
Co-construction of sulfur vacancies and carbon confinement in V5S8/CNFs to induce an ultra-stable performance for half/full sodium-ion and potassium-ion batteries†
Lihong
Xu‡
a,
Xiaochuan
Chen‡
ab,
Wenti
Guo
 fg,
Lingxing
Zeng
*acd,
Tao
Yang
h,
Peixun
Xiong
e,
Qinghua
Chen
ac,
Jianmin
Zhang
*fg,
Mingdeng
Wei
e and
Qingrong
Qian
*acd
fg,
Lingxing
Zeng
*acd,
Tao
Yang
h,
Peixun
Xiong
e,
Qinghua
Chen
ac,
Jianmin
Zhang
*fg,
Mingdeng
Wei
e and
Qingrong
Qian
*acd
aEngineering Research Center of Polymer Green Recycling of Ministry of Education, College of Environmental Science and Engineering, Fujian Normal University, Fuzhou, Fujian 35007, China. E-mail: zenglingxing@fjnu.edu.cn; qrqian@fjnu.edu.cn; Tel: +86 591 83465156
bKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, School of Chemistry, Sun Yat-Sen University, Guangzhou, China
cFujian Key Laboratory of Pollution Control & Resource Reuse, Fuzhou, Fujian 350007, China
dKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China
eFujian Provincial Key Laboratory of Electrochemical Energy Storage Materials, Fuzhou University, Fuzhou, Fujian 350002, China
fFujian Provincial Key Laboratory of Quantum Manipulation and New Energy Materials College of Physics and Energy, Fujian Normal University, Fuzhou, Fujian 350117, China. E-mail: jmzhang@fjnu.edu.cn; Tel: +86 591 22868141
gFujian Provincial Collaborative Innovation Center for Advanced High-Field Superconducting Materials and Engineering, Fuzhou, Fujian 350117, China
hTEMA-NRG, Mechanical Engineering Department University of Aveiro, 3810-193 Aveiro, Portugal
First published on 6th February 2021
Abstract
The construction of anode materials for sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs) with a high energy and a long lifespan is significant and still challenging. Here, sulfur-defective vanadium sulfide/carbon fiber composites (D-V5S8/CNFs) are designed and fabricated by a facile electrospinning method, followed by sulfuration treatment. The unique architecture, in which V5S8 nanoparticles are confined inside the carbon fiber, provides a short-range channel and abundant adsorption sites for ion storage. Moreover, enlarged interlayer spacings could also alleviate the volume changes, and offer small vdW interactions and ionic diffusion resistance to store more Na and K ions reversibly and simultaneously. The DFT calculations further demonstrate that sulfur defects can effectively facilitate the adsorption behavior of Na+ and K+ and offer low energy barriers for ion intercalation. Taking advantage of the functional integration of these merits, the D-V5S8/CNF anode exhibits excellent storage performance and long-term cycling stability. It reveals a high capacity of 462 mA h g−1 at a current density of 0.2 A g−1 in SIBs, while it is 350 mA h g−1 at 0.1 A g−1 in PIBs, as well as admirable long-term cycling characteristics (190 mA h g−1/17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles/5 A g−1 for SIBs and 165 mA h g−1/3000 cycles/1 A g−1 for PIBs). Practically, full SIBs upon pairing with a Na3V2(PO4)3 cathode also exhibit superior performance.
000 cycles/5 A g−1 for SIBs and 165 mA h g−1/3000 cycles/1 A g−1 for PIBs). Practically, full SIBs upon pairing with a Na3V2(PO4)3 cathode also exhibit superior performance.
Introduction
Commercialized lithium-ion batteries (LIBs) have occupied the energy storage market for quite a while. However, the safety issues and the cost of lithium precursors are increasing rapidly.1,2 Therefore, alternative battery systems such as sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs) are beginning to attract great interest.3–5 Compared with lithium, sodium and potassium resources are more abundant (Na: 2.36 wt%, K: 2.09 wt% and Li: 0.0017 wt%) on the Earth's crust. What is more, Na and K elements, which have similar physical–chemical properties to Li, are of lower cost as well. Therefore, SIBs and PIBs are becoming the most promising candidates for next generation energy storage systems.6–8 Unfortunately, the large radii of sodium ions (1.02 Å) and potassium ions (1.38 Å) versus lithium ions (0.76 Å) make some excellent LIB anode materials suffer from severely sluggish diffusion kinetics and cause large volume expansion during the sodium/potassium storage process, resulting in poor cycling life and limiting their practical applications. Therefore, exploring the state-of-art SIB and PIB anode materials and aiming for the same level of reversible capacity/power density and long-term cycling stability are both urgent and challenging for their practical applications today.9–11Many promising candidates have been extensively investigated as anode materials in recent years, including carbons (e.g., hard carbon, carbon fibers and carbon nanotubes), metals and alloys (Sb, Sn and SnSb), metal oxides, and transition metal dichalcogenides (TMDs such as MoS2 and VS2), etc.12,13 Among them, layered vanadium sulfide materials including VS2, V2S3, V3S4 and V5S8 have received great attention, due to their high theoretical capacity. More importantly, their unique structures are held together via weak van der Waals interactions, which permit the intercalation of sodium and potassium ions without significant structural distortion.14,15 V5S8, as a family member of vanadium sulfides, has been employed as the anode for the SIBs and PIBs due to its large interlayer structure and superior ion storage performance. For example, Yang et al. reported that V5S8–graphite hybrid nanosheets could deliver a high reversible capacity of 496 mA h g−1 after 500 cycles at 1 A g−1 for SIBs.16 Guo and co-workers synthesized few-layered V5S8 nanosheets wrapped on hollow carbon spheres as the PIB anode material, which exhibited a stable cycling capacity of 190 mA h g−1 at a current density of 2 A g−1 after 1000 cycles.17 Nevertheless, the full potential of vanadium sulfides as electrode materials in SIBs and PIBs remains elusive. It is hence highly desirable and paramount to explore appropriate vanadium sulfide electrode materials for SIBs/PIBs that can enable a superior long cycle life (e.g. >15000 cycles for SIBs and >3000 cycles for PIBs), high reversible capacity and fast Na+/K+ deintercalation.
To tackle the above-mentioned issue, extensive efforts have been devoted to optimizing the ion storage performance of vanadium sulfides through three typical strategies: (i) introducing the carbon component to engineer the nanostructure composites is considered as one of the effective methods to improve the conductivity. For instance, Mai's group fabricated layer-by-layer stacked VS2 nanosheets, which displayed a capacity of 204 mA h g−1 after 600 cycles at 5 A g−1 in SIBs.18 Wang et al. prepared a VS4-rGO composite and achieved a reversible capacity of ∼400 mA h g−1 after 300 cycles at 0.5 A g−1 in SIBs.19 All these electrodes exhibited an enhanced electrochemical performance with a high reversible capacity. (ii) Engineering surface defect vacancies to boost the number of active sites. The Na and K ions can be inserted into materials through the edge side, but through the surface defects. The formation of defects not only helps to improve the electronic conductivity of materials and redistribution of charge, but also remarkably enhances the insertion channels and decreases the diffusion pathway.20 (iii) Interlayer expansion is recently expected to be a new strategy to improve the storage performance, as it can reduce vdW interactions and ionic diffusion resistance and maintain the stability of materials, resulting in a reduced diffusion energy barrier and enhanced ionic diffusion kinetics. Until now, although some advances have been achieved in the development of vanadium-based sulfide anode materials for energy storage, less attention has been paid to the defect and spacing expansion structure relationship of vanadium sulfide application in SIBs and PIBs. Therefore, optimizing all the factors to prepare promising vanadium sulfide materials, and further elucidating the influence of defect structures on storage performances, and kinetic mechanisms at the atomic level, would be very intriguing.
The authors in this work tried to fabricate vanadium sulfide/carbon fiber composites with rich sulfur defects (D-V5S8/CNFs) via a facile electrospinning technology coupled with a one-step sulfuration–carbonization process (Scheme 1). The synergistic effect between the defects and V5S8/CNFs endows the electrode with a fast, highly reversible and long cycle life. When utilized as anodes for SIBs, the as-prepared D-V5S8/CNF electrode shows an excellent electrochemical storage performance. The anode delivers a high reversible capacity, which is much better than the perfect V5S8/C (P-V5S8/C), and a long-term cycling stability of over 17000 cycles. More specifically, the D-V5S8/CNFs can exhibit excellent potassium-ion storage in terms of a high specific capacity (350 mA h g−1 at a current density of 0.1 A g−1) and a prolonged cycling stability (3000 cycles). Practically, the V5S8/CNFs//NVP sodium full cell that uses Na3V2(PO4)3 as the cathode also demonstrates a desirable energy storage performance, which can power a commercial light-emitting diode.
