies. As shown in Fig. 5d, the merged yellow dyeing site demonstrated that the red emission of CD-PY@CB[8]@RhB@HA-ADA and green Mito-Tracker Green were in good co-localization. Furthermore, the cytotoxicity experiments of CD-PY@CB[8]@RhB@HA-ADA nanoparticles were conducted by CCK8 assays. As shown in Fig. S25,† the nanoparticles showed negligible cytotoxicity to A549 cells.
Conclusions
In summary, a novel supramolecular assembly was constructed to realize purely organic light-harvesting phosphorescence energy transfer (PET) in aqueous solution. By interacting with CB[8] to form pseudorotaxane, the phosphorescence of CD-PY is “turned on” with a green phosphorescence emission in aqueous solution. Interestingly, through the host–guest interaction between β-CD and RhB, efficient solution-state light-harvesting PET occurred between pseudorotaxane and RhB, giving high energy transfer efficiency and an ultrahigh antenna effect (36.42). Most importantly, its secondary assembly with HA-ADA that acts as a targeting agent for cancer cells to form nanoparticles can promote the DF of RhB by suppressing non-radiative relaxation processes and shielding quenchers. Additionally, such nanoparticles can target the mitochondria of A549 cancer cells. This supramolecular assembly strategy paves the way for realizing purely organic PET targeted imaging.Experimental
Instruments and methods
All reagents and solvents were commercially available and used without further purification unless otherwise noted. NMR spectra were recorded on a Brucker AV400 spectrometer. High-resolution mass (HR-MS) spectra were recorded on a Q-TOF LC-MS with an ESI mode. Low resolution mass spectra were recorded on an LCQ-Advantage. UV/vis spectra were recorded on a Thermo Fisher Scientific EVO300 PC spectrophotometer in a conventional rectangular quartz cell (10 × 10 × 45 mm) at 25 °C. Photoluminescence spectra, the lifetime and the phosphorescence quantum efficiency were obtained on an FSP920 and FSP980. TEM experiments were performed on an FEI Tecnai G2 F20 at 200 kV. Fluorescence spectra were measured in a conventional rectangular quartz cell (10 × 10 × 45 mm) on a JASCO FP-750 spectrometer equipped with a constant temperature water bath.Synthesis of CD-PY
Compound CD-PY was synthesized according to the literature.56 6-OTs-β-CD (2.0 g, 0.78 mmol, 2.0 eq.) and 4-(4-bromophenyl)-pyridine (0.36 g, 1.56 mmol, 2.0 eq.) were added into 35 ml dry DMF. The mixture was stirred at 90 °C for 48 h under a N2 atmosphere. The reaction mixture was concentrated and added into a large amount of acetone to precipitate. The crude product was collected and further purified by HPLC (reversed phase) with a water–ethanol (v/v = 90:10) eluent, and the collected fraction was freeze-dried to obtain a white powder in 30% yield. 1H NMR (400 MHz, D2O) δ 8.91 (d, J = 6.8 Hz, 2H), 8.39 (d, J = 6.4 Hz, 2H), 7.89 (dd, J = 19.2, 8.8 Hz, 4H), 7.68 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 7.6 Hz, 2H), 5.15–4.98 (m, 7H), 4.18–3.37 (m, 42H). 13C NMR (100 MHz, D2O) δ 156.25, 145.43, 141.83, 132.77, 129.72, 129.06, 126.79, 125.27, 124.98, 101.89, 83.09, 81.73, 81.08, 73.18, 72.53, 71.84, 70.71, 60.86, 60.25, 59.40, 20.48. HRMS (ESI) m/z: [M − OTs−]+ calcd for [C53H77NO34Br]+, 1350.3510; found, 1350.3510.
Synthesis of HA-ADA
Compound HA-ADA was synthesized according to the literature.61 Hyaluronic acid (0.5 g) was dissolved in 50 ml of DMSO. Then, triethylamine (0.92 ml, 4.0 mmol) was added into the reaction solution and stirred for 1 h at room temperature. At this moment, adamantyl ethylenediamine (146.6 mg, 0.66 mmol) was added and the mixture was stirred continuously at room temperature overnight. Afterwards, the solution was diluted with 50 ml of water and dialyzed (Mw cut off = 3500 Da) against deionized water for 7 days. After dialysis, the sample was freeze-dried as a white powder. From 1H NMR spectra, the degree of substitution (DS) was determined to be 12%.1H NMR (400 MHz, D2O) δ 4.51 (s, 2H), 4.10–3.18 (m, 10H), 1.98 (s, 3H), 1.86–1.56 (m, 1H).Cytotoxicity experiments
Human lung adenocarcinoma cells (A549 cells, purchased from the Cell Resource Center, China Academy of Medical Science Beijing, China) were seeded in 96-well plates at a density of 5 × 104 cells per well in 100 μL complete DMEM containing 10% fetal bovine serum for 24 h at 37 °C in 5% CO2. Then the cells were incubated with CD-PY@CB[8]@RhB@HA-ADA (1:0.5:0.02:0.31) for 24 h. Relative cellular viability was determined by the CCK8 assay.
