
Thermally induced silane dehydrocoupling on porous silicon nanoparticles for ultra-long-acting drug release†
Ji Hyeon
Oh‡
 a,
Rae Hyung
Kang‡
a,
Jaehoon
Kim
a,
Eun-Kyoung
Bang
bc and
Dokyoung
Kim
*acdef
a,
Rae Hyung
Kang‡
a,
Jaehoon
Kim
a,
Eun-Kyoung
Bang
bc and
Dokyoung
Kim
*acdef
aDepartment of Biomedical Science, Graduate School, Kyung Hee University, Seoul 02447, Republic of Korea. E-mail: dkim@khu.ac.kr
bCreative Research Center for Brain Science, Brain Science Institute, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea
cKHU-KIST Department of Converging Science and Technology, Kyung Hee University, Seoul 02447, Republic of Korea
dDepartment of Anatomy and Neurobiology, College of Medicine, Kyung Hee University, Seoul 02447, Republic of Korea
eCenter for Converging Humanities, Kyung Hee University, Seoul 02447, Republic of Korea
fMedical Research Center for Bioreaction to Reactive Oxygen Species and Biomedical Science Institute, Kyung Hee University, Seoul 02447, Republic of Korea
First published on 20th August 2021
Abstract
Here, we report an ultra-long-acting drug release nano-formulation based on porous silicon nanoparticles (pSiNPs) that are prepared by thermally induced silane dehydrocoupling and lipid-coating. This robust formulation offers the ability to release an anticancer drug, for up to 2 weeks, in various biological environments; pH 7.4 buffer, cancer cells, and tumor xenograft model.
 Dokyoung Kim | Prof. Dokyoung Kim received his Ph.D. from the Department of Chemistry at POSTECH (Pohang University of Science and Technology) in 2014. After a post-doctoral stay at the University of California, San Diego (2015–2017), he started his independent career at Kyung Hee University, College of Medicine, Seoul. He has been interested in the biomedical applications of nanomaterials. |
Introduction
In translational medicine, the development of nano-formulations that allows the delivery of drugs to a specific site or release of the drugs at a programmed rate has been highlighted to overcome the various drawbacks of single drugs.1–4 Many nano-formulations have been introduced focusing on the short-term drug release at a specific site as a drug-delivery system.5–7 In some cases, long-term releasable nano-formulations have shown advantages for drugs that are metabolized too rapidly and are excreted from the body shortly after administration.8,9As a research group within the translational medicine field, we have focused on the development of new nano-formulations based on porous silicon nanoparticles (pSiNPs) for disease-specific drug-delivery and new surface modification methods to reinforce chemical/physical/biological properties of pSiNPs.5,10–12 The pSiNPs have been considered as a promising material for biomedical applications due to their remarkable features such as (i) a high biocompatibility and fast clearance of non-toxic side-product (Si(OH)4) after degradation, (ii) a high loading yield of the substrates (drugs, peptides, DNA/RNA, proteins, antibodies) while maintaining their bioactivities, (iii) high applicability for the targeted delivery with the incorporation of targeting moieties, and (iv) an in vivo self-reporting ability via emitting fluorescence within the near-infrared region.13–20 Among the features, the high biocompatibility is useful for disease-specific drug-delivery system because the pSiNPs nano-formulation could be accumulated in the targeting site and release the encapsulated substrates in the short term.16–18,21,22 However, such a fast-degradation rate of pSiNPs within the biological system deters the development of long-term releasable nano-formulation that can serve specific purposes, including restoration of cartilage, long-term contraception, and intraocular medication.23–28 Micrometer-scale porous silicon formulations and hybrid formulation of pSiNPs with polymers have been applied in these fields described above, but the ultra-long-acting drug based on the single pSiNPs formulation and has yet to have been reported.29,30
In this present work, we report a pSiNPs-based ultra-long-acting (>2 weeks) drug release nano-formulation prepared by thermally induced silane dehydrocoupling with octadecylsilane (ODS) and a lipid-coating using an amphiphilic non-ionic copolymer surfactant Pluronic® F-127 (F127). For the first time, the thermally induced silane dehydrocoupling was applied to the nano-size porous silicon, and its long-term-acting drug release ability was systematically analyzed by monitoring the anticancer drug (model drug: SN-38) release, transmission electron microscope (TEM) image changes, and attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectra changes. In addition, the long-term-acting drug release property of pSiNPs was verified within the cancer cell line (HeLa; cervical cancer cell) and tumor xenograft mouse model. The present findings hold great promise for the application of the pSiNPs for use in long-term-acting drug release within clinical sites.
Results and discussion
To investigate the long-term-acting drug release properties of pSiNPs, we first considered the thermally induced silane dehydrocoupling on the nanostructured surface of pSiNPs.31 As-prepared pSiNPs mainly contain a silicon-hydride (Si–H) functionality on the surface, which could be oxidized to a silicon-oxide (Si–O–Si) or silicon-hydroxide (Si–OH) functionality in the presence of oxidants (oxygen, water, ozone, etc.).10,18,32 Such as-prepared and oxidized pSiNPs are generally used for the drug delivery system (payload release profile: <24 hours) after loading the payload and surface modifications (Fig. S1, ESI†).33 Various surface functionalization methods (oxidation, hydrosilylation, hydrolytic condensation, ring-opening click-chemistry) are adaptable on the surface of pSiNPs, depending on the purpose of use.10,34–45In terms of long-term-acting drug release, we can consider a formation of a hydrophobic layer on the surface of pSiNPs, and two surface functionalization methods are represented; (i) hydrosilylation: silicon–carbon (Si–C) bond formation from hydrogen-terminated pSi (Si–H) and unsaturated carbon (alkene, alkyne) under specific conditions (high temperature, irradiation of a specific light, reactive catalyst). This method is powerful and efficient to enhance the stability of pSi from the oxidant, but it is not efficient for the nano-sized pSi because such harsh reaction conditions can induce the generation of undesired aggregates, oxidized side-products, and particle degradation.10,37–44 (ii) Hydrolytic condensation: silicon–oxygen–silicon bond formation from hydroxyl-terminated pSi (Si–OH) and organoalkoxysilane reagents under a mild condition. If a long-alkyl chain containing organoalkyoxysilane reagent is used, this method is also powerful to make a hydrophobic pSiNPs formulation. However, the hydrolytic condensation also has drawbacks such as clogging of cross-linking, long reaction time, low surface modification efficiency, and loss of pSiNPs during the modification.10,44–46
In this study, a pSiNPs formulation (named pSiNPs(SN-38)-ODS/F127 system) was prepared by thermally induced silane dehydrocoupling31 and a lipid-coating method, which showed a long-term-acting drug release profile for up to 2 weeks (Fig. 1a). The adaptation of thermally induced silane dehydrocoupling on the silicon nanostructures was reported by our group in 2016,31 and it was based on the main reaction of hydrogen-terminated pSi (Si–H) and organic trihydridosilanes such as ODS in a mild thermal condition (80 °C) to form Si–Si bond and this reaction accompany some Si–O–Si bond formation between partially hydrogen oxide-terminated pSi (Si–OH) and ODS. In our previous work, we showed that the dehydrocoupling reaction was also found to proceed on pSiNPs, but its hydrophobic property has deterred further study on the resulting pSiNPs.31 As a back-to-back work, we have tried to find biological applications using such hydrophobic property of the ODS-grafted pSiNPs (named pSiNPs-ODS, Fig. 1b) and finally designed the pSiNPs(SN-38)-ODS/F127 system. At the designing stage of pSiNPs(SN-38)-ODS/F127, we assumed two key points; (i) Hydrophobic pSiNPs-ODS could have a high loading yield for hydrophobic drugs. (ii) Hydrophobic pSiNPs-ODS has low water dispersibility (slow degradation), which could have an opposite characteristic if lipid-coated with an amphiphilic copolymer.
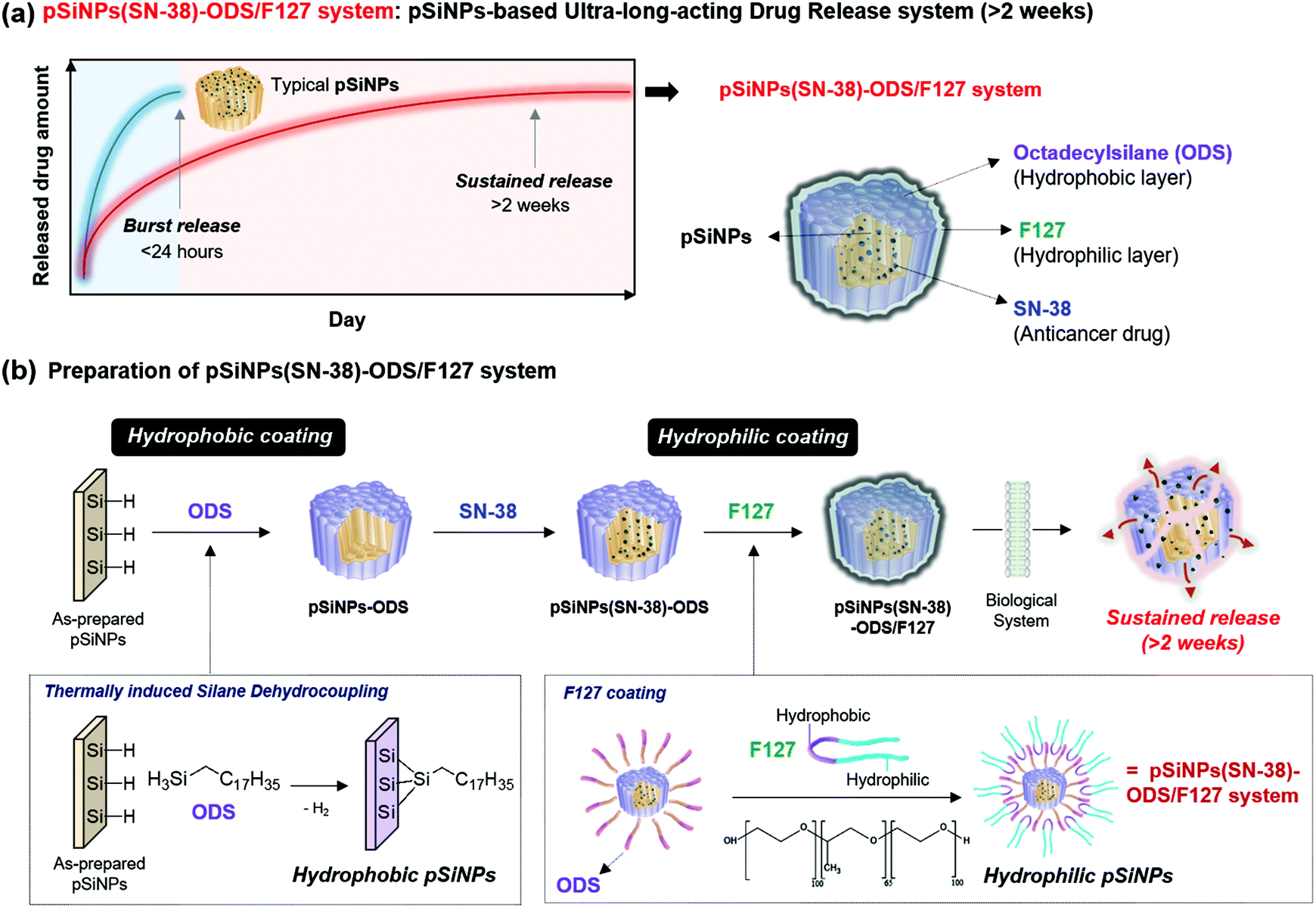 | ||
| Fig. 1 (a) Illustration of the pSiNPs(SN-38)-ODS/F127 system: pSiNPs-based ultra-long-acting drug release system. A newly developed pSiNPs(SN-38)-ODS/F127 system could release a model anticancer drug (SN-38 in this study) slowly and steadily over 2 weeks (336 h). The pSiNPs(SN-38)-ODS/F127 system consists of a hydrophobic layer (surface grafted with octadecylsilane (ODS)), a hydrophilic layer (surface coated with amphiphilic non-ionic copolymer surfactant (F127)), and SN-38 loaded pSiNPs. (b) Preparation scheme of the pSiNPs(SN-38)-ODS/F127 system. Step 1: thermally induced silane dehydrocoupling in a mild condition (80 °C, 16 h) (“Hydrophobic coating”). Step 2: SN-38 loading. 3rd step: F127 coating (“Hydrophilic coating”). | ||
The pSiNPs(SN-38)-ODS/F127 system was prepared considering the two key points above (Fig. 1b). First, hydrogen-terminated pSiNPs were fabricated by electrochemical etching of p++-type crystalline silicon wafers in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) aqueous HF:ethanol, as previously described.47,48 As-prepared pSiNPs were then modified by the ODS via thermally induced silane dehydrocoupling reaction at a mild condition (80 °C, 16 h stirring).31 The resulting nanoparticles (pSiNPs-ODS) were washed with n-hexane/ethanol, and then SN-38 (camptothecin derivatives, FDA-approved anticancer drug) was loaded into pSiNPs-ODS as a model drug to confirm the long-term-acting drug release ability of the ODS-functionalized the pSiNPs system.49 A solvent evaporation method (see details in the ESI†) was used for the loading of SN-38 into pSiNPs-ODS (Fig. S2, ESI†), and the loading yield was 42.4 ± 0.5%. pSiNPs also showed a similar loading yield (46.1 ± 0.1%) for SN-38, and this result indicated that the ODS-grafting does not interfere with the drug loading into the pore (Fig. S3–S5, ESI†).50 In the next step, amphiphilic F127 lipid-coating was introduced to the SN-38-loaded ODS-functionalized pSiNPs (named pSiNPs(SN-38)-ODS) using an ultrasonicator to enhance water dispersibility. The pSiNPs(SN-38)-ODS showed aggregated form in DI·H2O due to high hydrophobicity of ODS on the surface of the nanoparticle, while F127 coated nanoparticles (named pSiNPs(SN-38)-ODS/F127) showed high colloidal stability in DI·H2O because hydrophilic F127 covered the surface of nanoparticles (Fig. S6, ESI†). In order to verify that the loaded SN-38 was not released during the F127 coating step, the supernatant was analyzed by measuring the fluorescent intensity of SN-38 at 559 nm (Fig. S4 and S5, ESI†).51 Results indicate that a small amount of loaded SN-38 was released out during the F127 coating step (pSiNPs(SN-38)-ODS to pSiNPs(SN-38)-ODS/F127: 3.8% loss, pSiNPs(SN-38) to pSiNPs(SN-38)/F127: 4.3% loss). The morphological-/chemical-changes and the drug release profile of F127-coated pSiNPs(SN-38)-ODS (named pSiNPs(SN-38)-ODS/F127) were acquired for the comparison of other formulations, and we expected that the combination of ODS and F127 coating created a sustained-release system that continuously released a certain amount of SN-38.