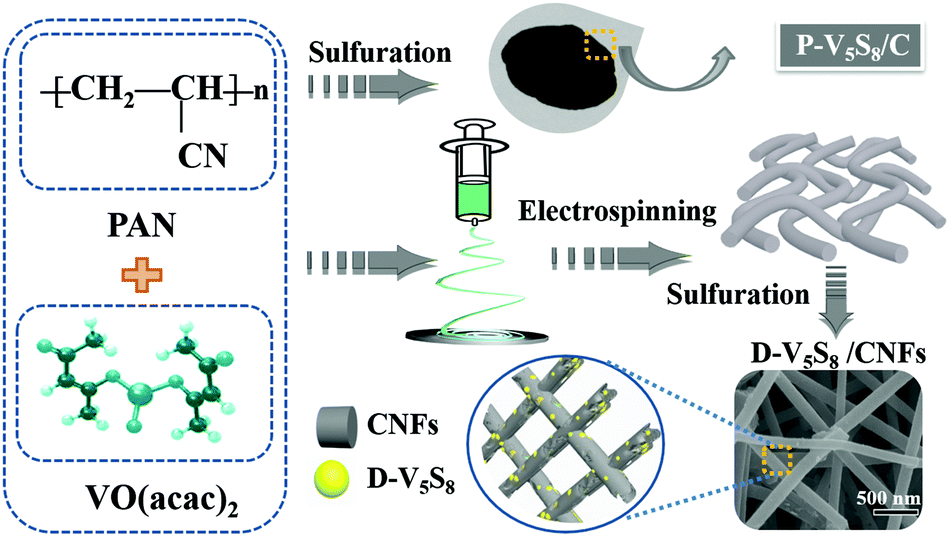 | ||
| Scheme 1 Schematic illustration for synthesizing the D-V5S8/CNF and P-V5S8/C composites. | ||
Experimental
All reagents were used without further purification. Polyacrylonitrile (PAN, MW = 150000) was purchased from Sigma-Aldrich (St Louis, MO, USA). Vanadyl acetylacetonate (VO(acac)2, 99%) was purchased from Aladdin (Shanghai, China). N,N-Dimethylformamide (DMF, AR) and sulfur powder (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Synthesis of sulfur defective V5S8/CNF composites (D-V5S8/CNFs)
Firstly, 0.5 g of PAN and 0.5 g of VO(acac)2 were added into 10 mL of DMF solution. The mixture was further magnetically stirred for 24 h at room temperature until a transparent solution was obtained. Electrospinning was carried out with a flow rate of 0.5 mL h−1 and an applied voltage of 22 kV. Then the as-spun VO(acac)2/PAN precursor fibers were collected on an aluminum current collector, which is located horizontally 15 cm from the tip-to-collector. Subsequently, the as-prepared VO(acac)2/PAN precursor fibers and sulfur powder (in mass ratios of 1:4) mixture were loaded into a ceramic boat and heated at 600 °C for 4 h under a mixed 5% H2/95% Ar atmosphere with a ramp rate of 2 °C min−1. The resultant product of sulfur-defective vanadium sulfide/carbon fiber composites was obtained and labeled as D-V5S8/CNFs.
Synthesis of pure V5S8/C materials (P-V5S8/C)
0.1 g of PAN and 0.1 g of VO(acac)2 were mixed with sulfur powder in a mass ratio of 1:4 and then ball milled for 3 h. Then, 2.4 g of sulfur powder and 0.6 g of the ball-milled raw material mixture were placed on two individual ceramic boats, and situated in the upstream and middle side of the tubular furnace, respectively (Scheme 1). Subsequently, the mixtures were annealed at 600 °C for 4 h with a ramp rate of 2 °C min−1 under a 5% H2/95% Ar atmosphere. The obtained product of perfect V5S8/C (containing some V2O3 impurities) was also obtained and labelled as P-V5S8/C for clarification.
Results and discussion
Characterization of the D-V5S8/CNF and P-V5S8/C composites
The microstructure and morphology of these two composites are characterized by SEM and TEM techniques. Obviously, the V5S8 phase and the carbon layer can be observed in the hybrid structure of the two samples (Fig. 1). In detail, as we can see, the as-prepared P-V5S8/C in Fig. 1a shows specific blocky structures. The corresponding high-resolution TEM (HRTEM) image reveals a lattice interplanar spacing of 0.2 nm, which is ascribed to the (−224) plane of V5S8 (Fig. 1d). As compared with P-V5S8/C, the nanofiber architectures in D-V5S8/CNFs can be clearly observed in Fig. 1b, and uniform nanofibers with a diameter of ca. 100 nm are interwoven together to form a network structure. Obviously, it can be observed that the V5S8 nanoparticles are freely distributed into/onto the entire carbon fibers (Fig. 1c). The corresponding high-resolution TEM images clearly reveal amorphous carbon structures with an interlayer spacing of 0.20 nm for the nanoparticles, matching well with the (−224) crystal plane of V5S8 (Fig. 1f). The energy-dispersive X-ray (EDX) spectrum (Fig. S1, ESI†) also shows a homogeneous distribution of the V and S elements throughout the fibers. It is worth noting that some broken lattices marked by blue circles and a well-resolved lattice fringe with an expanded spacing of 0.62 nm (0.565 nm of the pristine V5S8) can be observed in Fig. 1e, indicating the formation of rich defects.21,22 The above information suggests that sulfur-defective V5S8/CNF composites with an enhanced interlayer spacing probably exists under our conditions. | ||
| Fig. 1 SEM morphology of (a) P-V5S8/C and (b) D-V5S8/CNFs. (c) TEM image of D-V5S8/CNFs. The corresponding HRTEM images of (d) P-V5S8/C and (e and f) D-V5S8/CNFs. Note that the inset of (f) shows the relevant enlargement of the dashed box (red). | ||
The defect structures of the samples are also verified well by the Raman spectrum (Fig. 2a and b). For D-V5S8/CNFs (Fig. 2a), two characteristic peaks centered at about 1357 cm−1 and 1593 cm−1 are observed, which typically belong to the D-band and G-band of graphite, respectively.23,24 This manifests the low graphitization and high disorder of the derived carbon in the composite. The observed diffraction peaks at 994, 800, 692 and 515 cm−1 are characteristic of the V–S, C–S, V–O–V and O–3V bonds, respectively, which are in agreement with previous studies.25–28 Typically, the peaks at 280 cm−1 and 404 cm−1 correspond to A1g (out-of-plane), which are always observed in the 2D TMDs and stemmed from the curvature of the hexagonal VS2 and the opposite vibration of two S atoms with respect to the V atoms, respectively.26,27,29–31 Notably, compared to P-V5S8/C (Fig. 2b), D-V5S8/CNFs reveals a “shoulder peak” assigned to A1g at 423 cm−1. The presented weak shoulder peak of the Raman modes in the inset of Fig. 2a can be ascribed to the appearance of sulfur vacancies in the composite.20,32 Furthermore, the out-of-plane vibration mode A1g of D-V5S8/CNFs is blue-shifted in the corresponding modes of the P-V5S8/C sample. This change in the Raman vibration modes is considered to be related to the lattice distortion in the structure.33,34