Confocal laser scanning microscopy
The A549 cells (purchased from the Cell Resource Center, China Academy of Medical Science Beijing, China) were precultured for 24 h and then incubated with CD-PY@CB[8]@RhB@HA-ADA ([CD-PY] = 0.005 mM) for a further 24 h. The cells were stained with Mito-Tracker Green (100 nM), washed three times with PBS, and observed using a confocal laser scanning microscope.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21971127, 21672113, 21772099, and 21861132001).References
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- X. Zhen, C. Xie and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3938–3942 CrossRef CAS PubMed.
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef PubMed.
- Y. Su, Y. Zhang, Z. Wang, W. Gao, P. Jia, D. Zhang, C. Yang, Y. Li and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 9967–9971 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- W. Ratzke, L. Schmitt, H. Matsuoka, C. Bannwarth, M. Retegan, S. Bange, P. Klemm, F. Neese, S. Grimme, O. Schiemann, J. M. Lupton and S. J. Hoeger, J. Phys. Chem. Lett., 2016, 7, 4802–4808 CrossRef CAS PubMed.
- L. Xiao, S. Su, Y. Agata, H. Lan and J. Kido, Adv. Mater., 2009, 21, 1271–1274 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- H. Li, H. Li, W. Wang, Y. Tao, S. Wang, Q. Yang, Y. Jiang, C. Zheng, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2020, 59, 4756–4762 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- X.-Q. Liu, K. Zhang, J.-F. Gao, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 1–6 CrossRef.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- L. Xiao and H. Fu, Chem.–Eur. J., 2019, 25, 714–723 CrossRef CAS PubMed.
- S. Zheng, T. Zhu, Y. Wang, T. Yang and W. Z. Yuan, Angew. Chem., Int. Ed., 2020, 59, 10018–10022 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- H. Ma, H. Yu, Q. Peng, Z. An, D. Wang and Z. Shuai, J. Phys. Chem. Lett., 2019, 10, 6948–6954 CrossRef CAS PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS PubMed.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- K. Jinnai, R. Kabe and C. Adachi, Adv. Mater., 2018, 30, 1800365 CrossRef PubMed.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS PubMed.
- T. Ogoshi, H. Tsuchida, T. Kakuta, T.-a. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- H. Wu, Y. Zhou, L. Yin, C. Hang, X. Li, H. Agren, T. Yi, Q. Zhang and L. Zhu, J. Am. Chem. Soc., 2017, 139, 785–791 CrossRef CAS PubMed.
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734 CrossRef PubMed.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 CrossRef CAS PubMed.
- T. Ono, A. Taema, A. Goto and Y. Hisaeda, Chem.–Eur. J., 2018, 24, 17487–17496 CrossRef CAS PubMed.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS PubMed.
- L. Zhang, R. Zhang, P. Cui, W. Cao and F. Gao, Chem. Commun., 2013, 49, 8102–8104 RSC.
- D. Li, J. Qin and G. Yan, Sens. Actuators, B, 2018, 255, 529–535 CrossRef CAS.
- Z. Li, Y. Han and F. Wang, Nat. Commun., 2019, 10, 3735 CrossRef PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- R. Gao and D. Yan, Chem. Sci., 2017, 8, 590–599 RSC.
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773–7778 RSC.
- Y. Gong, H. Chen, X. Ma and H. Tian, ChemPhysChem, 2016, 17, 1934–1938 CrossRef CAS PubMed.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- Y. Chen and Y. Liu, Adv. Mater., 2015, 27, 5403–5409 CrossRef CAS PubMed.
- Y. Chen, F. Huang, Z.-T. Li and Y. Liu, Sci. China: Chem., 2018, 61, 979–992 CrossRef CAS.
- J.-J. Li, H.-Y. Zhang, Y. Zhang, W.-L. Zhou and Y. Liu, Adv. Opt. Mater., 2019, 1900589 CrossRef CAS.
- F.-F. Shen, Y.-M. Zhang, X.-Y. Dai, H.-Y. Zhang and Y. Liu, J. Org. Chem., 2020, 85, 6131–6136 CrossRef CAS PubMed.
- Z. Liu, W. Zhou, J. Li, H. Zhang, X. Dai, Y. Liu and Y. Liu, Chem. Sci., 2020, 11, 4791–4800 RSC.
- X. Yu, W. Liang, Q. Huang, W. Wu, J. J. Chruma and C. Yang, Chem. Commun., 2019, 55, 3156–3159 RSC.
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- R. Serra-Gómez, G. Tardajos, J. González-Benito and G. González-Gaitano, Dyes Pigm., 2012, 94, 427–436 CrossRef.
- Y. Yang, Y.-M. Zhang, D. Li, H.-L. Sun, H.-X. Fan and Y. Liu, Bioconjugate Chem., 2016, 27, 2834–2838 CrossRef CAS PubMed.
- F.-Q. Li, Q. Yu, Y.-H. Liu, H.-J. Yu, Y. Chen and Y. Liu, Chem. Commun., 2020, 56, 3907–3910 RSC.
- H. Chen, L. Xu, X. Ma and H. Tian, Polym. Chem., 2016, 7, 3989–3992 RSC.
- J. Yu, Y. Chen, Y.-H. Zhang, X. Xu and Y. Liu, Org. Lett., 2016, 18, 4542–4545 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization. See DOI: 10.1039/d0sc05343k |
| This journal is © The Royal Society of Chemistry 2021 |