
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA–π–A, D–π–D and D–π–A blue emitting fluorophores based on dispiro[fluorene-9,6′-indeno[1,2-b]fluorene-12′,9′′-fluorene]†
Damien
Thirion
 ,
Maxime
Romain
,
Joëlle
Rault-Berthelot
* and
Cyril
Poriel
*
,
Maxime
Romain
,
Joëlle
Rault-Berthelot
* and
Cyril
Poriel
*
Univ Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: Cyril.poriel@univ-rennes1.fr; joelle.rault-berthelot@univ-rennes1.fr
First published on 28th December 2020
Abstract
We report a series of blue fluorophores based on the dispiro[fluorene-9,6′-indeno[1,2-b]fluorene-12′,9′′-fluorene] (DSF-IF) scaffold. The DSF-IF scaffold, introduced in 2006 in the literature for organic electronic applications, is nowadays a very useful building block to construct functional materials. However, the functionalization of the DSF-IF scaffold has been poorly investigated to date. In this work, we focus on three different molecular architectures incorporating at each extremity of the central dihydroindenofluorenyl core either an electron accepting unit (A–π–A), an electron donating unit (D–π–D) or one of each (D–π–A). The different substitutions of the dihydroindeno[1,2-b]fluorenyl core has allowed the strong modulation of the properties of the dyes and the precise study of the effect of this fragment as a π-conjugated bridge.
Introduction
Designing, synthesizing and studying new molecular building blocks have been the pillars of the fantastic development of organic electronics over the last thirty years.1 Year after year, the basic fragments such as fluorene,2–5 spirobifluorene6–9 or carbazole10,11 have allowed the development of more sophisticated and more efficient molecular fragments. Nowadays, these fragments continue to draw real attention and have even been recently incorporated in a new generation of π-conjugated systems, i.e. nanorings.12–14The dispiro[fluorene-9,6′-indeno[1,2-b]fluorene-12′,9′′-fluorene] scaffold (DSF-IF) fragment has been particularly studied in the last ten years.15 DSF-IF is constructed by the assembly of a dihydroindeno[1,2-b]fluorene backbone5,15–22 (bridged terphenyl unit) flanked by two spiro linked fluorene units. This scaffold, introduced in 2006,23 displays many interesting properties (high thermal stability, high quantum yield etc.)24,25 and has been advantageously used in the design of highly efficient materials for OLEDs (violet to blue emitters24,26–30 and high triplet host materials for phosphors31) and for solar cells (as hole transporting materials in perovskite solar cells).32 DSF-IF is also found as a building block in fused extended π-conjugated systems.33–36 From an organic chemistry point of view, the formation of the two spiro bridges of substituted DSF-IF compounds has also been the purpose of several synthetic studies, which have led to a new generation of DSF-IF positional isomers.15,37,38 In addition, this scaffold is not restricted to organic electronic applications since its appealing 3D geometry has allowed the design of predictably ordered materials,39 liquid crystals,40 and electroactive polymers,24,28 showing its interesting versatility. However, despite the growing attention paid to this fragment, functionalization of the DSF-IF fragment (and more generally of the dihydroindenofluorenyl backbone) has only been poorly investigated to date30,41–47 mainly due to synthetic issues. This remains a drawback in DSF-IF chemistry. Indeed, the functionalization of a molecular scaffold, i.e. the incorporation of electron-donating and/or electron-accepting groups, is a widely known chemical strategy in electronics to obtain high efficiency materials. In this work, we report on three different molecular architectures incorporating at each extremity of the central dihydroindenofluorenyl core either an electron accepting unit, namely 1,2-diphenyl-1H-benzo[d]imidazole (A–π–A design-AA-DSF-IF), an electron donating unit, namely diphenylamine (D–π–D design-DD-DSF-IF) or one of each (D–π–A design-DA1-DSF-IF and DA2-DSF-IF). Note that DA2-DSF-IF has been previously reported in 2011 for its fluorescence properties in single-layer OLEDs.45
These different substitutions of the central dihydroindeno[1,2-b]fluorenyl core have allowed the strong modulation of the properties of the DSF-IF scaffold and the study of the effect of this fragment as a π-conjugated bridge. After consideration of their synthetic features, the 1H NMR, and the electrochemical and photophysical properties of the resulting four blue fluorophores are reported through a combined experimental and theoretical approach. Application as an emissive layer within non-doped blue OLEDs is finally reported. The present work provides a structure–property relationship study of the functionalization of the DSF-IF scaffold. As the performances of optoelectronic organic devices are directly linked to the properties of the active organic layer, such a type of study on barely studied building blocks may help to design, in the future, highly efficient and multifunctional materials.
Results and discussion
Synthesis
The four fluorophores investigated (AA-DSF-IF, DD-DSF-IF DA1-DSF-IF and DA2-DSF-IF) are presented in Scheme 1. They possess either two phenylbenzimidazolyl groups (AA-DSF-IF), two dimethoxyamino units (DD-DSF-IF) or one phenylbenzimidazolyl and either one diphenylamino or one dimethoxyphenylamino unit (DA1-DSF-IF and DA2-DSF-IF, respectively). These fragments are widely known in the field of organic electronics, particularly for the design of host materials for PhOLEDs48–50 or blue fluorophores for OLEDs51,52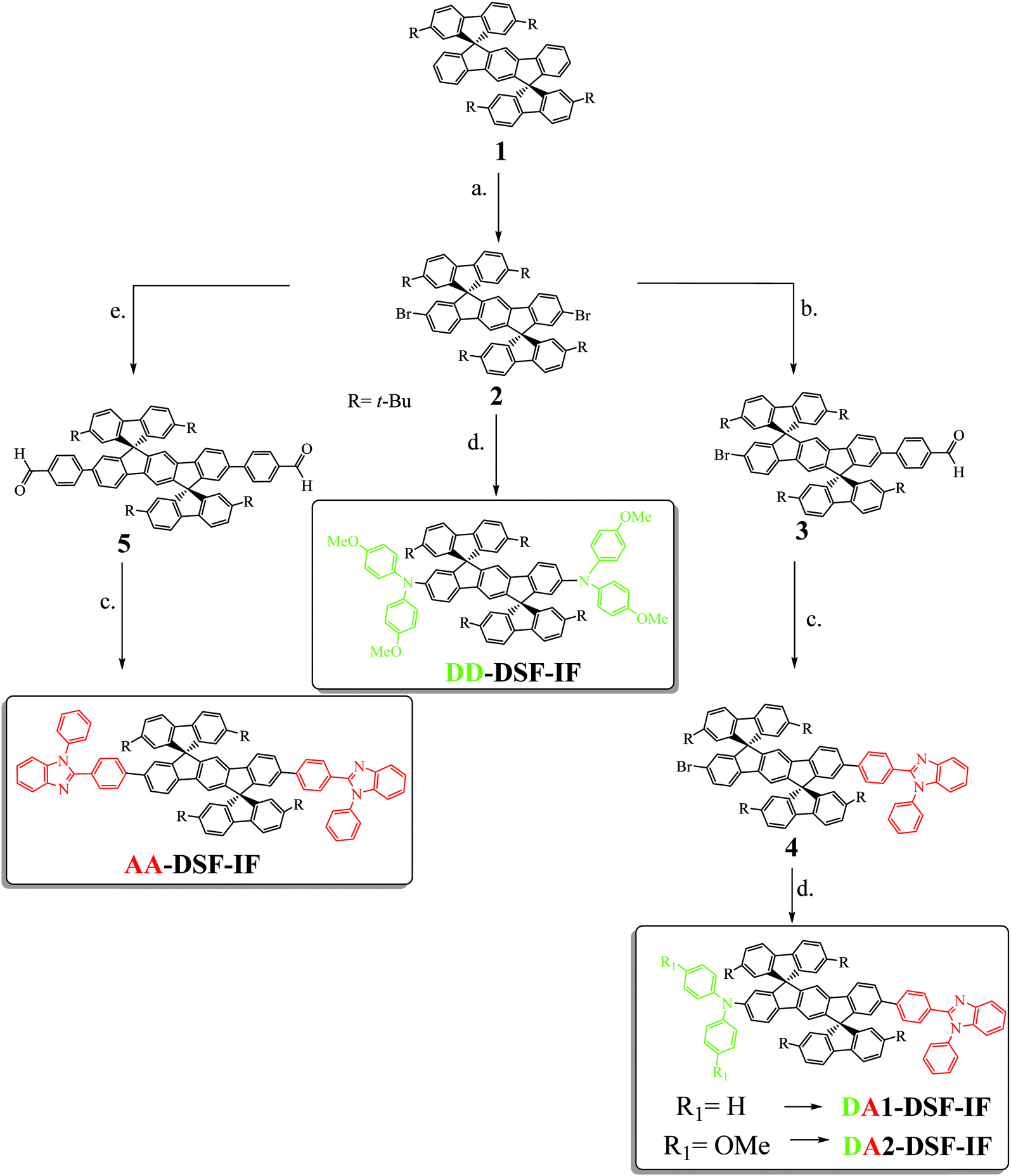 | ||
| Scheme 1 Synthesis of DA1-, DA2-, AA- and DD-DSF-IF. (a) Br2, Na2CO3, I2 (catalytic amount), CH2Cl2/Water, rt, (90%); (b) Pd2(dba)3/P(t-Bu)3, Na2CO3, 4-formylbenzeneboronic acid (1.4 eq. vs.2), toluene/water (6/1), 100 °C, (30%); (c) methoxyethanol, N-phenyl-o-phenylenediamine, 120 °C (70% for AA-DSF-IF and 65% for 4); (d) Pd(OAc)2, P(t-Bu)3, t-BuOK, diphenylamine for DA1-DSF-IF or 4,4′-dimethoxydiphenylamine for DA2-DSF-IF, toluene, 100 °C (<10% for DA1-DSF-IF, 50% for DA2-DSF-IF and 50% for DD-DSF-IF); (e) Pd2(dba)3, P(t-Bu)3, Na2CO3, 4-formylbenzeneboronic acid (2 eq. vs.2), toluene/water, 100 °C, (85%). | ||
The synthesis of the four targeted molecules (Scheme 1 and Scheme SI, ESI†) starts with the synthesis of the key DiSpiroFluorene-IndenoFluorene scaffold (1), prepared from 2,7-di-tert-butyl-fluoren-9-one and 2,2′′-diiodoterphenyl as previously reported.53 In addition, 1 will be used in this study as a relevant model compound to DA1-DSF-IF, DA2-DSF-IF, AA-DSF-IF, DD-DSF-IF, in order to study the influence of the incorporation of the electron donating and/or electron accepting units on the properties of the dihydroindenofluorenyl moiety.