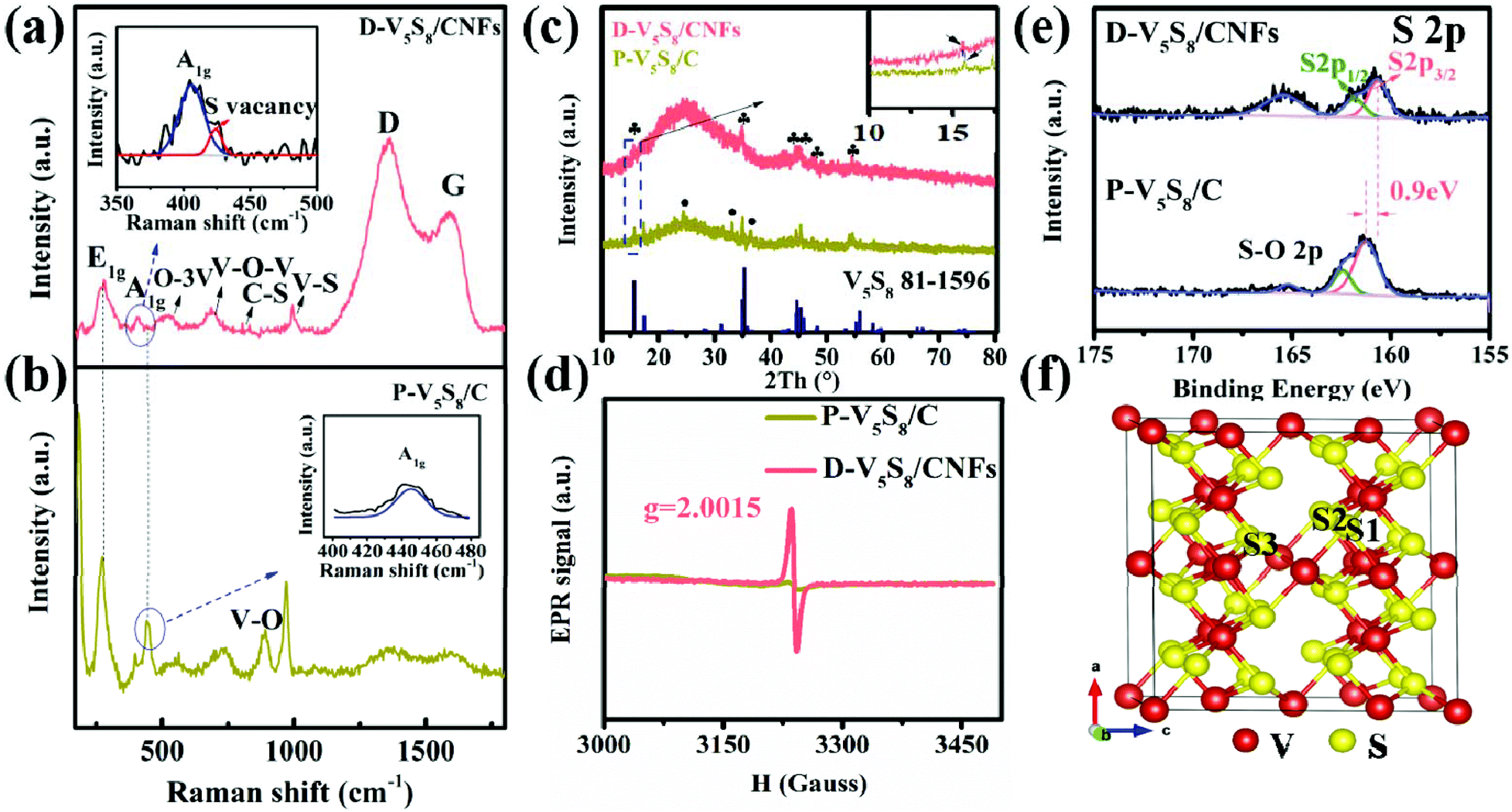 | ||
| Fig. 2 (a and b) Raman spectra, (c) XRD pattern, (d) EPR spectrum, and (e) XPS analysis of the P-V5S8/C and D-V5S8/CNF samples. (f) Schematic illustration of the atom arrangements in V5S8; note that S1, S2, and S3 represent the possible sulfur defects. | ||
Furthermore, the X-ray diffraction (XRD) patterns of the as-prepared D-V5S8/CNF and P-V5S8/C composites are presented in Fig. 2c. For D-V5S8/CNFs, the obvious diffraction peaks located at 15.6°, 35.2°, 44.7°, 45.3°, 48.3° and 55.8° can be indexed well to the (002), (222), (−224), (224), (006) and (620) crystal planes of V5S8, respectively (JCPDS card no. 81-1596). A hump peak of the sample at around 25° reveals the amorphous nature of carbon, elucidating that the V5S8/CNF composite has been successfully produced.35 Meanwhile, the XRD pattern of P-V5S8/C shows similar crystal structures and typical amorphous carbon spectra. However, compared to D-V5S8/CNFs, P-V5S8/C shows three extra diffraction peaks around 24.3°, 33.0° and 36.4°, which can be ascribed to the (012), (104) and (110) crystal planes of V2O3, respectively (JCPDS card no. 84-0317). This demonstrates the formation of a V5S8/C hybrid structure with some V2O3 impure crystalline phases in the P-V5S8/C sample, which may lead to inferior rate capability and rapid capacity decay. Furthermore, the (002) peak of D-V5S8/CNFs shifts to a lower angle compared to P-V5S8/C (in the inset of Fig. 2c), consistent with the magnified HRTEM image results in Fig. 1e.
Electron paramagnetic resonance (EPR) is an important technique to characterize the unpaired electrons of materials on coordinatively unsaturated defective sites and reveal the information of sulfur vacancies. Fig. 2d shows an EPR signal that arised from a sulfur vacancy. Unlike P-V5S8/C, D-V5S8/CNFs show a high intensity of the signal at the g-factor, g = 2.0015, suggesting increased exposed active centers that arise from the sulfur vacancy.20,36,37 Therefore, combined with the EPR results, the successful synthesis of sulfur-defective V5S8/CNF composites can be further confirmed.
The chemical binding environment of D-V5S8/CNFs and P-V5S8/C is further evaluated by an X-ray photoelectron spectroscopy (XPS) technique. The survey spectra successfully confirmed the presence of C, V, and S elements in the two materials, as shown in Fig. S2, ESI.†Fig. 2e shows the high-resolution S 2p XPS spectrum of the samples, in which the S 2p peaks of D-V5S8/CNFs exhibit three obvious binding energies at 160.5 eV, 161.7 eV and 165.3 eV, respectively, corresponding to the typical S 2p3/2, S 2p1/2 and oxidized S. The small amount of elemental O is due to the slight oxidation of the surface upon exposure to air.38,39 Importantly, comparable to the corresponding typical spectrum of P-V5S8/C, an apparent negative shift of ≈0.9 eV toward the lower binding energy can be observed in D-V5S8/CNFs. In addition, the high-resolution V 2p XPS spectrum of D-V5S8/CNFs and P-V5S8/C presented in Fig. S3 (ESI†) also reveals an obvious negative shift of ≈0.7 eV, which further shows the presence of sulfur vacancies.36,40,41 Taken together, all the above information suggests that sulfur-defective V5S8/CNF composites are successfully designed under our conditions. The total S contents in D-V5S8/CNFs are determined by elemental analysis to be 10.3 wt% (Table S1, ESI†). The V5S8 loading amounts in the obtained products D-V5S8/CNFs and P-V5S8/C are calculated to be 32.1 wt% and 30.1 wt%, respectively (Fig. S4, ESI†).16,17,42 The in situ constructed sulfur defects and carbide architecture engineering strategy may endow the D-V5S8/CNFs with a significantly enhanced electrochemical performance.
In order to further elucidate the functions of the sulfur defects in the V5S8 composite materials, we carried out the first-principles method to assess the different S-defective configurations via the formation energy calculation. All the possible positions (S1, S2, and S3) for the sulfur defects are calculated and shown in Fig. 2f and Fig. S5, ESI.† By comparing the formation energies of the three configurations of S-defective V5S8 in Table S2 (see the ESI†), it can be observed that V5S8 with an S2 vacancy defect has the lowest formation energy of −351.24 eV, suggesting that the defect prefers the S2 site for V5S8.
Electrochemical performance for SIBs
The electrochemical performance of the electrode for SIB anodes is investigated by assembling the CR2025-type coin half-cell with an electrolyte of 1 M NaClO4 in EC:DMC.
