
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInversion kinetics of some E/Z 3-(benzylidene)-2-oxo-indoline derivatives and their in silico CDK2 docking studies†‡
Hany S. Mansour,
Hend A. A. Abd El-wahab,
Ahmed M. Ali and
Tarek Aboul-Fadl *
*
Department of Medicinal Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt. E-mail: fadl@aun.edu.eg
First published on 17th February 2021
Abstract
The structure-based design of some CDK2 inhibitors with a 3-(benzylidene)indolin-2-one scaffold as potential anticancer agents was realized. Target compounds were obtained as E/Z mixtures and were resolved to corresponding E- and Z-diastereomers. In silico studies using MOE 2019.01 software revealed better docking on the targeted enzyme for the Z-diastereomer compared to the E-one. A time-dependent kinetic isomerization study was carried out for the inversion of E/Z diastereomers in DMSO-d6 at room temperature, and were found to obey the first order kinetic reactions. Furthermore, a determination of the kinetic inter-conversion rate order by graphical analysis method and calculation of the rate constant and half-life of this kinetic process were carried out. For the prediction of the stability of the diastereomer(s), a good multiple regression equation was generated between the reaction rates of isomerization and some QM parameters with significant p value.
Introduction
The 2-oxo-indoline scaffold is strongly associated with the anticancer activity through multiple targets as cyclin-dependent kinases (CDKs) and receptor tyrosine kinases (RTKs).1–4 Cyclin-dependent kinases (CDKs) are the key regulators of the cell cycle,5–8 the complex process by which cells divide. The recognition of the importance of CDKs to the process of cell division has stimulated an interest in them as potential targets for the management of cancer.9,10 Molecules having the 2-oxo-indoline scaffold usually compete with ATP for binding in the enzyme active site, and prevent phosphate transfer from ATP to the appropriate residue (Tyr, Ser, Thr). Thus, they block auto- and substrate phosphorylation, and therefore signal transduction and oncogenic activity.11 The selectivity of these classes of compounds to the subclasses of CDKs and RTKs is determined by other interactions between substituents on the scaffold and residues found around this hinge region at the interaction site.12,13 3-Arylidene-2-oxo-indoline derivatives are at the heart of a wide range of clinically, medicinally and biologically important compounds.14–17 A number of 3-arylidene-2-oxo-indolines have been approved for clinical application. Of particular significance is sunitinib, which is a clinically used receptor tyrosine kinase (RTK) inhibitor for the treatment of advanced renal cell carcinoma and gastrointestinal stromal tumours,14 in addition to semaxanib, toceranib phosphate, and SU9516.11,14 They possess the capacity to bind the receptor tyrosine kinase (RTK), CDK2 enzyme and Aurora B with high affinity.14 Orantinib is a receptor tyrosine kinase inhibitor that was approved as an antiproliferating agent, and nintedanib is an inhibitor of PDGF, FGF and VEGF receptors and was approved for the treatment of non-small-cell lung cancer,18,19 Fig. 1.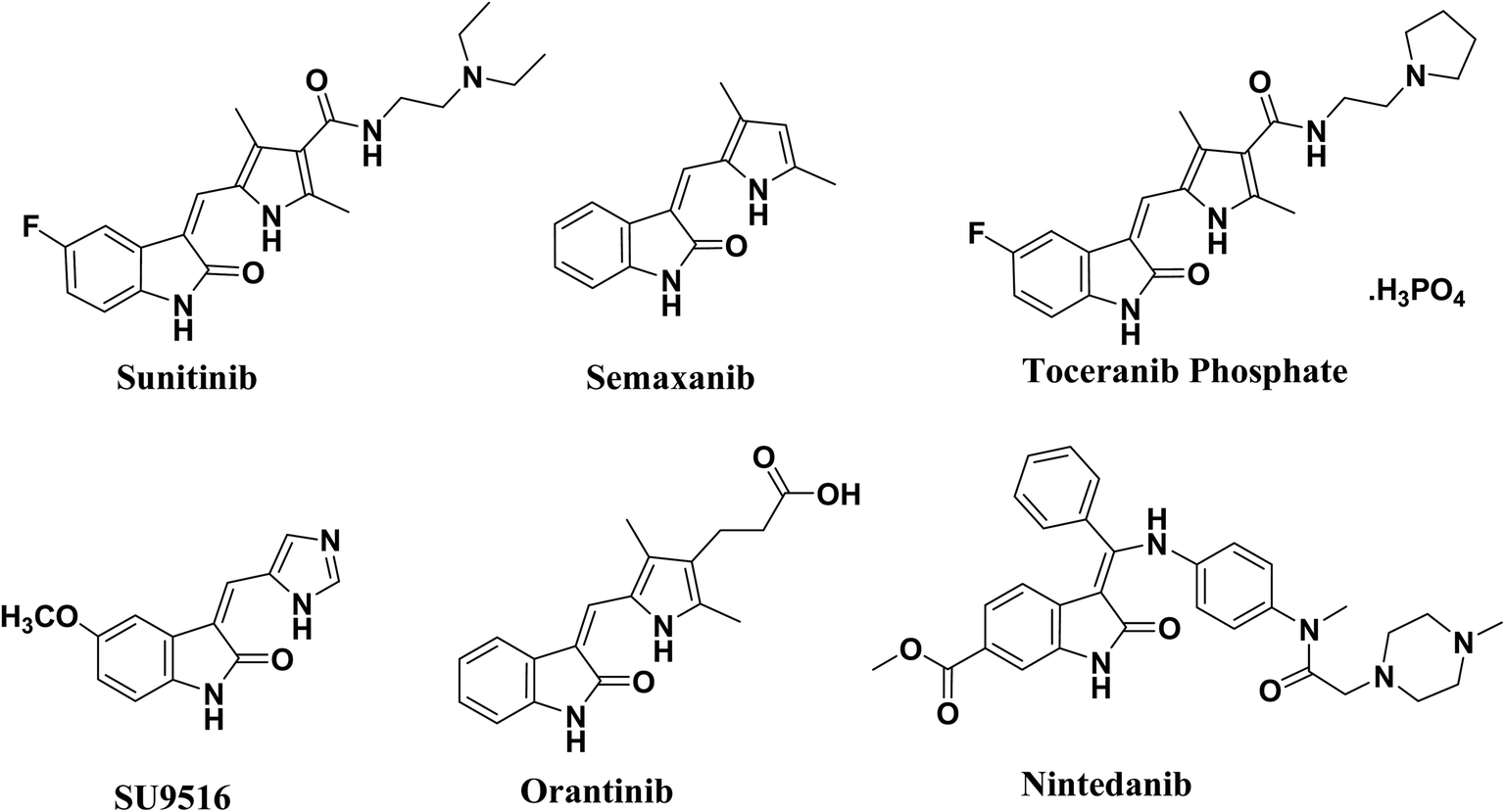 | ||
| Fig. 1 Clinically approved 2-oxo-indoline derivatives. | ||
It has been established for more than half a century that stereochemistry is fundamental in biological activities.20,21 The stereoselection, which has been extensively studied, was almost exclusively explained in terms of diastereoselective interactions for sunitinib and related compounds. Interestingly, these molecules were bound as the Z-diastereomer, even though they were the E-diastereomer in solution. The E form must be converted to the Z form before binding, as all E-diastereomers were inactive. Apparently, this conversion was shown to be mediated by acid, base or light.22 Removing such unsaturation and/or designing stereochemically stable compounds may be feasible, provided that activities are not compromised. This would avoid the issue of E/Z isomerization however, the vinyl proton was determined to be essential for activity.17,22
Molecular modeling revealed that the oxindole moiety of sunitinib is crucial for binding to the ATP binding site of RTKs and CDKs.23,24 Additionally, the pyrrole functionality and the C(5)-F element of (2-oxoindolin-3-ylidene)methylpyrrole in the sunitinib structure both enhance affinity by inducing further hydrophobic interactions.19,20,22 In contrast, the water-soluble diethyl amino ethyl tail of sunitinib lies on the protein surface. The RTK inhibitory activity and selectivity of (2-oxo-indolin-3-ylidene)methylpyrrole derivatives revealed strong relationships with structural modifications.25
In view of the above facts, and in continuation of our interest in the synthesis and in silico investigation of the CDK activities of 2-oxo-indoline derivatives,26 the current work describes the synthesis of a series of some 3-benzylidene substituted oxindole derivatives (Fig. 2).
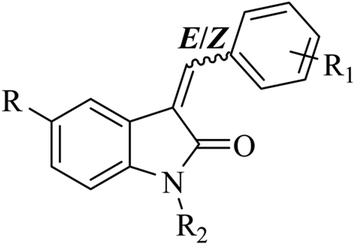 | ||
| Fig. 2 General structure for 3-benzylidene-substituted oxindoles. | ||
Recent studies have been made for its effective synthesis, and also for the stereoselective synthesis of (E) and (Z) diastereomers.27–31 Accordingly, the kinetics of the E/Z isomerization of the designed molecules seemed to be of interest.
Results and discussion
Chemistry
3-Benzylidenes 6(a–c) were obtained using the Wolff–Kishner-like reduction of indolin-2,3-dione, 1 (isatin), with hydrazine hydrate, followed by reaction with an appropriate benzaldehyde.32,33 5-Nitro derivatives 4(a–c) were obtained by nitration at position 5 of the 2-oxo-indoline moiety with potassium nitrate in cold concentrated sulfuric acid.34,35 N-Substituted derivatives [5(a–d) and 7(a–b)] were obtained by reacting [4(a–c) and 6(a–c)] with benzyl chloride or propyl bromide according to the reported procedures,26,27 Scheme 1.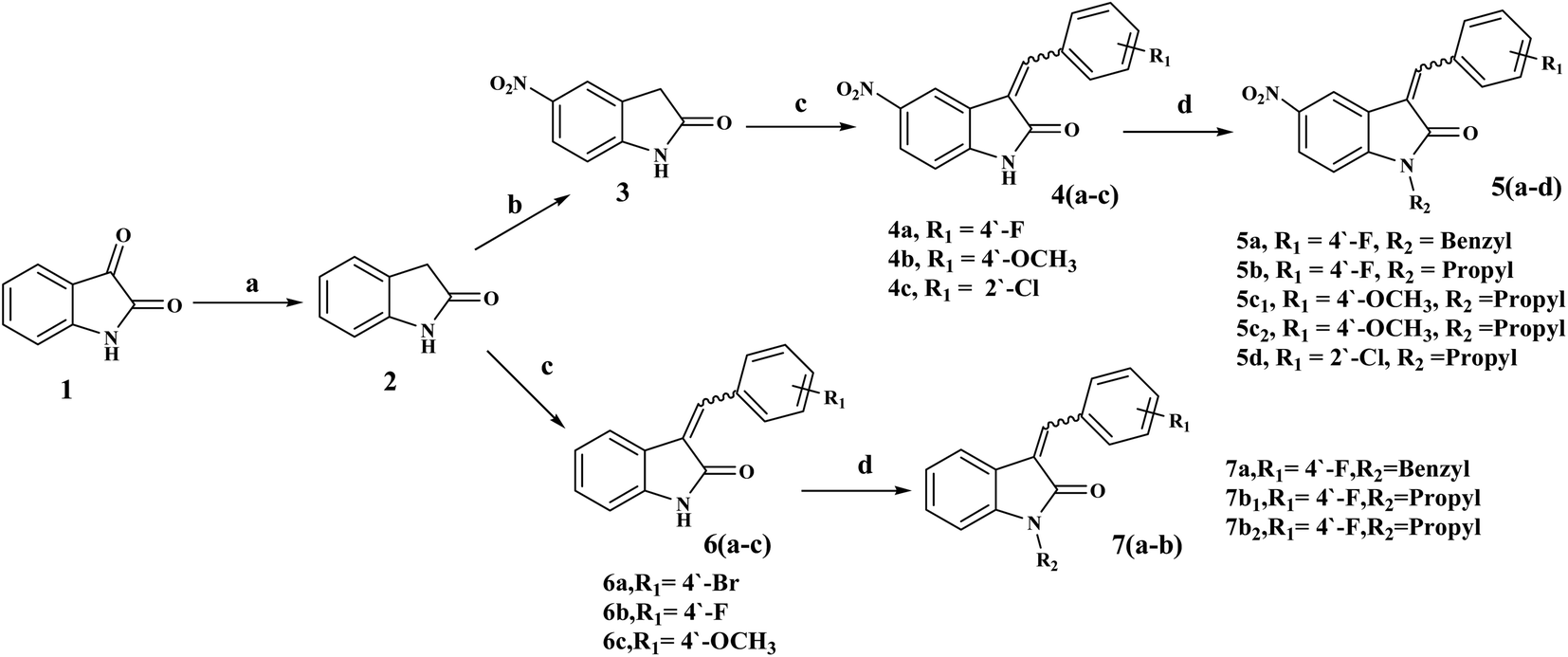 | ||
| Scheme 1 (a) NH2NH2·H2O, reflux at 140 °C/4 h; (b) KNO3/H2SO4, stir at 0–5 °C/1.5 h; (c) substituted benzaldehydes, piperidine, ethanol, reflux at 90 °C/4–5 h; (d) K2CO3/KI, DMF, alkyl halides/stir at 60–80 °C/4–12 h. | ||
All of the synthesized compounds 4(a–c), 5(a–d), 6(a–c) and 7(a–b) were obtained as mixtures of the E and Z diastereomers in different ratios, as identified with their vinyl-H and 2′ and 6′-Hs chemical shifts (ppm) of E and Z, Table 1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z diastereomeric ratios of the synthesized compounds 5(a–d), 6a, 6c and 7(a–b) and the yield (%)
:Z diastereomeric ratios of the synthesized compounds 5(a–d), 6a, 6c and 7(a–b) and the yield (%)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | Substituents | Chemical shifts (δ ppm) | Initial E:Z ratio |
Yield (%) | |||||
| R1 | R2 | R3 | vinyl H | 2′ and 6′-Hs | |||||
| E | Z | E | Z | ||||||
| 5a | NO2 | 4′-F | Benzyl | 8.02 | 8.38 | 7.91 | 8.60 | 6:94 |
67 |
| 5b | NO2 | 4′-F | Propyl | 7.93 | 8.29 | 7.87 | 8.58 | 17:83 |
77 |
| 5c1 | NO2 | 4′-OCH3 | Propyl | 7.86 | 8.19 | 7.78 | 8.56 | 30:70 |
45 |
| 5c2 | NO2 | 4′-OCH3 | Propyl | 7.86 | 8.19 | 7.78 | 8.56 | 65:35 |
45 |
| 5d | NO2 | 2′-Cl | Propyl | 7.87 | 8.30 | 7.71 | 8.06 | 93:7 |
62 |
| 6a | H | 4′-Br | H | 7.57 | 7.78 | 7.72 | 8.32 | 100:0 |
80 |
| 6c | H | 4′-OCH3 | H | 7.59 | 7.75 | 7.72 | 8.48 | 98:2 |
75 |
| 7a | H | 4′-F | Benzyl | 7.8 | 7.96 | 7.83 | 8.54 | 18:82 |
67 |
| 7b1 | H | 4′-F | Propyl | 7.7 | 7.89 | 7.79 | 8.51 | 2:98 |
55 |
| 7b2 | H | 4′-F | Propyl | 7.7 | 7.89 | 7.79 | 8.51 | 88:12 |
55 |
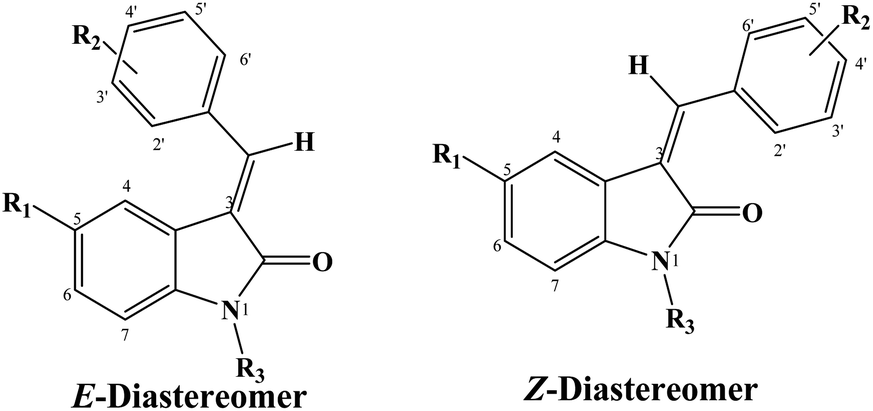
The configuration of the major diastereomer was confirmed by 2D NOE analysis, within the time of measurement. The E-configured compounds showed NOE between the proton at C4 and the protons at C-2′ and C-6′ (Fig. 3A), whereas the Z-configured compounds showed NOE between the proton at the C-4 and the vinyl proton (Fig. 3B).
 | ||
| Fig. 3 General configuration of E and Z diastereomers. (A) NOE interaction between H2′,6′ and H4 in E-diastereomer. (B) NOE interaction between vinyl-H and H4 in Z-diastereomer. | ||
The assignment of the E/Z diastereomers were confirmed by 1H-NMR, where the vinyl protons resonated at a slight downfield shift in the Z-diastereomer compared to the E-diastereomer due to the influence of the 2-carbonyl group. The ortho-benzylidene protons (H2′,6′) were found to be more shielded in the E-diastereomers for the same reason, as shown in Fig. 3. The 1H NMR spectra of 5c is shown in Fig. 4 as a representative example. The chemical shifts of the major diastereomer (Z) revealed signals of Hvinyl at 8.25 ppm, H4 at 8.72 ppm and H2′,6′ at 8.59 ppm, while those of the minor diastereomer (E) resonated at 7.9, 8.52 and 7.82 ppm, respectively.
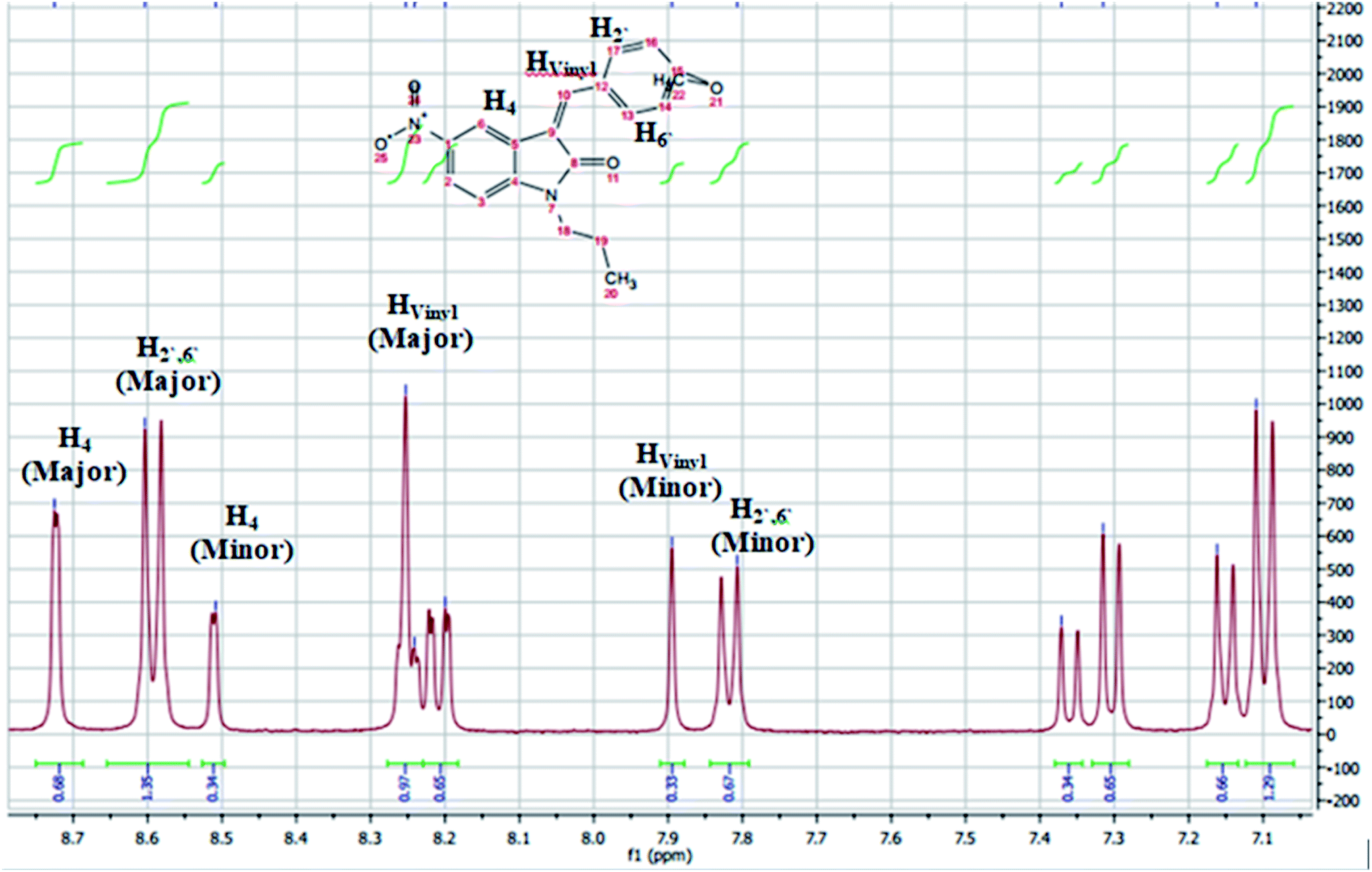 | ||
| Fig. 4 1H-NMR chart (aromatic region) of compound 5c1, mixture of the E/Z diastereomers with a ratio = 35:65. | ||
The configuration of the E/Z diastereomers of 5c was further confirmed by the NOESY 2D-1H NMR. The NOE interaction between Hvinyl and H4 was shown in the Z-diastereomer, Fig. 5A, while the NOE interaction between H2′,6′ and H4 was shown in the E-diastereomer, Fig. 5B.
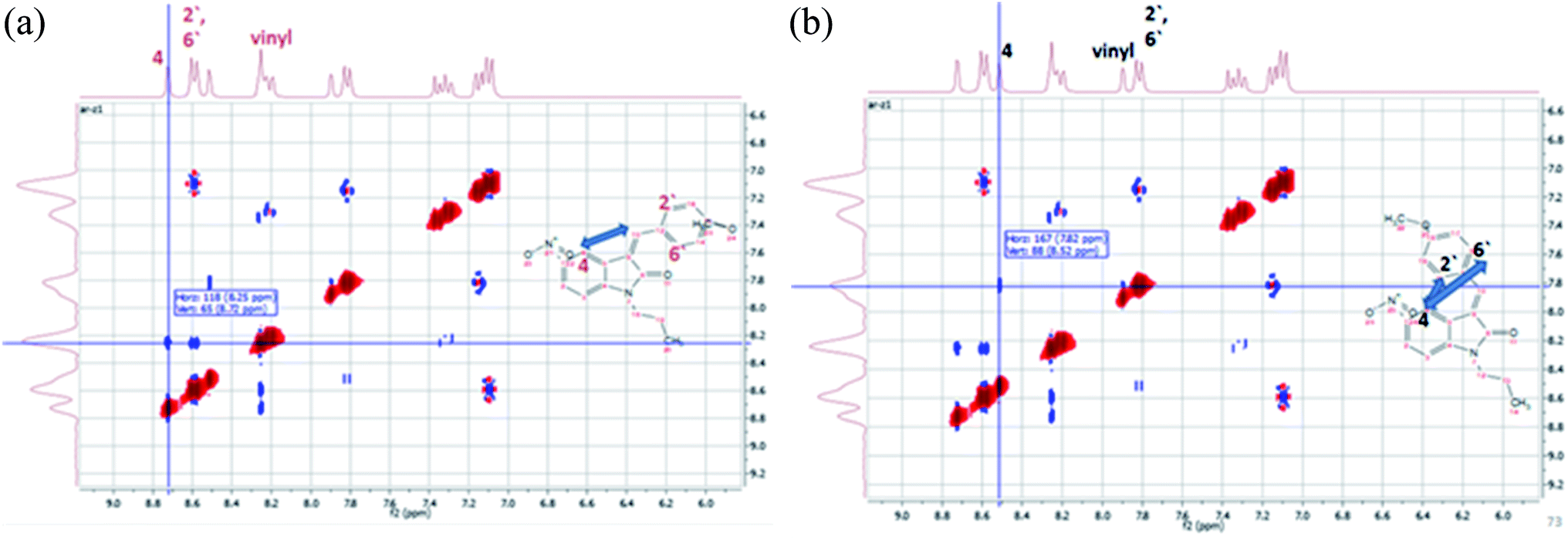 | ||
| Fig. 5 NOESY 1H–1H 2D NMR interactions of the E/Z-diastereomers of 5c. (A) The major diastereomer (Z). (B) The minor diastereomer (E). | ||
Kinetics of the isomerization study
It has been reported that the E/Z diastereomers of 3-benzylidene-2-oxo-indolines both can undergo isomerization to each other.28,29 This phenomenon is also observed in the change of the initial E:Z product ratios for the synthesized compounds listed in Table 1 during the measurement of the 1H NMR spectra in DMSO-d6. To acquire knowledge on the rates of the configurational isomerization of the E/Z diastereomers for compounds 5a, 5b, 5c1, 5c2, 5d, 6a, 6c, 7a, 7b1 and 7b2, the change of the initial diastereomeric ratios at time intervals was measured in DMSO-d6 at room temperature, and are listed in Table 2.
| Compd | Substituents | E:Z ratio |
E:Z ratio |
E:Z ratio |
E:Z ratio |
E:Z ratio |
Isomerization | ||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | Time: 0 h | 1 h | 2 h | 3 h | 5 h | ||
| 5a | NO2 | 4′-F | Benzyl | 5.7:94.3 |
8.3:91.7 |
12.3:87.7 |
13.0:87.0 |
17.4:82.6 |
From Z to E |
| 5b | NO2 | 4′-F | Propyl | 17:83 |
17:83 |
17:83 |
17:83 |
17:83 |
No isomeriz. |
| 5c1 | NO2 | 4′-OCH3 | Propyl | 30.1:69.9 |
34.6:65.4 |
33.8:66.2 |
35.1:64.9 |
35.1:64.9 |
From Z to E |
| 5c2 | NO2 | 4′-OCH3 | Propyl | 64.9:35.1 |
61.3:38.7 |
55.2:44.8 |
51.3:48.7 |
44.4:55.6 |
From E to Z |
| 5d | NO2 | 2′-Cl | Propyl | 92.6:7.4 |
88.5:11.5 |
84.0:16.0 |
82.0:18.0 |
80.6:19.4 |
From E to Z |
| 6a | H | 4′-Br | H | 100:0 |
96.2:3.8 |
94.3:5.7 |
91.7:8.3 |
90.1:9.9 |
From E to Z |
| 6c | H | 4′-OCH3 | H | 98.0:2.0 |
96.2:3.8 |
93.5:6.5 |
91.7:8.3 |
90.1:9.9 |
From E to Z |
| 7a | H | 4′-F | Benzyl | 18.0:82.0 |
30.1:69.9 |
35.1:64.9 |
38.3:61.7 |
42.9:57.1 |
From Z to E |
| 7b1 | H | 4′-F | Propyl | 2.0:98.0 |
13.0:87.0 |
15.3:84.7 |
20.6:79.4 |
29.1:70.9 |
From Z to E |
| 7b2 | H | 4′-F | Propyl | 87.7:12.3 |
87.0:13.0 |
87.0:13.0 |
87.0:13.0 |
87.0:13.0 |
From E to Z |
The diastereomeric ratios of the tested compounds were monitored using 1H NMR by measuring the integral height values of the vinyl and 2′,6′-protons of the respective E and Z diastereomers. With the exception of compounds 5b, 5c1 and 7b2 that revealed no or insignificant isomerization at the reaction conditions, the rate of the isomerization reactions of the other tested compounds were found to display first-order kinetics over the investigated solvent and temperature. Representative first-order kinetics plots for the isomerization of the tested compounds are shown in Fig. 6. From the slopes of the linear correlations, it can be seen that the potential for isomerization proceeds with varied rates, and the rate data obtained for the tested compounds are recorded in Table 3.
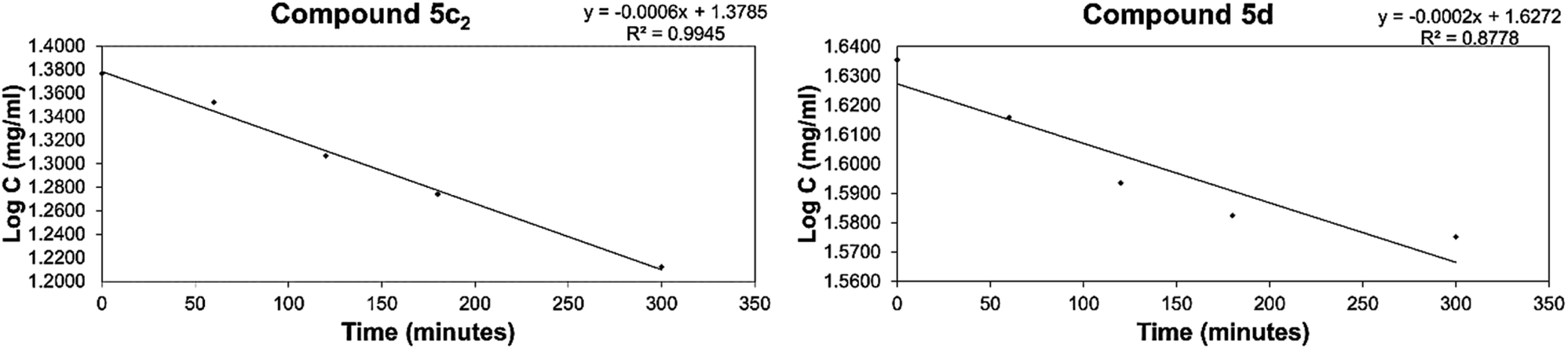 | ||
| Fig. 6 Graphical representation of first-order kinetic isomerization of the E/Z diastereomers for representative compounds 5c2 and 5d. | ||
| Compd | k × 103 (min−1) (r2, r) | t1/2 (h) |
|---|---|---|
| 5a | 0.461 (0.969, 0.984) | 25.08 |
| 5c2 | 1.382 (0.995, 0.997) | 8.37 |
| 5d | 0.461 (0.878, 0.937) | 25.08 |
| 6a | 0.230 (0.930, 0.964) | 50.15 |
| 6c | 0.230 (0.951, 0.975) | 50.15 |
| 7a | 0.415 (0.996, 0.998) | 27.85 |
| 7b1 | 0.921 (0.969, 0.984) | 12.53 |
No general behavior pattern for the isomerization of the tested compounds was observed. Compounds 5b and 5c showed no significant Z/E isomerization, but the rate of E/Z isomerization of 5c (t1/2 = 8.3 h) revealed more stability for the Z isomer. On the other hand, compound 7b showed no E/Z isomerization, but the rate of Z/E isomerization (t1/2 = 12.53 h) revealed that the E diastereomer is more stable. Attempts to carry out additional studies on these compounds using other solvents, such as CDCl3, failed due to solubility issues. No attempts were made to investigate the mechanism of isomerization; however, this observed phenomenon is consistent with previously reported ones.28,29,36,37
Molecular structure and modeling studies
Molecular orbital calculations were carried out to investigate the structure and stability of the E/Z-diastereomers of the subjected molecules. The investigated benzylidene oxindole structures were optimized in the ground state using AM1 and PM3, and MNDO semi-empirical methods with default Hartree–Fock (RHF) basis using MOE 2019.01 software.To determine which diastereomer is more stable ((E) or (Z)), the heat of formation (HF, kcal mol−1) was calculated for each diastereomer by three different methods, AM1, MNDO and PM3. The results are recorded in Table 4.
| Compd and config. | Heat of formation (kcal mol−1) | Total energy (kcal mol−1) | Potential energy (kcal mol−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AM1 | MNDO | PM3 | AM1 | MNDO | PM3 | AM1 | MNDO | PM3 | ||
| 5a | E | 37.256 | 31.578 | 11.693 | −112488.86 |
−112511.38 |
−103287 |
93.276 | 89.752 | 94.042 |
| Z | 37.340 | 30.934 | 6.627 | −112488.77 |
−112512.03 |
−103286.73 |
95.959 | 92.903 | 96.041 | |
| 5b | E | −1.852 | −7.613 | −21.439 | −100700.08 |
−100751.77 |
−92381.56 |
77.165 | 72.529 | 84.174 |
| Z | −1.635 | −8.351 | −31.127 | −100699.96 |
−100752.5 |
−92380.82 |
80.214 | 75.271 | 85.692 | |
| 5c | E | 5.382 | −0.8790 | −26.998 | −100804.24 |
−101053.02 |
−92797.19 |
92.313 | 91.605 | 92.963 |
| Z | 4.534 | −1.486 | −25.804 | −100803.59 |
−101053.63 |
−92795.82 |
94.633 | 93.192 | 95.918 | |
| 5d | E | 37.420 | 32.169 | 5.746 | −98132.34 |
−97872.59 |
−89533.23 |
80.254 | 73.658 | 88.894 |
| Z | 37.414 | 31.458 | 10.397 | −98132.13 |
−97873.28 |
−89532.20 |
85.874 | 77.747 | 90.804 | |
| 6a | E | 52.500 | 33.937 | 40.929 | −67732.13 |
−67673.79 |
−63169.75 |
59.044 | 60.917 | 64.915 |
| Z | 52.959 | 33.491 | 41.925 | −67731.67 |
−67674.24 |
−63168.77 |
60.866 | 61.366 | 65.561 | |
| 6c | E | 9.479 | −8.195 | −4.928 | −70874.54 |
−71014.01 |
−65587.71 |
69.970 | 72.328 | 74.772 |
| Z | 9.435 | −8.622 | −3.910 | −70874.59 |
−71014.44 |
−65586.70 |
71.709 | 73.687 | 75.343 | |
| 7a | E | 33.941 | 25.706 | 15.403 | −93328.44 |
−93289.73 |
−86419.95 |
86.114 | 79.532 | 97.061 |
| Z | 41.257 | 15.488 | 21.588 | −93326.40 |
−93290.25 |
−86418.98 |
89.310 | 80.142 | 97.613 | |
| 7b | E | −4.797 | −22.279 | −22.610 | −81539.30 |
−81529.20 |
−75514.30 |
65.373 | 62.142 | 79.378 |
| Z | −3.140 | −22.891 | −21.660 | −81537.63 |
−81529.82 |
−75513.34 |
67.784 | 62.867 | 81.496 | |
From the results recorded in Table 4, the AM1 Hamiltonian shows that the (E) diastereomers are more stable than (Z) diastereomers for compounds 5a, 5b, 6a, 7a and 7b. However, the (Z) diastereomers are more stable than (E) diastereomers for compounds 5c, 5d and 6c. This is consistent with the observed rate of isomerization of the tested compounds (5c2 and 7b1 compared to the other tested compounds). The MNDO model showed that the (Z) diastereomers are slightly more stable, while the PM3 model revealed that the (E) diastereomers are more stable, except for compounds 5a and 5b.
In order to disclose this inconsistency between the three methods and the additional calculations of the total energy and potential energy of each diastereomer, these calculations were also computed by three different methods (AM1, MNDO and PM3), and the results are recorded in Table 4. According to the total energy calculations, the (E) diastereomers are more stable than the (Z) diastereomers according to AM1 and PM3 for all compounds except compound 6c, as the AM1 calculations reveal that the (Z) diastereomer is slightly more stable. The MNDO calculations for the total energy still shows that the (Z) diastereomers are slightly more stable than the (E) diastereomer. However, based on the potential energy calculations by the three methods (AM1, MNDO and PM3), it is clear that the (E) diastereomers are more stable than (Z) isomers for all compounds.
In conclusion, the AM1 method has shown better performance compared to PM3 and MNDO methods for the molecular geometry optimization of the investigated 3-benzylidene oxindole molecules.
For a better understanding of the factors affecting the rate of E/Z or Z/E isomerization, the Spearmen correlation (2-tailed) was studied between t1/2 and 9 selected physicochemical parameters, Table 5. Data were recorded and analyzed with IBM SPSS V22, and the P-value was considered significant if it was equal to or less than 0.05.
| Compd | t1/2 (h) | Heat of formation | Dipole moment | Electrostatic energy | Strain energy | vdW energy | Topological polar surface area | vdW area | Electrostatic interaction energy | Torsion energy |
|---|---|---|---|---|---|---|---|---|---|---|
| AM1_H | AM1_dipole | E_ele | E_strain | E_vdW | TPSA | vdW_ area | E_rele | E_tor | ||
| a AM1_H: heat of formation, AM1_dipole: dipole moment, E_ele: electrostatic energy, E_strain: strain energy, E_vdW: vdW energy, TPSA: topological polar surface area, vdW_area: vdW area, E_rele: electrostatic interaction energy, E_tor: torsion energy. | ||||||||||
| 5c | 8.37 | 5.382 | 2.939 | −1.068 | 15.948 | 7.525 | 75.360 | 335.918 | 20118.777 |
0.914 |
| 5d | 25.08 | 37.420 | 3.400 | 9.667 | 14.098 | 5.785 | 66.130 | 321.263 | 19081.748 |
−0.052 |
| 6a | 50.15 | 52.500 | 2.349 | −13.894 | 10.470 | 4.959 | 29.100 | 243.157 | 10623.542 |
7.366 |
| 6c | 50.15 | 9.479 | 3.308 | −18.750 | 12.762 | 6.472 | 38.330 | 246.036 | 11923.694 |
7.411 |
| 5a | 25.08 | 37.340 | 3.895 | 7.004 | 15.010 | 9.655 | 66.130 | 344.125 | 21515.154 |
4.774 |
| 7a | 27.85 | 41.257 | 3.532 | 0.3197 | 14.470 | 7.020 | 20.310 | 315.987 | 12584.364 |
5.612 |
| 7b | 12.53 | −3.140 | 3.621 | −1.1060 | 12.974 | 4.445 | 20.310 | 279.953 | 10202.203 |
0.011 |
Accordingly, the cross correlation coefficient between t1/2 (E/Z isomerization) and E_strain, TPSA, vdW_area and electrostatic interaction energy was high (0.949) with a significant p value (0.05), Table 6. However, when studying the cross correlation between t1/2 (Z/E isomerization), no significant correlation was obtained between the t1/2 (Z/E isomerization) and calculated descriptors.
| Variable | t1/2 (E/Z) | AM1_H | AM1_dipole | E_ele | E_strain | E_vdW | TPSA | vdW_area | E_rele | E_tor |
|---|---|---|---|---|---|---|---|---|---|---|
| a Significant p value (0.05). | ||||||||||
| t1/2 | 1.000 | |||||||||
| AM1_H | 0.632 | 1.000 | ||||||||
| AM1_dipole | −0.211 | 0.200 | 1.000 | |||||||
| E_ele | −0.736 | 0.000 | 0.400 | 1.000 | ||||||
| E_strain | −0.949a | 0.800 | 0.400 | 0.600 | 1.000 | |||||
| E_vdW | 0.632 | −1.000 | 0.200 | 0.000 | 0.800 | 1.000 | ||||
| TPSA | −0.949a | 0.800 | 0.400 | 0.600 | 1.000 | 0.800 | 1.000 | |||
| vdW_area | −0.949a | 0.800 | 0.400 | 0.600 | 1.000 | 0.800 | 1.000 | 1.000 | ||
| E_rele | −0.948a | 0.800 | 0.400 | 0.600 | 1.000 | 0.000 | 1.000 | 1.000 | 1.000 | |
| E_tor | 0.738 | 0.000 | −0.400 | −1.000 | −0.600 | −1.000 | −0.600 | −0.600 | −0.600 | 1.000 |
A good multiple regression equation was generated between the half-life (t1/2) of the isomerization and the QM parameters with significant p value, eqn (1): multiple regression equation between t1/2 and significant QM parameters.
| t1/2 = 187.5378 + 1.542499 × E_strain − 1.63343 × TPSA − 1.0012 × vdW-area + 0.012667 × E_rele, n = 7, R2 = 0.8536 | (1) |
The isomerization rate and stability of the diastereomers can be estimated using this equation. Matching the experimental data with the estimated ones, Table 7 and Fig. 7, revealed a quite good relationship.
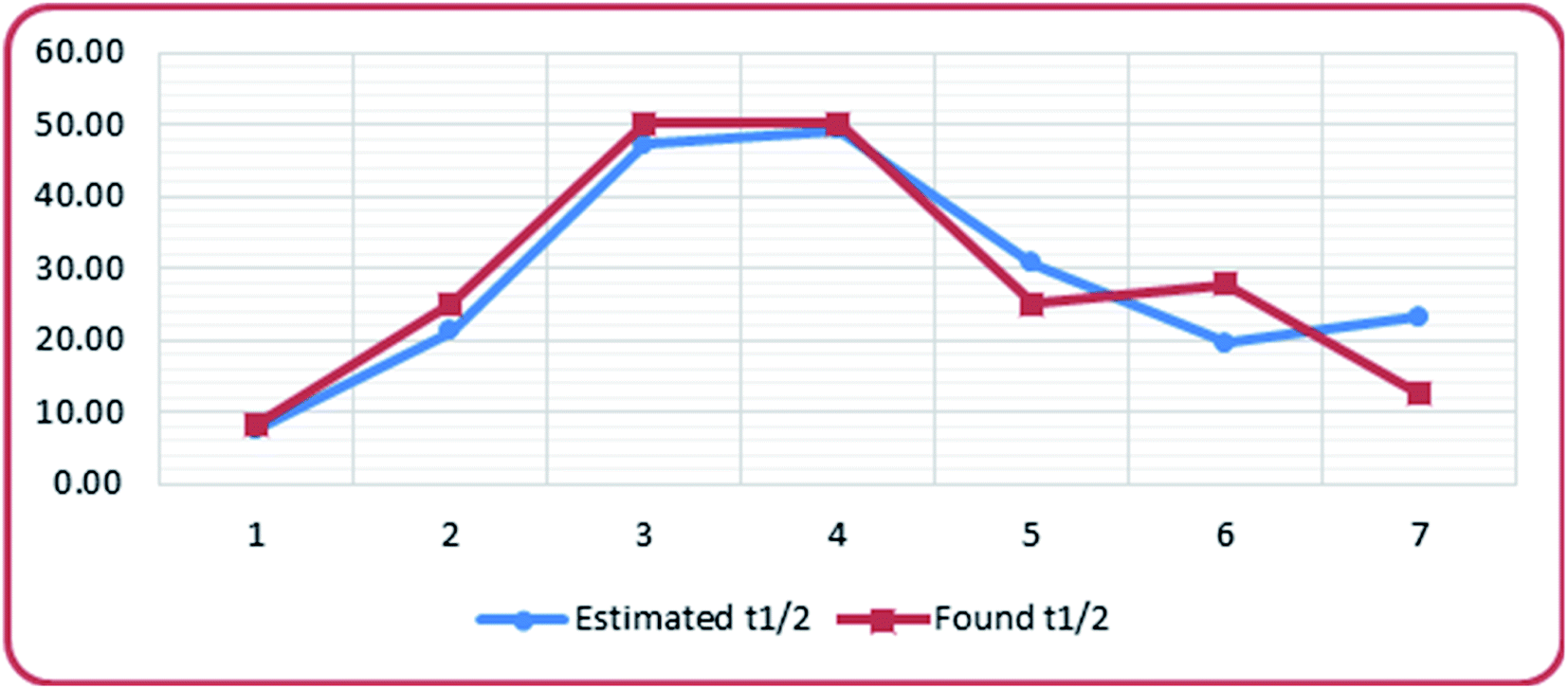 | ||
| Fig. 7 Correlation between found t1/2 and estimated t1/2. | ||
Molecular docking study on CDK2
The molecular docking study was performed to predict the binding and effect of the investigated molecules on the CDK2 enzyme, and the results are recorded in Table 8. As a general pattern, it is clear from the obtained data that the Z-diastereomers revealed better binding affinity to the active site of CDK2. In most of the tested compounds, the binding scores are quite close to that of the reference drug sunitunib [redocking rmsd of sunitinib = 2]. Further computational studies together with biological activity are currently in progress. Fig. 8 illustrates the 2D and 3D interactions of the co-crystallized sunitinib, the Z-diastereomer of 5a, and the E-diastereomer 6c.| Compd | Score (S, kcal mol−1) | Compd | Score (S, kcal mol−1) |
|---|---|---|---|
| Sunitinib | −7.8956 | ||
| 5a-E | −7.8976 | 5a-Z | −8.0125 |
| 5b-E | −7.3031 | 5b-Z | −7.2988 |
| 5c-E | −7.5347 | 5c-Z | −7.1837 |
| 5d-E | −6.7651 | 5d-Z | −7.1422 |
| 6a-E | −6.3125 | 6a-Z | −6.6100 |
| 6c-E | −6.3683 | 6c-Z | −6.7596 |
| 7a-E | −6.5126 | 7a-Z | −7.7525 |
| 7b-E | −6.9255 | 7b-Z | −6.8315 |
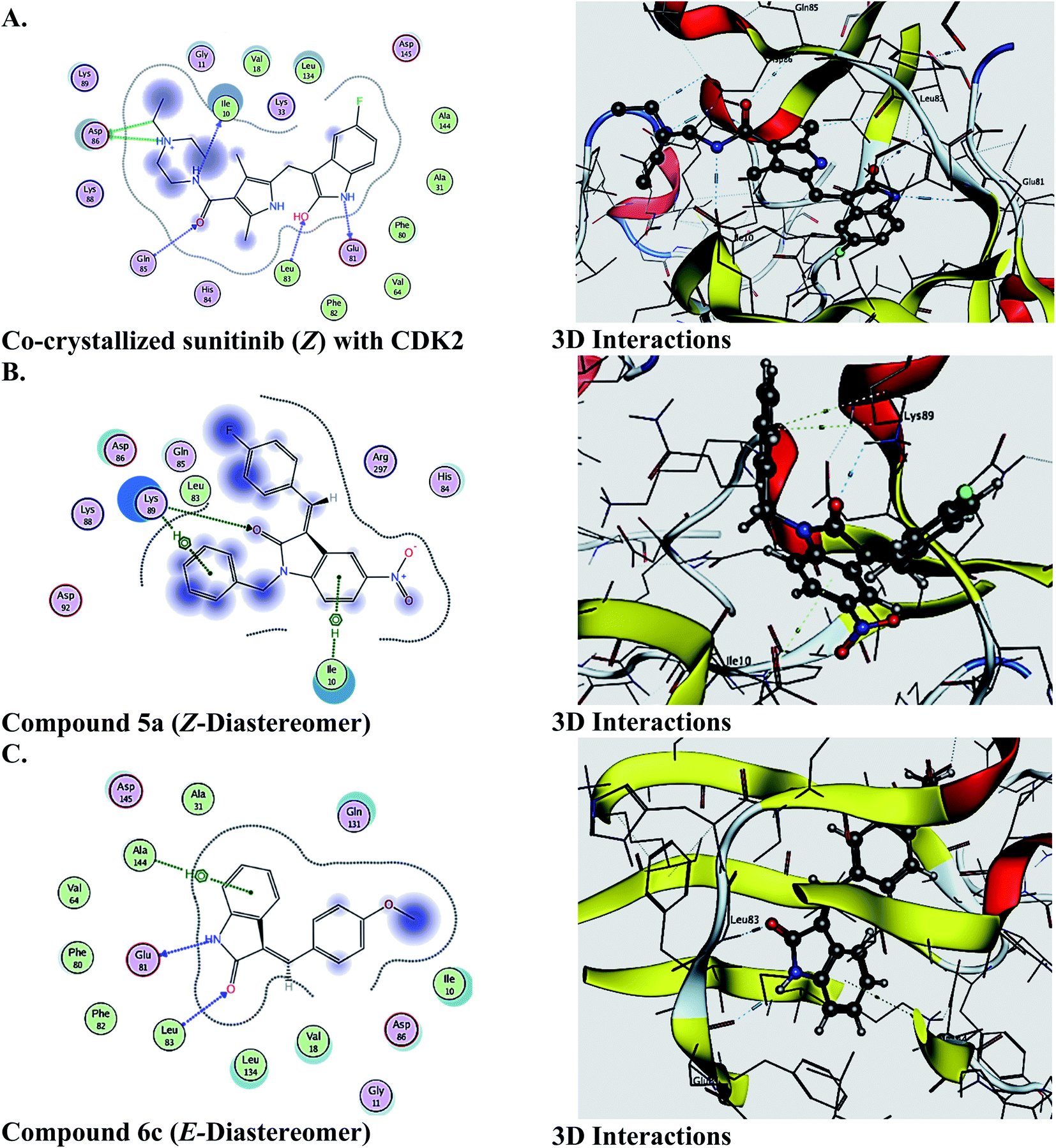 | ||
| Fig. 8 2D and 3D ligand interactions of (A) co-crystallized sunitinib (Z) with CDK2, (B) 5a (Z-diastereomer), (C) compound 6c (E-diastereomer). All ligands are docked into the active site of CDK2 (PDB ID: 3TI1). | ||
Conclusion
A series of 3-benzylideneindolin-2-ones and 5-nitro 3-benzylidene indolin-2-ones have been synthesized and obtained as E/Z-diastereomers. The compounds have been assigned as Z or E configurations by 1H NMR analysis. The configuration of the separated E- and Z-diastereomers were confirmed by 2D NOE. A kinetic study of the E/Z isomerization was carried out in DMSO-d6 at room temperature, and they were found to obey first-order kinetic reactions. To justify the stability of (E) or (Z), the heat of formation (HF, kcal mol−1) was calculated for each diastereomer by three different methods, AM1, MNDO and PM3. No clear justification could be obtained; however, the AM1 method has shown better performance compared to PM3 and MNDO. Furthermore, a good multiple regression equation was generated between the reaction rates of isomerization and some QM parameters with significant p value for prediction of stability of the diastereomer(s). In silico docking studies using MOE 2019.01 software revealed better docking on the targeted CDK2 enzyme for the Z-diastereomer compared to the E-one, and comparable docking scores to sunitinib as a reference drug.Experimental section
Chemicals were obtained commercially, and were used as received. TLC was carried out on Merck 0.2 mm pre-coated silica gel (60 F-254) aluminum sheets. The spots were visualized by UV. Synthesized compounds were purified by column chromatography using methanol/CH2Cl2 as the mobile phase. Melting points were measured with a capillary melting point SMP1 apparatus (Stuart Scientific), and are uncorrected. 1H and 13C NMR spectra were recorded on a Brucker-400 MHz at the NMR unit, Faculty of Pharmacy El-Mansoura University, El-Mansoura, Egypt. The chemical shifts are expressed in parts per million (δ) downfield relative to tetramethylsilane as an internal standard (0 ppm). Coupling constants are reported in hertz (Hz). Elemental analyses were performed on a Perkin Elmer 2400 CHN elemental analyzer at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt.All calculations based on semi-empirical molecular orbital theory have been carried out using Molecular Operating Environment (MOE 2019.01, 2019; Chemical Computing Group, Canada) software at the Medicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut, Egypt.
Synthesis of indolin-2-one (oxindole) (2)
A stirred solution of isatin 1 (0.01 mol; 1.47 g) in hydrazine hydrate (15 ml) was heated to 140 °C for 4 h. The reaction was cooled to room temperature, poured into ice-cold water, then acidified to pH 2 with 6 N hydrochloric acid and kept at room temperature for 2 days. The solution was extracted with CH2Cl2 (5 × 20), dried over anhydrous sodium sulfate, filtered, and then re-crystallized from water to give the pure compound. Yield = 80% (1.18 g), mp: 126–128 °C (lit.,38 127–129 °C).1H-NMR (DMSO-d6, 60 MHz) δ:
9.15 (bs, 1H, N![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) ), 7.4–6.7 (m, 5H, 4,5,6,7), 3.48 (s, 2H, 3,3) ppm.
), 7.4–6.7 (m, 5H, 4,5,6,7), 3.48 (s, 2H, 3,3) ppm.
Synthesis of 5-nitroindolin-2-one (3)
To a stirred solution of 2-oxo-indoline 2 (30 mol; 4 g) in cold concentrated sulfuric acid at 0 °C (28 ml), a cold solution of potassium nitrate (30 mmol; 3.87 g) in concentrated sulfuric acid (28 ml) was added in one portion. Stirring was continued for 1.5 h in an ice bath (the temperature of the mixture must not exceed 5 °C), and the mixture was then added to 250 ml of crushed ice. The buffy-colored precipitate was collected by filtration, washed with water, and dried. The raw product was purified by recrystallization from acetic acid (50%) to give the pure product. Yield: 70% (3.75 g), mp: 247–250 °C (lit.,34 249–254 °C).1H-NMR (DMSO-d6, 60 MHz) δ:
11.25 (bs, 1H, N), 8.3–8 (m, 2H, 4,6), 7.01 (d, J = 7.7 Hz, 1H, 7), 3.66 (s, 2H, 3,3) ppm.
General procedure for the synthesis of target compounds (6a and 6c) as reported33,39
Piperidine (0.01 mmol) was added to a solution of compound 2 or 3 (10 mmol) and appropriate substituted benzaldehyde (11 mmol) in ethanol (15 ml). The mixture was refluxed for 3–5 h, and then the reaction mixture was cooled to room temperature and left overnight. The precipitate was filtered, washed with ethanol, and dried at room temperature to give the titled compounds. The obtained crude products were purified by column chromatography.IR: 3472 and 1708.2 cm−1.
1H-NMR (DMSO-d6, 400 MHz) δ:
E-Diastereomer: 10.65 (s, 1H, N), 7.72 (d, J = 8.3 Hz, 2H, 2′,6′), 7.66 (d, J = 8.3 Hz, 2H, 3′,5′), 7.57 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–), 7.49 (d, J = 7.7 Hz, 1H, 4), 7.24 (t, J = 7.7 Hz, 1H, 5), 6.87 (m, 2H, 6,7) ppm.
C–), 7.49 (d, J = 7.7 Hz, 1H, 4), 7.24 (t, J = 7.7 Hz, 1H, 5), 6.87 (m, 2H, 6,7) ppm.
Z-Diastereomer: 10.69 (s, 1H, N), 8.32 (d, J = 8.3 Hz, 2H, 2′,6′), 7.78 (s, 1H, C–), 7.71 (m, 1H, 4), 7.68 (m, 2H, 3′,5′), 7.22 (t, J = 7.7 Hz, 1H, 5), 6.99 (t, J = 7.7 Hz, 1H, 6), 6.82 (m, 1H, 7) ppm.
IR: 3452.5 and 1703.5 cm−1.
1H-NMR (DMSO-d6, 400 MHz) δ:
E-Diastereomer: 10.58 (s, 1H, NH), 7.72 (d, J = 8.5, 2H, 2′,6′), 7.66 (d, J = 7.8 Hz, 1H, 4), 7.59 (s, 1H, C–), 7.22 (t, J = 7.7 Hz, 1H, 5), 7.09 (d, J = 8.5 Hz, 2H, 3′,5′), 6.87 (m, 2H, 6,7), 3.84 (s, 3H, –OC3) ppm.
Z-Diastereomer: 10.61 (s, 1H, NH), 8.48 (d, J = 8.5, 2H, 2′,6′), 7.75 (s, 1H, C–), 7.66 (d, J = 7.8 Hz, 1H, 4), 7.18 (t, J = 7.7 Hz, 1H, 5), 7.04 (d, J = 8.5 Hz, 2H, 3′,5′), 6.98 (t, J = 7.7 Hz, 1H, 6), 6.82 (d, J = 8.0 Hz, 1H, 7), 3.84 (s, 3H, –OC3) ppm.
General procedure for the synthesis of target compounds [5(a–d) & 7(a–b)]
The appropriate compound [4(a–c) and 6(a–c)] (10 mmol.) was dissolved in anhydrous DMF (5–10 ml), and cooled on ice with stirring. Solid K2CO3 (12 mmol) was added in one portion, and the dark colored suspension was brought to room temperature and stirred for an additional 1 h. Alkyl chloride or bromide (11 mmol) and KI (2 mmol) were added, and the reaction mixture was stirred at 60–80 °C for 5–24 h, until the starting material had been consumed (monitored by TLC). The reaction mixture was poured into HCl (0.5 M, 50 ml) and extracted with ethyl acetate (2 × 25 ml). The ethyl acetate portion was washed with brine and dried over anhydrous Na2SO4. The crude product was purified by column chromatography using CH2Cl2.:Z ratio = 6:94.Yellow solid powder; yield: 67%; mp: 218–221 °C;
1H-NMR (DMSO-d6, 400 MHz) δ:
Z-Diastereomer: 8.77 (d, J = 2.2 Hz, 1H, 4), 8.60 (dd, J = 8.9, 1.8 Hz, 2H, 2′,6′), 8.38 (s, 1H, C–), 8.20 (dd, J = 8.7, 1.8 Hz, 1H, 6), 7.43–7.23 (m, 7H, benzylic Hs, 3′,5′), 7.20 (d, J = 9.4 Hz, 1H, 7), 5.09 (s, 2H, –C2–) ppm.
E-Diastereomer: 8.34 (d, J = 2.2 Hz, 1H, 4), 8.22 (dd, J = 8.7, 2.2 Hz, 1H, 6), 8.02 (s, 1H, C-), 7.91 (dd, J = 8.9,1.8 Hz, 2H, 2′,6′), 7.47–7.25 (m, 7H, benzylic Hs, 3′,5′), 7.22 (d, J = 9.4 Hz, 1H, 7), 5.10 (s, 2H, –C2–) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
Z-Diastereomer: 166.2, 165.5, 146.3, 143.1, 140.9, 136.5, 136, 135.9, 130.7, 129.2 (2C), 128, 127.8 (2C), 125.6, 123, 117.6, 116.2, 116, 115.9, 109.5, 43.6 ppm.
E-Diastereomer: 168.1, 162.9, 148.6, 142.6, 140.1, 136.2, 132.8, 132.7, 130.6, 129.3 (2C), 128.1, 127.7 (2C), 126.9, 125.2, 123.1, 121.1, 116.7, 116.5, 110, 43.5 ppm.
Elemental analysis calculated/found for C22H15FN2O3: C 70.58/70.41; H 4.04/4.23; N 7.48/7.29.
:Z ratio = 17:83.Bright yellow solid powder; yield: 77%; mp: 177–180 °C;
1H-NMR (DMSO-d6, 400 MHz) δ:
Z-Diastereomer: 8.72 (d, J = 2.2 Hz, 1H, 4), 8.58 (dd, J = 8.8, 1.8 Hz, 2H, 2′,6′), 8.29 (s, 1H, C–), 8.23 (dd, J = 1.8, 8.8 Hz, 1H, 6), 7.37 (t, J = 9.2 Hz, 2H, 3′,5′), 7.31 (d, J = 9.2 Hz, 1H, 7), 3.80 (t, 2H, –C2–CH2–CH3), 1.65 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
E-Diastereomer: 8.68 (d, J = 2.2 Hz, 1H, 4), 8.37 (dd, J = 1.8, 8.8 Hz, 1H, 6), 7.93 (s, 1H, C–), 7.87 (dd, J = 8.8, 1.8 Hz, 2H, 2′,6′), 7.53 (d, J = 8.9 Hz, 1H, 7), 7.44 (t, J = 9.2 Hz, 2H, 3′,5′), 3.80 (t, 2H, –C2–CH2–CH3), 1.65 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
Z-Diastereomer: 167.9, 166.1, 146.8, 142.8, 140.3, 135.9, 135.8, 130.8, 125.7, 123.3, 117.5, 116.1, 115.9, 115.8, 109.1, 41.7, 21, 11.5 ppm.
E-Diastereomer: 165.4, 162.9, 149.1, 142.3, 139.5, 132.7, 132.6, 130.6, 126.8, 125.1, 123.3, 117.5, 116.7, 116.5, 109.7, 41.8, 20.8, 11.6 ppm.
Elemental analysis calculated/found for C18H15FN2O3: C 66.25/66.47; H 4.63/4.75; N 8.58/8.8.
:Z ratio = 30:70.Yellow solid powder; yield: 45%; mp: 150–153 °C;
1H-NMR (DMSO-d6, 400 MHz) δ:
Z-Diastereomer: 8.67 (d, J = 1.3 Hz, 1H, 4), 8.56 (d, J = 8.9, 2H, 2′,6′), 8.19 (s, 1H, C–), 8.17 (dd, J = 8.7 Hz, 2.2 Hz, 1H, 6),7.25 (d, J = 8.8 Hz, 1H, 7), 7.06 (d, J = 8.9 Hz, 2H, 3′,5′), 3.86 (s, 3H, –OC3), 3.78 (t, 2H, –C2–CH2–CH3), 1.63 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
E-Diastereomer: 8.47 (d, J = 1.3 Hz, 1H, 4), 8.21 (dd, J = 8.7 Hz, 2.2 Hz, 1H, 6), 7.86 (s, 1H, C–), 7.78 (d, J = 8.8, 2H, 2′,6′), 7.31 (d, J = 8.8 Hz, 1H, 7),7.13 (d, J = 8.9 Hz, 2H, 3′,5′), 3.87 (s, 3H, –OC3), 3.78 (t, 2H, –C2–CH2–CH3), 1.63 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
Z-Diastereomer: 166.3, 162.6, 146.2, 142.7, 141.6, 135.8 (2C), 127, 125.6, 124.8, 120.7, 115.1, 114.4 (2C), 108.8, 56, 41.6, 21.1, 11.6 ppm.
E-Diastereomer: 168.3, 161.9, 148.7, 142.2, 140.8, 132.6 (2C), 126.3, 126.2, 122.8, 121.3, 117.2, 114.9 (2C), 109.4, 56, 41.7, 21, 11.6 ppm.
Elemental analysis calculated/found for C19H18N2O4: C 67.44/67.71; H 5.36/5.49; N 8.28/8.57.
:Z ratio = 65:35.Yellow solid powder; yield: 77%; mp: 147–150 °C;
1H-NMR (DMSO-d6, 400 MHz) δ:
E-Diastereomer: 8.47 (d, J = 1.3 Hz, 1H, 4), 8.21 (dd, J = 8.7 Hz, 2.2 Hz, 1H, 6), 7.86 (s, 1H, C–), 7.78 (d, J = 8.8, 2H, 2′,6′), 7.33 (d, J = 8.8 Hz, 1H, 7),7.14 (d, J = 8.9 Hz, 2H, 3′,5′), 3.88 (s, 3H, –OC3), 3.79 (t, 2H, –C2–CH2–CH3), 1.63 (m, 2H, –CH2–C2–CH3), 0.90 (t, 3H, –CH2–CH2–C3) ppm.
Z-Diastereomer: 8.68 (d, J = 1.3 Hz, 1H, 4), 8.57 (d, J = 8.9, 2H, 2′,6′), 8.19 (s, 1H, C–), 8.18 (dd, J = 8.7 Hz, 2.2 Hz, 1H, 6), 7.27 (d, J = 8.8 Hz, 1H, 7),7.08 (d, J = 8.9 Hz, 2H, 3′,5′), 3.88 (s, 3H, –OC3), 3.79 (t, 2H, –C2–CH2–CH3), 1.63 (m, 2H, –CH2–C2–CH3), 0.90 (t, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
E-Diastereomer: 168.3, 161.9, 148.7, 142.2, 140.8, 132.6 (2C), 126.3, 126.2, 122.8, 121.3, 117.2, 114.9 (2C), 109.4, 56, 41.7, 21, 11.6 ppm.
Z-Diastereomer: 166.3, 162.6, 146.2, 142.7, 141.6, 135.8 (2C), 127, 125.6, 124.8, 120.7, 115.1, 114.4 (2C), 108.8, 56, 41.6, 21.1, 11.6 ppm.
Elemental analysis calculated/found for C19H18N2O4: C 67.44/67.71; H 5.36/5.49; N 8.28/8.57.
:Z ratio = 93:7.Yellow solid powder; yield: 62%; mp: 136–139 °C;
1H-NMR (DMSO-d6, 400 MHz) δ:
E-Diastereomer: 8.25 (dd, J = 7.8, 2.2 Hz, 1H, 6), 7.97 (d, J = 2.2 Hz, 1H, 4), 7.87 (s, 1H, C–), 7.85 (d, J = 7.8 Hz, 1H, 3′), 7.71 (d, J = 8.8 Hz, 1.1 Hz, 1H, 6′), 7.62 (td, J = 7.5 Hz, 0.8 Hz, 1H, 4′), 7.55 (t, J = 2.4 Hz, 1H, 5′), 7.38 (d, J = 8.8 Hz, 1H, 7), 3.80 (t, J = 2.4 Hz, 2H, –C2–CH2–CH3), 1.65 (m, 2H, –CH2–C2–CH3), 0.91 (t, J = 2.4 Hz, 3H, –CH2–CH2–C3) ppm.
Z-Diastereomer: 8.76 (d, J = 2.2 Hz, 1H, 4), 8.30 (s, 1H, C–), 8.25 (dd, J = 7.8, 2.2 Hz, 1H, 6), 8.06 (dd, J = 8.0, 1.2 Hz, 1H, 6′), 7.59–7.52 (m, 1H, 3′), 7.48 (t, J = 7.5 Hz, 1H, 5′), 7.39 (t, J = 7.5 Hz, 1H, 4′), 7.31 (d, J = 8.8 Hz, 1H, 7), 3.72 (t, J = 2.4 Hz, 2H, –C2–CH2–CH3), 1.61 (m, 2H, –CH2–C2–CH3), 0.87 (t, J = 2.4 Hz, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
E-Diastereomer: 167.5, 149.4, 142.3, 136.1, 133.6, 132.7, 132.5, 130.9, 130.6, 128.1, 127.3, 127.2, 120.6, 117.9, 109.9, 41.9, 21, 11.6 ppm.
Z-Diastereomer: 165.6, 147.6, 142.9, 136.8, 134, 132.8, 132.2, 131.9, 129.6, 126.9, 126.6, 126.2, 123.8, 116.7, 109.2, 41.6, 21, 11.6 ppm.
Elemental analysis calculated/found for C18H15ClN2O3: C 63.07/62.89; H 4.41/4.58; N 8.17/8.45.
:Z ratio = 18:82.Yellow semisolid; yield: 67%;
1H-NMR (DMSO-d6, 400 MHz) δ:
Z-Diastereomer: 8.54 (dd, J = 8.5, 1.8 Hz, 2H, 2′,6′), 7.96 (s, 1H, C–), 7.79 (d, J = 8.5 Hz, 1H, 4), 7.42–7.22 (m, 8H, Benzylic Hs, 5, 3′,5′), 7.06 (t, J = 8.7 Hz, 1H, 6), 6.98 (d, J = 8.9 Hz, 1H, 7), 5.00 (s, 2H, –C2–) ppm.
E-Diastereomer: 7.83 (dd, J = 8.5, 1.8 Hz, 2H, 2′,6′), 7.80 (s, 1H, C–), 7.56 (d, J = 8.5 Hz, 1H, 4), 7.42–7.22 (m, 8H, Benzylic Hs, 5, 3′,5′), 6.99 (d, J = 8.9 Hz, 1H, 7), 6.92 (t, J = 8.7 Hz, 1H, 6), 5.00 (s, 2H, –C2–) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
Z-Diastereomer: 165.7, 164.9, 162.4, 143.5, 136.5, 135.3, 132.4, 131.2, 130.6, 129.1 (2C), 127.9, 127.8 (2C), 125.4, 122.7, 122.3, 120.2, 116.5, 115.7, 109.5, 43.1 ppm.
E-Diastereomer: 167.8, 164.4, 161.9, 141.3, 137.2, 137, 135.2, 132.3, 129.4, 129.2 (2C), 127.9, 127.7 (2C), 126.9, 124.4, 122.4, 102.7, 116.3, 115.9, 110.1, 43 ppm.
Elemental analysis calculated/found for C22H16FNO: C 80.23/79.97; H 4.90/5.12; N 4.25/4.48.
:Z ratio = 2:98.Yellow semisolid; yield: 55%;
1H-NMR (DMSO-d6, 400 MHz) δ:
Z-diastereomer: 8.51 (dd, J = 7.8, 1.8 Hz, 2H, 2′,6′), 7.89 (s, 1H, C-), 7.77 (d, J = 8.5 Hz, 1H, 4), 7.36–7.26 (m, 3H, 3′,5′, 6), 7.07 (m, 2H, 5,7), 3.72 (t, 2H, –C2–CH2–CH3), 1.64 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
E-Diastereomer: 7.79 (dd, J = 7.8, 1.8 Hz, 2H, 2′,6′), 7.7 (s, 1H, C–), 7.54 (d, J = 8.5 Hz, 1H, 4), 7.40–7.28 (m, 3H, 3′,5′, 6), 7.11 (d, J = 8.8 Hz, 1H, 7), 6.92 (t, J = 8.9 Hz, 1H, 5), 3.72 (t, 2H, –C2–CH2–CH3), 1.64 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
Z-Diastereomer: 167.6, 165.7, 162.3, 141.8, 136.4, 135.2, 135.1, 129.5, 125.7, 122.6, 122, 120.1, 115.8, 115.6, 109.1, 41.2, 21, 11.7 ppm.
E-Diastereomer: 164.8, 164.3, 161.8, 143.9, 135.9, 132.4, 132.3, 130.7, 127.1, 124.3, 122.1, 120.6, 116.5, 116.2, 109.6, 41.3, 20.9, 11.7 ppm.
Elemental analysis calculated/found for C18H16FNO: C 76.85/76.59; H 5.73/5.89; N 4.98/5.13.
:Z ratio = 88:12.Yellow semisolid; yield: 55%; 1H-NMR (DMSO-d6, 400 MHz) δ:
E-Diastereomer: 7.79 (dd, J = 7.8, 1.8 Hz, 2H, 2′,6′), 7.7 (s, 1H, C–), 7.54 (d, J = 8.5 Hz, 1H, 4), 7.40–7.28 (m, 3H, 3′,5′, 6), 7.11 (d, J = 8.8 Hz, 1H, 7), 6.92 (t, J = 8.9 Hz, 1H, 5), 3.71 (t, 2H, –C2–CH2–CH3), 1.64 (m, 2H, –CH2–C2–CH3), 0.90 (t, 3H, –CH2–CH2–C3) ppm.
Z-Diastereomer: 8.51 (dd, J = 7.8, 1.8 Hz, 2H, 2′,6′), 7.89 (s, 1H, C–), 7.77 (d, J = 8.5 Hz, 1H, 4), 7.36–7.26 (m, 3H, 3′,5′, 6), 7.07 (m, 2H, 5,7), 3.69 (t, 2H, -C2–CH2–CH3), 1.64 (m, 2H, –CH2–C2–CH3), 0.89 (t, 3H, –CH2–CH2–C3) ppm.
13C-NMR (DMSO-d6, 100 MHz) δ:
E-Diastereomer: 164.8, 164.3, 161.8, 143.9, 135.9, 132.4, 132.3, 130.7, 127.1, 124.3, 122.1, 120.6, 116.5, 116.2, 109.6, 41.3, 20.9, 11.7 ppm.
Z-Diastereomer: 167.6, 165.7, 162.3, 141.8, 136.4, 135.2, 135.1, 129.5, 125.7, 122.6, 122, 120.1, 115.8, 115.6, 109.1, 41.2, 21, 11.7 ppm.
Elemental analysis calculated/found for C18H16FNO: C 76.85/76.59; H 5.73/5.89; N 4.98/5.13.
Kinetic measurements
Isomerization rates of the tested compounds (5a, 5b, 5c1, 5c2, 5d, 6a, 6c, 7a, 7b1, 7b2) in DMSO-d6 were determined at room temperature. The reactions were initiated by dissolving 10 mg of the tested compound to 0.3 ml of DMSO-d6 in NMR tubes. At appropriate intervals, the 1H-NMR spectrum for each sample was measured, and the integral height values of the vinyl and 2′,6′-protons chemical shifts of the respective E and Z diastereomers were determined. The residual heights displayed a first-order rate of kinetics. The results of the kinetic measurements are given in Tables 2 and 3.Molecular modeling studies
The molecular docking studies were performed (Medicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Egypt) using Molecular Operating Environment (MOE 2019.01, 2019; Chemical Computing Group, Canada) as the computational software. The three-dimensional structures and conformations of the enzyme, cyclin-dependent kinase enzyme (CDK2) in complex with sunitinib (PDB ID: 3TI1), was acquired from the Protein Data Bank (PDB) web site.40 The binding affinity of the docked molecules was expressed as binding score (S, kcal mol−1). Briefly, docking of the investigated molecules was performed in four steps: preparation of the 3D structure of the target, preparation of the ligands, running docking, and interpretation of the results. Docking was performed using the default settings of the MOE program [Placement, triangular Matcher; Rescoring 1, London dG with retain = 30; Refinement, Forcefield; Rescoring 2, GBVI/WSA dG with retain = 30]. The key amino acids involved in the interaction of sunitinib with CDK2 in the co-crystalized target are Ile10, Glu81, Leu83, Gln85 and Asp86. Re-docking of sunitinib was performed to accurately evaluate the efficiency of the method (rmsd = 2).Conflicts of interest
The authors declare no conflict of interest.References
- M. Tutone and A. M. Almerico, Eur. J. Med. Chem., 2017, 142, 300–315 CrossRef CAS.
- H. N. Bramson, W. D. Holmes, R. N. Hunter, K. E. Lackey, B. Lovejoy, M. J. Luzzio, V. Montana, W. J. Rocque, D. Rusnak, L. Shewchuk, J. M. Veal, J. Corona, D. H. Walker, L. F. Kuyper, S. T. Davis, S. H. Dickerson, M. Edelstein, S. V. Frye, R. T. Gampe, P. A. Harris and A. Hassell, J. Med. Chem., 2001, 44, 4339–4358 CrossRef CAS.
- P. R. Murthi, N. Suresh, C. S. Rani, M. V. B. R. Rao and M. Pal, Lett. Drug Des. Discovery, 2015, 12, 109–116 CrossRef CAS.
- J. Ai, M. Lv, X. Li, Z. Chen, G. Hu and Q. Li, Med. Chem. Res., 2018, 27, 161–170 CrossRef CAS.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS.
- T. A. Chohan, A. Qayyum, K. Rehman, M. Tariq and M. S. H. Akash, Biomed. Pharmacother., 2018, 107, 1326–1341 CrossRef CAS.
- X. Grana and E. P. Reddy, Oncogene, 1995, 11, 211–219 CAS.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, Nat. Rev. Drug Discovery, 2015, 14, 130–146 CrossRef CAS.
- İ. Doğan, G. Bölek and B. Kahveci, Synthesis of Some New Isatin Derivatives and Identification of Their Structures, issue: Special [en] Süleyman Demirel Üniversitesi Fen Bilimleri Enstitüsü Dergisi, 2019, vol. 23, pp. 67–70, DOI:10.19113/sdufenbed.432261.
- N. E. A. A. El-sattar, E. H. K. Badawy, W. H. Abdel-hady, M. I. Abo-alkasem, A. A. Mandour and N. S. M. Ismail, Chem. Pharm. Bull., 2021, 69, 106–117 CrossRef.
- P. Pakravan, S. Kashanian, M. M. Khodaei and F. J. Harding, Pharmacol. Rep., 2013, 65, 313–335 CrossRef CAS.
- G. M. Šekularac, J. B. Nikolić, P. Petrović, B. Bugarski, B. Durović and S. Z. Drmanić, J. Serb. Chem. Soc., 2014, 79, 1347–1354 CrossRef.
- Y. Ding, L. Zhao, Y. Fu, L. Hao, Y. Fu, Y. Yuan, P. Yu and Y. Teng, Molecules, 2021, 26, 176 CrossRef CAS.
- X. Chen, T. Yang, A. Deivasigamani, M. K. Shanmugam, K. M. Hui, G. Sethi and M. L. Go, ChemMedChem, 2015, 10, 1548–1558 CrossRef CAS.
- T. T. L. Huong, D. T. M. Dung, N. Van Huan, L. Van Cuong, P. T. Hai, L. T. T. Huong, J. Kim, Y. G. Kim, S. B. Han and N. H. Nam, Bioorg. Chem., 2017, 71, 160–169 CrossRef CAS.
- D. T. M. Dung, P. T. P. Dung, D. T. K. Oanh, P. T. Hai, L. T. T. Huong, V. D. Loi, H. Hahn, B. W. Han, J. Kim, S.-B. Han and N.-H. Nam, Med. Chem., 2015, 11, 725–735 CrossRef CAS.
- W. Chu, D. Zhou, V. Gaba, J. Liu, S. Li, X. Peng, J. Xu, D. Dhavale, D. P. Bagchi, A. Avignon, N. B. Shakerdge, B. J. Bacskai, Z. Tu, P. T. Kotzbauer and R. H. Mach, J. Med. Chem., 2015, 58, 6002–6017 CrossRef CAS.
- H. S. Al-Salem, M. Arifuzzaman, H. M. Alkahtani, A. N. Abdalla, I. S. Issa, A. Alqathama, F. S. Albalawi and A. F. M. Motiur Rahman, Molecules, 2020, 25, 1–16 CrossRef.
- L. Wollin, E. Wex, A. Pautsch, G. Schnapp, K. E. Hostettler, S. Stowasser and M. Kolb, Eur. Respir. J., 2015, 45, 1434–1445 CrossRef CAS.
- F. Dufrasne and M. Galanski, Curr. Pharm. Des., 2007, 13, 2781–2794 CrossRef CAS.
- K. Suman, Y. Bharath, V. Anuradha, V. Mandava and M. Pal, Mini-Rev. Med. Chem., 2018, 18, 1498–1505 CrossRef CAS.
- J. J. Haddad, Saudi Pharm. J., 2012, 20, 103–123 CrossRef.
- R. Carbó-Dorca and P. Bultinck, J. Math. Chem., 2004, 36, 231–239 CrossRef.
- K. I. Ramachandran, G. Deepa and K. Namboori, Computational Chemistry and Molecular Modeling, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- T. H. Yang, C. I. Lee, W. H. Huang and A. R. Lee, Molecules, 2017, 22, 1–19 Search PubMed.
- M. A. Yousef, A. M. Ali, W. M. El-Sayed, W. Saber, H. H. A. Farag and T. Aboul-Fadl, Bioorg. Chem., 2020, 105, 104366 CrossRef CAS.
- S. Varun and R. Kakkar, MedChemComm, 2019, 10, 351–368 RSC.
- W. J. Lin, K. S. Shia, J. S. Song, M. H. Wu and W. T. Li, Org. Biomol. Chem., 2015, 14, 220–228 RSC.
- H. Cheng, X. Yao, S. Yin, T. Wang and Z. Zhang, J. Org. Chem., 2020, 85, 5863–5871 CrossRef CAS.
- C. Vila, S. Slack, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2019, 361, 1902–1907 CrossRef CAS.
- S. Su, N. Wang, C. Li, B. Song, X. Jia and J. Li, Asian J. Org. Chem., 2014, 3, 269–272 CrossRef CAS.
- C. Crestini and R. Saladino, Synth. Commun., 1994, 24, 2835–2841 CrossRef CAS.
- S. Mokhtari, Med. Chem., 2015, 5, 242–252 Search PubMed.
- T. Kniess, M. Kuchar and F. Wuest, Synth. Commun., 2008, 38, 3017–3022 CrossRef CAS.
- R. P. Sonawane and R. R. Tripathi, Int. Lett. Chem., Phys. Astron., 2013, 12, 30–36 Search PubMed.
- C. Raji Reddy, V. Ganesh and A. K. Singh, RSC Adv., 2020, 10, 28630–28634 RSC.
- H. Ankati, S. K. Akubathini, S. Kamila, C. Mukherjee, S. R. D. Mello and E. R. Biehl, Open Org. Chem. J., 2009, 2, 1–10 CrossRef.
- F. J. D. Carlo, J. Am. Chem. Soc., 1944, 66, 1420 CrossRef.
- K. R. Senwar, T. S. Reddy, D. Thummuri, P. Sharma, V. G. M. Naidu, G. Srinivasulu and N. Shankaraiah, Eur. J. Med. Chem., 2016, 118, 34–46 CrossRef CAS.
- https://www.rcsb.org/structure/3TI1, accessed 6 October 2020.
Footnotes |
| † Presented in 18th Austrian Chemistry Days, Linz – Austria, September 24 to 27, 2019. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10672k |
| This journal is © The Royal Society of Chemistry 2021 |