With 1 in hand, the next step was to introduce the bromine atoms on the dihydroindenofluorenyl core (step a. Scheme 1), prior to the Pd-catalyzed cross-coupling reactions. Since the 2,7-positions of the fluorenyl moieties of 1 had been protected and as electrophilic bromination of dihydroindenofluorene derivatives had been previously reported in the literature,54–56 we believed that the introduction of bromine atoms would be an easy task. However, it turned out that the different reported conditions only led to very poor yields of 2 due to either over-bromination reactions on the position C4 of the fluorene units or no bromination, highlighting the different reactivities of the spirofluorene-bridged dihydroindenofluorenyl unit compared to that of the alkyl-bridged dihydroindenofluorenyl unit previously reported in the literature.54,56 We thus hypothesized that the electron-withdrawing character of the spirolinked fluorene units found in 1 deactivates the dihydroindenofluorenyl core towards such electrophilic bromination. After intensive scouting, we finally found that a mixture of bromine and sodium carbonate and a catalytic amount of iodine cleanly lead to the dibromination of the dihydroindenofluorenyl core with 90% yield.
The key desymmetrization of 2 was then carried out through a selective Pd-catalyzed Suzuki–Miyaura cross-coupling reaction between 2 and 4-formylbenzeneboronic acid. The best conditions found to synthesize 3 (30% yield)‡ was to use Pd2(dba)3/P(t-Bu)3 as the catalytic system,57 and sodium carbonate as the base in a mixture of toluene and water (6/1) at 100 °C. It should be mentioned that decreasing the temperature of the reaction or switching to other Pd catalysts (Pd(dppf)Cl2, Pd(PPh3)4 or Pd(OAc)2) only leads to lower yields, highlighting the very poor reactivity of the bromine atoms attached to the dihydroindenofluorene core. The benzimidazolyl group was then easily constructed through the condensation of 3 with N-phenyl-o-phenylenediamine in methoxyethanol leading to 4 with 70% yield. The Hartwig Pd-catalyzed C–N coupling reaction was finally adopted to introduce either the diphenylamine or the dimethoxydiphenylamine units providing DA1-DSF-IF and DA2-DSF-IF with overall yields of 10%§ and 50%, respectively.
A similar synthetic strategy has been carried out to obtain the symmetric compounds AA-DSF-IF and DD-DSF-IF. AA-DSF-IF is obtained in two steps from compound 2. The Pd-catalyzed cross-coupling reaction between 2 and 4-formylbenzeneboronic first readily provides bisphenylaldehyde 5 with high yields (85%). The benzimidazolyl fragments are then constructed through the condensation of 5 with N-phenyl-o-phenylenediamine in methoxyethanol leading to AA-DSF-IF with 70% yield. Similarly, DD-DSF-IF is directly obtained in one step from 2 by the above described Hartwig Pd-catalyzed C–N coupling reaction between 2 and dimethoxydiphenylamine (50% yield). These routes are efficient to symmetrically functionalize the DSF-IF platform.
1H NMR spectroscopy studies
Detailed 1H NMR characterization of all compounds has been performed in order to study the influence of the attachment of a donor and an acceptor group (and their respective electron withdrawing/donating strength) on the dihydroindenofluorenyl core. In assigning 1H NMR spectra of all the molecules, the following numbering is used, in which H1–H4 and H1′–H4′ belong to the fluorene moieties and H5–H9 and H5′–H9′ belong to the dihydroindenofluorenyl unit. Complete assignments have been performed by 2D NMR spectroscopy experiments: HMBC (Heteronuclear Multiple Bond Correlation), HMQC (Heteronuclear Multiple Quantum Coherence) and 1H/1H COSY (Correlation Spectroscopy).As DA1-DSF-IF and DA2-DSF-IF display similar behaviour, DA2-DSF-IF will be used as a representative example.
Compared to their constitutive building block 1, the hydrogen atoms of the fluorene units H1–H4 of the functionalized molecules, DA2-DSF-IF, AA-DSF-IF and DD-DSF-IF, present almost identical chemical shifts. Indeed, due to the spiro-bridges, the hydrogen atoms of the fluorene units H1–H4 are only weakly affected by the shielding/deshielding effects of the arylamine and phenylbenzimidazolyl groups. H1 being the closest to the spiro-bridge, it is more influenced by the substitution of the dihydroindenofluorenyl core. The chemical shifts of the hydrogen atoms H5–H9 of the dihydroindenofluorenyl core are obviously more affected by the donor/acceptor substitution than those of the fluorenyl units. Thus, due to the withdrawing effect of the benzimidazolyl group, an intense deshielding of H5 and H7 is observed for AA-DSF-IF, respectively, by 0.23 ppm and 0.27 ppm compared to 1. These deshielding effects then decrease from H5 to H9 as the distance to the benzimidazolyl group increases leading, compared to 1, to almost identical chemical shifts for H8 and H9 (Table 1). These features highlight the moderate acceptor effect of the phenylbenzimidazolyl group. In the case of DD-DSF-IF, the electronic influence of the substitution is stronger than that observed in AA-DSF-IF. Indeed, in DD-DSF-IF, the signals of H5 and H7 in the α-position of the amine-substituted carbon are shielded by 0.37 ppm (H5) and 0.51 ppm (H7) compared to those of 1. In contrast to AA-DSF-IF, the shielding induced by the arylamines also affects the more distant hydrogens H8 and H9, respectively, by 0.25 ppm and 0.23 ppm. Thus, the strength of the electron donating/withdrawing character of the diarylamine/phenylbenzimidazole group can be hence correlated to the resulting shielding/deshielding effects of the hydrogen atoms of the dihydroindenofluorenyl backbone. The case of DA2-DSF-IF is more complicated to unravel due to the dissymmetry and hence to the splitting of the whole set of signals. On each side of the dihydroindenofluorenyl core, the shielding and deshielding effects induced by the amine and by the phenylbenzimidazole on the hydrogen atoms H5′–H8′ and H5–H8 in DA2-DSF-IF are similar to those observed in the symmetrical molecules AA-DSF-IF and DD-DSF-IF. Note that the chemical shift of H5 perfectly reflects the different strengths of the two arylamine units (with or without methoxy groups). Indeed, both signals are shielded compared to non-substituted 1, with H5 in DA2-DSF-IF being more shielded than its homologue in DA1-DSF-IF (δH5 = 6.42 ppm for DA1-DSF-IF, δH5 = 6.30 ppm for DA2-DSF-IF and δH5 = 6.65 ppm for 1). The central hydrogen atoms H9 and H9′ are simultaneously affected by both effects and present therefore intermediate chemical shifts between those of AA-DSF-IF and DD-DSF-IF. However, H9 is clearly more influenced by the amine than by the benzimidazole unit. Indeed, the chemical shift of H9 in both 1 and AA-DSF-IF is almost identical (7.21 vs 7.22 ppm) but appears strongly shielded in DA2-DSF-IF (7.04 ppm, one pendant amine) and even more shielded in DD-DSF-IF (6.98 ppm, two pendant amines). This highlights the stronger electron-donating effect of the amine on the dihydroindenofluorenyl core compared to the electron-withdrawing effect of the phenylbenzimidazolyl fragment.
|
|
||||
|---|---|---|---|---|
| 1 | DA2-DSF-IF | AA-DSF-IF | DD-DSF-IF | |
| a Overlapped with another singal. | ||||
| H1 | 6.74 | 6.82a | 6.77 | 6.81a |
| H1′ | 6.74 | 6.76a | 6.77 | 6.81a |
| H3 | 7.45 | 7.45a | 7.41 | |
| H3′ | 7.45 | 7.45a | 7.41 | |
| H4 | 7.81 | 7.82 | 7.82 | 7.71 |
| H4′ | 7.81 | 7.82 | 7.82 | 7.71 |
| H5 | 6.65 | 6.30 | 6.88 | 6.28 |
| H5′ | 6.65 | 6.86a | 6.88 | 6.28 |
| H7 | 7.26 | 7.53 | 6.75a | |
| H7′ | 7.26 | 7.50 | 7.53 | 6.75a |
| H8 | 7.60 | 7.66 | 7.35 | |
| H8′ | 7.60 | 7.62 | 7.66 | 7.35 |
| H9 | 7.21 | 7.04 | 7.22 | 6.98 |
| H9′ | 7.21 | 7.14 | 7.22 | 6.98 |
Electrochemical properties
The electrochemical behaviour of AA-, DD- and DA2-DSF-IF has been investigated using cyclic voltammetry (CV) and is summarized in Table 2. As for NMR studies, the behaviour of DA2-DSF-IF is used as a representative example of the D–π–A design.| E ox (V) | E red (V) | E oxonset (V) | E redonset (V)b | HOMO (eV) | LUMO (eV) | ΔEEl (eV) | |
|---|---|---|---|---|---|---|---|
| a Reduction not detected in THF. b E redonset obtained from the onset reduction potential recorded in CH2Cl2. | |||||||
| 1 28,53 | 1.33, 1.61, 1.79, 2.03 | −2.6 | 1.21 | −2.47 | −5.61 | −1.93 | 3.68 |
| AA-DSF-IF | 1.26, 1.56, 1.63, 1.78, 1.89 | −2.20, −2.50 | 1.15 | −1.96 | −5.55 | −2.44 | 3.11 |
| DD-DSF-IF | 0.51, 0.67, 1.34, 1.42, 1.62 | Not detecteda | 0.41 | −2.38b | −4.81 | −2.02 | 2.79 |
| DA2-DSF-IF | 0.57, 1.22, 1.61, 1.89, 2.14 | −2.20, −2.39 | 0.47 | −2.05 | −4.87 | −2.35 | 2.52 |
It has been previously reported29 that 1 is oxidized along three successive one-electron oxidations with maxima at 1.33, 1.61 and 1.79 V leading to a stable tri-radical cation with one radical cation on each π-system. A third bielectronic oxidation process is observed for 1 at a potential higher than 2 V leading to 15+. whose high reactivity allows carbon–carbon coupling between indenofluorenyl units inducing a weak electrodeposition process (see additional CVs and DPV in the ESI†).
Oxidation of AA-DSF-IF occurs also along three successive one-electron oxidation processes with maxima at 1.26, 1.56 and 1.63 V followed by a multielectronic wave peaking at 1.9 V (Fig. 1, top right). There is a 50 mV shift between the first oxidation of AA-DSF-IF and that of 1 (1.26 V vs. 1.31 V, respectively). Indeed, compared to 1, two different electronic effects should be considered in AA-DSF-IF: (i) the extension of the π-conjugation induced by the presence of two additional phenyl units at C3 and C8 of the central dihydroindenofluorenyl core, which should decrease its first oxidation potential and (ii) the electron-withdrawing effects of the two benzimidazolyl units, which should lead to an increase of the first oxidation potential. One can hence conclude that the effect of the π-conjugation extension in AA-DSF-IF (the fragment to consider is the 3,8-biphenyl-dihydroindenofluorene) is not totally erased by the withdrawing effect of the two benzimidazolyl units.