Fig. 3a presents the cyclic voltammetry (CV) curve of the electrode at a scan rate of 0.2 mV s−1 for the initial five cycles in a voltage window of 0.01–3.0 V (vs. Na/Na+). A broad cathodic peak appears at around 0.23 V and disappears in the subsequent cycles, which is attributed to the formation of the irreversible solid electrolyte interphase (SEI) layer.43 Meanwhile, the two weak reduction peaks observed at 1.94 V and 0.6 V are related to the insertion of Na+ into V5S8 and accompanied by the formation of amorphous Na2S and V (V5S8 + xNa+ + xe− ↔ NaxV5S8 ↔ Na2S + V),16 and the two oxidation peaks in the curves at around 1.65 V and 2.2 V are assigned to the formation of NaxV5S8 and V5S8via the reverse reaction between Na2S and V, respectively. In order to reveal the above CV dis(charge) reaction mechanism, ex situ XRD is employed to analyze the phase change during the charge and discharge processes of the electrode (Fig. S6, ESI†). In the full discharge to 0.01 V, a peak at 23.5° assigned to (111) Na2S is observed, which is caused by the insertion of Na+ into V5S8. Conversely, after charging to 3 V, a weak diffraction peak centered at 15.6° gradually appears, which is attributed to the (002) peak of V5S8. A diffraction peak at 23.5° assigned to Na2S is also observed even in the fully charged state, which may be related to the fact that 100% reversible efficiency is not absolutely obtained due to the V5S8 dispersion in carbon materials.44 These results are in agreement with the CV analysis. Impressively, in the subsequent cycles, the curves almost superpose with each other, indicative of good stability and reversibility during the repeatable desodiation/sodiation process.45
 | ||
| Fig. 3 Electrochemical performances of the D-V5S8/CNF and P-V5S8/C composites in SIBs. (a) CV curves of D-V5S8/CNFs at a scan rate of 0.2 mV s−1 between 0.01 and 3.0 V. (b) Galvanostatic charge–discharge profiles of D-V5S8/CNFs at a current density of 0.2 A g−1. (c) Cycling performance of the D-V5S8/CNF and P-V5S8/C samples at 0.2 A g−1. (d) Rate performance. Long-term cycling performance and coulombic efficiency at (e) 2 A g−1 and (f) 5 A g−1 of the as-prepared D-V5S8/CNF electrode. | ||
Next, the galvanostatic charge–discharge (GCD) profiles of the D-V5S8/CNF electrode (Fig. 3b) are similar to the CV results. Specifically, the anode delivers initial charge and discharge capacities of 950 and 462 mA h g−1, respectively, with an undesirable initial coulombic efficiency (ICE) of 49%. The capacity loss arises from the formation of an SEI layer and the side reactions between the materials and the electrolyte.46,47 The cycling performances of the activated electrodes at a current density of 0.2 A g−1 are shown in Fig. 3c. Owing to the structural advantage of V5S8, CNFs, and the sulfur defective structures, the D-V5S8/CNF electrode exhibits a high and stable reversible capacity of 462 mA h g−1 after 70 cycles. Notably, in the first few cycles, the electrode delivers a lower charge specific capacity in comparison with the relative discharge capacity, attributed to the gradual insertion process of the Na ion deep inside the carbon lattice of the thick carbon layers.48 After the insertion continues in the first several cycles, the coulombic efficiency (CE) of the electrode gradually reaches 100%. In contrast, the specific capacity of P-V5S8/C maintains a relatively stable value with a low discharge capacity of ≈200 mA h g−1 after 70 cycles at the same current density, demonstrating the preferable electrochemical properties of D-V5S8/CNFs.49,50
The rate performances of the D-V5S8/CNF and P-V5S8/C electrodes are also shown in Fig. 3d. At a low current density of 0.1 A g−1, the D-V5S8/CNF electrode can exhibit a reversible capacity of 500 mA h g−1. When the current density reaches 0.2, 0.5, 1.0, and 2.0 A g−1, the specific capacities are sustained at high values of 464, 448, 418, and 352 mA h g−1, respectively, and when the current density is restored back to 0.1 A g−1, a stable reversible capacity of around 470 mA h g−1 can be achieved, indicating the significant storage reversibility of this composite electrode. However, the rate capacities of the P-V5S8/C electrode are inferior, only showing capacities of 248, 247, 225, 200 and 176 mA h g−1 at 0.1, 0.2, 0.5, 1 and 2 A g−1, respectively. In addition to the excellent rate performance, an outstanding cycling stability at a high current density is also observed for D-V5S8/CNFs. As depicted in Fig. 3e, this electrode can maintain a high cycling capacity of ≈340 mA h g−1 at a current density of 2 A g−1 even over 4000 cycles, with a CE of approximately 100%. Meanwhile, the long-term cycling stabilities of the D-V5S8/CNF electrode are further evaluated at a higher current density of 5 A g−1, as shown in Fig. 3f. Impressively, the activated electrode delivers an ultralong cycling retention for up to 17000 cycles, and retained a capacity of ca. 190 mA h g−1 without obvious capacity loss. The average capacity fading is only 0.1% per cycle. Both the capacity and cycling retention of D-V5S8/CNFs are superior to those of the P-V5S8/C electrode (Fig. S7, ESI†). Such an excellent long-term cycling stability has not been reported in previous vanadium-based sulfide materials to the best of our knowledge (Table S3, ESI†).
To further explain the origin of the excellent rate stability in the D-V5S8/CNF electrode, the kinetic properties are studied by CV at different scan rates in the voltage range of 0.01–3.0 V vs. Na+/Na, as shown in Fig. S8 (ESI†). Fig. S8a† shows the CV profiles of D-V5S8/CNFs, recorded at scan rates of 0.2–2 mV s−1. With the increase of the scan rate, the curves of the electrode exhibit two redox peaks, which are marked as reduction (peak R1) and oxidation (peak O1) peaks. Specifically, the capacitive behavior can be calculated according to the equation, i = a × νb.51 By plotting log(i) against log(ν), the tunable parameter, b value, can be achieved from the slope, where i is the peak current and ν is the scan rate. The ideal b value approaches 0.5, revealing that the faradaic insertion/extraction is an ion diffusion dominant process, while the b value close to 1 indicates the surface-induced capacitive step, which makes a major contribution to the capacitive response, and the b value in the range of 0.5 and 1 indicates the coexistence of two mixed mechanisms. In our case, as shown in Fig. S8b,† the two redox peaks, namely the reduction (peak R1) and oxidation (peak O1) peaks, deliver b values of about 0.72 and 0.89, respectively, indicating that the charge storage in this electrode is a mixed-control process.52 Quantitatively, the mixed mechanism at a given sweep rate can be further divided into a capacitive-controlled process (k1ν) and an ion diffusion dominant process (k2ν1/2). The fraction of the two processes can be studied by calculating k1 and k2 according to the equation, i(υ) = k1 × ν + k2 × ν1/2. To be specific, Fig. S8c† shows the CV curve at a scan rate of 0.6 mV s−1 for the capacitive current (blue region) compared with the total current, in which about 80% of the dominating capacitive contribution is quantified. The pseudocapacitive contribution percentage gradually enlarged upon increasing the scanning speed (Fig. S8d†). When the scan rate increased from 0.2 to 2 mV s−1, the contribution percentage of the capacitive-controlled process becomes higher and finally reaches 88%, which can enhance the Na-ion migration capability and facilitate the fast insertion/extraction behavior.53,54
In addition, the impedance changes during the charge and discharge processes are also investigated by electrochemical impedance spectroscopy (EIS). In Fig. S9a,† a depressed semicircle located at the high frequency area and an inclined line in the low-frequency area are presented in the result of Nyquist plots, which can be fitted with an equivalent electrical circuit (Fig. S9b†). The parameters in the equivalent electrical circuit model include the ohmic resistance Rs (the resistances of the electrolyte, separator, and electrical contacts), the charge transfer resistance Rct, the interfacial resistance of the constant phase element (CPE) and the Warburg impedance Zw obtained from Na+ insertion into the active electrode. Note that after cycling, an additional semicircle in the middle frequency region can be observed, which refers to the resistance of the SEI film.34,53 The result demonstrates a stable SEI film formed on the surface of D-V5S8/CNFs, which is consistent with the electrochemical analysis. Meanwhile, all the fitting results are listed in Table S4.† Specifically, as seen from the smaller semicircle at the medium-high frequency and the greater slope at the low frequency, the electrode after cycling shows a lower charge transfer resistance than that of the pristine electrode (the value of Rct went down from 205.9 Ohm to 175.2 Ohm), indicating the excellent charge-transfer kinetics during the cycling process with the optimal vacancy defective composition structure.55–57 The SEM data of the electrode after cycling with integrated fiber morphologies are also shown in Fig. S10,† which are further beneficial for maintaining the mechanical stability and improving the cycling performance. Such fast charge–discharge mechanisms and stable structures provide evidence for setting forth superior sodium storage properties.
Then, to better understand the enhanced Na-storage performance of the sulfur defects in V5S8/CNFs, the density of states (DOS) and partial density of states (PDOS) of the perfect V5S8 and S-defective V5S8 structures have been calculated, as shown in Fig. S11.† The band structures along the high symmetry lines in the perfect V5S8 and S-defective V5S8 Brillouin zones are presented in Fig. S11a and d.† Clearly, S-defective V5S8 is predicted to have metallic properties due to having many electronic states across the Fermi level, which is in good agreement with the previous calculations performed.17 Furthermore, Fig. S11b and e† show the DOS of the perfect V5S8 and S-defective V5S8, respectively. It is clearly shown that the contribution of vanadium near the Fermi level is more significant. More notably, V5S8 with S vacancy defects has a smaller state density than the pure case. It can be found that the proportion of the vanadium element decreases near the Fermi level, while the DOS of the sulfur element increases slightly. This means that the introduction of S-vacancy defects breaks the space symmetry of the original system, thus making the DOS near the Fermi level gentle. The result further verifies that the presence of defects in V5S8 can make it a stronger electron-deficient system, which favorably enhances the electronic conductivity and the Na+/K+ adsorption capability.17,58,59 The corresponding PDOS projected over S displays that the valence band and the conduction band originate from the perfect-V5S8 and S-defective V5S8, respectively, from the S s and the S p orbitals, see Fig. S11c and f.†
Meanwhile, the charge-density differences of the perfect V5S8 and S-defective V5S8 models are calculated as shown in Fig. 4. The red charge density represents an electron accumulation region and the blue one represents the opposite. From these results, the re-distribution of the charge characteristics with the existence of S vacancies can be clearly observed. It can be clearly seen from the charge density that the system forms ionic bonds. In Fig. 4c and f, we can find that the vacancy defect of the sulfur atom induces a change in the charge transfer, that is, the surrounding vanadium atoms contribute to the electrons (the red part). Specifically, the charge density difference between the perfect and rich defect structures also indicates the advantage of boosting Na+ or K+ ion adsorption. The charge-density differences of 0 × d and 0.75 × d distances from the XOY plane (001) are shown in Fig. S12 (see the ESI†). These results verify that the defects induce the changes in the electronic-state distribution, acting as effective recombination centers. The rich defect structure is more energetically beneficial for the adsorption of Na ions, which can contribute to more active sites and have better reaction kinetics.60 This matches well with the results of the abovementioned electrochemical and kinetic analyses.
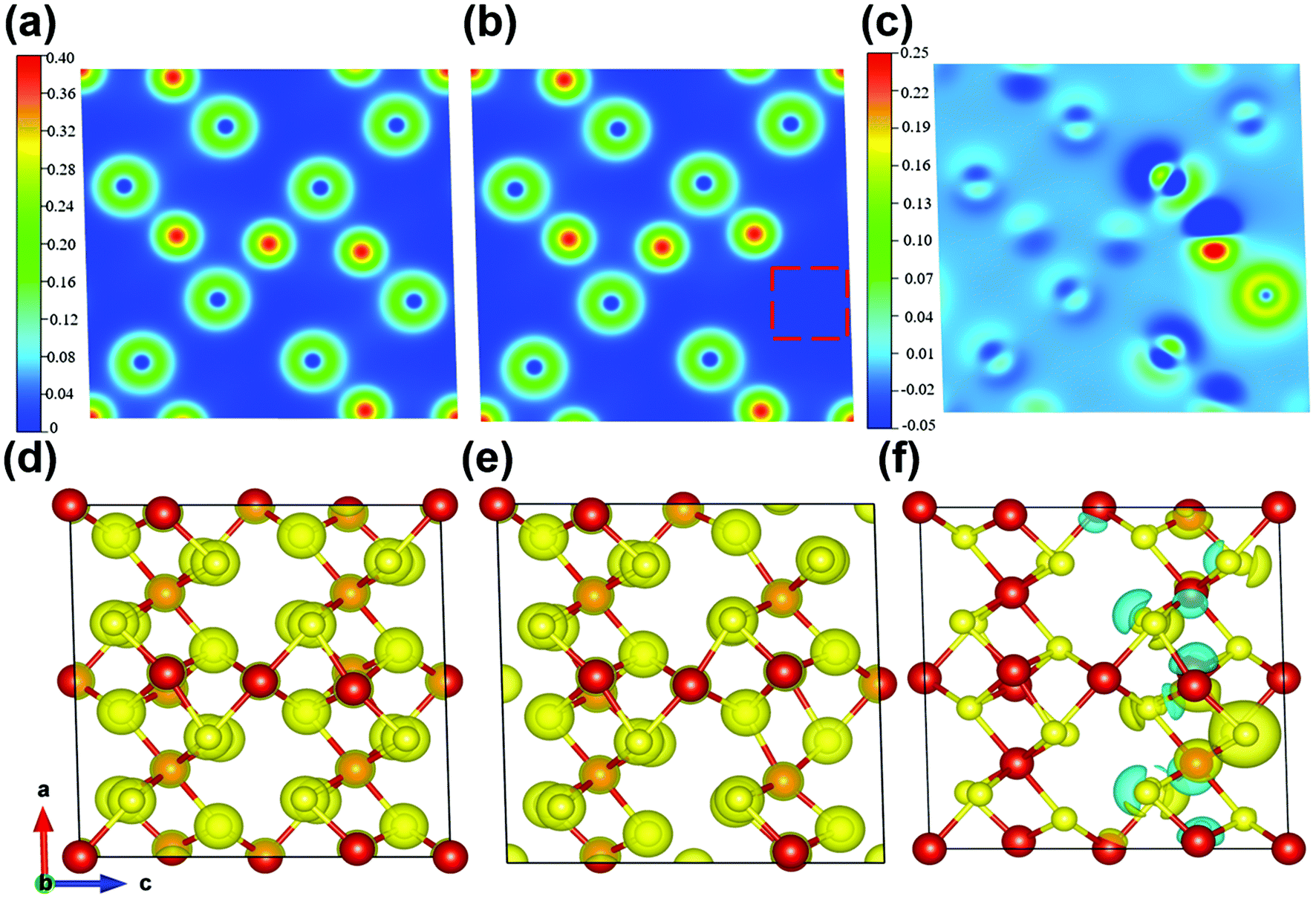 | ||
| Fig. 4 Charge density on the (010) surface of (a) P-V5S8 and (b) D-V5S8, and (c) the charge-density difference between (a) and (b). (d–f) Relaxed structures of the 3D structure view corresponding to (a), (b) and (c), respectively. The isosurface values are set to be 0.14 e per bohr3 and 0.045 e per bohr3 for the charge density and charge-density difference, respectively. For the 2D charge density figures, the blue and red colors represent the charge depletion regions and accumulation regions, respectively. The values of saturation are marked on a scale in figures. | ||
In view of the abovementioned calculation results, it can be concluded that the formation of sulfur defects is favorable to enhance the electronic conductivity by promoting faster transport of electrons. Besides, the intrinsic defect characteristics can further optimize the distribution of electron density and the formation of stronger electron deficiency characteristics, ensuring significant Na-ion storage.
Moreover, the high capacitance and excellent cycling stability properties of the S-defective V5S8/CNF anode composite encourage us to further evaluate its practicability. Specifically, the D-V5S8/CNFs//NVP sodium-ion full cell, which is composed of a D-V5S8/CNF anode and a coupled cathode Na3V2(PO4)3 (NVP), is assembled. The current density and specific capacity of the full battery are calculated based on the mass of the D-V5S8/CNF anode. The working mechanism of the full cell is shown in Fig. 5a. The full cell reveals two flat voltage plateaus at 0.8 and 1.8 V, respectively, consistent with the voltage differences between the NVP//Na half cell (NVP cathodes are assembled in the half cell using Na metal as the anode) at a voltage of about 3.4 V and the D-V5S8/CNF anode at about 2.6 V and 1.6 V (Fig. 5b). Moreover, as shown in Fig. 5c, the full cell presents nearly overlapping CV curves and a similar charge–discharge voltage platform during the charge/discharge processes, which originated from the outstanding electrochemical reversibility and reaction kinetics. Additionally, the electrochemical performance of the full SIB at 500 mA g−1 is shown in Fig. 5d. It exhibits a desirable reversible capacity of 312 mA h g−1 after 70 cycles with a CE close to 95%, showing the excellent reversibility of the SIB full cell. More interestingly, as shown in the inset of Fig. 5d, the as-assembled D-V5S8/CNFs//NVP full battery can power a commercial light-emitting diode (LED, with a working voltage of 2 V) after being charged to 3.35 V. Such an excellent full sodium-ion battery performance makes it promising for application in portable electronics.61
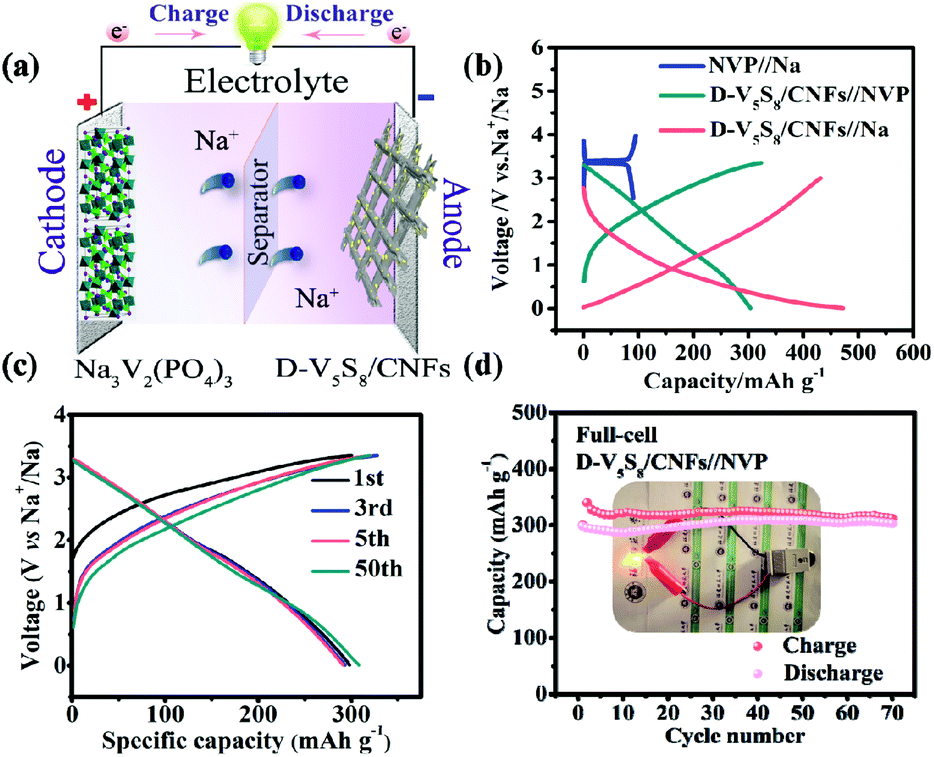 | ||
| Fig. 5 The energy storage performance of a D-V5S8/CNF full cell. (a) Working mechanism of the full cell. (b) Typical charge–discharge curves of the NVP cathode, the D-V5S8/CNF anode and the D-V5S8/CNFs//NVP full cell. (c) Galvanostatic charge/discharge profiles. (d) Cycling performance of the D-V5S8/CNFs/CNFs//NVP full cell at a current density of 500 mA g−1. Inset of (d): an LED lightened by the flexible D-V5S8/CNFs//NVP full battery. | ||
Electrochemical performance in PIBs
The potassium storage performances of S-defective D-V5S8/CNFs and P-V5S8/C are also studied from 0.01 V to 3 V.In Fig. 6a, the CV profiles of D-V5S8/CNFs, recorded at a scan rate of 0.2 mV s−1, exhibit less obvious redox peaks in contrast to those in SIBs. Two poor reduction peaks appear at about 0.8 V and 0.01 V, corresponding to K+ insertion into the materials and subsequently accompanied by the conversion reaction. Specifically, the process could be related to the intercalation of K ions into V5S8 to form different KxV5S8 phases and transform into V finally, according to the literature (V5S8 + K+ + e− ↔ K2S3 + V).17 Furthermore, the observed reduction peak at around 0.65 V disappeared in the subsequent cycles, which is ascribed to the formation of SEI layers during the repeatable charge and discharge processes. From the 2nd scan cycle, it is seen that the CV profiles almost coincided well with each other, indicating the superior reversibility of the electrode.62,63
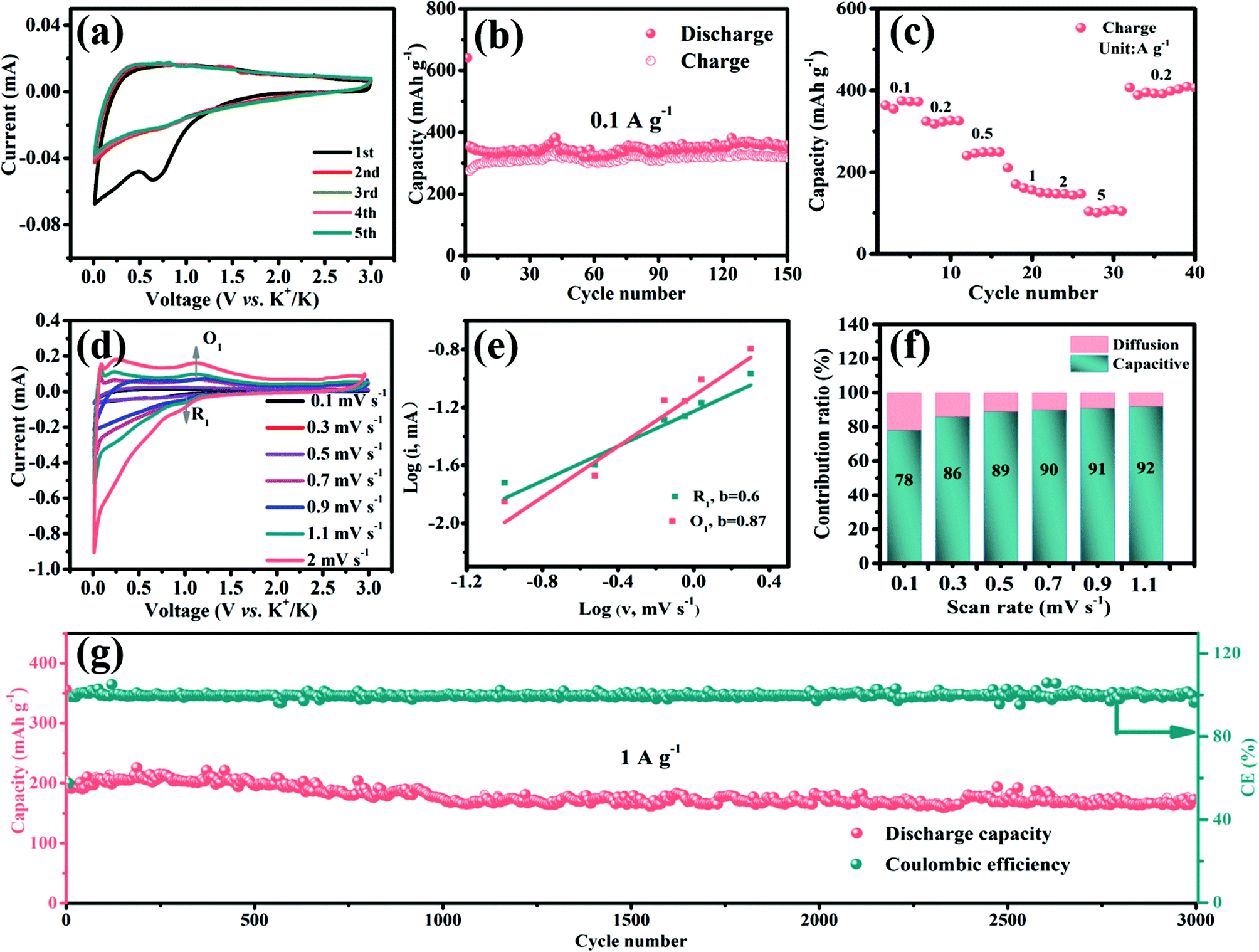 | ||
| Fig. 6 Electrochemical performance of the as-prepared D-V5S8/CNF composite in PIBs. (a) CV curves at a scan rate of 0.2 mV s−1. (b) Cycling performance of the D-V5S8/CNF electrode at a current density of 0.1 A g−1. (c) Rate performance. (d) CV curves at various scan rates from 0.1 to 2 mV s−1. (e) b value determination. (f) Contribution percentages of diffusion (red) and capacitance (blue) at different rates. (g) Long-term cycling performance at a current density of 1 A g−1 of the D-V5S8/CNF electrode. | ||
Then, the cycling performances of D-V5S8/CNFs and P-V5S8/C are also investigated at 0.1 A g−1, as shown in Fig. 6b and Fig. S13 (ESI†). As expected, D-V5S8/CNFs display an initial discharge capacity of 641 mA h g−1 at a current density of 0.1 A g−1, and still sustain a high reversible capacity of 350 mA h g−1 after 150 cycles (Fig. 6b). However, the P-V5S8/C electrode (Fig. S13a†) only shows a reversible capacity of 111 mA h g−1 after 100 cycles under the same conditions.
Besides that, the rate capacities at various current densities from 0.1 A g−1 to 5 A g−1 of the D-V5S8/CNF and P-V5S8/C electrodes are also studied. Benefiting from the advantages in the structure, the reversible charge capacities of D-V5S8/CNFs (Fig. 6c) are 372, 326, 249, 157, 144, and 108 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, and 5 A g−1, respectively, which are superior to those of the P-V5S8/C electrode as shown in Fig. S13b.† Moreover, when the current density is switched back to 0.1 A g−1, a high charge capacity of 406 mA h g−1 can be obtained after 40 cycles, showing the excellent reversibility.
The CV curves of the D-V5S8/CNF electrode at various scan rates from 0.1 mV s−1 to 2.0 mV s−1 are also recorded in the voltage range of 0.01–3.00 V vs. K+/K. Fig. 6d shows the various scan rates of the CV experiments. Correspondingly, the b-values based on the reduction peak (R1) and oxidation peak (O1) are found to be 0.6 and 0.87, respectively, suggesting a mixed capacitive-controlled and diffusion-controlled behavior during the reversible depotassium and potassium processes (Fig. 6e). Furthermore, the capacitive contribution percentage of the electrode gradually increases from 78% (0.1 mV s−1) to 92% (1.1 mV s−1), suggesting a dominant contribution from the pseudocapacitive behavior of the designed D-V5S8/CNF electrode in PIBs.53,64–67
As expected, the obtained D-V5S8/CNFs show a superior long-term cycling performance for potassium storage as well. Remarkably, the D-V5S8/CNF anode can still maintain a specific discharge capacity of 165 mA h g−1, with a negligible attenuation tendency even after prolonged cycling for 3000 cycles at 1 A g−1 and the CE approximately approaches 100% (Fig. 6g). The capability and stability are much higher than those of the P-V5S8/C anode (41 mA h g−1 at 1 A g−1, Fig. S13†). Thus, the resultant S-defective V5S8/CNFs exhibit one of the best electrochemical performances with respect to the reversible capacity and long-term cycling when compared with other sulfide anode materials for PIBs (Table S3†), elucidating their excellent potential as ion storage systems.68
Minimum energy pathway (MEP) and NEB methods are further used to estimate the immigration of intercalated K ions and energy barriers in detail. According to related reports, the K ion diffusion pathways of perfect V5S8 along the b-direction are employed as the optimal migration diffusion pathways.17Fig. 7(a–c) shows the minimum energy pathway in perfect V5S8 and S-defective V5S8. It can be found that the barrier energy is 0.155 eV along the b-direction on S-defective V5S8, which is lower than 0.204 eV on perfect V5S8 (Fig. 7d). The results demonstrate that K shows much faster ion diffusion in D-V5S8 than in P-V5S8 at room temperature. Moreover, the relatively low energy barriers will make the diffusion of K easier and consequently enhance the rate capabilities of the D-V5S8/CNFs.26
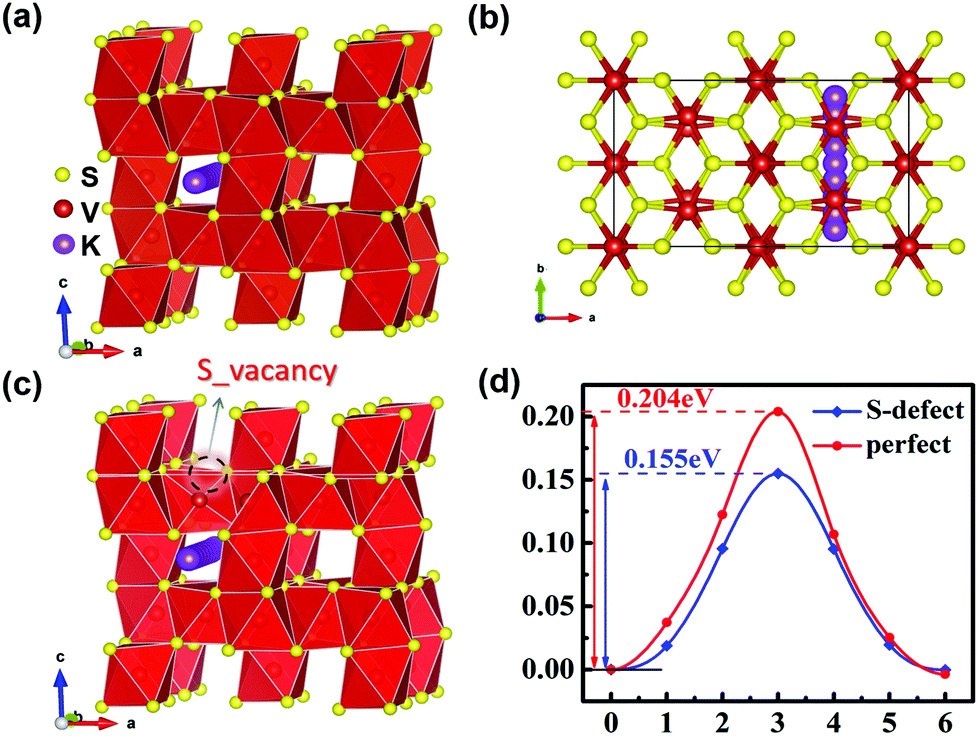 | ||
| Fig. 7 The minimum energy path for the K atom diffusion pathways in (a and b) P-V5S8 and (c) S-defective V5S8.17 (d) The corresponding barrier energy. The yellow, red and purple balls represent the S, V and K atoms, respectively. | ||
Conclusion
A facile and efficient method has been designed and demonstrated to synthesize sulfur-defective vanadium sulfide/carbon fiber composites (D-V5S8/CNFs). The confirmed outstanding electrochemical performance of D-V5S8/CNFs can be attributed to the following facts: (1) the nanosized V5S8 particles generated during the conversion and cycling process can minimize the entropic barrier for the reversible conversion reaction, which leads to high electrochemical utilization and good cycling stability. (2) First-principles calculations also illuminate that the presence of S defects in the structure is important for the reversible adsorption of Na and K ions. Both Na and K reversibly bind to the heteroatom, therefore the S, and the associated defects would have an important influence on sodium and potassium storage, which can provide ample binding sites for Na and K ions. (3) The carbon fibers (CNFs) as a flexible support frame can provide adsorption-type active sites and shorten the diffusion pathway of Na and K ions, accompanied by superior cycling stability. This route reported in this work can and will be extended to other advanced transition metal dichalcogenide (TMD) based compounds, implying a great prospect in the future for energy storage applications.Conflicts of interest
There are no conflicts to declare.Author contributions
Lihong Xu and Xiaochuan Chen have performed the experiments, analyzed the results and written the manuscript. Wenti Guo has performed the analysis of DFT calculations. Lingxing Zeng has come up with the idea, guided the work and corrected the manuscript. Tao Yang has edited the manuscript. Peixun Xiong has participated in the synthesis of materials and resources. Qinghua Chen has guided the work. Jianmin Zhang has performed the analysis of DFT calculations. Mingdeng Wei has edited the manuscript. Qingrong Qian has guided the work and supplied funding acquisition.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC 51502036, 21875037 and 11874113), the National Key R&D Program of China (2019YFC1904500), the New Century Talent Project of Fujian Province and the Natural Science Foundation of Distinguished Young Scholars for Fujian Province (2019J06015).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- J. Xia, L. Liu, S. Jamil, J. Xie, H. Yan, Y. Yuan, Y. Zhang, S. Nie, J. Pan, X. Wang and G. Cao, Energy Storage Mater., 2019, 17, 1–11 CrossRef.
- Q. Xu, J.-K. Sun, J.-Y. Li, Y.-X. Yin and Y.-G. Guo, Energy Storage Mater., 2018, 12, 54–60 CrossRef.
- Q. Zhang, J. Mao, W. K. Pang, T. Zheng, V. Sencadas, Y. Chen, Y. Liu and Z. Guo, Adv. Energy Mater., 2018, 8, 1703288 CrossRef.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- S. Wang, L. Xia, L. Yu, L. Zhang, H. Wang and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- F. Li, Z. Wei, A. Manthiram, Y. Feng, J. Ma and L. Mai, J. Mater. Chem. A, 2019, 7, 9406–9431 RSC.
- S. Guo, J. Yi, Y. Sun and H. Zhou, Energy Environ. Sci., 2016, 9, 2978–3006 RSC.
- T. Wu, M. Jing, Y. Tian, L. Yang, J. Hu, X. Cao, G. Zou, H. Hou and X. Ji, Adv. Funct. Mater., 2019, 29, 1900941 CrossRef.
- Y. L. Bai, Y. S. Liu, C. Ma, K. X. Wang and J. S. Chen, ACS Nano, 2018, 12, 11503–11510 CrossRef CAS.
- C. Yang, J. Feng, F. Lv, J. Zhou, C. Lin, K. Wang, Y. Zhang, Y. Yang, W. Wang, J. Li and S. Guo, Adv. Mater., 2018, 30, 1800036 CrossRef.
- B. Cao, Q. Zhang, H. Liu, B. Xu, S. Zhang, T. Zhou, J. Mao, W. K. Pang, Z. Guo, A. Li, J. Zhou, X. Chen and H. Song, Adv. Energy Mater., 2018, 8, 1801149 CrossRef.
- W. Wang, J. Zhou, Z. Wang, L. Zhao, P. Li, Y. Yang, C. Yang, H. Huang and S. Guo, Adv. Energy Mater., 2018, 8, 1701648 CrossRef.
- Y. Zhang, Q. Zhou, J. Zhu, Q. Yan, S. X. Dou and W. Sun, Adv. Funct. Mater., 2017, 27, 1702317 CrossRef.
- C. Yang, X. Ou, X. Xiong, F. Zheng, R. Hu, Y. Chen, M. Liu and K. Huang, Energy Environ. Sci., 2017, 10, 107–113 RSC.
- L. Li, W. Zhang, X. Wang, S. Zhang, Y. Liu, M. Li, G. Zhu, Y. Zheng, Q. Zhang, T. Zhou, W. K. Pang, W. Luo, Z. Guo and J. Yang, ACS Nano, 2019, 13, 7939–7948 CrossRef CAS.
- R. Sun, Q. Wei, J. Sheng, C. Shi, Q. An, S. Liu and L. Mai, Nano Energy, 2017, 35, 396–404 CrossRef CAS.
- S. Wang, F. Gong, S. Yang, J. Liao, M. Wu, Z. Xu, C. Chen, X. Yang, F. Zhao, B. Wang, Y. Wang and X. Sun, Adv. Funct. Mater., 2018, 28, 1801806 CrossRef.
- J. Lu, F. Lian, Y. Zhang, N. Chen, Y. Li, F. Ding and X. Liu, J. Mater. Chem. A, 2020, 8, 6532–6538 RSC.
- J. Zhang, C. Zhang, Z. Wang, J. Zhu, Z. Wen, X. Zhao, X. Zhang, J. Xu and Z. Lu, Small, 2018, 14, 1703098 CrossRef.
- Y. Xu, F. Bahmani, M. Zhou, Y. Li, C. Zhang, F. Liang, S. H. Kazemi, U. Kaiser, G. Meng and Y. Lei, Nanoscale Horiz., 2019, 4, 202–207 RSC.
- T. Jin, H. Li, Y. Li, L. Jiao and J. Chen, Nano Energy, 2018, 50, 462–467 CrossRef CAS.
- W. Li, J. Huang, L. Feng, L. Cao, Y. Feng, H. Wang, J. Li and C. Yao, J. Mater. Chem. A, 2017, 5, 20217–20227 RSC.
- J. Ding, H. Zhang, H. Zhou, J. Feng, X. Zheng, C. Zhong, E. Paek, W. Hu and D. Mitlin, Adv. Mater., 2019, 31, 1900429 CrossRef.
- Y. Liu, Z. Sun, X. Sun, Y. Lin, K. Tan, J. Sun, L. Liang, L. Hou and C. Yuan, Angew. Chem., Int. Ed., 2020, 59, 2473–2482 CrossRef CAS.
- R. Sun, Q. Wei, Q. Li, W. Luo, Q. An, J. Sheng, D. Wang, W. Chen and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 20902–20908 CrossRef CAS.
- W. Yang, L. Dong, W. Yang, C. Xu, G. Shao and G. Wang, Small Methods, 2019, 4, 1900670 CrossRef.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao, Y. Li and Y. Li, Adv. Mater., 2017, 29, 1702061 CrossRef.
- D. Yu, Q. Pang, Y. Gao, Y. Wei, C. Wang, G. Chen and F. Du, Energy Storage Mater., 2018, 11, 1–7 CrossRef CAS.
- J. Wang, N. Luo, J. Wu, S. Huang, L. Yu and M. Wei, J. Mater. Chem. A, 2019, 7, 3691–3696 RSC.
- C. Lee, B. G. Jeong, S. J. Yun, Y. H. Lee, S. M. Lee and M. S. Jeong, ACS Nano, 2018, 12, 9982–9990 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 364 CrossRef CAS.
- M. K. Jaiswal, J. K. Carrow, J. L. Gentry, J. Gupta, N. Altangerel, M. Scully and A. K. Gaharwar, Adv. Mater., 2017, 29, 1702037 CrossRef.
- L. Xu, P. Xiong, L. Zeng, Y. Fang, R. Liu, J. Liu, F. Luo, Q. Chen, M. Wei and Q. Qian, Nanoscale, 2019, 11, 16308–16316 RSC.
- S. Jiao, Z. Yao, F. Xue, Y. Lu, M. Liu, H. Deng, X. Ma, Z. Liu, C. Ma, H. Huang, S. Ruan and Y.-J. Zeng, Appl. Catal., B, 2019, 258, 117964 CrossRef CAS.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao and T. Hu, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS.
- D. Wu, W. Zhang, Y. Feng and J. Ma, J. Mater. Chem. A, 2020, 8, 2618–2626 RSC.
- W. Li, J. Huang, L. Feng, L. Cao, Y. Liu and L. Pan, J. Power Sources, 2018, 398, 91–98 CrossRef CAS.
- Q. Liu, W. Yao, L. Zhan, Y. Wang and Y.-A. Zhu, Electrochim. Acta, 2018, 261, 35–41 CrossRef CAS.
- X. Ou, X. Liang, F. Zheng, Q. Pan, J. Zhou, X. Xiong, C. Yang, R. Hu and M. Liu, Chem. Eng. J., 2017, 320, 485–493 CrossRef CAS.
- L. Zeng, X. Chen, R. Liu, L. Lin, C. Zheng, L. Xu, F. Luo, Q. Qian, Q. Chen and M. Wei, J. Mater. Chem. A, 2017, 5, 22997–23005 RSC.
- P. Xiong, P. Bai, A. Li, B. Li, M. Cheng, Y. Chen, S. Huang, Q. Jiang, X. H. Bu and Y. Xu, Adv. Mater., 2019, 31, 1904771 CrossRef CAS.
- D. Xu, H. Wang, R. Qiu, Q. Wang, Z. Mao, Y. Jiang, R. Wang, B. He, Y. Gong, D. Li and X. Hu, Energy Storage Mater., 2020, 28, 91–100 CrossRef.
- Q. Deng, F. Chen, S. Liu, A. Bayaguud, Y. Feng, Z. Zhang, Y. Fu, Y. Yu and C. Zhu, Adv. Funct. Mater., 2020, 30, 1908665 CrossRef CAS.
- G. D. Park, J. H. Kim, S. K. Park and Y. C. Kang, ACS Appl. Mater. Interfaces, 2017, 9, 10673–10683 CrossRef CAS.
- P. Lu, X. Wang, L. Wen, X. Jiang, W. Guo, L. Wang, X. Yan, F. Hou, J. Liang, H. M. Cheng and S. X. Dou, Small, 2019, 15, 1805064 CrossRef.
- A. Mahmood, S. Li, Z. Ali, H. Tabassum, B. Zhu, Z. Liang, W. Meng, W. Aftab, W. Guo, H. Zhang, M. Yousaf, S. Gao, R. Zou and Y. Zhao, Adv. Mater., 2019, 31, 201805430 Search PubMed.
- T. Liu, X. Zhang, M. Xia, H. Yu, N. Peng, C. Jiang, M. Shui, Y. Xie, T.-F. Yi and J. Shu, Nano Energy, 2020, 67, 104295 CrossRef CAS.
- T. Liu, N. Peng, X. Zhang, R. Zheng, M. Xia, H. Yu, M. Shui, Y. Xie and J. Shu, Nano Energy, 2021, 79, 105460 CrossRef CAS.
- Y. Wang, Y. Zhang, J. Shi, X. Kong, X. Cao, S. Liang, G. Cao and A. Pan, Energy Storage Mater., 2018, 18, 366–374 CrossRef.
- F. Luo, X. Feng, L. Zeng, L. Lin, X. Li, B. Kang, L. Xiao, Q. Chen, M. Wei and Q. Qian, Chem. Eng. J., 2021, 404, 126430 CrossRef CAS.
- Y. Liu, Y. Xiao, F. Liu, P. Han and G. Qin, J. Mater. Chem. A, 2019, 7, 26818–26828 RSC.
- G. Xia, C. Wang, P. Jiang, J. Lu, J. Diao and Q. Chen, J. Mater. Chem. A, 2019, 7, 12317–12324 RSC.
- L. Wang, J. Sun, R. Song, S. Yang and H. Song, Adv. Energy Mater., 2016, 6, 1502067 CrossRef.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- S. Niu, Z. Wang, T. Zhou, M. Yu, M. Yu and J. Qiu, Adv. Funct. Mater., 2017, 27, 1605332 CrossRef.
- W. Xu, C. Lv, Y. Zou, J. Ren, X. She, Y. Zhu, Y. Zhang, S. Chen, X. Yang, T. Zhan, J. Sun and D. Yang, J. Power Sources, 2019, 442, 227184 CrossRef CAS.
- R. Guo, C. Lv, W. Xu, J. Sun, Y. Zhu, X. Yang, J. Li, J. Sun, L. Zhang and D. Yang, Adv. Energy Mater., 2020, 10, 1903652 CrossRef CAS.
- H. He, D. Huang, Q. Gan, J. Hao, S. Liu, Z. Wu, W. K. Pang, B. Johannessen, Y. Tang, J. L. Luo, H. Wang and Z. Guo, ACS Nano, 2019, 13, 11843–11852 CrossRef CAS.
- Q. Zhang, H. Chen, L. Luo, B. Zhao, H. Luo, X. Han, J. Wang, C. Wang, Y. Yang, T. Zhu and M. Liu, Energy Environ. Sci., 2018, 11, 669–681 RSC.
- Y. S. Xu, S. Y. Duan, Y. G. Sun, D. S. Bin, X. S. Tao, D. Zhang, Y. Liu, A. M. Cao and L. J. Wan, J. Mater. Chem. A, 2019, 7, 4334–4352 RSC.
- Y. An, Y. Tian, Y. Li, S. Xiong, G. Zhao, J. Feng and Y. Qian, J. Mater. Chem. A, 2019, 7, 21966–21975 RSC.
- M. S. Balogun, W. Qiu, J. Jian, Y. Huang, Y. Luo, H. Yang, C. Liang, X. Lu and Y. Tong, ACS Appl. Mater. Interfaces, 2015, 7, 23205–23215 CrossRef CAS.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 RSC.
- L. Zeng, F. Luo, X. Xia, M. Q. Yang, L. Xu, J. Wang, X. Feng, Q. Qian, M. Wei and Q. Chen, Chem. Commun., 2019, 55, 3614–3617 RSC.
- L. Zeng, Y. Fang, L. Xu, C. Zheng, M. Yang, J. He, H. Xue, Q. Chen, M. Wei and Q. Qian, Nanoscale, 2019, 11, 6766–6775 RSC.
- B. Kang, X. Chen, L. Zeng, F. Luo, X. Li, L. Xu, M. Yang, Q. Chen, M. Wei and Q. Qian, J. Colloid Interf. Sci., 2020, 574, 217–228 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08788b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |