
A tetrahedral DNA nanorobot with conformational change in response to molecular trigger†
Fengyu
Liu
 a,
Xiaoming
Liu
*a,
Qing
Shi
a,
Christopher
Maffeo
b,
Masaru
Kojima
c,
Lixin
Dong
d,
Aleksei
Aksimentiev
b,
Qiang
Huang
a,
Toshio
Fukuda
a and
Tatsuo
Arai
a
a,
Xiaoming
Liu
*a,
Qing
Shi
a,
Christopher
Maffeo
b,
Masaru
Kojima
c,
Lixin
Dong
d,
Aleksei
Aksimentiev
b,
Qiang
Huang
a,
Toshio
Fukuda
a and
Tatsuo
Arai
a
aKey Laboratory of Biomimetic Robots and Systems, Ministry of Education, State Key Laboratory of Intelligent Control and Decision of Complex System, Beijing Advanced Innovation Center for Intelligent Robots and Systems and School of Mechatronical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: liuxiaoming555@bit.edu.cn
bDepartment of Physics, University of Illinois at Urbana Champaign, Urbana IL 61802, USA
cDepartment of Materials Engineering Science, Osaka University, Osaka 560-8531, Japan
dDepartment of Biomedical Engineering, City University of Hong Kong, Hong Kong 999077, China
First published on 9th August 2021
Abstract
Dynamic DNA origami nanostructures that respond to external stimuli are promising platforms for cargo delivery and nanoscale sensing. However, the low stability of such nanostructures under physiological conditions presents a major obstacle for their use in biomedical applications. This article describes a stable tetrahedral DNA nanorobot (TDN) programmed to undergo a controlled conformational change in response to epithelial cell adhesion molecule (EpCAM), a molecular biomarker specifically expressed on the circulating tumor cells. Multiresolution molecular dynamics simulations verified the overall stability of the folded TDN design and characterized local distortions in the folded structure. Atomic force microscopy and gel electrophoresis results showed that tetragonal structures are more stable than unfolded DNA origami sheets. Live cell experiments demonstrated the low cytotoxicity and target specificity of TDN. In summary, the proposed TDN can not only effectively resist nuclease catalysis but also has the potential to monitor EpCAM-positive cells precisely.
Introduction
From pioneering two-dimensional (2D) folds,1,2 to complex polyhedra3–6 and three-dimensional (3D) structures,7–10 programmed self-assembly of DNA has been used to engineer various nanosystems11–14 of high biocompatibility and programmability.15–18 Recently, multiple 3D DNA nanostructures were reported to achieve gene silencing, environment sensing, and drug delivery.19–23 Dynamic DNA nanorobots assembled with 3D DNA nanostructures have been designed to undergo the programmed responses upon encountering the specific biomolecular triggers.24,25 However, the practical utility of dynamic nanostructures is limited by unanticipated distortions of their local structure and gradual unraveling under physiological conditions.26Benefiting from the small external surface and enclosed internal environment, 3D DNA nanostructures have been shown to possess excellently targeted delivery capability24,25,27 and structural stability.11,27–30Among those, tetrahedral DNA nanostructures have emerged as particularly robust systems because of their enhanced structural stability.11,27 Ke and colleagues have shown that a DNA tetrahedron can be assembled from a 2D DNA origami sheet through a multistep hierarchical folding procedure.31 This procedure, nevertheless, relies on precision folding of the DNA nanostructure at every single folding step. Unanticipated stochastic distortions of the local structure along the multistep folding pathway can make the high-yield fabrication of such 3D nanostructures difficult. The likelihood and severity of local distortions in a folded structure can be examined through all-atom32,33 and coarse-grained26,32,33 molecular dynamics simulations. However, the microscopic simulations of sparse, dynamic nanostructures have until recently been limited by the length and time scales involved.
Herein, we propose an efficient strategy to fold a 2D DNA origami sheet (DOS) into a 3D tetrahedral DNA nanostructure (TDN) using multiple parallel folding elements. Utilizing a multiresolution simulation method,34 we confirm the feasibility of the folding strategy by examining the stability of the folded tetrahedral nanostructure. Specifically, we aid the folding of DOS by five pairs of DNA molecules containing the SYL3C aptamer35 sequence. We tune the complementary lengths of the molecules to stabilize the folded structure of the TDN and separate upon exposure to the SYL3C aptamer target—epithelial cell adhesion molecule (EpCAM).36,37 Following the previous works,38,39 we choose the HT29 cell line with a high expression level of EpCAM as a circulating tumor cells (CTCs) model. We then employ atomic force microscopy (AFM), gel electrophoresis, and fluorescence imaging to characterize the conformational change and the serum stability change of TDN after being activated by EpCAM. In short, not only to enhance the stability of DNA nanostructures, but our work also provides a new idea for assembling dynamic DNA nanorobots that can identify EpCAM-positive cells.
Results and discussion
According to the multiple parallel folding, five pairs of DNA duplexes containing SYL3C aptamer are added to the assembled DOS sample (Fig. 1A left side). During the slow annealing process, these SYL3C aptamer duplexes are precisely anchored to the specific locations of the DOS and bend the DOS into a tetrahedral nanostructure along the potential folding axes (Fig. 1A mid-side). Considering the established dependence of the in vivo intake capacity on the size of the nanostructures,40–42 we chose the size of the unfolded DOS plate to be 35 nm × 36 nm × 2 nm. To enable fluorescence characterization of the DNA nanostructures, we strategically assembled multiple DNA-conjugated fluorophores on the DOS. The TDN was designed to undergo a conformation transition upon activated by EpCAM, resulting in the separation between the closely adjacent fluorophore and quencher (Fig. 1B). This separation means that the spatial location of TDN can be captured by fluorescence microscopy to inspect further whether the TDN recognizes target cells. A detailed candnano43 schematic was presented in ESI Fig. S1.† Tables S1 and S2† listed the nucleotide sequences of the DNA strands.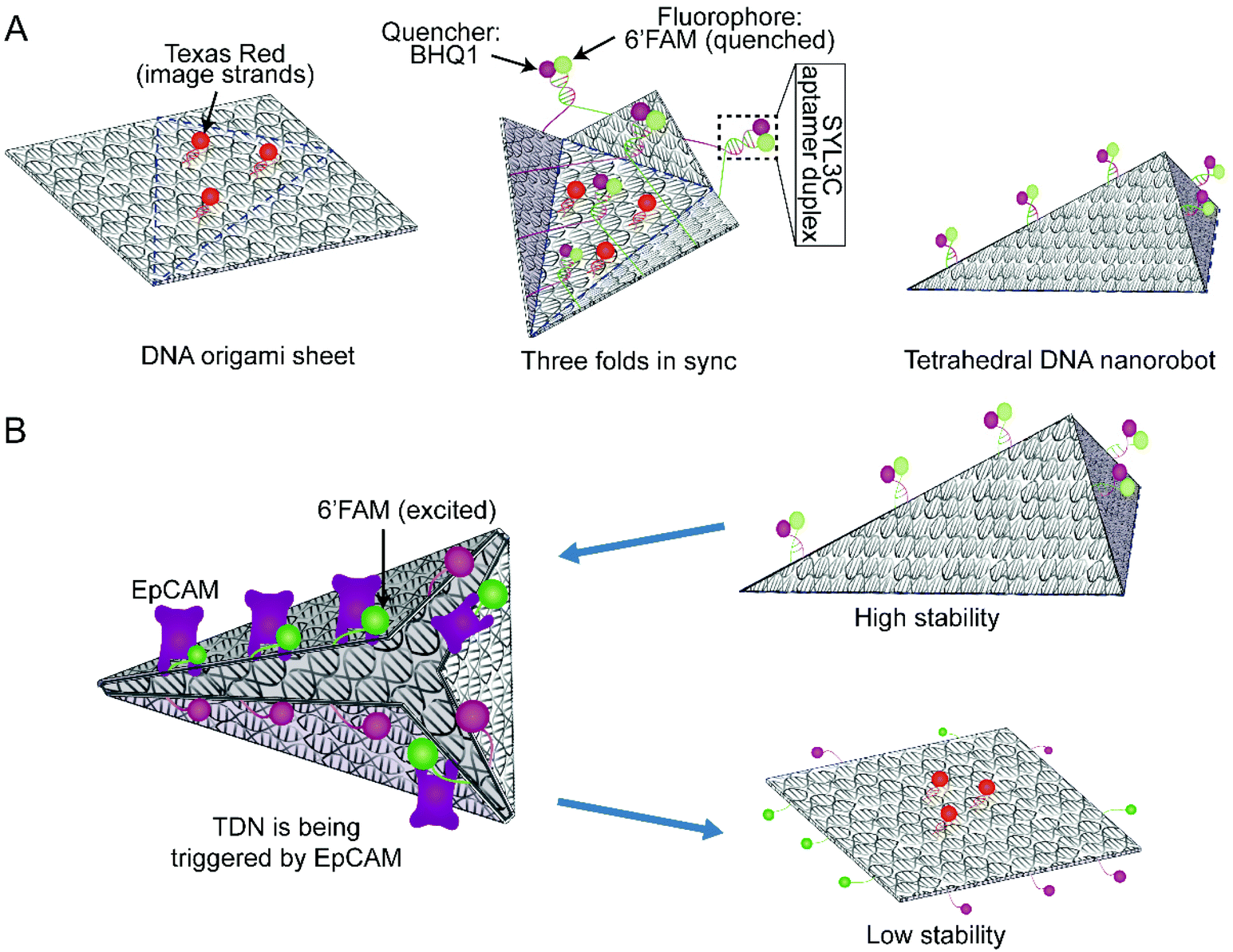 | ||
| Fig. 1 Schematic diagram of the synthesis and conformational change of TDN. (A) Schematic of 2D rectangular DOS (left side); driven by the five SYL3C aptamer duplexes, DOS folds synchronously along the three blue dotted lines (mid-side); the shape of the successfully folded TDN is shown on the right side. Blue dotted lines refer to the potential folding axes. (B) The conformational change of TDN is triggered by EpCAM, leading to the structural stability change. | ||
Our folding strategy requires DOS to bend into a tetrahedral object along the potential folding axes (Fig. 1A). However, such sharp bending would introduce significant stress into the nanostructure that may affect the final folded geometry and possibly limit the folding yield. To examine the expected equilibrium shape, assuming complete hybridization of all staple strands, we performed multiresolution simulations of the tetrahedral nanostructure using the mrdna framework.34 The mrdna model represents DNA helices using a variable-resolution bead-based polymer model calibrated to reproduce the mechanical properties of DNA. Starting from an idealized conformation (Fig. 2A), the object was simulated using mrdna at 4 bp/bead resolution for 20 μs, during which the object maintained its intended tetrahedral shape (Fig. 2B). Subsequently, a pair of short mrdna simulations at the 2 beads/bp level introduced enough detail to allow the generation of an oxDNA44–46 model of the object. Unlike the mrdna model, where DNA helices cannot kink or melt, the oxDNA model allows base pairing and base stacking interactions to rupture. Importantly, the oxDNA model was parameterized to reproduce experimentally-known thermodynamic and mechanical properties of DNA, including its melting temperature. During the 750 ns oxDNA simulation of the object, the overall geometry of the object was seen to remain stable (Fig. 2C, Fig. S2 and ESI Animations 1–5†). The largest configurational change involved kinking of the three adjacent helices at the scaffold crossovers located near the crease for the “lid” of the object. Without the loss of base stacking at this site, these helices would have been highly curved. Most base pairs remained intact during the simulation, although several were seen to melt (Fig. 2D, Fig. S2 and ESI Animations 1–5†), mostly near the ends of the DNA and occasionally at crossovers.
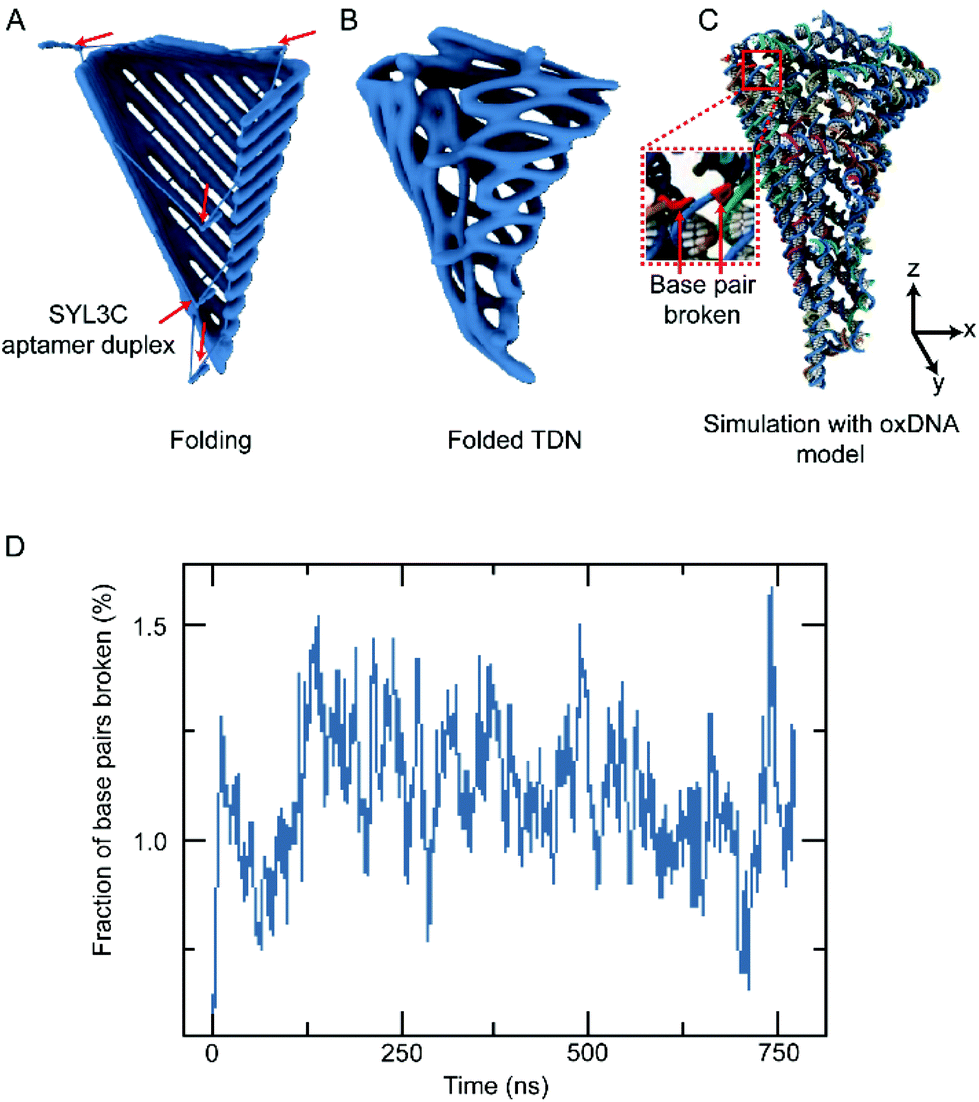 | ||
| Fig. 2 Multiresolution simulations of a tetrahedral object. (A) Initial idealized configuration obtained from the cadnano design. The red arrows indicate the location of the SYL3C aptamer duplexes. (B) Average configuration of the objects during a 20 μs mrdna simulation performed at 4 bp/bead resolution. (C) Typical configuration of the tetrahedral object during a 750 ns oxDNA simulation started from the conformation obtained at the end of the mrdna simulation. (D) The fraction of broken base pairs relative to the idealized design during the oxDNA simulation. Similar simulation outcomes were observed in three independent runs. | ||
To experimentally assemble TDN, we first fabricated a single-layer DOS of a simple 2D structure (Fig. 1A). Before the fabrication of DOS, two additional strands (Help strands in Table S1†) were introduced to provide cutting sites for the restriction endonucleases Bsp1286 I and Kpn I. The addition of the endonucleases cleaved the M13 bacteriophage DNA, producing 1498nt fragments of single-stranded (ss) DNA (p1498). The p1498 ssDNA was reacted with the staple strands in a thermocycler for more than 10 h (Fig. S1 and Table S1†). Fig. S1† specifies the detailed configuration of the scaffold and staple strands, along with the location of the strands intended for capture of the fluorescence probes, and of the duplex strands (6′FAM-modified; BHQ1-modified) used for driving the folding process. The assembled DOS had a size of 35 nm × 36 nm × 2 nm (l × w × h), as confirmed by AFM and gel electrophoresis (Fig. 3A and Fig. S3A†).
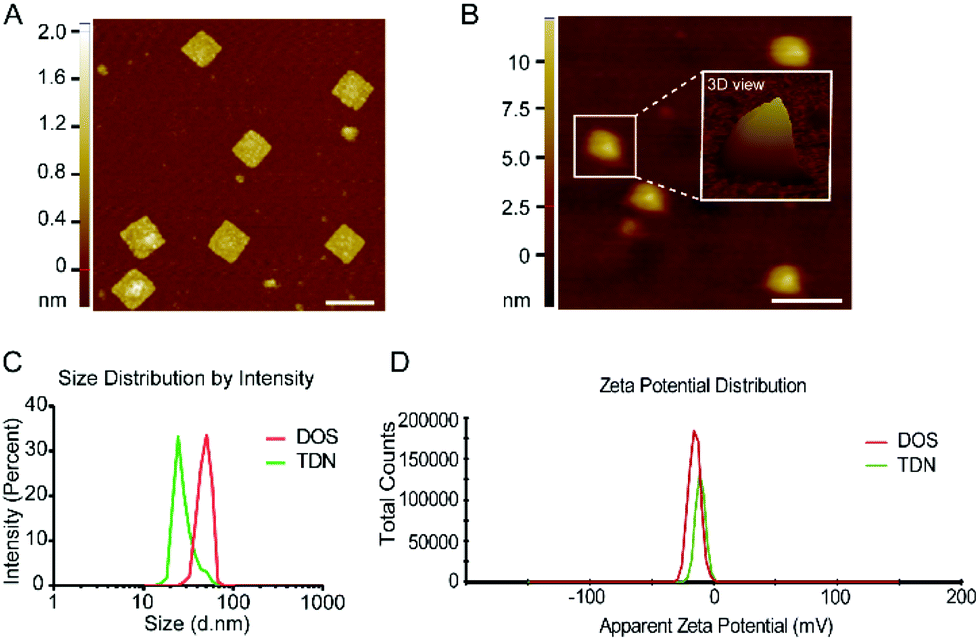 | ||
| Fig. 3 Characterization of DNA nanostructures. (A) The AFM image of a rectangular DOS. The data are representative of three independent experiments. Scale bar: 50 nm. (B) The AFM image is indicative of the tetrahedral morphology of a folded TDN. The data are representative of three independent experiments. Scale bar: 50 nm. (C, D) A Malvern analyzer measured the hydrodynamic size distributions and apparent zeta potentials of DOS and TDN. The data shown represent the mean from three independent experiments. | ||
Following that, we introduced the locking/unlocking mechanism that could be triggered by exposure to EpCAM, enabling controllable conformational change in TDN (Fig. 4A). As a biomarker protein, EpCAM expresses explicitly on the surface of CTCs.36,37 The previous study35 found DNA SYL3C aptamer to exhibit high specificity for binding EpCAM from multiple CTCs. Inspired by these advances, we used SYL3C aptamer to synthesize SYL3C aptamer duplex with specific complementary lengths. It drives the synthesized DOS folding into the TDN (Fig. 1A right side) and disassociates upon exposure to EpCAM (Fig. 1B). The critical factor for DNA aptamers to recognize specific proteins is the stem-loop structure,25,47 or G-quadruplex.48 Tang and co-workers confirmed that a duplex length shorter than 10 bp could not form a stable double-stranded structure.47 Conversely, when a duplex length was longer than 26 bp, the probability of the duplex being triggered to open was significantly reduced.25 Considering the stability of the SYL3C aptamer duplex, we changed the spatial configuration of the stem-loop structure by adjusting the complementary length of the SYL3C aptamer duplex. Here, we explored the opening efficiency of four duplexes with different complementary lengths (13 bp, 17 bp, 22 bp, 29 bp) after being triggered by EpCAM. The optimization was done by varying the length of the complementary fragments in the SYL3C aptamer, which was 6′FAM-modified, whereas the other strand was BHQ1-labeled (Table S2†). The SYL3C aptamer duplexes of various lengths of the complementary sequence were incubated with HT29 cells, and the fluorescence intensity was monitored. Higher fluorescence intensity indicates that SYL3C aptamer duplexes are dissociated by EpCAM activation, whereas free duplexes emitted only weak fluorescence. The relative fluorescence kinetics study showed that the fluorescence intensity increased the fastest when the duplex length was 17 bp (Fig. 4B). That is, after 2 h of incubation, the fluorescence intensity from the 17 bp duplex sample reaches more than 80% of the positive control sample. To verify whether the SYL3C aptamer duplex can be recognized and triggered only by EpCAM, we pretreated the HT29 cells with anti-EpCAM antibody, which should have prevented EpCAM from triggering the duplex. The relative fluorescence ratio in the Ab group was significantly lower than that in the positive control group (Fig. 4C), indicating that the SYL3C aptamer duplex with a complementary length of 17 bp was explicitly activated by EpCAM. Additional fluorescence images confirmed that the 17 bp duplexes were triggered mainly by HT29 cells (Fig. 4D). These results demonstrate that the highest triggering efficiency is achieved when the complementary length of the SYL3C aptamer duplex is set to 17 bp.
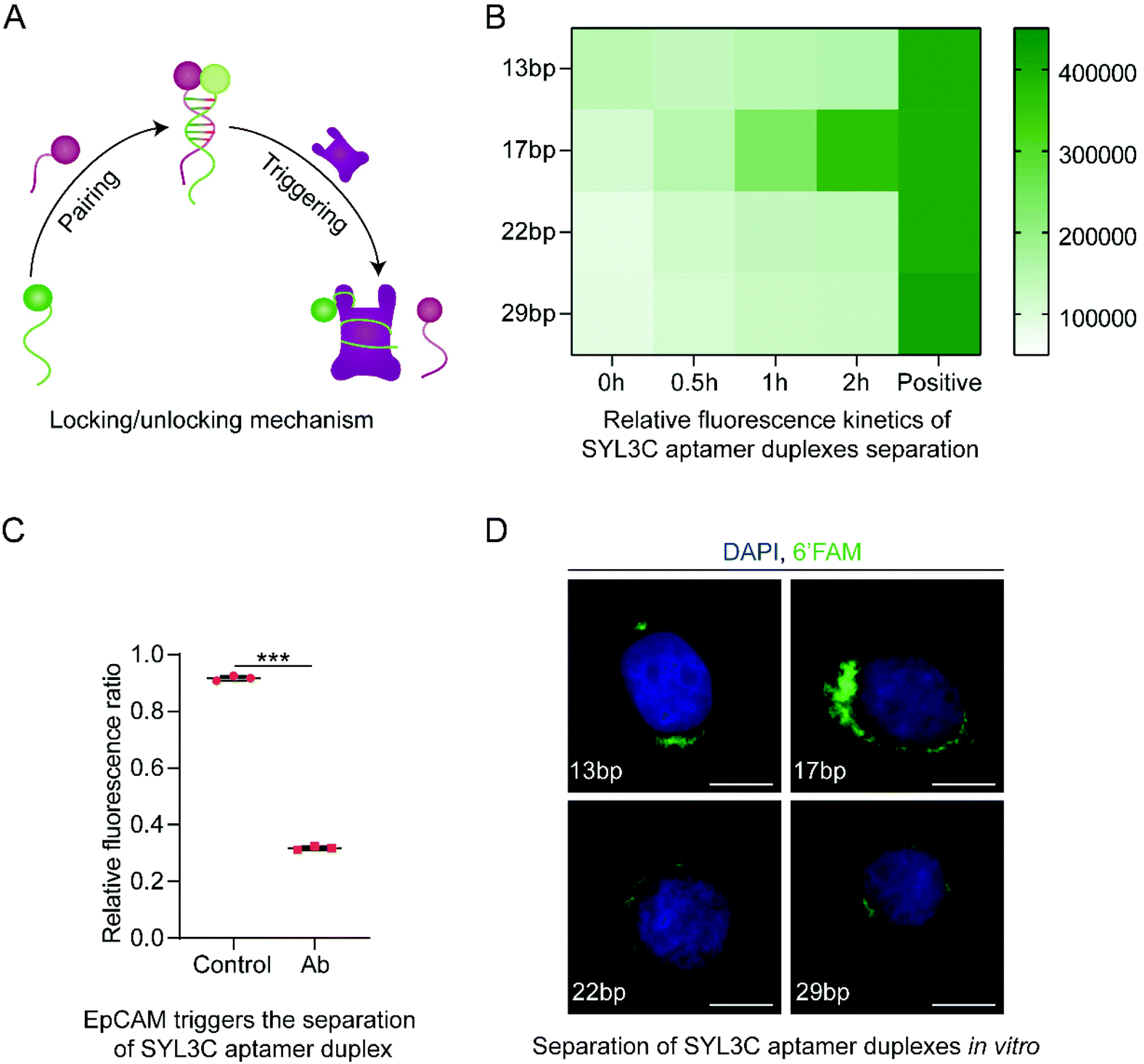 | ||
| Fig. 4 Screening analysis of the complementary length of SYL3C aptamer duplex. (A) Synthesis and separation mechanism of SYL3C aptamer duplex. The SYL3C aptamer serves both as the biosensors to recognize the epithelial cell adhesion molecule (EpCAM) on the surface of CTCs and as the molecular locking/unlocking mechanism of the TDN. (B) Relative fluorescence kinetics study indicating that the SYL3C aptamer duplex with a complementary length of 17 bp had the highest activation efficiency by the HT29 cells. The positive control sample refers to the A1 (Table S2†) modified only by 6′FAM. The data shown represent the mean ± SD from three independent experiments. (C) The SYL3C aptamer duplex with a complementary length of 17 bp was specifically activated and separated by EpCAM. Both groups used 17 bp SYL3C aptamer duplex. The HT29 cells in the Ab group were preincubated with anti-EpCAM antibody for 2 h to block EpCAM on the cell membrane. Data shown represent mean ± SD from three independent experiments. Comparisons were made using Student's t-test. ***p < 0.001. (D) Fluorescent images showed the binding of SYL3C aptamer duplex with different complementary lengths to HT29 cells after incubation for 1 h at 37 °C. The images are representative of three independent experiments. Scale bars: 10 μm. | ||
To fold a single layer of DOS into TDN, we added five 17 bp SYL3C aptamer duplexes into the DOS sample (Fig. S1 and Table S1†). Five duplexes drive a single layer of DOS to fold along the potential creases (Fig. 1A mid-side). The tetrahedral nanostructures of TDN were observed in the results of AFM (Fig. 3B). Furthermore, we used the Malvern analyzer to characterize the size distribution of DOS and TDN. The size distribution data showed that the peak value of DOS was 50.7 nm, and the TDN was 24.4 nm (Fig. 3C). According to the measurement results, the apparent zeta potential of TDN was higher than that of DOS (Fig. 3D). This phenomenon may be caused by the internal space of the tetrahedral TDN. The yields of TDN are an essential indicator to measure the feasibility of the folding strategy. The gel electrophoresis results (Fig. S3B†) of DOS and TDN with the same initial loading were counted. The comparison of the gray value between DOS and TDN indicated that the average yield of TDN was 93.7%. We also acquired and analyzed the parameters in the AFM images of TDN, such as the area, perimeter, and peak-to-valley ratio. On average, 91.2% of the nanostructures in the AFM images have the shape of a tetrahedron. In summary, we can conclude that with the help of multiple parallel folding, more than 90% of DOS have been successfully assembled into TDN. The break of base pairs at the maximum folding stress and the unstable anchoring of duplexes may be the reasons for the unsuccessful folding of a few DOS (less than 10%).
By placing TDN and DOS in a cell culture medium containing 10% fetal bovine serum (FBS), we investigated the serum stability of these nanostructures in a cell culture environment. After different treatment periods, the mixture of cell culture medium (10% FBS) and DNA nanostructures were filtered (pore size: 0.45 μm) to remove impurities. Finally, the harvested DNA nanostructures were analyzed by polyacrylamide gel electrophoresis (PAGE). The gel electrophoresis results (Fig. S4A†) demonstrated that more than 83.9% TDN can remain stable for 12 h under the conditions of mimicking the physiological cell environment. The analysis of the gray value indicated that only 61.9% of DOS can remain stable in the cell culture environment (Fig. S4B†). As the treatment time increased to 24 h, more than 55.7% of DOS were degraded by nucleases on average. The more compact local structure49 of the tetrahedron is the primary factor that rigid TDN has more robust serum stability than DOS. Compared with DOS, the encapsulated TDN may have fewer chances to interact with nucleases.50,51
Apart from the gel electrophoresis analysis, we also employed AFM to characterize the serum stability of DOS and TDN. We first used AFM to perform contact scanning of DNA nanostructures harvested from DOS treated with 10% FBS for 12 h at 37 °C. Compared with the configuration of untreated DOS (Fig. 3A), the morphology of all DOS has changed to varying degrees (Fig. S5A†). The tearing of irregular rectangles and the cutting of incomplete structures may be caused by the nuclease catalysis. To explore how many TDN changed their conformation during the 12 h treatment with 10% FBS. We then incubated TDN in 10% FBS for 12 h at 37 °C. The relevant samples were then recovered and scanned by AFM. As shown in Fig. S5B,† only a quarter of the TDN configuration has changed. The gel electrophoresis analysis report (Fig. S4B†) also pointed out that compared with the TDN without 10% FBS treatment, 83.9% DNA nanostructures still existed in the TDN (10% FBS, 12 h) group. These results suggest that TDN exhibits better structural stability than DOS in resisting nuclease erosion.
Using confocal microscopy analysis, we examined whether the configuration of TDN would change after encountering EpCAM on the HT29 cell membrane. To eliminate the false positives caused by image strands falling from the DNA nanostructures and being uptake by HT29 cells, we first used the free Texas red-ssDNA (FTR) treated group as a negative control. To further investigate the relative position and conformational change of DNA nanostructures when recognizing HT29 cells for guiding subsequent experiments. We employed the free 6′FAM-17 bp duplex (FFD) group as a positive control. After 1 h of incubation, only a tiny amount of FTR was taken up by HT29 cells (Fig. 5A top row), while a large amount of green fluorescence (Fig. 5A bottom row) confirmed that FFD was recognized and activated by HT29 cells.
 | ||
| Fig. 5 Results of TDN targeting HT29 cells. (A) FTR (1 nM) labeled with Texas Red as the negative control, FFD (1 nM) labeled with 6′FAM as the positive control. These images are representative of three independent experiments. Scale bars: 15 μm. (B) Confocal images revealed the results of BDT (top row), TDOS (middle row), and TDN (bottom row) being captured by HT29 cells after incubation for 1 h at 37 °C. The BDT, TDOS, and TDN were visualized with the Texas Red. The random strands of BDT and the SYL3C aptamer strands of TDOS or TDN were modified with the 6′FAM. The data are representative of three independent experiments. Scale bars: 15 μm. (C) Comparative analysis of the relative Texas Red fluorescence ratio of HT29 cells treated with the same molar concentration (1 nM) of BDT, TDOS and TDN. Relative Texas Red fluorescence ratio = (red fluorescence area/total image area) × 100%. Data shown represent mean ± SD from three independent experiments. Comparisons were made using one-way ANOVA. *p < 0.05, ***p < 0.001. (D) Co-localization analysis of Texas Red and 6′FAM in the white dotted area of Fig. 5B. | ||
To observe the conformational change of TDN more directly, in addition to the SYL3C aptamer modified by the 6′FAM, we also introduced image strands modified by Texas Red, which hybridize with the image capture strands (Fig. S1 and Table S1†). Driven by 6′FAM/BHQ1 modified random duplexes (Fig. S1†), DOS was folded into the bare DNA tetrahedron (BDT). Unlike the SYL3C aptamer duplex, the two complementary sequences of random duplex do not contain SYL3C aptamer (Table S1†). As a control group, although BDT has the same tetrahedral structure as TDN, the structural configuration of BDT cannot be changed by the trigger of EpCAM. As another control group, the targeted DNA origami sheet (TDOS) is a layer of DNA origami sheet with the ability to target EpCAM. TDOS has Texas Red modified image strands and 6′FAM/BHQ1 modified SYL3C aptamer strands/random strands. We incubated the same amount (1 nM) of BDT, TDOS, or TDN with HT29 cells at 37 °C for 1 h and evaluated their intake, respectively. According to confocal images (Fig. 5B), many TDN were found inside the HT29 cells. Comparative analysis of the relative texas red fluorescence ratio from BDT, TDOS, and TDN samples showed that TDN had a more vital ability to recognize HT29 cells than BDT and TDOS (Fig. 5C). To further reveal the unfolding of TDN in HT29 cells, we selected the white dotted area in Fig. 5B for co-localization analysis. As shown in Fig. 5D, the gray value distributions of Texas Red and 6′FAM were highly consistent, indicating that the cellular intake was accompanied by the conformational change of TDN in this area.
To determine whether TDN could specifically recognize HT29 cells in the presence of other cells, we used a co-culture model of BJ cells and HT29 cells. The result of the positive control group presented that FFD specifically aggregated on the membrane of the HT29 cells (white arrows in Fig. S6A† bottom row) rather than on the BJ cells (black arrows in Fig. S6A† bottom row). We then cultivated the same amount of BDT or TDN with the co-culture model at 37 °C for 1 h. Using confocal microscopy to visualize DNA nanostructures, we found that only a few BDT were nonspecifically captured by HT29 cells or BJ cells (Fig. S6B†). More importantly, plentiful TDN were appeared in the cytoplasm of HT29 cells but not in BJ cells. These results indicate that the dynamic TDN can be activated specifically by HT29 cells, and hence that TDN can be considered as a new nanorobot for monitoring EpCAM-positive cells. The analysis results of flow cytometry also showed that the total amount of fluorescence carried by HT29 cells was significantly higher than BJ cells (Fig. S7†), suggesting that if we define a minimum fluorescence threshold, the TDN should have the potential to screen HT29 cells in the presence of BJ cells. To further examine the biocompatibility of TDN, we chose multiple concentrations of TDN to incubate with HT29 cells or BJ cells for 24 h. The cell viability data (Fig. S8†) of all HT29 cells and BJ cells remained above 94.5%, with no significant differences between the two groups, indicating that TDN has low cytotoxicity. Together, all the above results demonstrate that TDN can be used as an excellent nanorobot to resist nuclease catalysis effectively and recognize EpCAM-positive cells specifically.
Conclusions
To conclude, we have demonstrated a strategy for folding a DNA origami sheet into a hollow 3D object through the hybridization of DNA strands located at the edges of the sheet. Multiresolution simulations verified the feasibility of the folded structure and characterized an ensemble of possible microscopic conformations. The screened 17 bp SYL3C aptamer duplexes that can target EpCAM were introduced into the folding process to construct dynamic TDN that responds to EpCAM triggers. As a result, the TDN has been proved to possess better serum stability and specific recognition in live cell culture experiments. Our research provides a novel strategy for designing and folding relevant DNA nanodevices, and develops a new method for enhancing the serum stability of traditional DNA origami sheets. Notably, the conformational change of TDN triggers the serum stability change. The serum stability change means that TDN has a more stable tetrahedral shape before identifying target cells, which may help control the initial dose of TDN used for monitoring objects. On the contrary, the serum stability of TDN after recognizing the target cells is weakened, which contributes to its rapid degradation to further reduce cytotoxicity. Therefore, we optimistically speculate that TDN would have excellent potential to detect EpCAM-positive cells, whether used in microfluidic chips or circulation in vivo.Author contributions
F.L. and X.L. conceived the concept. T.A., T.F., Q.H., and X.L. supervised the studies. F.L. and X.L. prepared and characterized the DNA nanostructures. C.M. and A.A. designed and performed simulation analysis. F.L. performed cell experiments. F.L. and C.M. drew the schematic illustrations. F.L., X.L., and C.M. interpreted data. F.L. and X.L. wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China under grants (61873037, 61903039), China Postdoctoral Science Foundation (BX20190035, 2020M680015), National Science Foundation USA under the grant (DMR-1827346), and the Grant-in-Aid for Scientific Research (JP19H02093, JP19H02097) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.Notes and references
- H. Yan, T. H. LaBean, L. Feng and J. H. Reif, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8103–8108 CrossRef CAS PubMed.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS PubMed.
- R. Veneziano, S. Ratanalert, K. Zhang, F. Zhang, H. Yan, W. Chiu and M. Bathe, Science, 2016, 352, 1534 CrossRef CAS PubMed.
- W. M. Shih, J. D. Quispe and G. F. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS PubMed.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- H. Zhang, G. S. Demirer, H. Zhang, T. Ye, N. S. Goh, A. J. Aditham, F. J. Cunningham, C. Fan and M. P. Landry, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7543–7548 CrossRef CAS PubMed.
- J. Liu, L. Song, S. Liu, Q. Jiang, Q. Liu, N. Li, Z.-G. Wang and B. Ding, Nano Lett., 2018, 18, 3328–3334 CrossRef CAS PubMed.
- G. Chen, D. Liu, C. He, T. R. Gannett, W. Lin and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 3844–3851 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed.
- S. Jiang, Z. Ge, S. Mou, H. Yan and C. Fan, Chem, 2021, 7, 1156–1179 CAS.
- H. Ijäs, I. Hakaste, B. Shen, M. A. Kostiainen and V. Linko, ACS Nano, 2019, 13, 5959–5967 CrossRef PubMed.
- A. M. Maier, C. Weig, P. Oswald, E. Frey, P. Fischer and T. Liedl, Nano Lett., 2016, 16, 906–910 CrossRef CAS PubMed.
- E. Kopperger, J. List, S. Madhira, F. Rothfischer, D. C. Lamb and F. C. Simmel, Science, 2018, 359, 296–301 CrossRef CAS PubMed.
- M. Liu, S. Jiang, O. Loza, N. E. Fahmi, P. Šulc and N. Stephanopoulos, Angew. Chem., Int. Ed., 2018, 57, 9341–9345 CrossRef CAS PubMed.
- A. Keller and V. Linko, Angew. Chem., Int. Ed. Engl., 2020, 59, 15818–15833 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson and J.-Y. Han, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS PubMed.
- F. M. Anastassacos, Z. Zhao, Y. Zeng and W. M. Shih, J. Am. Chem. Soc., 2020, 142, 3311–3315 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- J. Hahn, S. F. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed.
- Y. X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström and B. Högberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS PubMed.
- D. Bhatia, S. Surana, S. Chakraborty, S. P. Koushika and Y. Krishnan, Nat. Commun., 2011, 2, 339 CrossRef PubMed.
- Y. Ke, J. Sharma, M. Liu, K. Jahn, Y. Liu and H. Yan, Nano Lett., 2009, 9, 2445–2447 CrossRef CAS PubMed.
- J. Yoo and A. Aksimentiev, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20099–20104 CrossRef CAS PubMed.
- M. D. E. Jepsen, R. S. Sørensen, C. Maffeo, A. Aksimentiev, J. Kjems and V. Birkedal, Nanoscale, 2019, 11, 18475–18482 RSC.
- C. Maffeo and A. Aksimentiev, Nucleic Acids Res., 2020, 48, 5135–5146 CrossRef CAS PubMed.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- S. V. Litvinov, W. van Driel, C. M. van Rhijn, H. Bakker, H. van Krieken, G. J. Fleuren and S. O. Warnaar, Am. J. Pathol., 1996, 148, 865–875 CAS.
- C. Patriarca, R. M. Macchi, A. K. Marschner and H. Mellstedt, Cancer Treat. Rev., 2012, 38, 68–75 CrossRef CAS PubMed.
- Y. Gao, S. Gu, Y. Zhang, X. Xie, T. Yu, Y. Lu, Y. Zhu, W. Chen, H. Zhang, H. Dong, P. J. Sinko and L. Jia, Small, 2016, 12, 2595–2608 CrossRef CAS PubMed.
- U. Dharmasiri, S. K. Njoroge, M. A. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2011, 83, 2301–2309 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- C. Wiraja, Y. Zhu, D. C. S. Lio, D. C. Yeo, M. Xie, W. Fang, Q. Li, M. Zheng, M. Van Steensel and L. Wang, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2018, 119, 6459–6506 Search PubMed.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS PubMed.
- T. E. Ouldridge, A. A. Louis and J. P. Doye, J. Chem. Phys., 2011, 134, 085101 CrossRef PubMed.
- P. Šulc, F. Romano, T. E. Ouldridge, L. Rovigatti, J. P. Doye and A. A. Louis, J. Chem. Phys., 2012, 137, 135101 CrossRef PubMed.
- B. E. Snodin, F. Randisi, M. Mosayebi, P. Šulc, J. S. Schreck, F. Romano, T. E. Ouldridge, R. Tsukanov, E. Nir, A. A. Louis and J. P. Doye, J. Chem. Phys., 2015, 142, 234901 CrossRef PubMed.
- M. S. L. Tang, S. C. Shiu, M. Godonoga, Y. W. Cheung, S. Liang, R. M. Dirkzwager, A. B. Kinghorn, L. A. Fraser, J. G. Heddle and J. A. Tanner, Nanomedicine, 2018, 14, 1161–1168 CrossRef CAS PubMed.
- K. Tsukakoshi, K. Abe, K. Sode and K. Ikebukuro, Anal. Chem., 2012, 84, 5542–5547 CrossRef CAS PubMed.
- S. Ramakrishnan, B. Shen, M. A. Kostiainen, G. Grundmeier, A. Keller and V. Linko, ChemBioChem, 2019, 20, 2818–2823 CrossRef CAS PubMed.
- J. W. Conway, C. K. McLaughlin, K. J. Castor and H. Sleiman, Chem. Commun., 2013, 49, 1172–1174 RSC.
- J.-W. Keum and H. Bermudez, Chem. Commun., 2009, 7036–7038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, supporting figures, supporting tables and animations. See DOI: 10.1039/d1nr02757c |
| This journal is © The Royal Society of Chemistry 2021 |