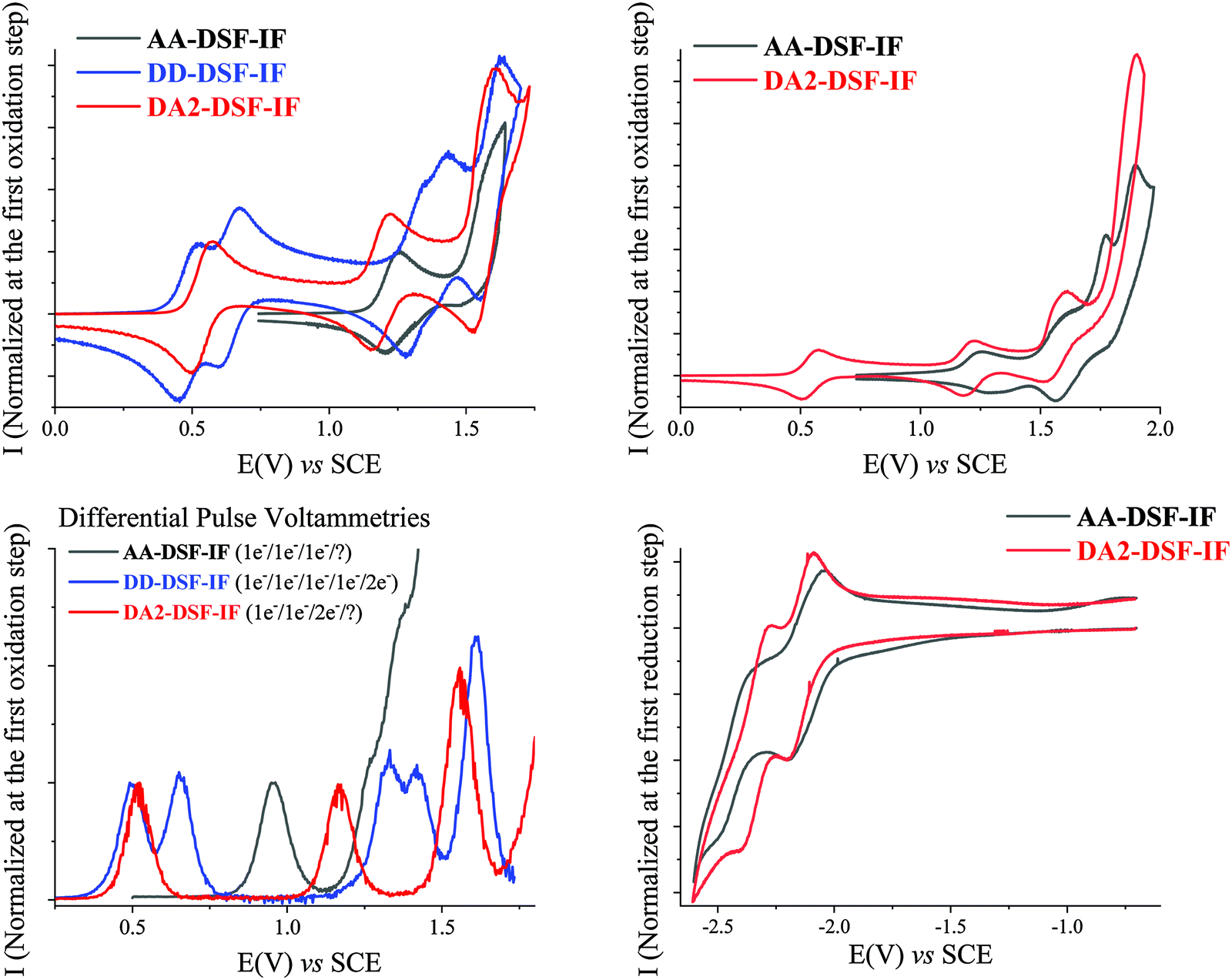 | ||
| Fig. 1 Electrochemical studies of AA-DSF-IF (black), DD-DSF-IF (blue) and DA2-DSF-IF (red). Cyclic voltammetries recorded in Bu4NPF6 0.2 M in CH2Cl2 (top) and in THF (bottom right). Working platinum disk electrode. Sweep-rate 100 mV s−1. Differential pulse voltammetry recorded in Bu4NPF6 0.2 M in CH2Cl2 (Bottom Left). Working platinum disk electrode. Pulse height: 50 mV, Scan-rate: 10 mV s−1. | ||
Oxidation of DD-DSF-IF shows a drastically different behaviour with two first reversible oxidation waves recorded at 0.5 and 0.67 V followed by three reversible waves with maxima at 1.35, 1.42 and 1.62 V (Fig. 1, top left). Compared to 1, we note that the two first oxidations occur at much lower potentials (0.5/0.67 V vs. 1.31 V) clearly indicating that the electron transfers do not occur on the same fragment (see theoretical calculation in Fig. 2 for the nature of the orbitals involved). The two first electron transfers in DD-DSF-IF are mainly centred on the diarylamine units, whereas the other three are involved in the central DSF-IF core. The first four oxidations are monoelectronic, whereas the fifth is bielectronic as pointed out by DPVs (Fig. 1, bottom left). Remarkably, the DD-DSF-IF fifth oxidation leads to a highly charged (6+) and stable species (each successive oxidation step is reversible, see Fig. 1, top left) with almost certainly a dicationic charge on the central dihydroindenofluorenyl core and on the two fluorenyl units. Highly stable charged species have been previously observed for other DSF-IF derivatives with up to five successive electron oxidations.28DD-DSF-IF is even more charged with a 6+ charge on the whole molecule.
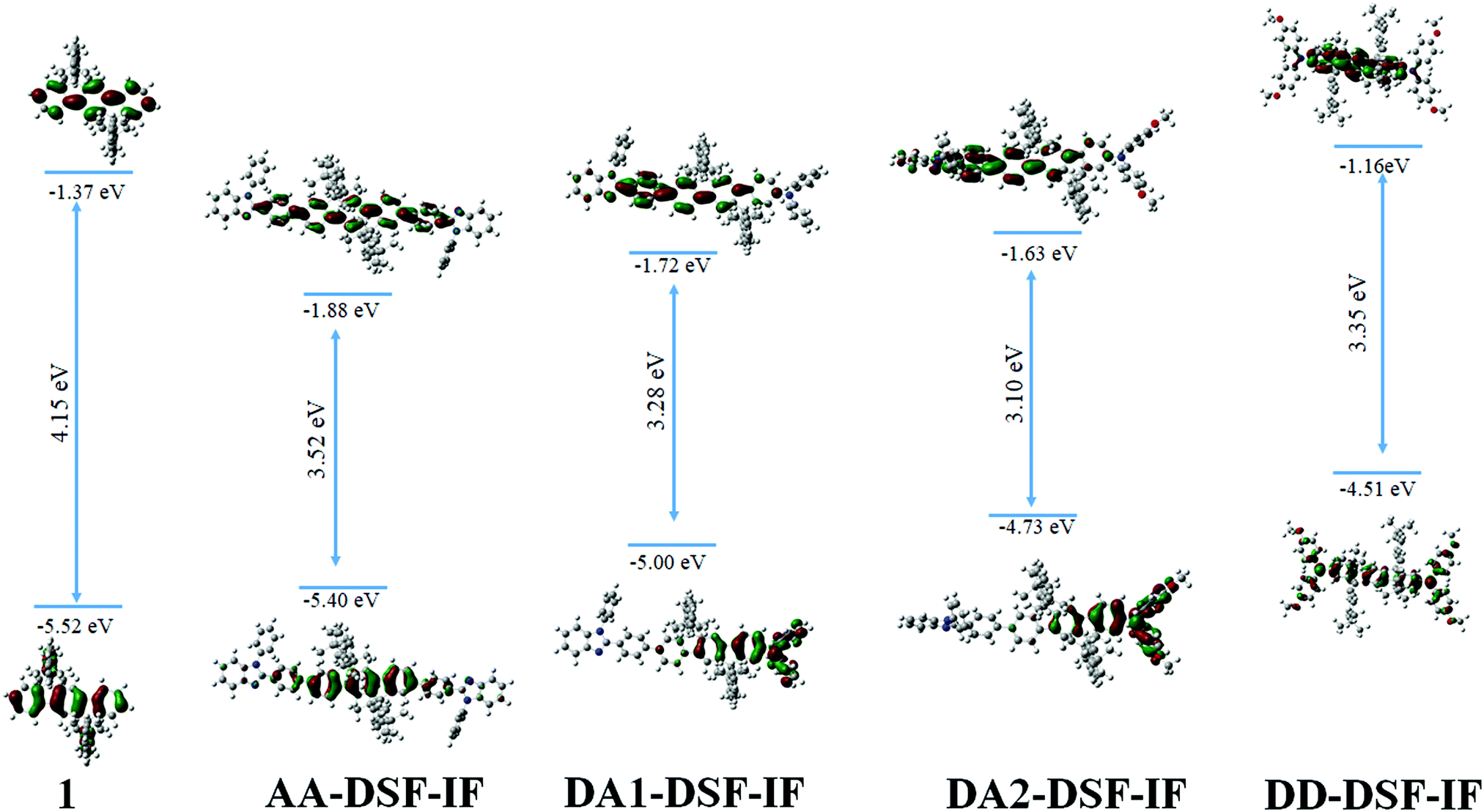 | ||
| Fig. 2 Representation of frontier molecular orbitals, HOMO–LUMO difference and electron density contour obtained from DFT calculations for 1, AA-DSF-IF, DA1-DSF-IF, DA2-DSF-IF and DD-DSF-IF (isovalue: 0.03 [e bohr−3]1/2). | ||
CV recorded along DA2-DSF-IF oxidation presents three successive waves with maxima at 0.57, 1.2 V and 1.61 V (Fig. 1, top left) followed by a fourth oxidation with a maximum at 1.89 V (Fig. 1, top right). The two first oxidations are monoelectronic, whereas the third is bielectronic as pointed out by DPVs (Fig. 1, bottom left). The first oxidation of DA2-DSF-IF (0.57 V) is anodically shifted compared to that of 1 but is in the same potential range than that of DD-DSF-IF (0.5–0.67 V) (Fig. 1, top left). The first electron transfer in DA2-DSF-IF can be hence assigned to the oxidation of the di(p-methoxyphenyl)amine units (with nevertheless a contribution of the phenyl ring of the dihydroindenofluorenyl core, see Fig. 2 for the nature of the orbitals involved) and not anymore on the dihydroindenofluorenyl core as in 1. Oxidation at higher potentials leads to the oxidation of the DSF-IF core.
As the cathodic behaviour of the target molecules was not observed in DCM, they have been recorded in THF. Reduction of 1 occurs in a single wave with a maximum at −2.6 V (see the ESI†). The reduction of DD-DSF-IF was not observed in THF and hence determined in CH2Cl2.
The reduction of AA- and DA2-DSF-IF (Fig. 1, bottom right) shows two successive reversible reduction waves with maxima at −2.2/−2.5 V for AA-DSF-IF and −2.2/−2.39 V for DA2-DSF-IF. The first reduction recorded for DA2-DSF-IF is clearly ascribed to the reduction of the whole central core phenylbenzimidazole-dihydroindenofluorenyl unit(s) as confirmed by molecular modelling (Fig. 2). Concerning AA-DSF-IF, its lowest reduction potential (−2.2 V) compared to that of 1 (−2.6 V) is due to the electron-withdrawing effect of benzimidazole on the central dihydroindenofluorenyl core.
The onset oxidation and reduction potentials have been determined to evaluate the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels and the corresponding electrochemical gap ΔEEl (Table 2). DD-DSF-IF and AA-DSF-IF respectively possess the highest HOMO and lowest LUMO in the series (−4.81 eV and −2.44 eV, respectively). DA2-DSF-IF with HOMO/LUMO levels respectively recorded at −4.87/−2.35 eV possesses the electronic properties of both DD-DSF-IF and AA-DSF-IF.
This shows the efficiency of attaching both donor and acceptor units at each extremity of the dihydroindenofluorenyl core in order to finely tune the HOMO/LUMO energy levels.
ΔEEl of AA-DSF-IF (3.11 eV) is 0.57 eV shorter than that of 1 (3.68 eV) mainly due to its low LUMO level induced by the presence of the electron withdrawing benzimidazolyl unit and also owing to the fact that AA-DSF-IF possesses an almost identical HOMO level compared to that of 1 (−5.55 vs. −5.61 eV, respectively). Thus, the gap contraction of AA-DSF-IF is explained by a balance between the extension of π-conjugation due to the presence of connected phenylbenzimidazolyl groups (mainly inducing a higher HOMO) and the withdrawing effect of the latter on the dihydroindenofluorenyl core (inducing lower LUMO and HOMO levels). Interestingly, DD-DSF-IF possesses a ΔEEl of 2.79 eV, contracted by nearly 0.9 eV compared to that of 1. This is ascribed to the very high HOMO level of DD-DSF-IF compared to that of 1 (−4.81 eV vs. −5.61 V) caused by the easy oxidation of its two amino-aryl units. Finally and as mentioned above, DA2-DSF-IF with a gap of 2.52 eV takes advantage of both the higher HOMO (oxidation of the arylamine) and lower LUMO (reduction of the phenylbenzimidazole) leading to a large ΔEEl contraction of 1.16 eV compared to that of 1. This strategy is widely used in the field of organic electronics to contract the HOMO/LUMO gap.58,59
Molecular modelling (density functional theory-DFT, Fig. 2) has been performed in order to gain insight into the nature of the molecular orbitals. The optimized geometry and frontier molecular orbitals (HOMO and LUMO) of the ground state were obtained at the B3LYP/6-31-G level. The calculated HOMO–LUMO gap for DD-DSF-IF and AA-DSF-IF (3.35 eV/3.52 eV) well reproduced the main characteristics of these molecules, that is, a high HOMO and a low LUMO level, respectively (see electrochemistry above). Theoretical calculations confirm the gap contraction observed between DA1-DSF-IF (3.28 eV) and DA2-DSF-IF (3.10 eV), attributed to the increase of both the HOMO and LUMO levels (by 0.26 eV and 0.08 eV, respectively) due to the electron donating effect of the methoxy groups. These values are qualitatively consistent with the electrochemical and photophysical data (vide supra) although the HOMO and especially the LUMO energies are overestimated by the DFT method (ca. 0.3–0.5 and 0.8–1.1 eV, respectively). The nature of the different frontier orbitals shed light on the experimental results obtained by electrochemistry. Non-substituted 1 possesses HOMO/LUMO energy levels exclusively spread out on the dihydroindenofluorenyl core due to the different conjugation lengths between fluorene (bridged biphenyl) and dihydroindenofluorene (bridged terphenyl).53AA-DSF-IF presents quite similar behaviour with both HOMO and LUMO centred on the dihydroindenofluorenyl backbone with, however, a slight contribution of the phenylbenzimidazolyl units. The molecular frontier orbitals of DD-DSF-IF are differently spread out as the HOMO is dispersed on the arylamine/dihydroindenofluorene fragment, whereas its LUMO is exclusively distributed on the dihydroindenofluorenyl fragment. In the case of DA1-DSF-IF, the HOMO is mainly dispersed on the diphenylamine with nevertheless an important contribution of the dihydroindenofluorenyl unit, whereas the presence of the methoxy groups in DA2-DSF-IF leads to an HOMO more dispersed on the arylamine and less on the dihydroindenofluorene. In contrast, the LUMO appears similar for both DA1-DSF-IF and DA2-DSF-IF, with dispersion on the phenyl-dihydroindenofluorenyl/benzimidazole fragment. One can note that the LUMO of both DA1-DSF-IF and DA2-DSF-IF is more spread out on the phenylbenzymidazolyl unit than that of AA-DSF-IF, which is more dispersed on the dihydroindenofluorenyl unit.
Photophysical studies
The phostophysical data are gathered in Table 3.| λ abs liq (nm) | λ em liq (nm) | λ abs film (nm) | λ em film (nm) | ΔEopt (eV)a | Quantum yield θb | |
|---|---|---|---|---|---|---|
| a ΔEopt has been calculated from the absorption edge of the UV-vis absorption spectrum with ΔEopt (eV) = hc/λ, λ being the absorption edge (in meters). With h = 6.6 × 10−34 J s (1 eV = 1.6 × 10−19 J) and c = 3.0 × 108 m s−1, this equation may be simplified as: ΔEopt (eV) = 1237.5/λ (in nm). b Compared to quinine sulphate. | ||||||
| 1 | 313, 329, 337, 345 | 349, 367 | 316, 332, 340, 349 | 355, 375 | 3.49 | 56 |
| AA-DSF-IF | 313, 333, 370 | 413, 437, 468 (sh) | 316, 337, 377 | 423, 454, 480 | 3.03 | 76 |
| DD-DSF-IF | 368 (sh), 391 (sh), 410 | 424, 450 | 312, 398 (sh), 414 | 439, 466 | 2.90 | 86 |
| DA1-DSF-IF | 313, 377, 394 (sh) | 420, 445 | 316, 382 | 513 | 2.93 | 70 |
| DA2-DSF-IF | 313, 352, 395, 409 | 435, 461 | 316, 410 | 476 | 2.83 | 97 |
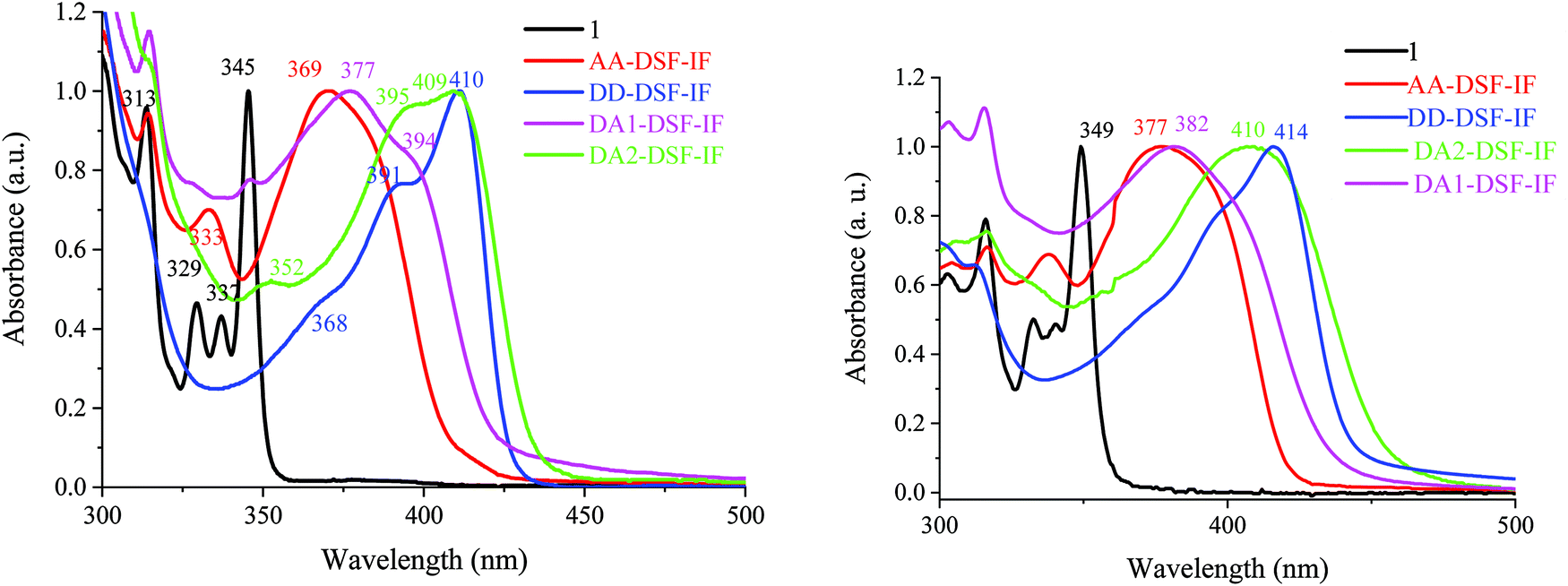 | ||
| Fig. 3 UV-vis absorption spectra in solution in cyclohexane (left) and as a solid thin film (right) of 1, AA-DSF-IF, DD-DSF-IF, DA1-DSF-IF and DA2-DSF-IF. | ||
In solution in cyclohexane, AA-DSF-IF presents several absorption bands with a large and intense band at 370 nm (Fig. 3, left, red line). The first bands (313 and 333 nm) are similar to those observed for 1 (313, 329, and 337 nm, black line) showing that the fluorene units are not affected by the substitution of the dihydroindenofluorenyl unit. The lowest energy band of AA-DSF-IF (370 nm) is bathochromatically shifted by 25 nm compared to that of 1, indicating the extension of the π-conjugation in the former due to the presence of the two phenylbenzimidazolyl units (in accordance with electrochemical conclusions). Moreover, the spectrum of AA-DSF-IF appears much less resolved than that of 1 due to its higher flexibility, induced by the rotation of the phenylbenzimidazolyl units.
The DD-DSF-IF absorption spectrum presents a main large band with maxima at 391 and 410 nm (Fig. 3, left, blue line) showing that the π-conjugation is impressively extended compared to 1 and AA-DSF-IF due to the two di-methoxyphenyl-amine units. The pendant arylamine units also endow the molecule with a higher flexibility compared to 1, leading to a less structured spectrum. It should be noted that the maximum recorded at 410 nm is in accordance with that recorded for a structurally related compound diphenylamino-dihydroindeno[1,2-b]fluorene (λ = 407 nm).55
Similarly, DA1-DSF-IF and DA2-DSF-IF, respectively, present maxima at 377 nm (Fig. 3, left, pink line) and at 409 nm (Fig. 3, left, green line), significantly red-shifted compared to that recorded for 1 (345 nm), clearly indicating an extension of the π-conjugation once the donor and acceptor groups were connected. Both compounds present a transition at 313 nm, indicating that the fluorenyl units are not affected by the substitution of the dihydroindenofluorenyl unit due to the spiro bridges. Thus, DA1-DSF-IF and DA2-DSF-IF present an optical gap ΔEopt of 2.93 eV and 2.83 eV, respectively, in the same range as those of DD-DSF-IF (ΔEopt = 2.90 eV) and AA-DSF-IF (ΔEopt = 3.03 eV) but strongly contracted compared to that of 1 (ΔEopt = 3.49 eV). It should be noted that there is a slight difference between the energy gap determined through electrochemical experiments ΔEel and UV-vis absorption spectra ΔEopt due to the different processes involved.
Absorption spectra in the thin solid film (Fig. 3, right) appear to be very similar to the respective solution ones with nevertheless a very small bathochromic shift indicating the absence of strong aggregation in the solid state. This is caused by the spirofluorene units, which efficiently avoid intermolecular interactions in the solid state.60,61 The emission properties will provide different results (see below, Fig. 4).
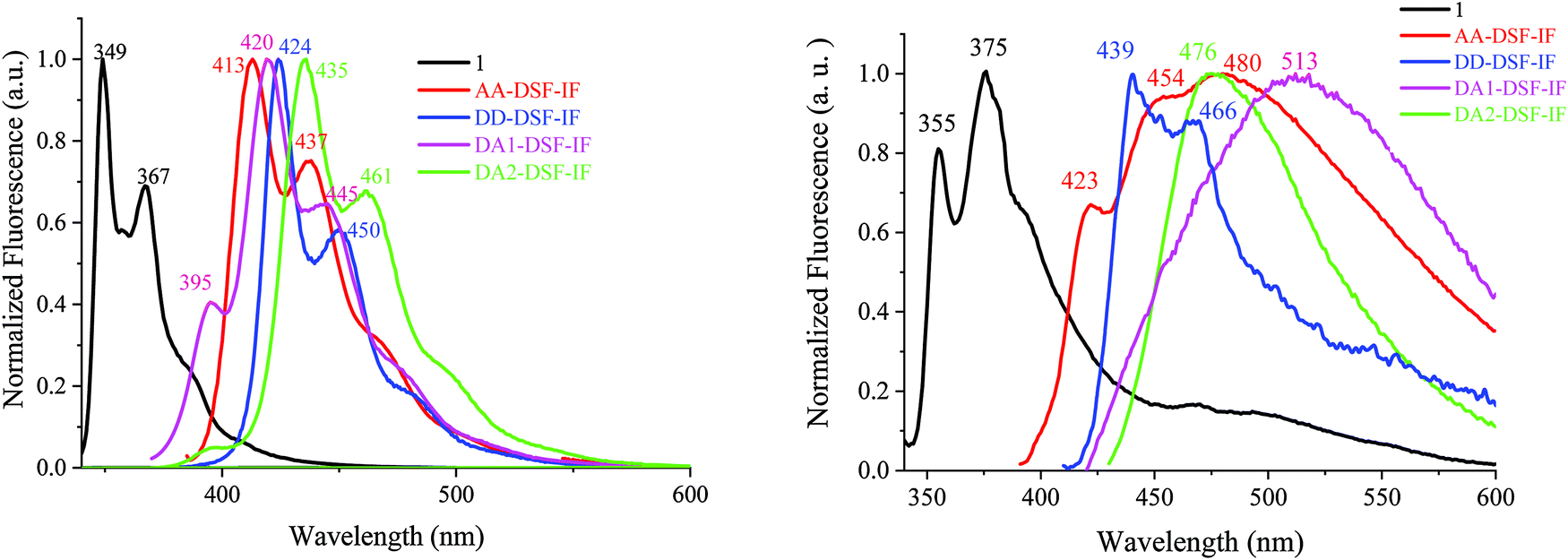 | ||
| Fig. 4 Emission spectra in cyclohexane (left) and in the thin film (right) of 1 (λexc = 310 nm, black), AA-DSF-IF, DD-DSF-IF, DA1-DSF-IF and DA2-DSF-IF, λexc = 350 nm. | ||
The symmetrical AA-DSF-IF and DD-DSF-IF present well resolved spectra with maxima at 413/437 and at 424 nm, respectively. The Stokes shift (SS), measured as the difference (in nm) between the lowest energy absorption band and the highest energy emission band, is larger than that of 1 (43 nm for AA-DSF-IF, 14 nm for DD-DSF-IFvs. 4 nm for 1), clearly indicating more nuclear rearrangements in the excited state leading to less rigid structures, which usually activate non radiative deactivation pathways. However, the quantum yields of AA-DSF-IF and DD-DSF-IF in solution in cyclohexane remain very high, ca. 76 and 86%, respectively, and even higher than that of 1 (ca. 56%),62 indicative of very efficient blue fluorophores. Despite the pendant donor/acceptor groups, both DA1-DSF-IF (λmax = 420/445 nm) and DA2-DSF-IF (λmax = 435/461 nm) also present well resolved spectra with maxima in the deep blue region, red-shifted by 71 and 86 nm compared to 1 (λmax = 349 nm). Both molecules DA1-DSF-IF and DA2-DSF-IF also appear as very efficient fluorophores with high quantum yields of 70 and 97%, respectively, in cyclohexane.
The fluorescence decay curves of AA-DSF-IF, DD-DSF-IF, DA1-DSF-IF and DA2-DSF-IF measured in cyclohexane provide very short lifetimes, around 1 ns for all the fluorophores (0.66, 0.96, 0.90 and 1.1 ns, see Figure in the ESI†). In the thin film, the lifetimes measured are also below 1 ns for all the compounds.
In general, the control of the emission wavelength in D–π–A fluorophores is not an easy task as the control of the charge transfer is difficult. In these examples, we manage to retain the fluorescence (in cyclohexane) in the blue region. Despite fantastic progresses in the last twenty years, stable and efficient blue emission arising from organic materials still continues to hold the attention of many research groups worldwide.63–67 However, what is really informative before OLED applications is the emission color in the solid state. For AA-DSF-IF, the emission spectrum in the solid state (Fig. 4, right) is large and unstructured with a maximum at 480 nm and a long emission tail up to 600 nm, very different from its solution spectrum. Such a feature may indicate the existence of intermolecular π–π interactions in the solid state probably between phenylbenzimidazolyl units due to a planar excited state which may favor interactions between these fragments. The spectrum of DD-DSF-IF presents two main bands at 439/466 nm, both red shifted by 15 nm compared to its solution spectrum surely due to the different dielectric constants of the media. The emission of D–π–A fluorophores appears very different. DA2-DSF-IF possesses a λmax in the blue region (476 nm), close to its solution spectrum, whereas DA1-DSF-IF possesses a λmax in the green region (513 nm), strongly red shifted compared to its solution spectrum. Thus, the methoxy groups borne by the phenylamine fragments of DA2-DSF-IF seem to play a key role in the solid state arrangement, keeping the emission in the blue region. Thus, one can note that spiroconnected fluorenes, known to hinder intermolecular interactions,60,61 have a different influence depending on the molecule substitution. The dihydroindenofluorenyl core has a preponderant role and drive the intermolecular interactions.
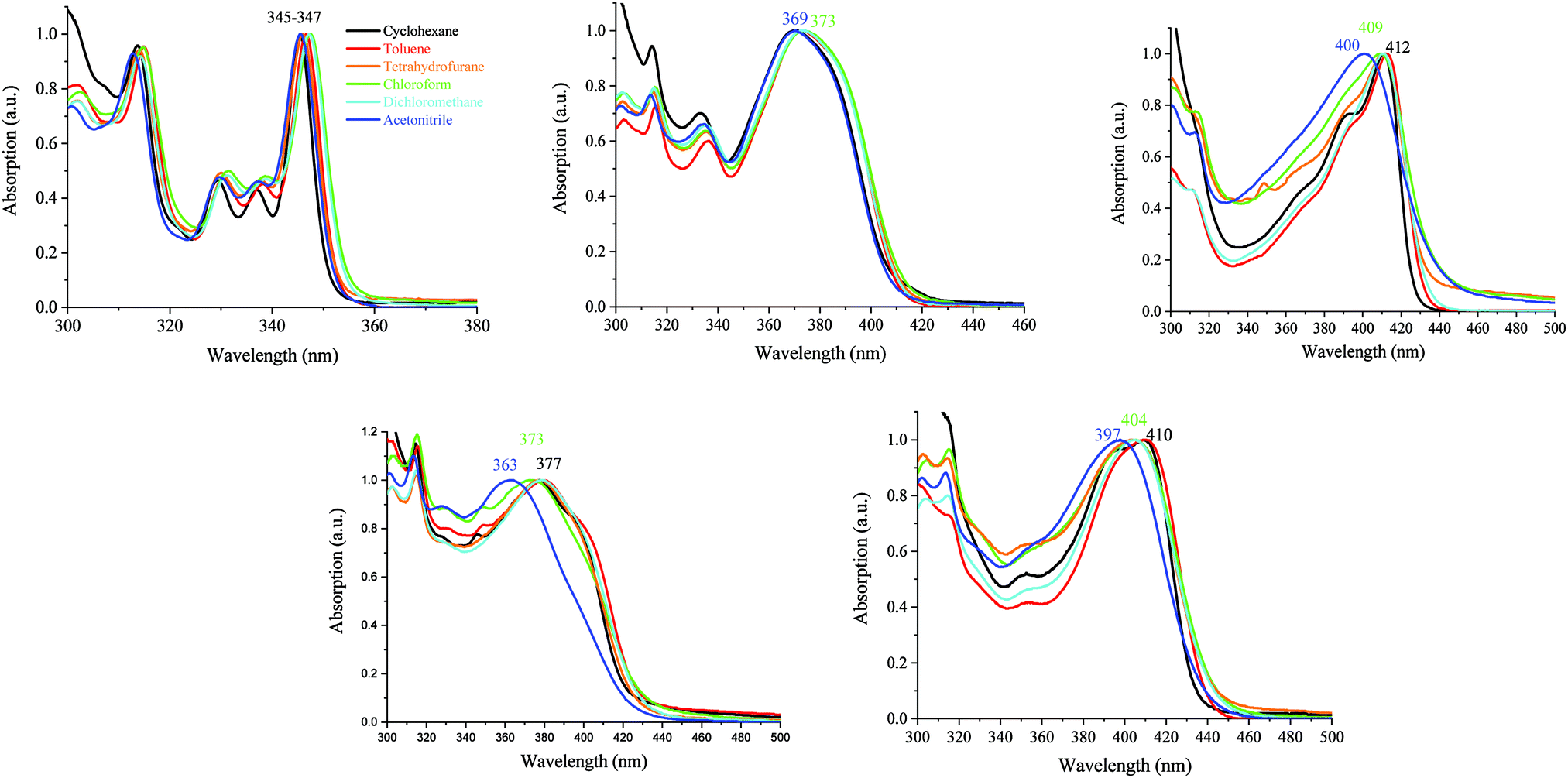 | ||
| Fig. 5 Absorption spectra of 1 (top left) and AA-DSF-IF (top middle) DD-DSF-IF (top right), DA1-DSF-IF (bottom left) and DA2-DSF-IF (bottom right) in different solvent polarity media: cyclohexane (black line), toluene (red line), chloroform (green line), THF (orange line), dichloromethane (cyan line), and acetonitrile (blue line). | ||
It should be mentioned that the shape of the DD-DSF-IF, DA1-DSF-IF and DA2-DSF-IF UV-vis absorption spectra is almost identical in all the solvents with (i) several vibronic-structured absorption bands around 300 nm characterising the absorption of the fluorene moieties68 and (ii) a main broad band around 400 nm attributed to the HOMO–LUMO transition possessing an intramolecular charge transfer (ICT, see below) character.
In contrast, the excited state is highly more sensitive to the polarity of the solvent and the luminescence is hence strongly modified (Fig. 6 and 7).
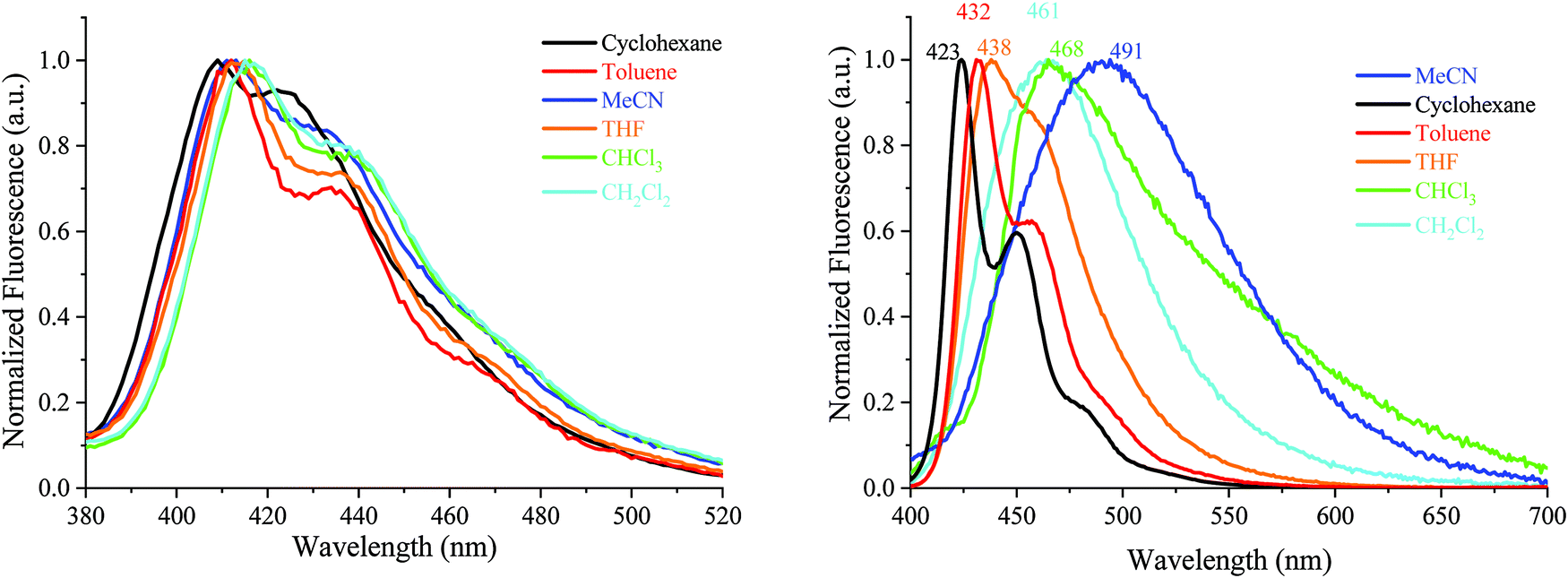 | ||
| Fig. 6 Emission spectra of AA-DSF-IF (λexc = 300 nm) and DD-DSF-IF (λexc = 390 nm) in different solvents. | ||
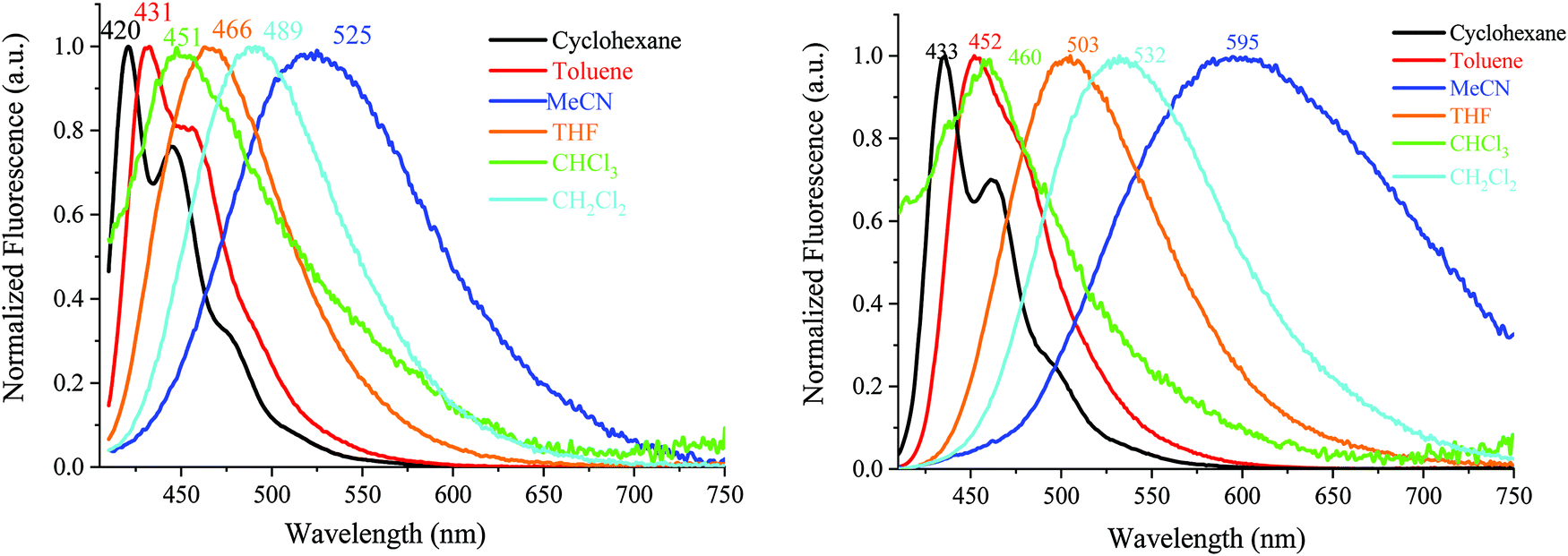 | ||
| Fig. 7 Emission spectra of DA1-DSF-IF (left) and DA2-DSF-IF (right), λexc = 400 nm, in different solvents. | ||
For 1, there is no modification of the shape of the emission band and almost no solvatochromic effect, the emission maximum being shifted from 349 nm in cyclohexane to 352 nm in acetonitrile. This indicates that the ground state dipole moment of 1 is almost identical to that in the excited state and there is hence no dipole–dipole interaction between 1 and the polar solvent molecules. In this molecule, the emission arises from the dihydroindenofluorenyl backbone.62
For AA-DSF-IF (Fig. 6, left) the solvatochromic effect is more intense than that observed for 1 but remains nevertheless modest. Indeed, as the polarity of the solvent increases, the main emission is less and less structured and slightly red-shifted. This is due to the dipole–dipole interactions in the excited state between polar solvents molecules and the phenylbenzimidazolyl fragment.
For DD-DSF-IF (Fig. 6, right) and despite its symmetric D–π–D arrangement, the polarity of the solvent seems to have a significant effect on the emission spectra. Indeed, a gradual shift of emission is observed from λ = 423 nm in apolar cyclohexane to λ = 491 nm in acetonitrile.
The two D–π–A dyes present the characteristics of both AA-DSF-IF and DD-DSF-IF and are even more sensitive than DD-DSF-IF to the polarity of the medium leading to considerable bathochromic shifts of the PL maxima (Fig. 7). In cyclohexane (λexc = 400 nm), both DA1-DSF-IF and DA2-DSF-IF have well-structured fluorescence bands at 420/445 nm for DA1-DSF-IF and 433/464 nm for DA2-DSF-IF. In a slightly more polar solvent, i.e., toluene, the fine vibronic structure is almost lost and a red shift of the maxima is observed for both compounds, being, however, still in the blue region. Then, when increasing the polarity from toluene to THF, chloroform, dichloromethane and acetonitrile, the main fluorescence bands become broader, completely lose their fine vibronic structure and a large bathochromic shift is observed. Thus, the main emission in acetonitrile is centered at 525 nm for DA1-DSF-IF and at 595 nm for DA2-DSF-IF, i.e. respectively, red-shifted by 105 nm and 162 nm compared to their corresponding emissions in cyclohexane. Due to the presence of the methoxy groups on the arylamines, the electron donating character of the donor group is stronger in DA2-DSF-IF compared to that in DA1-DSF-IF and a much larger solvatochromic effect is detected for the former. The huge solvent-dependence observed for both D–π–A dyes, DA1-DSF-IF and DA2-DSF-IF and the D–π–D dye DD-DSF-IF can be clearly related to the stabilization of the excited state due to dipole–dipole interactions. As the polarity of the solvent seems to have very little effect on the absorption spectra, the solvatochromic effect is clearly caused by a photoinduced ICT. Despite its symmetric structure, the charge transfer character of DD-DSF-IF can be easily visualized with the nature of the orbitals involved (Fig. 2), the HOMO being dispersed on the amine/dihydroindenofluorene fragment and the LUMO being exclusively dispersed on the dihydroindenofluorenyl core.
The dipole moment difference between the S0 and S1 states (Δμ = μ* − μ) of 63.0 and 73.8 D has been evaluated (see the ESI†) using the Lippert–Mataga formalism (the dipole moments in the ground state obtained through DFT calculations were 5.21 D for both DA1-DSF-IF and DA2-DSF-IF), translating the different polarities of the two dyes in the excited state (μ* = 68.2 and 79 D, for DA1-DSF-IF and DA2-DSF-IF, respectively). Thus, the strong solvatochromic effect and the corresponding high μ* observed are indicative of a significant photoinduced ICT.
The quantum yields (θ) of all the fluorophores have been also determined (with quinine sulfate as a reference) (see Table S1 in the ESI†) and it was seen that the compounds, which do present any solvatochromic effect, i.e., 1 and AA-DSF-IF, present a similar quantum yield, not dependent of the solvent polarity. Thus, 1 possesses a quantum yield of ca. 52–56% in cyclohexane, THF and acetonitrile, not affected by the polarity of the medium in accordance with the fact that no solvatochromic effect is observed. Similarly, the quantum yield of AA-DSF-IF recorded at 76% in cyclohexane remains stable in THF and acetonitrile. The three other compounds display large solvatochromic effects, and their quantum yields are significantly affected by the polarity of the media. This behaviour is often observed in fluorophores displaying strong ICT.69–71DD-DSF-IF displays a high quantum yield in cyclohexane, 86%, which drops down to 56 and to 26% in THF and acetonitrile, respectively, with the stabilization of the excited state. The two D–π–A fluorophores display an identical behaviour in accordance with the solvatochromic experiments presented above. For DA1-DSF-IF/DA2-DSF-IF, the quantum yield is measured at 70/97% in cyclohexane, 56/55% in THF and 26/22% in acetonitrile.
TD-DFT calculations have been carried out and some data are gathered in the ESI† (RB3LYP/6-31G(d)). For all the compounds studied, the main transition is attributed to a HOMO–LUMO transition and the value of the main absorption wavelength follows a similar order to the experimental spectra (from the shortest to the longest wavelength: 1 (335.5 nm), AA-DSF-IF (399.7 nm), DD-DSF-IF/DA1-DSF-IF (421.6/422.91 nm) and DA2-DSF-IF (444.45 nm)). Thus, as the HOMO/LUMO of DA1-DSF-IF and DA2-DSF-IF are mainly spread out on the diphenylamine and the phenyl-dihydroindenofluorenyl/benzimidazole, respectively, the corresponding transitions clearly undergo an ICT character as pointed out by the solvatochromic effect observed in fluorescence spectroscopy (this behaviour is very often observed72). Similarly and despite its D–π–D symmetry, the charge transfer character of DD-DSF-IF can also be pointed out with the nature of the orbitals involved. Indeed, the HOMO of DD-DSF-IF is dispersed on the amine/dihydroindenofluorenyl fragment, whereas its LUMO is exclusively dispersed on dihydroindenofluorenyl with no contribution of the amines.
These features also show that the electronic influence of the donor is much more pronounced than that of the acceptor in the present D–π–A dyes and that the fluorenyl units are not involved in the frontier orbitals.
Finally, as a first application for these materials, DA2-DSF-IF has been used as an emissive layer (EML) in simple OLEDs. The structure of the unoptimized OLED was ITO as the anode, poly(3,4-ethylenedioxythiophene) doped with poly(styrene-sulfonate) (PEDOT:PSS) as the hole injecting layer, N,N′-di(naphthyl)-N,N′-diphenyl-[1,1′-biphenyl]-4,4′-diamine (NPB) as the hole transporting layer, DA2-DSF-IF as the EML, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP) as the hole blocking layer and calcium as the cathode.
First, the electroluminescent (EL) spectrum of the OLED exhibits a nice structureless band in the blue region presenting maxima at ca. 465–472 nm (chromatic coordinates of 0.18; 0.28) (Fig. 8, left). The EL spectrum is in accordance with the DA2-DSF-IF thin-solid film fluorescence spectrum undoubtedly indicating that the charge recombination takes place in the EML without any recombination in other layers. In addition, no sign of troublesome parasite green emission26,73 (arising from intermolecular excimers, keto-defects or exciplexes) was detected, clearly indicating that the molecules of DA2-DSF-IF in the thin-film are isolated enough to avoid any strong π–π intermolecular interactions. In terms of performance, the turn on voltage of the device is detected below 5 V (4.8 V), translating a good charge injection within the device due to the high/low HOMO/LUMO energy level (−4.87/−2.35 eV). The other performances were modest with a luminance of ca. 1000 Cd m−2 (Fig. 7, right) for a current efficiency of 0.2 Cd A−1. Compared to 1 and AA-DSF-IF, DA2-DSF-IF displays nevertheless significantly higher performance (see the ESI†). By modification of the cathode (e.g. LiF/Al) and/or the corresponding blocking/transporting layers, these devices performances should be surely improved.
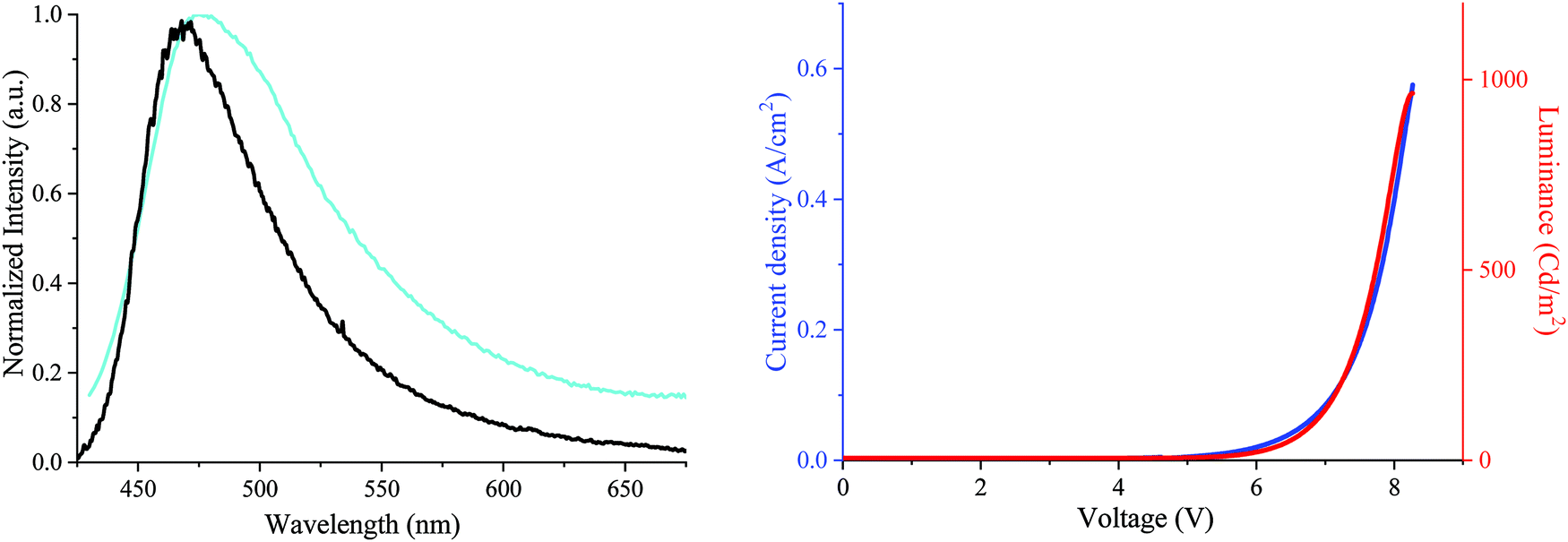 | ||
| Fig. 8 Left: EL spectrum of the DA2-DSF-IF-based device (black line) and the PL spectrum of the DA2-DSF-IF thin film (cyan line). Right, IVL characteristic (luminance: red line, current density: blue line). | ||
Conclusions
To conclude, this work reports a series of blue fluorophores constructed by the association of a dispiro[fluorene-9,6′-indeno[1,2-b]fluorene-12′,9′′-fluorene] scaffold and donor (diphenylamine) and acceptor (benzimidazole) units. Different substitutions of the dihydroindeno[1,2-b]fluorenyl core have allowed the modulation of the electronic properties of the DSF-IF scaffold and the study of the effect of the dihydroindeno[1,2-b]fluorenyl fragment as a π-conjugated bridge. The different functionalizations drastically tune the molecular orbital energy levels, maintaining, nevertheless, a high quantum yield. In addition, spiro-connected fluorenes, known to hinder intermolecular interactions, have a different influence depending on the dihydroindenofluorene substitution. As a first application, DA2-DSF-IF has been used as a fluorescent emitter in an OLED providing a stable blue emission (in accordance with its fluorescence spectrum) with a luminance of ca. 1000 cd m2 and a threshold voltage of 4.8 V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. T. and M. R. thank the Région Bretagne for a studentship. MR thanks Dr Bruno Lafitte and the Agence de l'Environnement et de la Maîtrise de l'Energie (ADEME) for a studentship. This work was granted access to the HPC resources of CINES under the allocation 2020-A0080805032 made by GENCI. We wish to thank the ANR (SPIROQUEST N°19-CE05-024) for financial support. Dr Cassandre Quinton (RENNES) is warmly thanked for the measurements of the lifetimes. The OLED fabrication and characterization have been performed by DT at the ENSCBP (IMS/UMR CNRS 5218-Bordeaux) under the guidance of Pr Laurence Vignau who is acknowledged.References
- Z.-Q. Jiang, C. Poriel and N. Leclerc, Mater. Chem. Front., 2020, 4, 2497–2498 RSC.
- Z. Ma, P. Sonar and Z.-K. Chen, Curr. Org. Chem., 2010, 18, 2039–2069 Search PubMed.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- S. Becker, C. Ego, A. C. Grimsdale, E. J. W. List, D. Marsitzky, A. Pogantsch, S. Setayesh, G. Leising and K. Müllen, Synth. Met., 2002, 125, 73–80 CrossRef CAS.
- L. J. Sicard, H.-C. Li, Q. Wang, X.-Y. Liu, O. Jeannin, J. Rault-Berthelot, L.-S. Liao, Z.-Q. Jiang and C. Poriel, Angew. Chem., Int. Ed., 2019, 58, 3848–3853 CrossRef CAS.
- C. Poriel, L. Sicard and J. Rault-Berthelot, Chem. Commun., 2019, 55, 14238–14254 RSC.
- R. Pudzich, T. Fuhrmann-Lieker and J. Salbeck, Adv. Polym. Sci., 2006, 199, 83–142 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS.
- X.-D. Zhu, Y.-L. Zhang, Y. Yuan, Q. Zheng, Y.-J. Yu, Y. Li, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2019, 7, 6714–6720 RSC.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS.
- L. Sicard, O. Jeannin, J. Rault-Berthelot, C. Quinton and C. Poriel, ChemPlusChem, 2018, 83, 874–880 CrossRef CAS.
- F. Lucas, L. Sicard, O. Jeannin, J. Rault-Berthelot, E. Jacques, C. Quinton and C. Poriel, Chem. – Eur. J., 2019, 25, 7740–7748 CrossRef CAS.
- L. Sicard, F. Lucas, O. Jeannin, P.-A. Bouit, J. Rault-Berthelot, C. Quinton and C. Poriel, Angew. Chem., Int. Ed., 2020, 59, 11066–11072 CrossRef CAS.
- C. Poriel and J. Rault-Berthelot, Acc. Chem. Res., 2018, 51, 1818–1830 CrossRef CAS.
- H. Reisch, U. Wiesler, U. Scherf and N. Tuytuylkov, Macromolecules, 1996, 29, 8204–8210 CrossRef CAS.
- S. Setayesh, D. Marsitzky and K. Müllen, Macromolecules, 2000, 33, 2016–2020 CrossRef CAS.
- A. C. Grimsdale, P. Leclère, R. Lazzaroni, J. D. Mackenzie, C. Murphy, S. Setayesh, C. Silva, R. H. Friend and K. Müllen, Adv. Funct. Mater., 2002, 12, 729–733 CrossRef CAS.
- G. Heimel, M. Daghofer, J. Gierschner, E. J. W. List, A. C. Grimsdale, K. Müllen, D. Beljonne, J. L. Brédas and E. Zojer, J. Chem. Phys., 2005, 122, 054501–054511 CrossRef.
- D. Hertel, S. Setayesh, H. G. Nothofer, U. Scherf, K. Müllen and H. Bässler, Adv. Mater., 2001, 13, 65–70 CrossRef CAS.
- S. Merlet, M. Birau and Z. Y. Wang, Org. Lett., 2002, 4, 2157–2159 CrossRef CAS.
- Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394–5404 CrossRef CAS.
- D. Horhant, J.-J. Liang, M. Virboul, C. Poriel, G. Alcaraz and J. Rault-Berthelot, Org. Lett., 2006, 8, 257–260 CrossRef CAS.
- C. Poriel, J.-J. Liang, J. Rault-Berthelot, F. Barrière, N. Cocherel, A. M. Z. Slawin, D. Horhant, M. Virboul, G. Alcaraz, N. Audebrand, L. Vignau, N. Huby, G. Wantz and L. Hirsch, Chem. – Eur. J., 2007, 13, 10055–10069 CrossRef CAS.
- S. Liu, D. Xia and M. Baumgarten, ChemPlusChem, 2021, 8, 36–48 CrossRef.
- C. Poriel, N. Cocherel, J. Rault-Berthelot, L. Vignau and O. Jeannin, Chem. – Eur. J., 2011, 17, 12631–12645 CrossRef CAS.
- M. Romain, D. Tondelier, J.-C. Vanel, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2013, 52, 14147–14151 CrossRef CAS.
- C. Poriel, J. Rault-Berthelot and D. Thirion, J. Org. Chem., 2013, 73, 886–898 CrossRef.
- F. Barrière, C. Poriel and J. Rault-Berthelot, Electrochim. Acta, 2013, 110, 735–740 CrossRef.
- K. H. Lee, S. O. Kim, J. N. You, S. Kang, J. Y. Lee, K. S. Yook, S. O. Jeon, J. Y. Lee and S. S. Yoon, J. Mater. Chem., 2012, 22, 5145–5154 RSC.
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2015, 54, 1176–1180 CrossRef CAS.
- W. Yu, J. Zhang, X. Wang, X. Liu, D. Tu, J. Zhang, X. Guo and C. Li, Solar RRL, 2018, 2, 1800048 CrossRef.
- Y. Wu, J. Zhang and Z. Bo, Org. Lett., 2007, 9, 4435–4438 CrossRef CAS.
- D. Xia, C. Duan, S. Liu, D. Ding, M. Baumgarten, M. Wagner, D. Schollmeyer, H. Xu and K. Müllen, New J. Chem., 2019, 43, 3788–3792 RSC.
- B. Kobin, J. Schwarz, B. Braun-Cula, M. Eyer, A. Zykov, S. Kowarik, S. Blumstengel and S. Hecht, Adv. Funct. Mater., 2017, 27, 1704077 CrossRef.
- R. Schlesinger, F. Bianchi, S. Blumstengel, C. Christodoulou, R. Ovsyannikov, B. Kobin, K. Moudgil, S. Barlow, S. Hecht, S. R. Marder, F. Henneberger and N. Koch, Nat. Commun., 2015, 6, 6754 CrossRef CAS.
- C. Poriel, F. Barrière, J. Rault-Berthelot and D. Thirion, Chem. – Eur. J., 2019, 25, 10689–10697 CrossRef CAS.
- C. Poriel, F. Barrière, D. Thirion and J. Rault-Berthelot, Chem. – Eur. J., 2009, 15, 13304–13307 CrossRef CAS.
- D. Beaudoin, J.-N. Blair-Pereira, S. Langis-Barsetti, T. Maris and J. D. Wuest, J. Org. Chem., 2017, 82, 8536–8547 CrossRef CAS.
- S. Thiery, B. Heinrich, B. Donnio, C. Poriel and F. Camerel, J. Mater. Chem. C, 2014, 2, 4265–4275 RSC.
- D. Thirion, M. Romain, J. Rault-Berthelot and C. Poriel, J. Mater. Chem., 2012, 22, 7149–7157 RSC.
- T.-C. Lin, C.-S. Hsu, C.-L. Hu, Y.-F. Chen and W.-J. Huang, Tetrahedron Lett., 2009, 50, 182–185 CrossRef CAS.
- S. Chaurasia, Y.-C. Chen, H.-H. Chou, Y. S. Wen and J. T. Lin, Tetrahedron, 2012, 68, 7755–7762 CrossRef CAS.
- M. M. Elmahdy, G. Floudas, L. Oldridge, A. C. Grimsdale and K. Müllen, ChemPhysChem, 2006, 7, 1431–1441 CrossRef CAS.
- D. Thirion, J. Rault-Berthelot, L. Vignau and C. Poriel, Org. Lett., 2011, 13, 4418–4421 CrossRef CAS.
- Z. P. Fan, X. Y. Li, X. E. Luo, X. Fei, B. Sun, L. C. Chen, Z. F. Shi, C. L. Sun, X. Shao and H. L. Zhang, Adv. Funct. Mater., 2017, 27, 1702318 CrossRef.
- M. Ozdemir, D. Choi, G. Kwon, Y. Zorlu, H. Kim, M.-G. Kim, S. Seo, U. Sen, M. Citir, C. Kim and H. Usta, RSC Adv., 2016, 6, 212–226 RSC.
- S.-y. Takizawa, V. A. Montes and P. Anzenbacher, Chem. Mater., 2009, 21, 2452–2458 CrossRef CAS.
- J.-J. Huang, Y.-H. Hung, P.-L. Ting, Y.-N. Tsai, H.-J. Gao, T.-L. Chiu, J.-H. Lee, C.-L. Chen, P.-T. Chou and M.-K. Leung, Org. Lett., 2016, 18, 672–675 CrossRef CAS.
- S. Gong, Y. Chen, J. Luo, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Funct. Mater., 2011, 21, 1168–1178 CrossRef CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- C.-H. Chen, W.-S. Huang, M.-Y. Lai, W.-C. Tsao, J. T. Lin, Y.-H. Wu, T.-H. Ke, L.-Y. Chen and C.-C. Wu, Adv. Funct. Mater., 2009, 19, 2661–2670 CrossRef CAS.
- C. Poriel, J. Rault-Berthelot, F. Barrière and A. M. Z. Slawin, Org. Lett., 2008, 10, 373–376 CrossRef CAS.
- H. Usta, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 8580–8581 CrossRef CAS.
- S.-Y. Ku, L.-C. Chi, W.-Y. Hung, S.-W. Yang, T.-C. Tsai, K.-T. Wong, Y.-H. Chen and C.-I. Wu, J. Mater. Chem., 2009, 19, 773–780 RSC.
- T. Hadizad, J. Zhang, Z. Y. Wang, T. C. Gorjanc and C. Py, Org. Lett., 2005, 7, 795–797 CrossRef CAS.
- F. Moreau, N. Audebrand, C. Poriel, V. Moizan-Baslé and J. Ouvry, J. Mater. Chem., 2011, 21, 18715–18722 RSC.
- F. Lucas, C. Quinton, S. Fall, T. Heiser, D. Tondelier, B. Geffroy, N. Leclerc, J. Rault-Berthelot and C. Poriel, J. Mater. Chem. C, 2020, 8, 16354–16367 RSC.
- F. Lucas, O. A. Ibraikulov, C. Quinton, L. Sicard, T. Heiser, D. Tondelier, B. Geffroy, N. Leclerc, J. Rault-Berthelot and C. Poriel, Adv. Opt. Mater., 2020, 8, 1901225 CrossRef CAS.
- J.-D. Peltier, B. Heinrich, B. Donnio, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2017, 23, 17290–17303 CrossRef CAS.
- S. Thiery, C. Declairieux, D. Tondelier, G. Seo, B. Geffroy, O. Jeannin, R. Métivier, J. Rault-Berthelot and C. Poriel, Tetrahedron, 2014, 70, 6337–6351 CrossRef CAS.
- D. Thirion, C. Poriel, R. Métivier, J. Rault-Berthelot, F. Barrière and O. Jeannin, Chem. – Eur. J., 2011, 17, 10272–10287 CrossRef CAS.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-K. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- J. Tagare, R. Boddula, S. S. Sudheendran, D. K. Dubey, J.-H. Jou, S. Patel and S. Vaidyanathan, J. Mater. Chem. C, 2020, 8, 16834–16844 RSC.
- M. Romain, C. Quinton, T. Roisnel, E. Jacques, J. Rault-Berthelot and C. Poriel, J. Org. Chem., 2018, 83, 1891–1897 CrossRef CAS.
- J. Tagare, D. K. Dubey, J.-H. Jou and S. Vaidyanathan, ChemistrySelect, 2019, 4, 6458–6468 CrossRef CAS.
- J. Tagare, R. Boddula, R. A. K. Yadav, D. K. Dubey, J.-H. Jou, S. Patel and S. Vaidyanathan, Dyes Pigm., 2021, 185, 108853 CrossRef CAS.
- S. Thiery, D. Tondelier, C. Declairieux, G. Seo, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, J. Mater. Chem. C, 2014, 2, 4156–4166 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Third Edition, Springer, 2006 Search PubMed.
- J. Shao, Z. Guan, Y. Yan, C. Jiao, Q.-H. Xu and C. Chi, J. Org. Chem., 2011, 76, 780–790 CrossRef CAS.
- J. Chen, W. Liu, J. Ma, H. Xu, J. Wu, X. Tang, Z. Fan and P. Wang, J. Org. Chem., 2012, 77, 3475–3482 CrossRef CAS.
- C. Poriel, J. Rault-Berthelot, S. Thiery, C. Quinton, O. Jeannin, U. Biapo, B. Geffroy and D. Tondelier, Chem. – Eur. J., 2016, 22, 17930 CrossRef CAS.
- A. C. Grimsdale, Curr. Org. Chem., 2010, 14, 2196–2217 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00875c |
| ‡ Under these conditions, this reaction also yields the disubstituted compound 5 (25%) further used for the synthesis of AA-DSF-IF. In addition, the starting material 2 is recovered (40%) and can be then recycled. |
| § It should be mentioned that this last step in the case of DA1-DSF-IF was a very low yield reaction due to the difficulty of purification. |
| This journal is © The Royal Society of Chemistry 2021 |