:1 (v:v) aqueous HF:ethanol, as previously described.47,48 As-prepared pSiNPs were then modified by the ODS via thermally induced silane dehydrocoupling reaction at a mild condition (80 °C, 16 h stirring).31 The resulting nanoparticles (pSiNPs-ODS) were washed with n-hexane/ethanol, and then SN-38 (camptothecin derivatives, FDA-approved anticancer drug) was loaded into pSiNPs-ODS as a model drug to confirm the long-term-acting drug release ability of the ODS-functionalized the pSiNPs system.49 A solvent evaporation method (see details in the ESI†) was used for the loading of SN-38 into pSiNPs-ODS (Fig. S2, ESI†), and the loading yield was 42.4 ± 0.5%. pSiNPs also showed a similar loading yield (46.1 ± 0.1%) for SN-38, and this result indicated that the ODS-grafting does not interfere with the drug loading into the pore (Fig. S3–S5, ESI†).50 In the next step, amphiphilic F127 lipid-coating was introduced to the SN-38-loaded ODS-functionalized pSiNPs (named pSiNPs(SN-38)-ODS) using an ultrasonicator to enhance water dispersibility. The pSiNPs(SN-38)-ODS showed aggregated form in DI·H2O due to high hydrophobicity of ODS on the surface of the nanoparticle, while F127 coated nanoparticles (named pSiNPs(SN-38)-ODS/F127) showed high colloidal stability in DI·H2O because hydrophilic F127 covered the surface of nanoparticles (Fig. S6, ESI†). In order to verify that the loaded SN-38 was not released during the F127 coating step, the supernatant was analyzed by measuring the fluorescent intensity of SN-38 at 559 nm (Fig. S4 and S5, ESI†).51 Results indicate that a small amount of loaded SN-38 was released out during the F127 coating step (pSiNPs(SN-38)-ODS to pSiNPs(SN-38)-ODS/F127: 3.8% loss, pSiNPs(SN-38) to pSiNPs(SN-38)/F127: 4.3% loss). The morphological-/chemical-changes and the drug release profile of F127-coated pSiNPs(SN-38)-ODS (named pSiNPs(SN-38)-ODS/F127) were acquired for the comparison of other formulations, and we expected that the combination of ODS and F127 coating created a sustained-release system that continuously released a certain amount of SN-38.
Next, we analyzed the particle size and surface charge changes of all the pSiNPs formulations (pSiNPs, pSiNPs-ODS, pSiNPs(SN-38), pSiNPs(SN-38)-ODS, pSiNPs(SN-38)/F127, and pSiNPs(SN-38)-ODS/F127, Fig. 2a). Each nanoparticle was measured in quintuplicate to determine the average values with standard deviation. SN-38-loaded pSiNPs (pSiNPs(SN-38)) and SN-38-loaded/F127-coated pSiNPs (pSiNPs(SN-38)/F127) were used as control formulations that had no ODS layer on the surface of pSiNPs. As-prepared pSiNPs displayed a homogeneous average hydrodynamic diameter of 276.0 ± 128.1 nm (poly-dispersity index; PDI: 0.251) and zeta-potential of −23.3 ± 11.2 mV, measured by dynamic light scattering (DLS, Fig. 2b, c and Table S1, ESI†). The negative zeta-potential was derived from the generation of some silicon hydroxide (Si–OH) functionality on the surface of pSiNPs during preparation steps. The ODS grafted particle (ethanol-dispersible), pSiNPs-ODS, showed a slight increase in size (314.5 ± 134.5 nm, PDI: 0.237) with a negative surface charge (−35.7 ± 11.9 mV). The SN-38-loaded particle, pSiNPs(SN-38)-ODS and pSiNPs(SN-38) also showed a slight increment in size (pSiNPs(SN-38)-ODS: 367.1 ± 108.4 nm, PDI: 0.387, pSiNPs(SN-38): 281.2 ± 100.8 nm, PDI: 0.436) whilst maintaining the negative surface charge (pSiNPs(SN-38)-ODS: −32.1 ± 12.8 mV, pSiNPs(SN-38): −31.6 ± 14.9 mV) compared with pristine pSiNPs. pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 (water-dispersible) showed a significant increment of size compared to the other formulations because of the light scattering from the polymer (pSiNPs(SN-38)/F127: 442.1 ± 76.35 nm, PDI: 0.270, pSiNPs(SN-38)-ODS/F127: 364.4 ± 69.54 nm, PDI: 0.118) with a negative surface charge of −24.3 ± 6.01 mV and −26.3 ± 6.08 mV respectively (Fig. 2b, c and Table S2, ESI†). It appears that a lipid coating of F127 was more compactly formed on the surface of ODS-grafted nanoparticles (pSiNPs(SN-38)-ODS/F127) than the non-grafted control (pSiNPs(SN-38)/F127), and each DLS data has corresponded to this. The transmission electron microscope (TEM) images of as-prepared pSiNPs showed a relatively homogeneous particle size and porous structure (Fig. 2d). The uniform size and overall morphology of the porous nanostructures were both maintained after modification with octadecylsilane, drug loading, and F127 coating processes. The attenuated total reflectance Fourier-transform infrared spectrum (ATR-FTIR) of as-prepared pSiNPs displayed three characteristic bands corresponding to stretching vibration of ν(O–H) at 3200–3500 cm−1, ν(Si–H) at 2087 cm−1, and stretching mode of ν(Si–O) at 1033 cm−1 (Fig. 2e, left, Fig. S7, ESI†).46 The ODS-grafted pSiNPs (pSiNPs-ODS, pSiNPs(SN-38)-ODS, pSiNPs(SN-38)-ODS/F127) showed bands at 2800–3000 cm−1 and 1467 cm−1 associated with ν(C–H) stretching vibration and δ(C–H) bending vibration of the aliphatic chain, which confirmed the successful modification with ODS on the surface of nanoparticles (Fig. 2e, left).31,46 The side reaction between pSiNPs and ethanol52 in the given condition was not observed, and the dehydrocoupling reaction yield was higher in the ethanol than in other solvents such as chloroform (Fig. S8, ESI†). The drug-loaded formulations (pSiNPs(SN-38)-ODS, pSiNPs(SN-38), pSiNPs(SN-38)-ODS/F127, pSiNPs(SN-38)/F127) showed signature bands in the region of 3200–3500 cm−1 associated with ν(O–H), 1750 cm−1 associated with ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 1170 cm−1 corresponding to ν(C–O), which derived from SN-38 (Fig. 2e, middle, Fig. S7, ESI†).46,53 The F127 coated formulation (pSiNPs(SN-38)-ODS/F127, pSiNPs(SN-38)/F127) showed a band at 2800–3000 cm−1 associated with ν(C–H) stretching vibration and 1100 cm−1 corresponding to ν(C–O) of F127 (Fig. 2e, right, Fig. S7, ESI†).46,54 To verify the F127 is coated on the surface of nanoformulations, we confirmed the colloidal stability of pSiNPs(SN-38)-ODS/F127 before and after removing the F127 in DI·H2O. The pSiNPs(SN-38)-ODS/F127 was washed/dispersed with cold deionized water (4 °C DI·H2O, 5 times) using centrifugation (14 000 rpm, 4 °C). In this condition, the F127 was liquified (the sol–gel transition temperature of F127: 20 °C)55 and the stacked F127 on the surface of pSiNPs(SN-38)-ODS/F127 was removed. Before removing the F127, the pSiNPs(SN-38)-ODS/F127 was well dispersed in DI·H2O (25 °C) because of hydrophilic F127 coated on the surface of nanoparticles. After removing the F127, the nanoparticles aggregated and settled down on the bottom due to the increased hydrophobicity. From these results, we conclude that the F127 is coated on the surface of nanoparticles (Fig. S9, ESI†).
O), and 1170 cm−1 corresponding to ν(C–O), which derived from SN-38 (Fig. 2e, middle, Fig. S7, ESI†).46,53 The F127 coated formulation (pSiNPs(SN-38)-ODS/F127, pSiNPs(SN-38)/F127) showed a band at 2800–3000 cm−1 associated with ν(C–H) stretching vibration and 1100 cm−1 corresponding to ν(C–O) of F127 (Fig. 2e, right, Fig. S7, ESI†).46,54 To verify the F127 is coated on the surface of nanoformulations, we confirmed the colloidal stability of pSiNPs(SN-38)-ODS/F127 before and after removing the F127 in DI·H2O. The pSiNPs(SN-38)-ODS/F127 was washed/dispersed with cold deionized water (4 °C DI·H2O, 5 times) using centrifugation (14 000 rpm, 4 °C). In this condition, the F127 was liquified (the sol–gel transition temperature of F127: 20 °C)55 and the stacked F127 on the surface of pSiNPs(SN-38)-ODS/F127 was removed. Before removing the F127, the pSiNPs(SN-38)-ODS/F127 was well dispersed in DI·H2O (25 °C) because of hydrophilic F127 coated on the surface of nanoparticles. After removing the F127, the nanoparticles aggregated and settled down on the bottom due to the increased hydrophobicity. From these results, we conclude that the F127 is coated on the surface of nanoparticles (Fig. S9, ESI†).
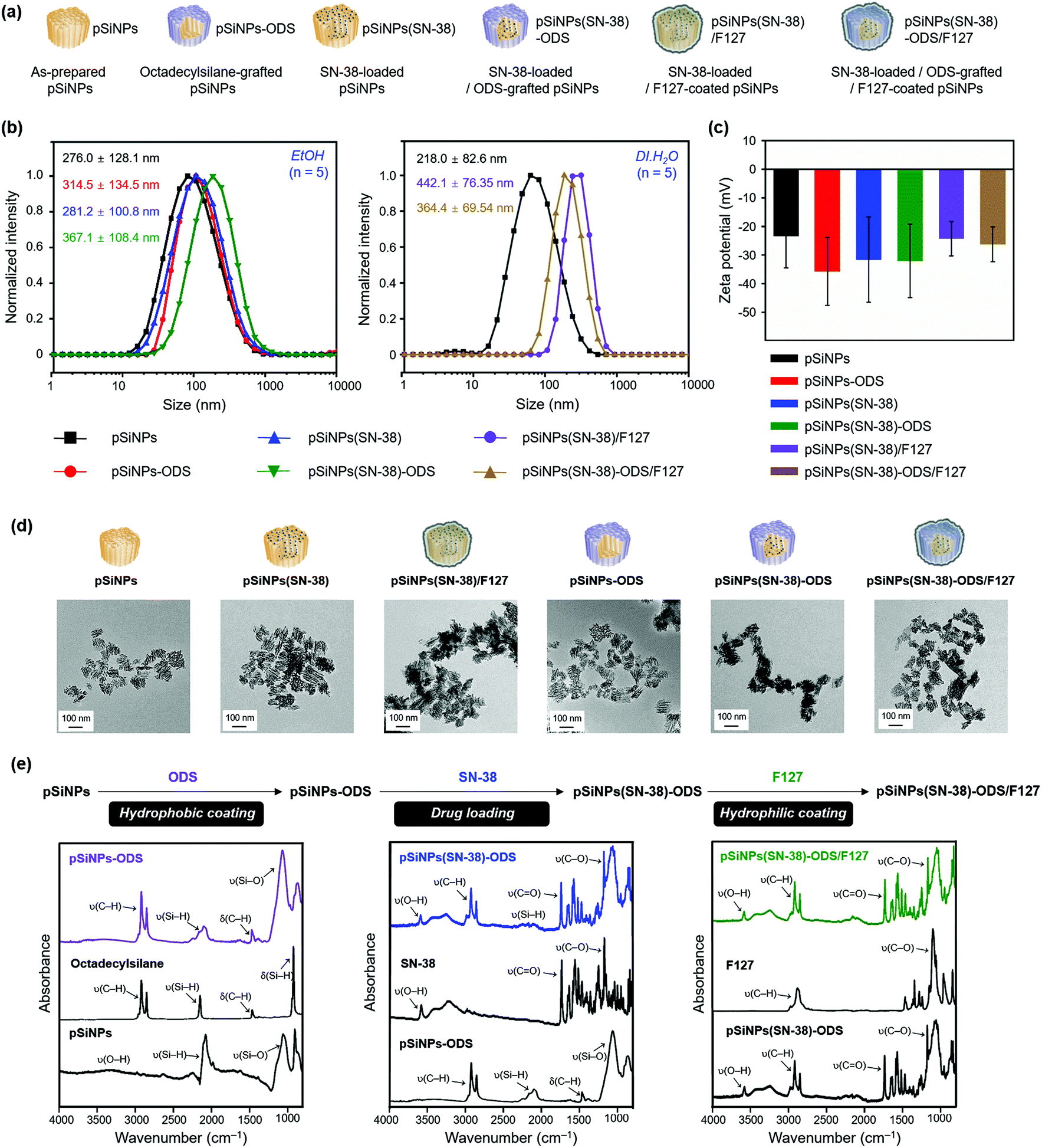 | ||
| Fig. 2 Characterization results of the pSiNPs formulations. (a) Schematic illustration of the pSiNPs formulations. pSiNPs: as-prepared pSiNPs, pSiNPs-ODS: ODS-grafted pSiNPs, pSiNPs(SN-38): SN-38-loaded pSiNPs, pSiNPs(SN-38)-ODS: SN-38-loaded/ODS-grafted pSiNPs, pSiNPs(SN-38)/F127: SN-38-loaded/F127-coated pSiNPs, and pSiNPs(SN-38)-ODS/F127: SN-38-loaded/ODS-grafted/F127-coated pSiNPs. (b) Average hydrodynamic diameter intensity distribution in EtOH (left) and deionized water (DI·H2O, right), and (c) Zeta-potential value of the pSiNPs formulations measured by dynamic light scattering (DLS). Means and standard deviations (S.D.) were calculated from quintuplicate measurements. The polydispersity index (PDI): <0.44. The zeta-potential value was measured in EtOH. (d) Transmission electron microscope (TEM) images of the pSiNPs formulations (scale bar: 100 nm). (e) Attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectra of the pSiNPs formulations. Symbols: ν = stretching, δ = bending. Broadband wavenumber for each peak (cm−1): ν(O–H) at 3200–3500 cm−1, ν(C–H) at 2800–3000 cm−1, ν(Si–H) at 2087 cm−1, ν(CO) at 1750 cm−1, δ(C–H) at 1467 cm−1, ν(C–O) at 1170 cm−1, and ν(Si–O) at 1033 cm−1. | ||
After the characterization of the pSiNPs formulations, we investigated the drug release profiling (Fig. 3 and Fig. S10, ESI†). The SN-38-loaded and water-dispersible pSiNPs formulations (pSiNPs(SN-38), pSiNPs(SN-38)/F127, pSiNPs(SN-38)-ODS/F127, 0.5 mg mL−1) were incubated in phosphate-buffered saline (PBS, pH 7.4) at 37 °C, and the drug release profiling was acquired by the fluorescence signal measurement of SN-38 in their supernatant at a certain time interval (0–48 h, 0–14 days). As a result, the formulations without the ODS layer (pSiNPs(SN-38), pSiNPs(SN-38)/F127) showed an initial burst release of SN-38 within 24 h (Fig. 3a). The amounts of SN-38 from pSiNPs(SN-38) and pSiNPs(SN-38)/F127 reached half-levels (>50%) within 10 h and max-levels (almost 100%) within 48 h (light blue background in Fig. 3a). However, pSiNPs(SN-38)-ODS/F127 only released 33.5% of SN-38 within 48 h, which implied a sustained release behavior, and the half-level and max-level of the released SN-38 were monitored around at 5 days and 14 days, respectively (light red background in Fig. 3a). This result indicates that the ODS layers on the surface of nanoparticles improved the drug retention property. We then analyzed the morphological changes (incubation: 0 h, 6 h, 24 h, 7 days, 14 days) of pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 using TEM (Fig. 3b). The TEM images revealed that pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 initially exhibited a porous structure with homogeneous morphology. After 6 h incubation, significant particle morphology changes (pore collapse, aggregation, degradation) were observed for pSiNPs(SN-38)/F127, whereas pSiNPs(SN-38)-ODS/F127 maintained its structure. By day 7, the porous structure of pSiNPs(SN-38)/F127 was not preserved, but pSiNPs(SN-38)-ODS/F127 showed some degradation whilst preserving their porous structure. At 14 days, pSiNPs(SN-38)/F127 was observed as a mostly aggregated form (no porous structure), but pSiNPs(SN-38)-ODS/F127 still maintained its original structure. We then further conducted the FTIR spectrum analysis paired with the TEM images to observe the surface functional group changes of the formulations (Fig. 3c). Within FTIR analysis, the characteristic peak of pSiNPs(SN-38)/F127 at 2087 cm−1 (ν(Si–H)) significantly decreased within 6 h and became negligible within 24 h (Fig. 3c, top, right).46 However, the characteristic region of pSiNPs(SN-38)-ODS/F127 at 2800–3000 cm−1 (ν(C–H)), which is derived from the ODS layer, remained after 14 days of incubation (Fig. 3c, bottom, right).46 We further analyzed the Raman spectrum and X-ray diffraction (XRD) of pSiNPs/F127 and pSiNPs-ODS/F127 (without SN-38 for Raman spectrum and XRD analysis) to identify the structural crystallinity stability of pSiNPs-ODS/F127 (Fig. S11, ESI†). The nanoparticles were incubated within 37 °C PBS for 24 h, and the crystallinity of the silicon frame was analyzed in dry powder condition. Before the incubation, both formulations showed distinct Si lattice mode peak at 515 cm−1.31 After the 24 h incubation, the peak intensity of the pSiNPs/F127 was significantly decreased, while silicon-crystallinity of the pSiNPs-ODS/F127 was maintained (Fig. S11, top, ESI†). In XRD analysis, as-prepared pSiNPs/F127 and pSiNPs-ODS/F127 clearly showed 2θ angles of characteristic peaks of Si; (111) at 28°, (220) at 47°, and (311) at 56°.31 After the incubation, the Si peaks of pSiNPs/F127 were significantly decreased, and a broad diffraction pattern of silica (SiO2) was observed at 22° representing significant oxidation of the pSiNPs.56,57 However, the pSiNPs-ODS/F127 maintained the peak intensity, which corresponds with the Raman analysis result (Fig. S11, bottom, ESI†). Taken together, these results indicated that the ODS-functionalization of pSiNPs played a key role in the long-term sustained drug-releasing properties of pSiNPs(SN-38)-ODS/F127.
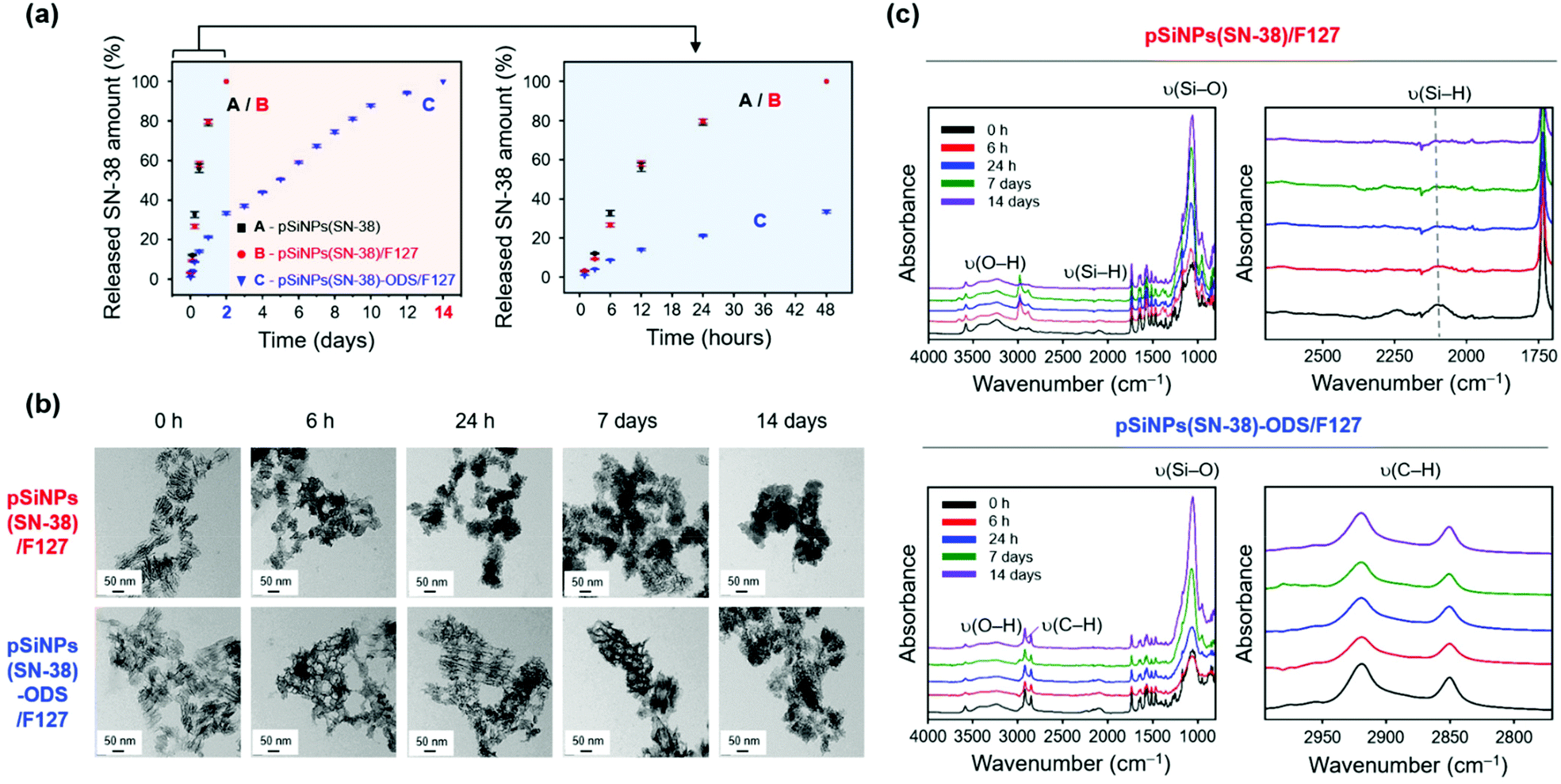 | ||
| Fig. 3 Drug release profiles of the pSiNPs formulations. (a) Plotted amounts of drug release (SN-38) from the pSiNPs formulations (0.5 mg mL−1, pSiNPs(SN-38), pSiNPs(SN-38)/F127, and pSiNPs(SN-38)-ODS/F127) as a function of time (time scale: days (left), hours (right)) at 37 °C incubated in PBS buffer (pH 7.4). The amount of SN-38 released was calculated by measuring the fluorescent intensity (560 nm) of SN-38 in the supernatant of the pSiNPs formulations. The excitation wavelength for the monitoring of SN-38: 364 nm. (b) TEM images of pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 at specific time points (scale bar: 50 nm). (c) ATR-FTIR spectra of pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 at specific time points. Left: full spectrum. Right: magnified spectrum for the ν(Si–H) and ν(C–H) regions. Symbols: ν = stretching vibration. | ||
With the promising in vitro drug release profiling results, we moved to the study in the cells (HeLa cell line, Fig. 4a and b) and animals (xenograft mouse model, Fig. 4c and d).
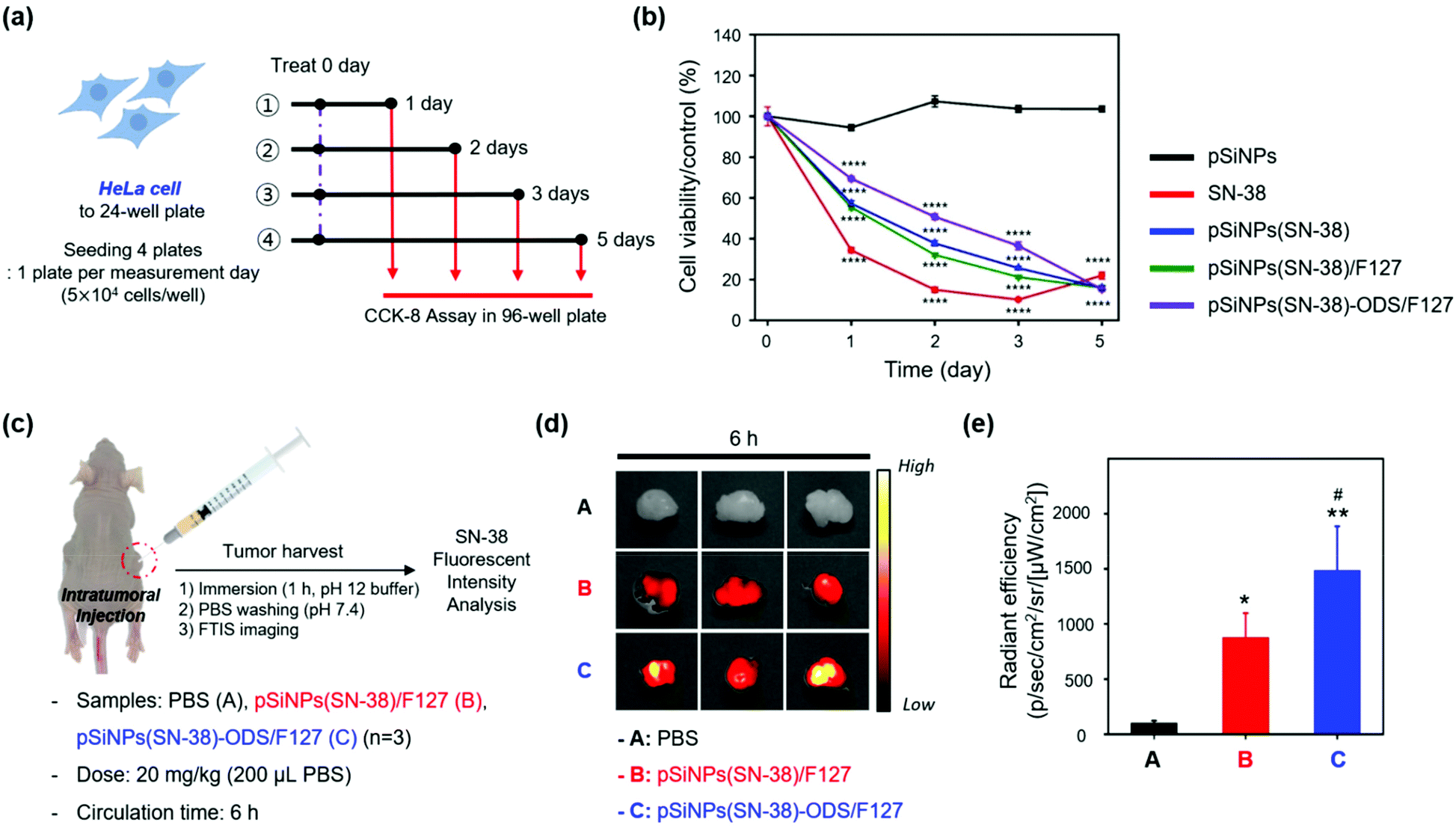 | ||
| Fig. 4 Drug release profiles of the pSiNPs formulations in cells and a tumor xenograft mouse model. (a) Scheme of the cell cytotoxicity experiment set-up in HeLa cells treated with the pSiNPs formulations (pSiNPs, pSiNPs(SN-38), pSiNPs(SN-38)/F127, pSiNPs(SN-38)-ODS/F127) and SN-38. HeLa cells were seeded on 24-well plates and incubated with the pSiNPs formulations (concentration of SN-38: 10 μM) and SN-38 (10 μM) for 1–5 days at 37 °C. The cytotoxicity was measured by CCK-8 assay in 96-well plates. (b) Cell cytotoxicity (%) curves of HeLa cells. Experimental results were expressed as mean ± standard deviation (S.D.). Data were analyzed by Significance-test using one-way ANOVA (for multiple comparisons) followed by Newman–Keuls's multiple comparison test. All statistical analyses were performed with Prism 8.0.1 software (GraphPad, La Jolla, CA, USA). ****p < 0.0001 compared with the pSiNPs. (c) Scheme of the drug release monitoring set-up within a tumor xenograft mouse model (cancer cell line: HeLa). PBS (control), pSiNPs(SN-38)/F127, and pSiNPs(SN-38)-ODS/F127 were injected intratumorally at a dose of 20 mg kg−1. (d) FTIS images of harvested tumor tissues. Excitation filter (λex: 390–490 nm) and emission filter (λem: 500–550 nm) were used for the signal collection. (e) Relative radiant efficiency plot of tumor from panel (d) (n = 3 per group). The data is presented as the mean ± S.D. The results of the FTIS data were analyzed by one-way analysis of variance (ANOVA) with the Newman–Keuls's multiple comparison test (*p < 0.05, **p < 0.01, compared with the PBS group) (#p < 0.05, compared with the pSiNPs(SN-38)/F127 group) [F (2, 6) = 20.60, P = 0.0021]. | ||
[In cells] We evaluated the cell viability of the HeLa cells for 5 days after treatment of SN-38 and nano-formulations (pSiNPs, pSiNPs(SN-38), pSiNPs(SN-38)/F127, pSiNPs(SN-38)-ODS/F127) (Fig. 4a). It was expected that (i) pSiNPs: there was no cellular toxicity for 5 days, (ii) SN-38: rapid and high cellular toxicity within 1–2 days, (iii) pSiNPs(SN-38) and pSiNPs(SN-38)/F127: lower cellular toxicity than an SN-38 for 1–4 days, (iv) pSiNPs(SN-38)-ODS/F127: lowest cellular toxicity for 1–4 days due to the slow release of SN-38. At the concentration of 10 μM SN-38 and its corresponded pSiNPs formulations (concentration of SN-38: 10 μM), similar cellular toxicity was observed (Fig. 4b), and the release of SN-38 maintained its activity (anticancer effect). On day 1, the cell viability of pSiNPs(SN-38)-ODS/F127 was 69.4%, pSiNPs(SN-38) was 57.2%, pSiNPs(SN-38)/F127 was 55.4%, SN-38 was 34.4%, and pSiNPs was 94.5%. On day 2, the viability of pSiNPs(SN-38)-ODS/F127: 50.8%, pSiNPs(SN-38): 37.8%, pSiNPs(SN-38)/F127: 32.1%, SN-38: 15.0%, and pSiNPs: 107.3%. On day 3, the viability of pSiNPs(SN-38)-ODS/F127: 36.5%, pSiNPs(SN-38): 25.6%, pSiNPs(SN-38)/F127: 21.3%, SN-38: 10.2%, and pSiNPs: 103.6%. On the last 5th day, cell viability of pSiNPs(SN-38)-ODS/F127: 15.3%, pSiNPs(SN-38): 15.9%, pSiNPs(SN-38)/F127: 16.0%, SN-38: 22.0%, and pSiNPs: 103.6%. From day 1 to day 3, pSiNPs showed no toxicity, and pSiNPs(SN-38)-ODS/F127 toxicity was lower compared to other groups. On the second day of the toxicity test, pSiNPs(SN-38)-ODS/F127 showed a cell viability of 1.6 times that of pSiNPs(SN-38)/F127, 1.3 times that of pSiNPs(SN-38), and 3.4 times that of SN-38 single treatment. Finally, pSiNPs(SN-38)-ODS/F127 was found to be the least toxic within 5 days, and the sustained-release system was found to work intracellularly. And on day 5, it was confirmed that almost all cells died in all groups including drugs, except pSiNPs. In a lower dose of SN-38 (1 μM) and SN-38-loaded pSiNPs formulations, they were also found a similar tendency (Fig. S12, ESI†). These results proved the ultra-long-acting drug release property of the pSiNPs(SN-38)-ODS/F127 system in HeLa cells.
[In animals] With the promising experimental results in cells, further analyses of pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 were conducted within the tumor xenograft mouse model (Fig. 4c). We prepared the tumor xenograft mouse model by subcutaneous injection of HeLa cells (4.5 × 106 cells) behind the flank of each mouse. When the tumor size of approximately 250 cm3 was reached, pSiNPs(SN-38)/F127 and pSiNPs(SN-38)-ODS/F127 (20 mg kg−1, 200 μL) were injected into the tumor site. After 6 h circulation, the mice were sacrificed, and the tumor was harvested and analyzed following the fluorescence signal of SN-38 remaining in the tumor tissues. The control set (PBS-injected group) showed no significant tissue auto-fluorescence at the given fluorescence monitoring condition (fluorescence tissue imaging set-up, excitation filter: 390–490 nm, emission filter: 500–550 nm) (Fig. 4d, top). In the nanoparticle-injected groups, the pSiNPs(SN-38)-ODS/F127-injected group (Fig. 4d, bottom) showed a higher radiant efficiency (1.7-folds) than the pSiNPs(SN-38)/F127-injected group (Fig. 4d, middle). This data represented that SN-38 released from the burst-releasing pSiNPs(SN-38)/F127-system was removed faster than the long-term releasing pSiNPs(SN-38)-ODS/F127-system, within a given circulation time (6 h, Fig. 4e). We also observed a significantly higher (1.3-folds) fluorescence intensity of SN-38 extracted from the pSiNPs(SN-38)-ODS/F127-injected tumor tissues than the pSiNPs(SN-38)/F127-injected set, which matched with the in vivo results (Fig. S13–S16, ESI†). These results indicated that hydrophobic coating with ODS and hydrophilic coating with F127 on pSiNPs could make an ultra-long-acting drug release system both in vitro and in vivo.
Conclusions
We have disclosed a new pSiNPs-based nano-formulation that is actualised by the hydrophobic layer formation via thermally induced silane dehydrocoupling and the hydrophilic layer formation via amphiphilic lipid coating. We introduced, for the first time, the thermally induced silane dehydrocoupling method on pSiNPs and have systematically analyzed their drug release profiling. The lipid-coated final formulation, pSiNPs(SN-38)-ODS/F127 system, is water dispersible for biomedical applications, and this robust formulation offers the ability to release an anticancer drug for up to 2 weeks in various biological environments; pH 7.4 buffer, cancer cells, and tumor xenograft model. This present strategy enables the release of drugs for an ultra-long-period whilst maintaining the drug activity, as an ultra-long-acting formulation, which we believe could be applied in translational medicine requiring an on-demand capacity for long-term drug release.Author contributions
J. H. O. was involved in the conceptualization of the project and performed the preparation and characterization of the materials. R. H. K. performed the preparation and characterization of the materials with J. H. O. The biological study was supported by J. K. D. K. initiated the project and drafted the manuscript with J. H. O., R. H. K., J. K., and E.-K. B. All authors have read and approved this manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): the authors are listed as inventors on a pending patent application related to the technology described in this work.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (NRF-2018-R1A6A1A03025124, NRF-2018-R1D1A1B07043383; D. K.) and the Bio & Medical Technology Development Program of the NRF of Korea funded by the Ministry of Science & ICT (NRF-2019-M3A9H1103783; D. K.). This research was also supported through a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI21C0239; D. K.) and the Bio & Medical Technology Development Program of the NRF of Korea (NRF-2021-M3A9I5030523; D. K.).Notes and references
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discovery Today, 2010, 15, 842–850 CrossRef CAS PubMed.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS PubMed.
- N. Ž. Knežević and G. N. Kaluđerović, Nanoscale, 2017, 9, 12821–12829 RSC.
- R. H. Kang, J.-E. Jang, E. Huh, S. J. Kang, D.-R. Ahn, J. S. Kang, M. J. Sailor, S. G. Yeo, M. S. Oh and D. Kim, Nanoscale Horiz., 2020, 5, 1213–1225 RSC.
- R. Luo, Z. Zhang, L. Han, Z. Xue, K. Zhang, F. Liu, F. Feng, J. Xue, W. Liu and W. Qu, Biomater. Sci., 2021, 9, 3718–3736 RSC.
- C. Su, X. Ren, F. Nie, T. Li, W. Lv, H. Li and Y. Zhang, RSC Adv., 2021, 11, 12915–12928 RSC.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36 CrossRef.
- R. H. Müller, M. Radtke and S. A. Wissing, Adv. Drug Delivery Rev., 2002, 54, S131–S155 CrossRef.
- S. H. Lee, J. S. Kang and D. Kim, Materials, 2018, 11, 2557 CrossRef CAS.
- R. H. Kang, Y. Kim, J. H. Kim, N. H. Kim, H. M. Ko, S.-H. Lee, I. Shim, J. S. Kim, H.-J. Jang and D. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 30359–30372 CrossRef CAS.
- D. Kim, J. Kang, T. Wang, H. G. Ryu, J. M. Zuidema, J. Joo, M. Kim, Y. Huh, J. Jung and K. H. Ahn, Adv. Mater., 2017, 29, 1703309 CrossRef.
- H. A. Santos, in Handbook of Porous Silicon, Springer International Publishing, 2016, pp. 1403–1417, DOI:10.1007/978-3-319-04508-5_128-1.
- H. A. Santos, E. Mäkilä, A. J. Airaksinen, L. M. Bimbo and J. Hirvonen, Nanomedicine, 2014, 9, 535–554 CrossRef CAS PubMed.
- C.-F. Wang, M. P. Sarparanta, E. M. Mäkilä, M. L. Hyvönen, P. M. Laakkonen, J. J. Salonen, J. T. Hirvonen, A. J. Airaksinen and H. A. Santos, Biomaterials, 2015, 48, 108–118 CrossRef CAS.
- Y. Jin, D. Kim, H. Roh, S. Kim, S. Hussain, J. Kang, C. G. Pack, J. K. Kim, S. J. Myung and E. Ruoslahti, Adv. Mater., 2018, 30, 1802878 CrossRef PubMed.
- W. Li, Z. Liu, F. Fontana, Y. Ding, D. Liu, J. T. Hirvonen and H. A. Santos, Adv. Mater., 2018, 30, 1703740 CrossRef.
- J.-H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- H. A. Santos, L. M. Bimbo, B. Herranz, M.-A. Shahbazi, J. Hirvonen and J. Salonen, J. Mater. Res., 2013, 28, 152–164 CrossRef CAS.
- S. Mariani, V. Robbiano, L. M. Strambini, A. Debrassi, G. Egri, L. Dähne and G. Barillaro, Nat. Commun., 2018, 9, 1–13 CrossRef PubMed.
- T. Kumeria, S. J. McInnes, S. Maher and A. Santos, Expert Opin. Drug Delivery, 2017, 14, 1407–1422 CrossRef CAS PubMed.
- S. Sinha, W. Y. Tong, N. H. Williamson, S. J. McInnes, S. Puttick, A. Cifuentes-Rius, R. Bhardwaj, S. E. Plush and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2017, 9, 42601–42611 CrossRef CAS PubMed.
- L. Cheng, E. Anglin, F. Cunin, D. Kim, M. Sailor, I. Falkenstein, A. Tammewar and W. Freeman, Br. J. Ophthalmol., 2008, 92, 705–711 CrossRef CAS.
- J. Fong, Z. Xiao and K. Takahata, Lab Chip, 2015, 15, 1050–1058 RSC.
- H. A. Nash, in Medical applications of controlled release, CRC Press, 2019, pp. 35–64, DOI:10.1201/9780429276620.
- U. Ripamonti, C. Ferretti and M. Heliotis, J. Anat., 2006, 209, 447–468 CrossRef CAS PubMed.
- H. Wang, S. C. Leeuwenburgh, Y. Li and J. A. Jansen, Tissue Eng., Part B, 2012, 18, 24–39 CrossRef CAS PubMed.
- D. Warther, Y. Xiao, F. Li, Y. Wang, K. Huffman, W. R. Freeman, M. Sailor and L. Cheng, Drug Delivery, 2018, 25, 1537–1545 CrossRef CAS PubMed.
- L. Vaccari, D. Canton, N. Zaffaroni, R. Villa, M. Tormen and E. di Fabrizio, Microelectron. Eng., 2006, 83, 1598–1601 CrossRef CAS.
- E. C. Wu, J.-H. Park, J. Park, E. Segal, F. Cunin and M. J. Sailor, ACS Nano, 2008, 2, 2401–2409 CrossRef CAS PubMed.
- D. Kim, J. Joo, Y. Pan, A. Boarino, Y. W. Jun, K. H. Ahn, B. Arkles and M. J. Sailor, Angew. Chem., 2016, 128, 6533–6537 CrossRef.
- E. J. Kwon, M. Skalak, A. Bertucci, G. Braun, F. Ricci, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Adv. Mater., 2017, 29, 1701527 CrossRef PubMed.
- L. M. Bonanno and E. Segal, Nanomedicine, 2011, 6, 1755–1770 CrossRef CAS PubMed.
- D. Kim, J. M. Zuidema, J. Kang, Y. Pan, L. Wu, D. Warther, B. Arkles and M. J. Sailor, J. Am. Chem. Soc., 2016, 138, 15106–15109 CrossRef CAS.
- A. F. Maddox, J. G. Matisons, M. Singh, J. Zazyczny and B. Arkles, Mater. Res. Soc. Symp. Proc., 2015, 1793, 35–40 CrossRef.
- Y. Ogata, H. Niki, T. Sakka and M. Iwasaki, J. Electrochem. Soc., 1995, 142, 1595 CrossRef CAS.
- R. Boukherroub, J. Wojtyk, D. D. Wayner and D. J. Lockwood, J. Electrochem. Soc., 2002, 149, H59 CrossRef CAS.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271–1308 CrossRef CAS PubMed.
- J. M. Buriak, Chem. Mater., 2014, 26, 763–772 CrossRef CAS.
- J. M. Buriak, M. P. Stewart, T. W. Geders, M. J. Allen, H. C. Choi, J. Smith, D. Raftery and L. T. Canham, J. Am. Chem. Soc., 1999, 121, 11491–11502 CrossRef CAS.
- V. R. Gonçales, J. Lian, S. Gautam, R. D. Tilley and J. J. Gooding, Annu. Rev. Anal. Chem., 2020, 13, 135–158 CrossRef.
- J. Salonen, M. Björkqvist, E. Laine and L. Niinistö, Appl. Surf. Sci., 2004, 225, 389–394 CrossRef CAS.
- M. P. Stewart and J. M. Buriak, Angew. Chem., Int. Ed., 1998, 37, 3257–3260 CrossRef CAS PubMed.
- Y. Jung, Y. Huh and D. Kim, Microporous Mesoporous Mater., 2020, 310, 110673 CrossRef.
- M. J. Sailor, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2014, pp. 355–380, DOI:10.1007/978-3-319-05744-6_37.
- M. J. Sailor, in Porous silicon in practice: preparation, characterization and applications, John Wiley & Sons, 2012, ch. 5, pp. 133–181, DOI:10.1002/9783527641901.
- I. N. Lees, H. Lin, C. A. Canaria, C. Gurtner, M. J. Sailor and G. M. Miskelly, Langmuir, 2003, 19, 9812–9817 CrossRef CAS.
- Z. Qin, J. Joo, L. Gu and M. J. Sailor, Part. Part. Syst. Charact., 2014, 31, 252–256 CrossRef CAS.
- M. Ramesh, P. Ahlawat and N. R. Srinivas, Biomed. Chromatogr., 2010, 24, 104–123 CrossRef PubMed.
- F. Olivieri, R. Castaldo, M. Cocca, G. Gentile and M. Lavorgna, Nanoscale, 2021, 13, 9091–9111 RSC.
- M. M. Mirhosseini, V. Haddadi-Asl and S. S. Zargarian, RSC Adv., 2016, 6, 80564–80575 RSC.
- R. Boukherroub, S. Morin, P. Sharpe, D. D. Wayner and P. Allongue, Langmuir, 2000, 16, 7429–7434 CrossRef CAS.
- H.-C. Lin, C.-H. Chuang, M.-H. Cheng, Y.-C. Lin and Y.-P. Fang, Pharmaceutics, 2019, 11, 569 CrossRef CAS PubMed.
- B. Karolewicz, M. Gajda, A. Górniak, A. Owczarek and I. Mucha, J. Therm. Anal. Calorim., 2017, 130, 383–390 CrossRef CAS.
- Y.-s. Jung, W. Park, H. Park, D.-K. Lee and K. Na, Carbohydr. Polym., 2017, 156, 403–408 CrossRef CAS PubMed.
- J. Joo, T. Defforge, A. Loni, D. Kim, Z. Li, M. J. Sailor, G. Gautier and L. T. Canham, Appl. Phys., 2016, 108, 153111 Search PubMed.
- G. Nallathambi, T. Ramachandran, V. Rajendran and R. Palanivelu, Mater. Res., 2011, 14, 552–559 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03263a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |