
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico study of natural compounds from sesame against COVID-19 by targeting Mpro, PLpro and RdRp†
Ahmed E.
Allam
 a,
Yhiya
Amen
b,
Ahmed
Ashour
b,
Hamdy K.
Assaf
a,
Heba Ali
Hassan
c,
Islam M.
Abdel-Rahman
d,
Ahmed M.
Sayed
e and
Kuniyoshi
Shimizu
*f
a,
Yhiya
Amen
b,
Ahmed
Ashour
b,
Hamdy K.
Assaf
a,
Heba Ali
Hassan
c,
Islam M.
Abdel-Rahman
d,
Ahmed M.
Sayed
e and
Kuniyoshi
Shimizu
*f
aDepartment of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Assiut, 71524, Egypt. E-mail: aallam81@yahoo.co.uk
bDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
cDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, Universities Zone, New Minia City 61111, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Deraya University, New-Minia, 61111, Egypt
eDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, Beni-Suef 62513, Egypt
fDepartment of Agro-Environmental Sciences, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, Fukuoka, Japan 819-0395
First published on 24th June 2021
Abstract
Natural products and traditional medicine products with known safety profiles are a promising source for the discovery of new drug leads. Natural products as sesame were reported to exhibit potential to protect from COVID-19 disease. In our study, the total methanolic extract of Sesamum indicum L. seeds (sesame) were led to isolation of seven known compounds, five lignan; sesamin 1, sesamolin 2, pinoresinol 3, hydroxymatairesinol 6, spicatolignan 7, together with two simple phenolic compounds; ferulic acid 4 and vanillic acid 5. All isolated compounds were evaluated in silico against three important SARS-CoV-2 protein targets; main protease (Mpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp) which possessed crucial role in replication and proliferation of the virus inside the human cell. The results revealed that compound 6 has the high affinity against the three main proteins, specially towards the SARS-CoV-2 Mpro that exceeded the currently used SARS-CoV-2 Mpro inhibitor darunavir as well as, exhibiting a similar binding energy at SARS CoV-2 PLpro when compared with the co-crystallized ligand. This activity continued to include the RdRp as it displayed a comparable docking score with remdesivir. Inferiorly, compounds 1 and 2 showed also similar triple inhibitory effect against the three main proteins while compound 7 exhibited a dual inhibitory effect against SARS CoV-2 PLPro and RdRp. Further molecular dynamic simulation experiments were performed to validate these docking experiments and to calculate their binding free energies (ΔGs). Compounds 1, 2, 3, 6, and 7 showed comparable binding stability inside the active site of each enzyme with ΔG values ranged from −4.9 to −8.8 kcal mol−1. All the compounds were investigated for their ADME and drug likeness properties, which showed acceptable ADME properties and obeying Lipinski's rule of five parameters. It can be concluded that the isolated compounds from sesame lignans could be an alternative source for the development of new natural leads against COVID-19.
1. Introduction
COVID-19, a newly emerged respiratory disease, is a worldwide pandemic which could cause death. The symptoms of COVID-19 range from mild to moderate including fever, headaches, dry coughing, dyspnea, diarrhea, loss of taste, fatigue, viral pneumonia, acute respiratory distress, and hypoxia. In severe cases, COVID-19 patients require oxygen therapy and mechanical ventilation.2 SARS-CoV-2 infection can activate innate and adaptive immune responses. However, uncontrolled inflammatory innate responses and impaired adaptive immune responses may lead to harmful tissue damage, both locally and systemically.2 Several antiviral drugs, including the nucleotide analogue remdesivir, are being actively tested; none has been specifically approved for COVID-19. Along with vaccine development and approaches that target the viral block, treatment therapies that address the immunopathology of the contagion have become a major objective.Sesame (Sesamum indicum L.) is one of the first crops processed for oil production. It is edible seeds and has a rich source of protein, as well as, one of the earliest condiments.3 Sesame seed contains about 50–60% of oil which is rich in different bioactive compounds including polyunsaturated fatty acids, tocopherol homologues, in addition to, lignans.4 These bioactive constituents enhance the stability and keeping quality of sesame oil along with numerous health benefits and have some important actions such as lowering cholesterol in humans, preventing high blood pressure and increasing vitamin E supplies in animals.5 Moreover, antioxidant and anti-carcinogenic activities of sesame seed have greatly increased its applications in health food products that assert for liver and heart protection and tumor prevention.6
A folk method expressed that adding sesame oil into the nostrils can prevent the spread of SARS-CoV-2. To find theoretical reasons to support this method for preventing viral infection with SARS-CoV-2, Fan et al. discussed from the perspective of colloid and interface science that sesame oil had a low surface tension and was incompatible with water.7–9 Recently, the Indian government gave similar recommendations that instilling two drops of sesame oil in each nostril every morning to prevent the new coronavirus.10 Sesame oil reported as having the ability to prevent the spread of SARS-CoV-2 due to the physical–chemical properties of sesame oil, such as low surface tension, high-boiling point, high viscosity, immiscible with water and antivirus activity.9
Several drug discovery approaches like quantitative structure–activity relationship (QSAR), virtual screening (VS), artificial intelligence and drug repositioning, are strongly required to help in discovering a treatment of the uncontrolled pandemic caused by SARSCoV-2.11 Numerous molecular studies were addressed to figure out the active sites of the SARS-CoV-2. Main protease (Mpro).12 Papain-like protease (PLpro).13 and RNA-dependent RNA polymerase (RdRp).14 of SARS-CoV-2 are some of the decisive factors in the infectious route of the virus, they have been reported as important targets for therapeutic strategies.
Main protease (Mpro) is a crystal structure was mainly used to elucidate the mechanism of inhibition by ligand N3.14–18 The recognition of the co-crystallized structure with inhibitor N3 (PDB 6LU7) interaction with human ACE2 (PDB 6M0J) provides a grasp of the active sites and the important amino acids that are responsible for the restraint of SARS-CoV-2.19 Moreover, it is having a precious role in enzymatic activity leading to its post-translational modification of replicase polyproteins.
Papain-like protease (PLpro) is a cysteine protease, proteolytically cleaves the viral polyprotein precursors, pp1a and pp1ab, at three sites to produce non-structural proteins Nsp1, Nsp2, and Nsp3.20 Hence, the two proteases Mpro and PLpro are equally important in the transcription/replication of the virus and for novel discovery of antiviral drug design.21
RNA-dependent RNA polymerase (RdRp), the core component of this complex is the catalytic subunit (Nsp12) of RdRp.22 This enzyme plays a vital role in a complex form with Nsp7 and Nsp8, in the replication of the viral RNA. It is considered as a target of different antiviral drugs encompassing remdesivir especially in its triphosphate form.23 Complex of Nsp12 with Nsp7, Nsp8 together with remdesivir triphosphate and a short RNA sequence have been crystallized (PDB 7BV2). This crystal structure was used to test the ability of the compounds isolated from sesame to be docked in the active site of RdRp, comparing with the position occupied by remdesivir triphosphate.
In the present study, the isolated compounds from sesame were investigated for the binding affinity with an in silico study against three important SARS-CoV-2 protein targets, main protease (Mpro), Papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp), to explore and development of drugs from natural food sources acting on pandemic COVID-19.
2. Results and discussion
2.1. Identification of the isolated compounds
The total methanolic extract of Sesamum indicum L. seeds were subjected to repeated open column chromatography techniques to afford seven known compounds, sesamin 1,24 sesamolin 2,25 pinoresinol 3,26 ferulic acid 4,27 vanillic acid 5,28 hydroxymatairesinol 6.29 which is first isolated from the species Sesamum indicum L. Spicatolignan 7, which is first isolated from the genus Sesamum,30Fig. 1. The structures of known compounds were identified by comparison of their spectroscopic data with those reported in the literature. | ||
| Fig. 1 Structure of the isolated compounds. | ||
2.2. Molecular docking results
The selected seven compounds were screened against the three important protein targets of SARS-CoV-2, i.e., Mpro, PLpro and RdRp by performing molecular docking using the computational program MOE 2019.010.The SARS-CoV-2 interacted first with the transmembrane protein of the human host cell receptor ACE.31 This process also internalizes the virus into the endosomes that allowed the virus to enter the human host cell. Thereafter, the RdRp facilitates the viral genome replication.32 The 3CLpro and PLpro act as proteases in the process of proteolysis of the viral polyprotein into functional units.33 Consequently, the Mpro, PLpro, RdRp responsible for translation of the genetic materials of the virus along with the replication inside the host cell ending with the proliferation of the virus into human cells. Therefore, these three proteins of SARS-CoV-2 play a pivotal role in viral life cycle and were considered as the therapeutic protein targets for the molecular docking with the selected seven compounds.
The X-ray crystallographic structure of Mpro complexed with N3 was obtained from the Protein Data Bank through the internet (http://www.rcsb.org/pdb/, code 6LU7). The substrate-binding site is in a cleft between domain I and II. A Michael acceptor inhibitor known as N3 was developed using computer-aided drug design. N3 is fitted inside the substrate-binding pocket of SARS-CoV-2 virus Mpro showing asymmetric units containing only one polypeptide.
To validate our study, the ligand was re-docked with the active pocket. N3 showed interactions with receptor having HB formations with Thr 190, Glu 166, Gln 189, His 163, His 164 residues (Fig. 2). The docking algorithm was able to predict the co-crystalized ligand pose with low RMSD value of 1.2 Å and binding affinity score of −8.45 kcal mol−1 (Fig. 2). The docking scores of the seven compounds against Mpro′ active site is summarized in Table 1. Five compounds showed docking scores higher than −6.4 kcal mol−1. Interestingly compound 6 (hydroxymatairesinol) showed binding energy score of −7.46 kcal mol−1, which is higher than the docking score of the currently used Mpro inhibitor darunavir (−7.1) kcal mol−1. It showed hydrogen bonding (HB) interaction with Met165 and Ser144 via the phenolic hydroxyl groups in both rings as a HB donor and with Gly143 as a HB acceptor through the aliphatic hydroxyl group. Compound 1 (sesamine) got a docking score of −6.71 kcal mol−1 and exhibited good binding mode with important interactions with Gln 189:HB donor and Thr 190:HB donor as shown in Table 1, similarly compound 2 (sesamolin) displayed the same key binding mode that resembles the binding mode in N3. Compound 7 (specatolignan) achieved an energy score of −6.74 kcal mol−1 forming HB binding via the carboxylic group entity with amino acid residues Glu 166 and Thr 190 as a HB donor and HB acceptor respectively. The order of docking scores was as follows: N3> 3 > darunavir > 2 > 6 > 1 > 4 > 5 > 7.
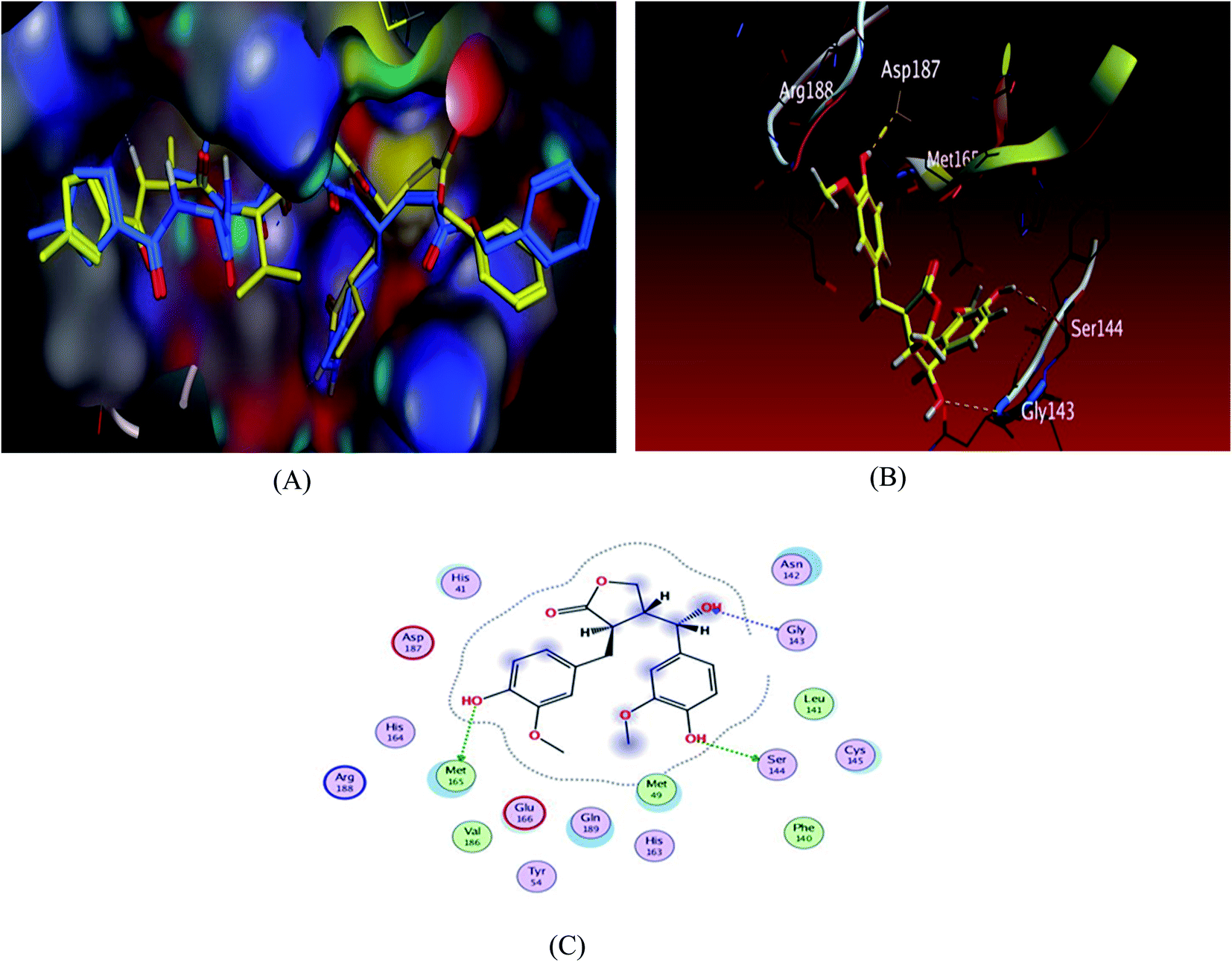 | ||
| Fig. 2 Docking results of compound 3 in the active site of SARS-CoV-2 main protease (6LU7). (A) Validation of docking procedure showing good matching between crystallized (yellow) and docked (blue) ligands. (B) Docking pose of compound 6. (C) 2D interactions of compound 6. | ||
| No. | Drug | S a kcal mol−1 | RMSD_refineb | Amino acid bond | Distance Å | E (kcal mol) |
|---|---|---|---|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket. b RMSD_refine: the root-mean-squared-deviation (RMSD) between the predicted pose and those of the crystal one (after and before refinement process, respectively). | ||||||
| 1 | Sesamin | −6.7157 | 1.2904 | Gln 189/H-donor | 3.25 | −0.7 |
| Thr 190/H-donor | 3.12 | −0.7 | ||||
| His 41/pi-H | 4.55 | −0.8 | ||||
| Gln 189/pi-H | 4.32 | −0.6 | ||||
| 2 | Sesamolin | −6.829 | 1.6894 | Gln 189/H-donor | 3.24 | −0.5 |
| Thr 190/H-donor | 3.21 | −1 | ||||
| Gln 189/pi-H | 4.42 | −0.6 | ||||
| 3 | Pinoresinol | −6.463 | 1.2304 | Thr 24/H-donor | 2.79 | −0.6 |
| Thr 26/pi-H | 4.51 | −1.2 | ||||
| 4 | Ferulic acid | −5.3292 | 0.9729 | Glu 199/H-donor | 2.89 | −4.2 |
| Thr 190/H-acceptor | 3.46 | −0.7 | ||||
| 5 | Vanillic acid | −4.8624 | 1.3641 | Glu 166/H-donor | 2.86 | −4.3 |
| Thr 190/H-acceptor | 3.31 | −1.2 | ||||
| Gln 189/pi-H | 3.55 | −0.5 | ||||
| 6 | Hydroxymatairesinol | −7.4674 | 1.348 | Met 165/H-donor | 3.21 | −1.6 |
| Ser 144/H-donor | 3.15 | −0.6 | ||||
| Gly 143/H-acceptor | 3.26 | −0.5 | ||||
| 7 | Spicatolignan | −6.7403 | 1.6428 | Glu 166/H-donor | 2.88 | −4.3 |
| Thr 190/H-acceptor | 3.42 | −0.8 | ||||
| 8 | Darunavir | −7.1034 | 1.607 | Gln 189/H-donor | 2.9 | −0.5 |
| Glu 166/H-donor | 3.33 | −0.6 | ||||
| Cys 145/H-donor | 4.09 | −0.8 | ||||
| 9 | N3 | −8.4526 | 1.6031 | Thr 190/H-donor | 2.97 | −2.3 |
| Glu 166/H-donor | 2.85 | −4.5 | ||||
| Gln 189/H-donor | 2.87 | −4.8 | ||||
| His 163/H acceptor | 2.71 | −4.3 | ||||
| His 164/H-donor | 2.94 | −3.7 | ||||
| His 41/pi-H | 3.89 | −0.8 | ||||
Despite huge research efforts on SARS-CoV-2 Mpro inhibitors, proteomic and structural biology works on SARS-CoV-2 PLpro and its inhibitors are very few. Nevertheless, Rut and co-workers utilised HyCoSuL (Hybrid Combinatorial Substrate Library) to scrutinize substrate specificity of SARS-CoV-2 PLpro enzymes.33 Two irreversible inhibitors namely VIR250 and VIR251 having high degree of PLpro selectivity over other proteases were identified. The same study reported their crystal structures bound to the enzyme's active site. Altogether, these crystal structures in complex with VIR250 (PDB 6WUU) provide a basic for rapid rational drug design against SARS-CoV-2 PLpro.
The designed ligands were docked into the active site of SARS CoV-2 papain-like protease. The results of the docking scores of the designed ligands and their RMSD values are reported in Table 2.
| No. | Compound | S a kcal mol−1 | RMSD_refineb |
|---|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket. b RMSD_refine: the root-mean-squared-deviation (RMSD) between the predicted pose and those of the crystal one (after and before refinement process, respectively). | |||
| 1 | Sesamin | −6.5524 | 1.5919 |
| 2 | Sesamolin | −6.454 | 1.3248 |
| 3 | Pinoresinol | −6.5131 | 1.3406 |
| 4 | Ferulic acid | −4.8177 | 1.4256 |
| 5 | Vanillic acid | −4.6805 | 1.3721 |
| 6 | Hydroxymatairesinol | −7.2085 | 1.348 |
| 7 | Spicatolignan | −6.6183 | 1.7336 |
Docking scores are comprised in the range −4.68 to −7.20 kcal mol−1, 5 compounds possessed docking scores above −6.4 kcal mol−1; compound 6 (hydroxymatairesinol) achieved a similar value of binding energy as the co-crystallized ligand VER 250 with S = −7.2085 kcal mol−1; the 2D interactions shown in Fig. 3 and Table 3, displayed HB binding mode with Met 208 and Asp 164 amino acid residues as H-donor while the binding with Arg166 as H-acceptor, followed by compound 7 with S value of −6.6183 kcal mol−1 also compound 1 and compound 3 revealed a comparable binding free energies with values −6.5524 and −6.5131 kcal mol−1, respectively.
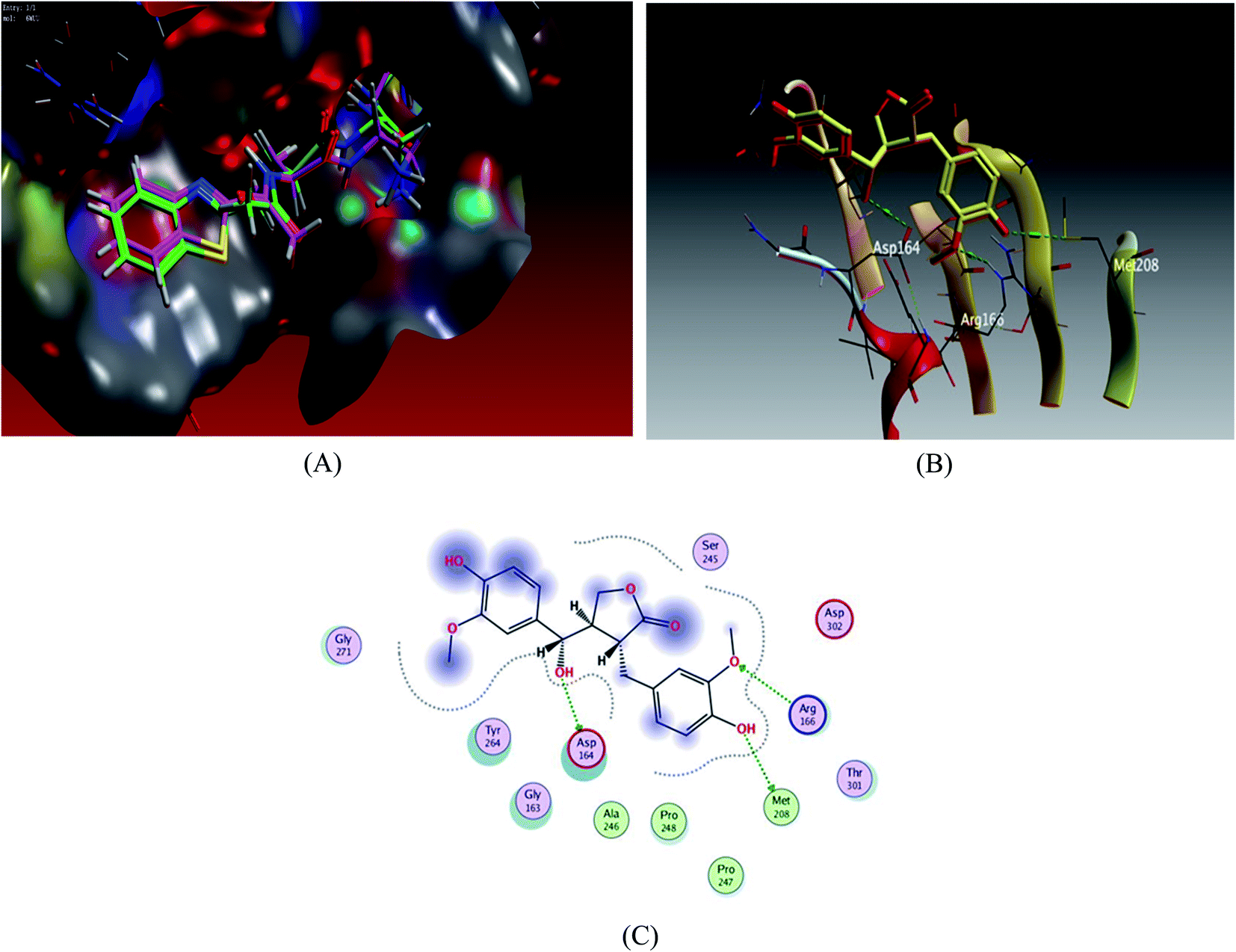 | ||
| Fig. 3 Docking results of compound 3 in the active site of SARS-CoV-2 papain-like protease (6WUU). (A) Validation of docking procedure showing good matching between crystallized and docked ligands. (B) Docking pose of compound 6. (C) 2D interactions of compound 6. | ||
| Compound | S a kcal mol−1 | Amino acid residue | Type of binding | Distance Å | E (kcal mol) |
|---|---|---|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket. | |||||
| Hydroxymatairesinol | −7.2085 | Met 208 | H-donor | 3.14 | −1.5 |
| Asp 164 | H-donor | 2.94 | −0.9 | ||
| Arg 166 | H-acceptor | 3.3 | −0.7 | ||
| VER 250 | −7.2103 | ASP 164 | H-donor | 2.84 | −7 |
| Tyr 268 | H-donor | 2.72 | −11.6 | ||
| Cys 270 | H-donor/acceptor | 3.37 | −0.5 | ||
| Gly 163 | H-donor/acceptor | 3.05 | −4.4 | ||
| Gly 271 | H-donor/acceptor | 2.84 | −0.8 | ||
| Pro247 | Pi-H | 4.5 | −0.6 | ||
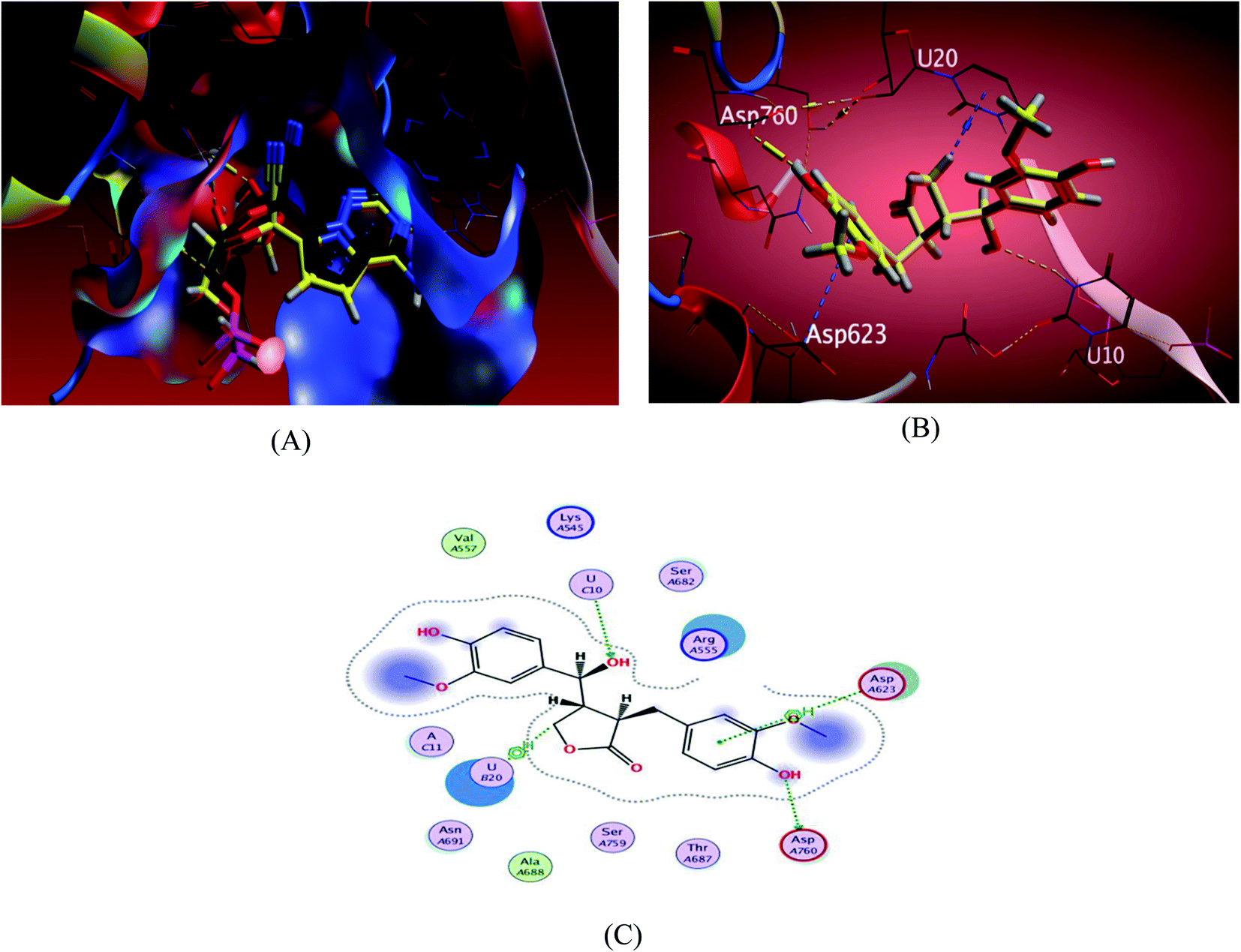 | ||
| Fig. 4 Docking results of compound 3 in the active site of SARS-CoV-2 RdRp (7BV2). (A) Validation of docking procedure showing good matching between crystallized and docked ligands. (B) Docking pose of compound 6. (C) 2D interactions of compound 6. | ||
The investigated sesame compounds were docked at the same active site that occupied by the remdesivir in the complex of Nsp12-Nsp7-Nsp8 and the template RNA, from the docking results Table 4, compound 6 achieved a binding energy score of −7.0179 kcal mol−1 that resembles the one obtained with the co crystallized ligand (remdesivir), the binding mode of the compound 6 showed a similarity with remdesivir in HB binding with nucleotide U10 as H-acceptor and as a H-donor with amino acid residue Asp760 both of the hydroxyl groups the alcoholic and the phenolic contributed in this interaction respectively. The binding with nucleotide U 20 was indicated as a type of H–Pi binding and with the residue Asp 623 as Pi–H interaction with the aromatic ring of the docked compounds, Table 5. It's noteworthy that, compound 1 possessed a dock score of −7.348 kcal mol−1 exceeding the score attained with remdesivir but no interaction obtained with the receptor with this selected pose, compound 2 also revealed a comparable free energy score of −6.9807 kcal mol−1 and good stability inside the pocket site but with no fundamental interactions.
| No. | Compound | S a kcal mol−1 | RMSD_refineb |
|---|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket. b RMSD_refine: the root-mean-squared-deviation (RMSD) between the predicted pose and those of the crystal one (after and before refinement process, respectively). | |||
| 1 | Sesamin | −7.348 | 1.4626 |
| 2 | Sesamolin | −6.9807 | 1.5327 |
| 3 | Pinoresinol | −6.3295 | 1.5142 |
| 4 | Ferulic acid | −5.3733 | 1.024 |
| 5 | Vanillic acid | −5.0486 | 0.6399 |
| 6 | Hydroxymatairesinol | −7.0179 | 1.4883 |
| 7 | Spicatolignan | −6.5373 | 1.1902 |
| Control | Remdesivir | −7.2069 | 1.0485 |
| Compound | S a kcal mol−1 | Amino acid residue | Type of binding | Distance Å | E (kcal mol) |
|---|---|---|---|---|---|
| a S: the score of a compound placement inside the protein binding pocket. | |||||
| Hydroxymatairesinol | −7.0179 | Asp760 | H-donor | 3.02 | −3 |
| U 10 | H-acceptor | 3.53 | −0.5 | ||
| U 20 | H-Pi | 3.92 | −0.9 | ||
| Asp 623 | Pi-H | 4.6 | −0.5 | ||
| Remdesivir | −7.2069 | Asp760 | H-donor | 3.31 | −0.9 |
| U 20 | H-donor | 2.95 | −1.8 | ||
| U 10 | H-donor/acceptor | 3.06 | −1.8 | ||
| Asn691 | H-acceptor | 3.1 | −1.5 | ||
| U 20 | Pi–Pi | 3.83 | 0 | ||
2.3. Drug-likeness properties and ADME prediction
Drug-likeness is a smooth balance among molecular properties affecting pharmacodynamics and pharmacokinetics of molecules, these molecular properties such as membrane permeability and bioavailability are always connected to some basic molecular descriptors such as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (partition coefficient), molecular weight (MW), topological polar surface area (TPSA), or hydrogen bond acceptors and donors count in a molecule. BOILED-egg diagram showing the possibility of absorption and penetration of inhibitors in the GI attract and brain using WLOGP and TPSA parameters, the basis of Egan's BOILED-Egg rule threshold values (WLOGP ≤ 5.88 and TPSA ≤ 131.6).
P (partition coefficient), molecular weight (MW), topological polar surface area (TPSA), or hydrogen bond acceptors and donors count in a molecule. BOILED-egg diagram showing the possibility of absorption and penetration of inhibitors in the GI attract and brain using WLOGP and TPSA parameters, the basis of Egan's BOILED-Egg rule threshold values (WLOGP ≤ 5.88 and TPSA ≤ 131.6).
Results of ADME prediction revealed that all investigated compounds exhibited acceptable ADME properties, compounds 2, 5 and 6 showed oral absorption, which is an important criterion for a drug that is intended for mass treatment. In addition, compounds 1, 3 and 4 can cross the blood brain barrier though within acceptable limits. Binding to P-glycoprotein (PGP+) was also studied and plotted, Fig. 5 and Table 6.
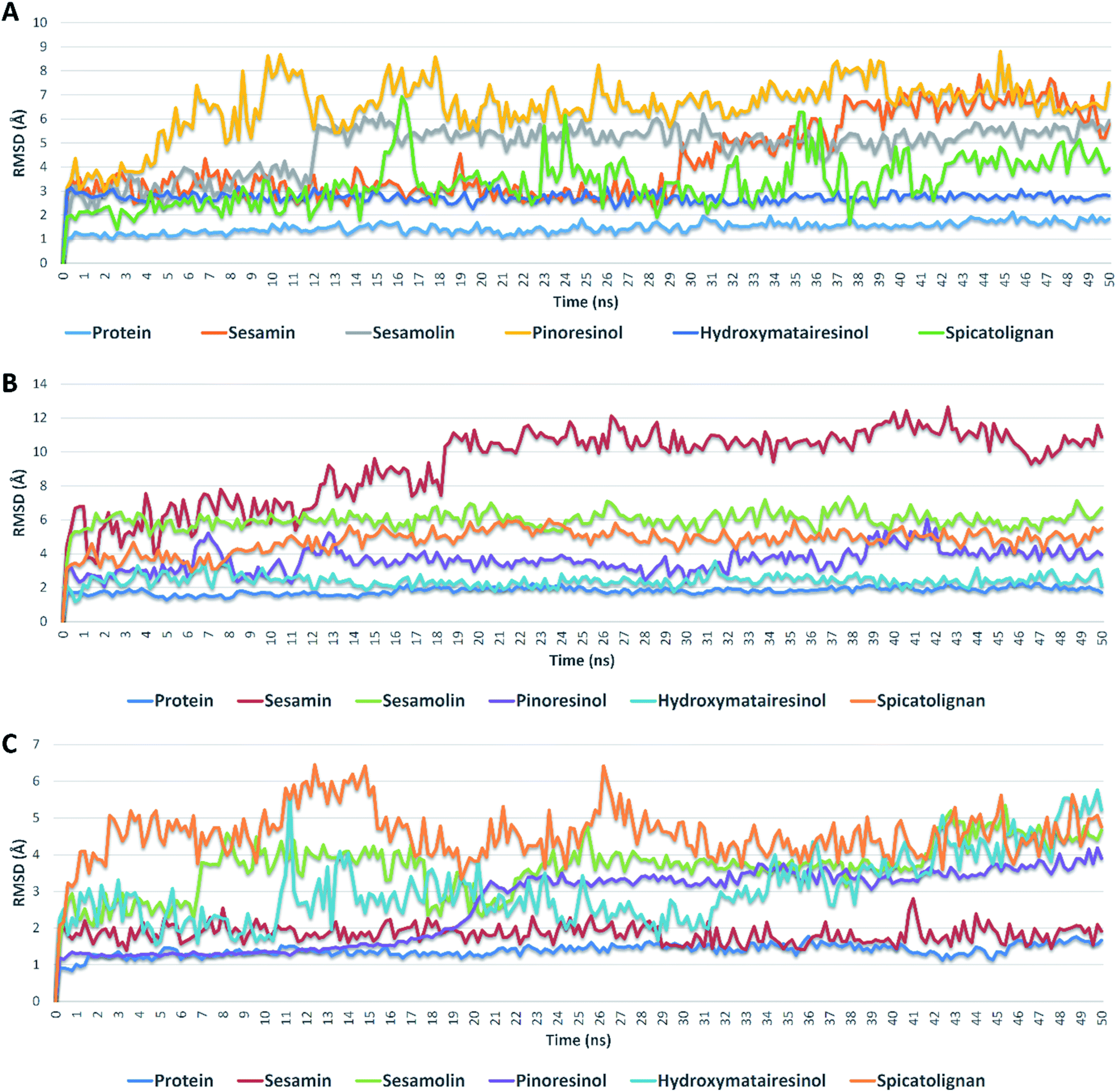 | ||
| Fig. 5 RMSDs of compounds 1, 2, 3, 6, and 7 inside the active sites of Mpro, PLpro, and RdRp during 50 ns of MDSs. | ||
| No | Name | Lipinski's rule of five | Drug-likeness | ||||
|---|---|---|---|---|---|---|---|
| Molecular weight (g mol−1) | Lipophilicity (logP) |
Hydrogen bond donors | Hydrogen bond acceptor | No. of rule violations | |||
| Less than 500 dalton | Less than 5 | Less than 5 | Less than 10 | Less than 2 violations | Lipinski's rule follows | ||
| 1 | Sesamine | 354.35 | 3.46 | 0 | 6 | 0 | Yes |
| 2 | Sesamoline | 386.35 | 3.48 | 1 | 8 | 0 | Yes |
| 3 | Pinoresinol | 358.39 | 2.67 | 2 | 6 | 0 | Yes |
| 4 | Ferulic acid | 194.18 | 1.62 | 2 | 4 | 0 | Yes |
| 5 | Vanillic acid | 168.15 | 1.4 | 2 | 4 | 0 | Yes |
| 6 | Hydroxymatairesinol | 374.38 | 2.53 | 3 | 7 | 0 | Yes |
| 7 | Spicatolignan | 372.37 | 2.58 | 3 | 7 | 0 | Yes |
All the investigated compounds obeying Lipinski's rule of five parameters.36 They have H-bond acceptor less than 10 and donor centres less than 5, these atoms favour H-bond formations and consequently develop water solubility. The obtained logP values and bioactivity scores (F > zero) revealed that they have a substantial bioavailability and cross the cell membrane efficiently.37 Also, all the compounds showed acceptable MW and convenient TPSA values. Table 7.
| No | Compound | MW (g mol−1) | WLOGP | TPSA | F | Water solubility silicos-IT class |
|---|---|---|---|---|---|---|
| a MW, molecular weight; TPSA, topological polar surface area; WLOGP, n-octanol/water partition coefficient (atomistic method); F, Abbott bioavailability scores. | ||||||
| 1 | Sesamine | 354.35 | 2.57 | 55.38 | 0.55 | Moderately soluble |
| 2 | Sesamoline | 386.35 | 1.67 | 84.84 | 0.55 | Soluble |
| 3 | Pinoresinol | 358.39 | 2.54 | 77.38 | 0.55 | Moderately soluble |
| 4 | Ferulic acid | 194.18 | 1.39 | 66.76 | 0.85 | Soluble |
| 5 | Vanillic acid | 168.15 | 1.1 | 66.76 | 0.85 | Soluble |
| 6 | Hydroxymatairesinol | 374.38 | 1.86 | 105.45 | 0.55 | Moderately soluble |
| 7 | Spicatolignan | 372.37 | 2.28 | 105.45 | 0.56 | Soluble |
Compound 6 satisfies all parameters of the Lipinski's rule, and complies to the BOILED-Egg approach showing no brain absorption and good GI tract absorption, which renders it a more suitable potential orally active drug and therapeutic inhibitor of SARS CoV-2 to be tested in clinical trials.
The structure of the SARS-CoV-2 RdRp in complex with a 50-base template–primer RNA and remdesivir. The double-stranded RNA template is inserted into the central channel of the RdRp where remdesivir, which considered as nucleoside analogue, is incorporated into the primer strand at the first replicated base pair and terminates chain elongation.38
Here the interaction between compound 6 methylene group and the uracil nucleotide 20 of the RNA in the form of H–Pi interaction resemble an interaction of the ligand remdesivir with it, which contribute with the other interactions either with the amino acid residues or the nucleotide in inhibition of the RdRp complex and stops the mRNA transcription and replication of SARS-CoV-2.
2.4. Molecular dynamic simulation
Further validation of the docking results was achieved via molecular dynamic simulations (MDS). Docking poses of the compounds with docking scores <−6 were subjected to a number of 50 ns MDS experiments to study their binding stabilities and to calculate their binding free energies (ΔGs).Compounds 1, 2, 3, 6 and 7 got docking scores <−6 kcal mol−1 upon docking against Mpro, PLpro and RdRp. As shown in Table 8 and Fig. 5, all of these compounds achieved comparable binding stability during the course of MDS.
| No. | Compound | ΔG(kcal mol−1) | Average RMSD (Å) | ||||
|---|---|---|---|---|---|---|---|
| Mpro | PLpro | RdRp | Mpro | PLpro | RdRp | ||
| 1 | Sesamin | −7.1 | −4.9 | −8.4 | 4.6 | 8.3 | 2.3 |
| 2 | Sesamolin | −6.8 | −5.0 | −7.3 | 5.2 | 6.2 | 3.9 |
| 3 | Pinoresinol | −5.2 | −6.9 | −8.8 | 7.4 | 3.9 | 2.9 |
| 6 | Hydroxymatairesinol | −8.3 | −8.5 | −7.3 | 2.8 | 2.2 | 3.8 |
| 7 | Spicatolignan | −8.0 | −6.5 | −6.6 | 4.1 | 4.9 | 4.8 |
Hydroxymatairesinol 6 was showed the least fluctuations and deviations from its starting binding pose with Mpro (RMSD ∼ 2.8 Å), and hence, the lowest ΔG. Sesamin 1 show an average RMSD (2.7 Å) similar to that of hydroxymatairesinol 6 until 29.6 ns, when its deviation started to increase gradually to end with 5.5 Å. Similarly, sesamolin 2 showed stable RMSD around 2.9 Å until 11.6 ns, when its RMSD suddenly increased to reach 5.6 Å and became equilibrated around this point until the end of MDS with low fluctuations. Spicatolignan 7 showed lowest RMSD (2.1 Å) until 15 ns. Starting from this point its fluctuations a round RMSD = 3.9 Å increased dramatically. Pinoresinol 3 showed the highst fluctuations and deviations inside the Mpro′s active site ending up the MSD with and average RMSD of 7.7 Å and ΔG = −5.2 kcal mol−1.
In case of PLpro, compounds 2, 3, 6 and 7 showed convergent steady binding stability with average RMSDs ranged from 2.2 to 6.2 Å showing minimal fluctuations (Table 8 and Fig. 5). Sesamin 1 was significantly less stable, where its RMSD was increasing gradually to reach 10.3 Å at 18.4 ns, and then became fluctuating around this point until the end of MDS.
Finally, binding stabilities of these compounds with RdRp's binding site were comparable showing relatively high fluctuations and RMSDs (∼5.1 Å), except for sesamin and sinoresinol (1 and 3) that were significantly more stable and steady (average RMSD = 2.5 Å) (Table 8 and Fig. 5), and got the lowest ΔG values (−8.4, −8.8 kcal mol−1, respectively).
From the previous reports on the molecular modelling-based screening of SARS CoV-2's molecular targets, particularly, Mpro, it can be concluded that: (i) ensemble docking protocols give more accurate results than simple ones as the former takes into account the active site's flexibility.39 (ii) Docking results should be validated by at least 50 ns of MDS as some times compounds with good docking scores did not grantee stable binding.40–42 (iii) Compounds that can achieve ΔG of −7 kcal mol−1 of lower have high potential to be active in vitro.43–45 (iv) Compounds with good scores and binding stability should also have good drug-like properties to be active in vitro.46–48
3. Experimental
3.1. Reagents and apparatus
Silica gel was purchased from Wako (Osaka, Japan). Thin-layer chromatography (TLC) silica gel G60 F254 and reversed phase preparative TLC were purchased from Merck (Darmstadt, Germany). All other ingredients used were of the highest grade available.The 1H, 13C-NMR and 2D spectra of the isolated compounds were recorded using a Bruker DRX 600 NMR spectrometer (Bruker Daltonics, Billerica, MA).
3.2. Preparation of extracts
5 kg of the fresh materials were freeze-dried and milled below 1 mm. Then, extracts were prepared by shaking (200 rpm) with solvent (methanol) at room temperature for 48 h to afford 59 g methanol extract. The extracted solution was filtered and evaporated using ADVANTEC no. 2 filter paper (Toyo Roshi Kaisha, Ltd., Tokyo).3.3. Fractionation and isolation
The methanol extract was suspended in distilled water and fractionated using liquid–liquid partition to afford n-hexane fraction (12 g), ethyl acetate fraction (17 g) and the remaining aqueous fraction (5 g). The n-hexane fraction was sub-fractionated on a silica gel column using n-hexane-ethyl acetate gradient elution (100%, 80% and 70%). The sub fractions eluted by 70% n-hexane:ethyl acetate (70–30) was crystallized to give some needles crystals, which washed with MeOH several times, then TLC was performed for checking purity to afford compound 1 (118 mg).The ethyl acetate fraction (17 g) was sub-fractionated on a silica gel column using n-hexane–ethyl acetate gradient elution (70%, 60%, 40% and 100%). The sub fractions eluted by n-hexane–ethyl acetate 40% (70:30) resulted in compound 2 (200 mg). The sub fractions eluted by n-hexane–ethyl acetate 40% (40:60) was subjected to reversed phase preparative TLC using 30% water in MeOH which resulted in compound 3 (15 mg), compound 4 (2.4 mg) and compound 5 (1.6 mg). The sub fractions eluted by 100% ethyl acetate was crystallized to give some needles crystals, which washed with MeOH several times, then TLC was performed for checking purity to afford compound 6 (1.6 mg). The remaining sub fractions which were eluted by 100% ethyl acetate was subjected to sub-column using n-hexane:ethyl acetate in gradient manner to afford compound 7 (3.8 mg). The structure of all compounds was elucidated by 1D and 2D NMR spectroscopy, including 1H, 13C, HSQC and HMBC experiments the ESI.†
3.4. Molecular docking study
The X-ray crystallographic structure of protein targets of SARS-CoV-2 Mpro (3CLpro), PLpro, and RdRp complexed with their ligands was obtained from the Protein Data Bank through the internet (http://www.rcsb.org/pdb/, code 6LU7, 6WUU and 7BV2). All molecular modelling calculations and docking studies were carried out using ‘Molecular Operating Environment 2019.0102’ software (MOE). The preparation of the protein included the removal of water molecules and uninvolved ligands then by using the quick preparation tool in MOE with applying the default options. Docking of the conformation database of the target compounds was done after preparation of the enzyme. The following methodology was generally applied: the enzyme active site was located by the site finder tool, and the docking tool was initiated. The program specifications were adjusted to ligand atoms as the docking site, alpha triangle as the placement methodology to be used and the post placement refinement were adjusted to make the receptor rigid. The scoring methodology London dG is used and was adjusted to its default values. The MDB file of the ligand to be docked was loaded and dock calculations were run automatically. Receptor–ligand interactions of the complexes were examined in 2D and 3D styles the poses that showed best ligand–enzyme interactions were selected and stored for energy calculations.The selection of poses was done according to their better obtained binding scores and RMSD refine values, especially most of them achieved very close binding modes compared to the native ligand. The obtained scores, RMSD refine values, and interactions with binding pocket site of the enzymes are discussed.
3.5. Drug-likeness properties and ADME prediction
Drug-likeness properties (Lipinski rule or rule of five) and Physicochemical properties (BOILED-Egg diagram or ADME prediction),49,50 were calculated by the free access website of the predictor Swiss ADME (http://www.swissadme.ch/).3.6. Molecular dynamic simulation
Molecular dynamic simulations (MDS) for the generated ligand–enzyme complexes were performed using the Nanoscale Molecular Dynamics (NAMD) 2.6 software,51 applying the CHARMM27 force field.52 Hydrogen atoms were added to the protein structures using the psfgen plugin included in the Visual Molecular Dynamic (VMD) 1.9 software.53 Afterward, the whole generated systems were solvated using water molecules (TIP3P) and 0.15 M NaCl. At first, the total energy of the generated systems was minimized and gradually heated to reach 300 K and equilibrated for 200 seconds. Subsequently, the MDS was continued for 50 ns, and the trajectory was stored every 0.1 ns and further analyzed with the VMD 1.9 software. The MDS output was sampled every 0.1 ns to calculate the root mean square deviation (RMSD). The parameters of compound 4 were prepared using the online software the VMD Force Field Toolkit (ffTK).53 Binding free energies (ΔG) were calculated using the free energy perturbation (FEP) method (Jo et al. 2013). The web-based software Absolute Ligand Binder54 was used to generate the input files for NAMD software which was performed the simulations required for ΔGs calculations.4. Conclusion
The isolated compounds from sesame seeds, particularly compounds 1, 2, 3, 6, and 7, were found to exhibit considerable binding affinity with three key SARS CoV's proteins; Mpro, PLpro and RdRp. This study pointed out the importance of using sesame as a source of naturally occurring compounds isolated from food sources in alleviating SARS-CoV-2, and hence an advice of using sesame seed as a part of food for COVID-19 patients. Additionally, it will help us to afford semisynthetic new scaffold with potential activity against this disease.Conflicts of interest
We declare that we have no conflict of interest.Acknowledgements
The authors would like to acknowledge OpenEye scientific software for providing the academic license. In addition, we acknowledge the Research and Education Support Center of the faculty of Agriculture, Kyushu University for supporting facilities for NMR and mass analysis.References
- Z. Xu, L. Shi, Y. Wang, J. Zhang, L. Huang, C. Zhang, S. Liu, P. Zhao, H. Liu, Y. Tai, C. Bai, T. Gao, J. Song, P. Xia, J. Dong and J. Zhao, Pathological findings of COVID-19 associated with acute respiratory distress syndrome, Lancet Respir. Med., 2020, 8(4), 420–422 CrossRef CAS PubMed.
- X. Cao, Nat. Rev. Immunol., 2020, 20(5), 269–270 CrossRef CAS PubMed.
- P. Carvalho, F. Borghetti, M. Buckeridge, L. Morhy and E. Filho, Rev. Bras. Fisiol. Veg., 2001, 13(2), 139–148 CrossRef.
- G. Brar and K. Ahuja, Annu. Rev. Plant Sci., 1979, 1, 245–313 CAS.
- N. Pathak, A. K. Rai, R. Kumari and K. V. Bhat, Pharmacogn. Rev., 2014, 8(16), 147–155 CrossRef PubMed.
- F. Cheng, T. Jinn, R. Hou and J. Tzen, Int. J. Biomed. Sci., 2006, 2(3), 284 CAS.
- S. Kang, W. Peng, Y. Zhu, S. Lu, M. Zhou, W. Lin, W. Wu, S. Huang, L. Jiang, X. Luo and M. Deng, Int. J. Antimicrob. Agents, 2020, 55(5), 105950 CrossRef CAS PubMed.
- W. Fan, B. Yang, Z. Wang and L. Wu, Sci. Adv., 2016, 2(8), e1600901 CrossRef PubMed.
- W. Fan, J. Zeng and Y. Xu, Antiviral, 2020, 10, 30–32 Search PubMed.
- G. V. Subhash, G. R. Kumar, A. Sapre and S. Dasgupta, Possible Prevention of COVID 19 by Using Linoleic Acid (C18) Rich Algae Oil, Int. J. Adv. Res., 2020, 36, 1–9 Search PubMed.
- A. E. Allam, H. K. Assaf, H. A. Hassan, K. Shimizu and Y. A. M. M. Elshaier, RSC Adv., 2020, 10(50), 29983–29998 RSC.
- H. A. Hassan, U. R. Abdelmohsen, O. M. Aly, S. Y. Desoukey, K. M. Mohamed and M. S. Kamel, Nat. Prod. Res., 2020, 1–5 Search PubMed.
- R. Arya, S. Kumari, B. Pandey, H. Mistry, S. C. Bihani, A. Das, V. Prashar, G. D. Gupta, L. Panicker and M. Kuma, J. Mol. Biol., 2020, 433(2), 166725 CrossRef PubMed.
- E. M. Zahran, A. Albohy, A. Khalil, A. H. Ibrahim, H. A. Ahmed, E. M. El-Hossary, G. Bringmann and U. R. Abdelmohsen, Mar. Drugs, 2020, 18(12), 645, DOI:10.3390/md18120645.
- Z. Linlin, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors, Science, 2020, 409–412 Search PubMed.
- R. Alexpandi, J. Freire De Mesquita, S. K. Pandian and A. V. Ravi, Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis, Front. Microbiol., 2020, 23(11), 1796 CrossRef PubMed.
- W. R. Ferraz, R. A. Gomes, A. L. Novaes and G. H. Trossini, Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study, Future Med. Chem., 2020, 12(20), 1815–1828 CrossRef CAS PubMed.
- S. S. K. A. Hossain, A. V. D. N. Kumar, A. G. D. Kollie, A. Nakhi and M. V. B. R. M. Palf, Ultrasound assisted synthesis of 3-alkynyl substituted 2-chloroquinoxaline derivatives: Their in silico assessment as potential ligands for N-protein of SARS-CoV-2, Tetrahedron Lett., 2020, 61(40), 152336 CrossRef PubMed.
- D. Kumar, G. Chauhan, S. Kalra, B. Kumar and M. S. Gill, Bioorg. Chem., 2020, 104, 104326 CrossRef CAS PubMed.
- B. H. Harcourt, D. Jukneliene, A. Kanjanahaluethai, J. Bechill, K. M Severson, C. M. Smith, P. A. Rota and S. C. Baker, J. Virol., 2004, 78(24), 13600–13612 CrossRef CAS PubMed.
- Sk. Abdul Amin, S. Banerjee, K. Ghosh, S. Gayen and T. Jha, Bioorg. Med. Chem., 2021, 1(29), 115860 CrossRef PubMed.
- W. Yin, C. Mao, X. Luan, D. D. Shen, Q. Shen, H. Su, X. Wang, F. Zhou, W. Zhao, M. Gao, S. Chang, Y. Xie, G. Tian, H. Jiang, S. Tao, J. Shen, Y. Jiang, H. Jiang, Y. Xu, S. Zhang, Y. Zhang and H. Xu, Science, 2020, 368(6498), 1499–1504 CrossRef CAS PubMed.
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res., 2020, 30(3), 269–271 CrossRef CAS PubMed.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11(11), 1117–1121 CrossRef CAS PubMed.
- P. Pusadkar, K. Eswaran, S. Bonde and N. Mohite, BioSci. Trends, 2015, 8(15), 3900–3906 Search PubMed.
- R. H. Mekky, E. Abdel-Sattar, A. Segura-Carretero and M. Contreras, Foods, 2019, 8(10), 432, DOI:10.3390/foods8100432.
- Y. Fukuda, T. Osawa, M. Namiki and T. Ozaki, Agric. Biol. Chem., 1985, 49(2), 301–306 CAS.
- L. Panzella, T. Eidenberger and A. Napolitano, Molecules, 2018, 23(3), 676 CrossRef PubMed.
- K. Prakash and S. Naik, J. Bioresour. Eng. Technol., 2014, 1, 48–66 Search PubMed.
- J. Zheng, G. Chen, H. Gao, B. Wu and L. Wu, J. Asian Nat. Prod. Res., 2007, 9(5), 431–435 CrossRef CAS PubMed.
- Y. Yuan, D. Cao, Y. Zhang, J. Ma, J. Qi, Q. Wang, G. Lu, Y. Wu, J. Yan, Y. Shi, X. Zhang and G. F. Gao, Nat. Commun., 2017, 8(1), 1–9 CrossRef PubMed.
- C. Liu, Q. Zhou, Y. Li, S. P. Watkins, L. J. Carter, J. Smoot, A. C. Gregg, A. C. Gregg, A. D. Daniels, S. Jervey and D. Albaiu, ACS Cent. Sci., 2020, 6(3), 315–331 CrossRef CAS PubMed.
- A. Kilianski, A. M. Mielech, X. Deng and S. C. Baker, J. Virol., 2013, 87(21), 11955–11962 CrossRef CAS PubMed.
- A. Kilianski, A. M. Mielech, X. Deng and S. C. Baker, Nature, 2020, 582(7811), 289–293 CrossRef PubMed.
- W. Rut, Z. Lv, M. Zmudzinski, S. Patchett, D. N. Scott, J. Snipas, F. Oualid, T. T. Huang, M. Bekes, M. Drag and S. K. Olsen, Sci. Adv., 2020, 6(42), eabd4596 CrossRef CAS PubMed.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43(20), 3714–3717 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, J. Chem. Inf. Model., 2014, 54(12), 3284–3301 CrossRef CAS PubMed.
- W. Yin, C. Mao, X. Luan, D. Shen, Q. Shen, H. Su, X. Wang, F. Zhou, W. Zhao, M. Gao, S. Chang, Y. Xie, G. Tian, H. Jiang, S. Tao, J. Shen, Y. Jiang, H. Jiang, Y. Xu, S. Zhang, Y. Zhang and H. Xu, Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir, Science, 2020, 368(6498), 1499–1504 CrossRef CAS PubMed.
- A. M. Sayed, H. A. Alhadrami, A. O. El-Gendy, Y. I. Shamikh, L. Belbahri, H. M. Hassan, R. A. Usama and M. E. Rateb, Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro), Microorganisms, 2020, 8(7), 970 CrossRef CAS PubMed.
- A. M. Sayed, A. M. Khalaf, M. E. Abdelrahim and M. O. Elgendy, Repurposing of some anti-infective drugs for COVID-19 treatment: a surveillance study supported by an in silico investigation, Int. J. Clin. Pract., 2020, 75(4), e13877 Search PubMed.
- N. Hisham Shady, K. A. Youssif, A. M. Sayed, L. Belbahri, T. Oszako, H. M. Hassan and U. R. Abdelmohsen, Sterols and Triterpenes: Antiviral Potential Supported by In-Silico Analysis, Plants, 2021, 10(1), 41 CrossRef PubMed.
- M. E. Abouelela, H. K. Assaf, R. A. Abdelhamid, E. S. Elkhyat, A. M. Sayed, T. Oszako, L. Belbahri, A. E. El Zowalaty and M. S. A. Abdelkader, Identification of Potential SARS-CoV-2 Main Protease and Spike Protein Inhibitors from the Genus Aloe: An In Silico Study for Drug Development, Molecules, 2021, 26(6), 1767 CrossRef CAS PubMed.
- H. A. Alhadrami, A. M. Sayed, A. M. Sharif, E. I. Azhar and M. E. Rateb, Olive-Derived Triterpenes Suppress SARS COV-2 Main Protease: A Promising Scaffold for Future Therapeutics, Molecules, 2021, 26(9), 2654 CrossRef CAS PubMed.
- S. A. Cuesta, J. R. Mora and E. A. Márquez, In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2, Molecules, 2021, 26(4), 1100 CrossRef CAS PubMed.
- Y. Zhu, J. Li and Z. Pang, Recent insights for the emerging COVID-19: drug discovery, therapeutic options and vaccine development, Asian J. Pharm. Sci., 2021, 16, 4–23 CrossRef PubMed.
- H. A. Alhadrami, A. M. Sayed, H. M. Hassan, K. A. Youssif, Y. Gaber, Y. Moatasim, O. Kutkat, A. Mostafa, M. A. Ali, M. E. Rateb, U. R. Abdelmohsen and N. M. Gamaleldin, Cnicin as an Anti-SARS-CoV-2: An Integrated In Silico and In Vitro Approach for the Rapid Identification of Potential COVID-19 Therapeutics, Antibiotics, 2021, 10(5), 542 CrossRef CAS PubMed.
- R. Orfali, M. E. Rateb, H. M. Hassan, M. Alonazi, M. R. Gomaa, N. Mahrous, M. GabAllah, A. Kandeil, S. Perveen, U. R. Abdelmohsen and A. M. Sayed, Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein, Antibiotics, 2021, 10(4), 420 CrossRef CAS PubMed.
- A. M. Sayed, A. R. Khattab, A. M. AboulMagd, H. M. Hassan, M. E. Rateb, H. Zaid and U. R. Abdelmohsen, Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale, RSC Adv., 2020, 10(34), 19790–19802 RSC.
- S. Katekhaye, R. L. Gavit and K. Laddha, Indian Drugs, 2011, 48, 54–58 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7(1), 1–13 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26(16), 1781–1802 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott Jr, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102(18), 3586–3616 CrossRef PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem., 2008, 29(11), 1859–1865 CrossRef CAS PubMed.
- S. Jo, W. Jiang, H. S. Lee, B. T. Roux and W. Im, CHARMM-GUI Ligand Binder for absolute binding free energy calculations and its application, J. Chem. Inf. Model., 2013, 53(1), 267–277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03937g |
| This journal is © The Royal Society of Chemistry 2021 |