
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A quantitative multiscale perspective on primary olefin formation from methanol
Toyin
Omojola†
 *ab,
Andrew J.
Logsdail
c,
André C.
van Veen
b and
Stefan Adrian F.
Nastase‡
*c
*ab,
Andrew J.
Logsdail
c,
André C.
van Veen
b and
Stefan Adrian F.
Nastase‡
*c
aDepartment of Chemical Engineering, Claverton Down, University of Bath, Bath BA2 7AY, UK. E-mail: toyin.omojola@cantab.net
bSchool of Engineering, Library Road, University of Warwick, Coventry CV4 7AL, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, UK. E-mail: stefan.nastase@kaust.edu.sa
First published on 18th August 2021
Abstract
The formation of the first C–C bond and primary olefins from methanol over zeolite and zeotype catalysts has been studied for over 40 years. Over 20 mechanisms have been proposed for the formation of the first C–C bond. In this quantitative multiscale perspective, we decouple the adsorption, desorption, mobility, and surface reactions of early species through a combination of vacuum and sub-vacuum studies using temporal analysis of products (TAP) reactor systems, and through studies with atmospheric fixed bed reactors. These results are supplemented with density functional theory calculations and data-driven physical models, using partial differential equations, that describe the temporal and spatial evolution of species. We consider the effects of steam, early degradation species, and product masking due to the inherent autocatalytic nature of the process, which all complicate the observation of the primary olefin(s). Although quantitative spectroscopic determination of the lifetimes, surface mobility, and reactivity of adspecies is still lacking in the literature, we observe that reaction barriers are competitive with adsorption enthalpies and/or activation energies of desorption, while facile diffusion occurs in the porous structures of the zeolite/zeotype catalysts. Understanding the various processes allows for quantitative evaluation of their competing energetics, which leads to molecular insights as to what governs the catalytic activity during the conversion of methanol to primary olefins over zeolite/zeotype catalysts.
1. Introduction
Methanol is an important intermediate in the conversion of renewable feedstock to liquid fuels and chemicals, and can be produced from syngas obtained via solid fuel gasification. Methanol can be converted to hydrocarbons (MTH), i.e., olefins (MTO), gasoline (MTG), and aromatics (MTA), over zeolite and zeotype catalysts.1–6 These zeolite and zeo-type catalysts are important as they offer a flexible yet tailored solution to the changing market demands of the chemical and petrochemical industry. A “hydrocarbon pool” mechanism has been widely established under atmospheric steady-state conditions for some of the most extensively studied catalysts, such as H-ZSM-5, H-SAPO-34, H-ZSM-22, H-ZSM-23, and H-Y.5,7–11Over H-ZSM-5 catalysts, the “hydrocarbon pool” is considered as being a “dual cycle” consisting of an olefin cycle and an aromatic cycle. At steady-state, ethylene forms via the aromatic cycle while propylene and other higher olefins form through the olefin cycle (within the steric limitations of the zeolite). Depending on the composition, activation conditions, and process conditions, including partial and total pressures, temperature,2,12 methanol concentration and loading, and reactor type such as batch,13,14 flow,15,16 or fluidised bed reactors,17,18 one cycle proceeds faster than the other during methanol to olefin conversion. The imbalance in the speed of the different reaction cycle allows the product distribution to be tuned according to market needs. Given the recent commercial importance of olefins, understanding the catalytic mechanisms by which valuable products such as ethylene and propylene are formed becomes significant. Although the “hydrocarbon pool” mechanism dominates under atmospheric conditions in a flow-reactor, the origin and evolution of this mechanism is not completely understood.
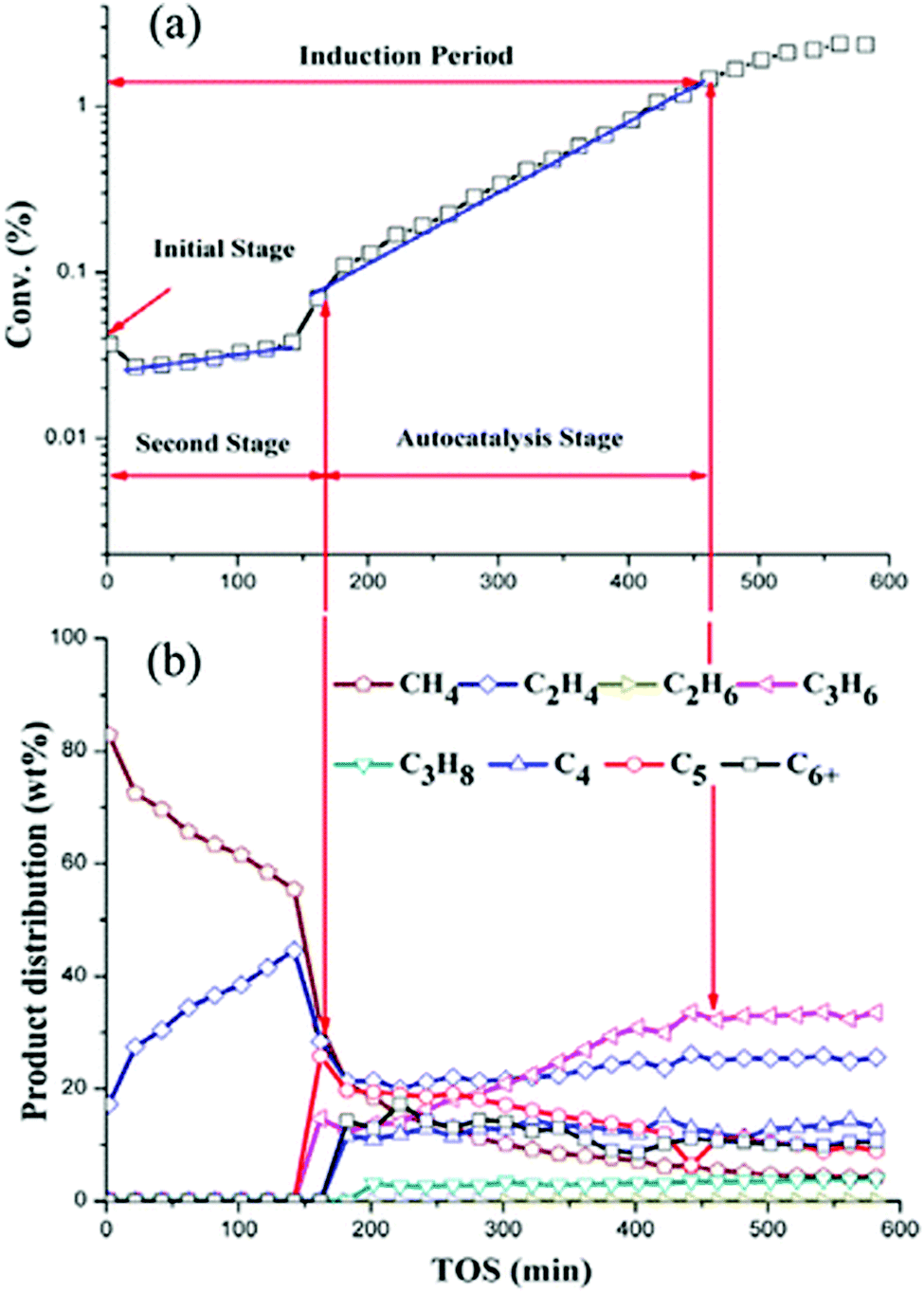
Hans Schulz19 used a capillary gas chromatography method to study H-ZSM-5 catalysts between 543–748 K. Methanol conversion at low temperatures (543–573 K) were grouped into several regimes: incubation period, autocatalysis (fast increase in rates), and retardation (inhibition through accumulation of organic compounds). At high temperatures (748 K), the identified regimes coincide with each other. Under atmospheric conditions at low temperatures, the catalyst attains a yellow colour which changes to pink after access to air, which is indicative of highly unsaturated organic compounds (Fig. 1). At 748 K and 15% lifetime (lifetime is defined as the time-on-stream until breakthrough of methanol at the catalyst-bed-end), a black zone of deactivated catalyst, a grey zone of main methanol conversion, and a zone of light-blue colour (in front of the reaction zone) can be distinguished. The front zone is still active, but “coke seeds” may have been formed on external catalyst sites.
 | ||
| Fig. 1 Photographs of the H-ZSM-5 catalyst bed after methanol conversion at high (above) and low (below) temperature. Reprinted from ref. 19 with permission from Elsevier. | ||
In Schulz's experimental work,19 however, it is important to note that steady-state was not achieved. Steady-state is defined here as conditions when gas phase species and adsorbed species do not change with respect to “clock” or “astronomical” time, as defined by Marin and Yablonsky.20 Astronomical or clock-time is the time on stream (TOS), and distinguishable from “intrinsic” time, which is the time scale at which a reaction occurs. The intrinsic time could be the reciprocal of its rate co-efficient and has the dimensions of s−1. Astronomical time is also different from the “residence time” or “transport time” of a chemical process. Finally, it is also important not to confuse “clock-time” or “astronomical-time” with “contact-time” or “space-time”. Steady-state can only be defined as no changes with respect to “clock-time” or “astronomical-time”. For Schulz, steady-state was not achieved during visual observations of MTO conversion over H-ZSM-5 catalysts at low temperatures (543–573 K) and up to 2 h. The reaction zone migrates slowly, and undesirable reactions occur before and behind the reaction zone.19
Operando time- and space-resolved high energy X-ray diffraction studies show the evolution of the morphology of H-ZSM-22 catalysts due to variation of the expansion and flattening of the unit cell with catalytic activity gradients.21 Wragg et al.22 used a kinetic model to describe time- and space-resolved high-energy operando X-ray diffraction studies over H-SAPO-34 catalysts for the formation of propylene, monocyclic aromatics, and polyaromatic coke molecules from methanol. The results of the kinetic model (Fig. 2) reproduce the observation that the hydrocarbon pool fraction is first formed from the middle of the bed and then expands backwards. The result agrees with a reaction scheme where the autocatalytic reaction mechanism allows for the concentration of the active hydrocarbon pool specie to slowly build up to a critical level in the direction of the reactor entrance, which gradually leads to a region of high activity. The kinetic model also accounts for the formation of a “coke front” as the hydrocarbon pool species react with methanol, as observed through X-ray diffraction.
 | ||
| Fig. 2 Formation of (a) aromatic HCP reaction intermediates, and (b) coke, from a kinetic model of the MTO process. The initial progress of hydrocarbon pool intermediates towards the bed inlet with TOS is clearly modelled, as is the formation of coke which spreads towards the outlet and causes the final expansion and deactivation. Reprinted from ref. 22, with permission from Wiley. | ||
The reaction zone is not always a “front” moving from inlet to the exit of the reactor, but can also grow against the flow direction, as observed by operando X-ray diffraction studies of H-SAPO-34 catalysts.21,22 The fixed bed reactor behaves as a “chromatographic reactor” where the movement of the front depends on where the coke stems from.
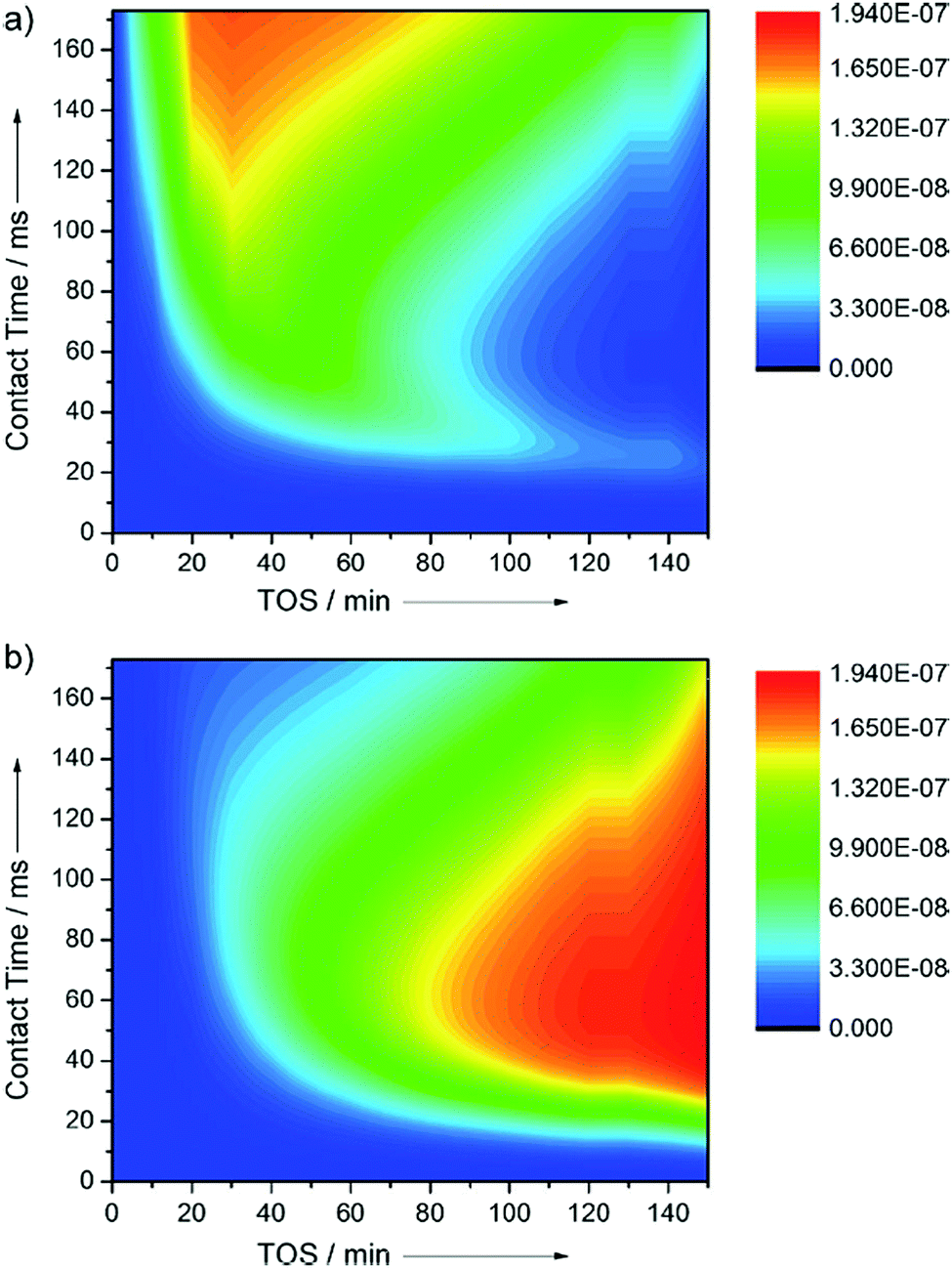
Recently, the formation of primary olefins was observed to go through three phases under atmospheric,23–25 and sub-vacuum26,27 conditions using “end-of-pipe” experiments in a temporal analysis of products (TAP) reactor:28 (a) an induction period (lag-phase), (b) a transition regime and (c) a plateau phase. The description of phases is purely based with respect to astronomical time, and the plateau phase (Fig. 3) is defined as steady-state.
 | ||
| Fig. 3 Step response of 5 vol% dimethyl ether at 300 °C over 10 mg of H-ZSM-5 (Si/Al = 25) catalysts in a temporal analysis of products reactor. Reprinted from ref. 27. Copyright 2019 Wiley Periodicals. | ||
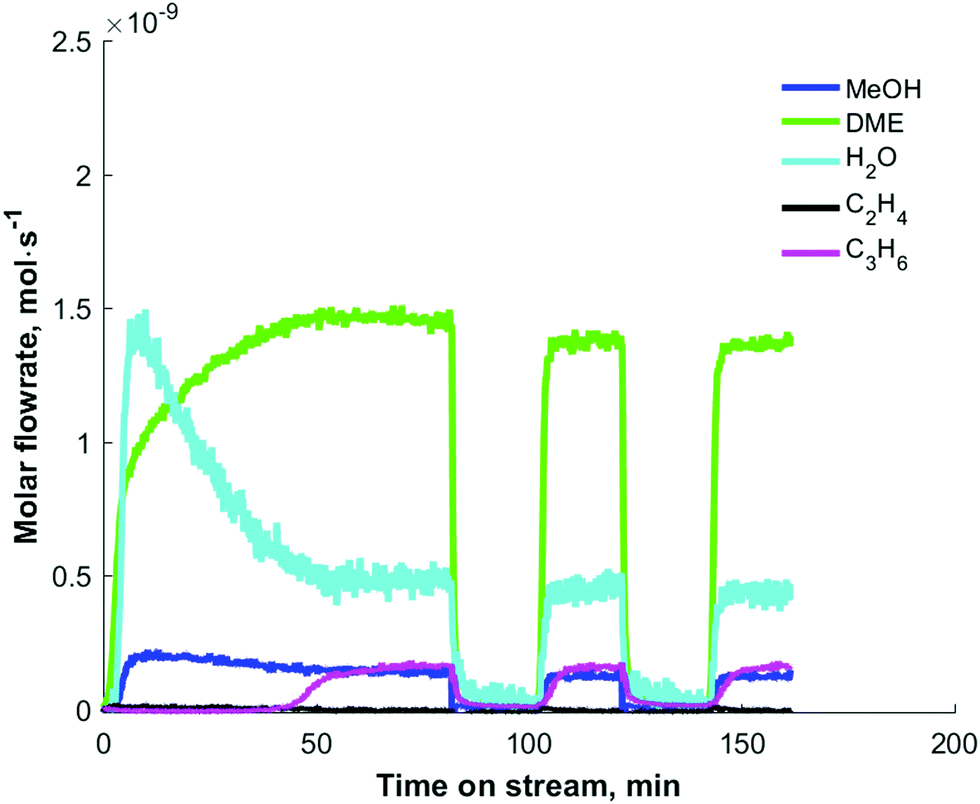
In Fig. 3, the induction period, observed during the step response of dimethyl ether over 10 mg H-ZSM-5 (Si/Al = 25) catalysts at 573 K and an inlet pressure of ca. 1000 Pa, is about 40 min. Qi et al.24 observed induction periods of about 2 h during the conversion of methanol to hydrocarbons at temperatures of 528 K (and below) and atmospheric pressure (Fig. 4). In their studies, methane, C2–C4 olefins and C2–C3 paraffins were observed. The major olefins observed were ethylene and propylene.
 | ||
| Fig. 4 Conversion of methanol (a) and product distribution (b) over H-ZSM-5 (Si/Al = 19) over H-ZSM-5 zeolite at 518 K as a function of time on stream (TOS). Reprinted from ref. 24. Copyright 2015 American Chemical Society. | ||
Process conditions, such as sub-vacuum pressures versus atmospheric pressures, temperatures, Si/Al ratio, and reactor feed, have a huge influence on the duration of the induction period and the initial species formed. Further comparative studies between H-ZSM-5 (Si/Al = 19, 21, 49, 99) and H-SAPO-34 (with two different Si contents) were conducted by Zhou et al.29 to understand the effect of initially retained organics, formed during the induction period, on the ensuing catalytic activity.
At this juncture, a clear distinction should be made between “primary” and “major” olefins. The primary olefin refers to the initial olefin formed. During MTO conversion, methylation is quite rapid30 due to the autocatalytic chemistry and with increasing temperatures, cracking over ZSM-5 catalysts as well as accompanying oligomerisation reactions (due to equilibration) occur. Consequently, what is usually observed from the gas chromatograph during steady-state kinetic studies in an “end-of-pipe” flow reactor is not the “primary olefin” but the “major” olefins as a result of these “masking effects”. In kinetic analysis, the primary products are then extrapolated using the Delplot technique.31,32 To merge both “primary” and “major” olefins, it is important to operate at conditions where the effects of these accompanying (side) chemistries and deactivation conditions are negligible. Steam and the formation of early degrading species during the induction period can mask the observation of the primary olefin. The operating conditions can be replicated via density functional theory calculations, which can then provide more evidence of the “primary” olefins.
The regeneration of catalysts during industrial MTO conversion is achieved through the use of fluidised bed reactors, due to their advantageous ability to transfer the catalyst between the main reactor, where the reaction occurs, and the regenerator, where the coke on the catalyst is burnt off.18 Fluidised bed reactors offer good heat-transfer properties and prevent runaway reactions, due to rapid mixing and thermal equilibration between the catalyst and the fluidising medium.18
Major insights into the formation of the first C–C bond, primary olefins, and other initial species produced during the induction period of MTO conversion are still lacking in the current literature. Adequate elucidation of the primary olefins requires the decoupling of active and degradation species, both of which may form in the initial stages. Furthermore, the separation of active sites from spectator sites, as well as characterisation of the active sites, is important. A macromolecular combination of a Brønsted acid site and active molecular species is required to form the active site of MTO chemistry.33 Energetics of the adsorption, desorption, surface reaction, and mobility of initial species over these active sites could provide quantitative insights into the formation of the first C–C bond and primary olefins.
1.1. Methods – kinetic and computational analysis
At the reactor level, experiments that investigate the formation of the first C–C bond during MTO conversion are conducted through: (a) transient methods12,27,34,35 such as step response measurements, temperature-programmed desorption, temperature-programmed surface reaction or pulse experiments, and (b) steady-state experiments conducted at low conversions. In the latter, information on the first C–C bond and primary olefins is obtained after the hydrocarbon pool has been established. The steady-state measurements are analysed kinetically as changing with respect to “space time” or “contact time” where, the former is defined based on conditions at the entrance of the reactor, and the latter is defined with respect to conditions based on the expansion of the fluid through the reactor incorporating effects of temperature, pressure, viscosity, and fluid density. The steady-state measurements (at low conversions) are usually measured after an astronomical time (or time-on-stream) of 7 min or higher. Extrapolations are then made from the finite contact time measurements (at low conversions) to zero contact time, in order to identify whether ethylene, propylene, or both, are primary and secondary products.31,32 Extrapolations should be regarded in highest scrutiny. It is important to note that within the initial 7 min to achieve steady-state, the “hydrocarbon pool” is formed and the concept of primary olefins formed “a priori” is eluded.On the other hand, transient experiments allow the catalyst to evolve from an initial state to a working state as starting molecules transform through the first C–C bond formation to primary olefins. A combination of observed reactor performance from both transient experiments and differential algebraic equations, which describe this performance, leads to the estimation of parameters that describe the reaction chemistry. Analysis of both transient and steady-state experiments occurs through the solution of partial differential or differential algebraic equations, respectively. Accompanying statistical analysis of the estimated parameters36 can help to obtain confidence intervals of the fitted parameters. The method complements first-principles microkinetic models applied to transient37 and steady-state experiments, and the scaling relations obtained therein.
Theoretical modelling provides insights into molecular-level properties that are otherwise difficult or inaccessible to experimental analysis. Ab initio static simulations offer a clear description of the potential energy surface, which can be used to determine molecular structures, charge distribution, and vibrational modes (with corresponding frequencies), together with kinetic and thermodynamic observables that can indicate reaction intermediates and pathways used to decipher experimental data.38–40 Molecular dynamics techniques have the ability to simulate real-time dynamics of atoms in zeolite pores, and has been used to investigate surface adsorption, proton transfers and diffusion effects,41,42 whilst positive bias meta-dynamic simulations can uncover rare-events and map out the free energy surface for pathways leading to meta- or un-stable intermediates.43,44 All of these techniques have been successfully applied to complement experimental analysis and improve the development of heterogeneous catalysts.45–47 In the existing studies of the first C–C bond and primary olefins, emphasis has been placed separately on quantitative information obtained either via density functional theory (DFT) calculations, hybrid quantum mechanics/molecular mechanics (QM/MM), and molecular dynamics (MD) calculations or data-driven simulations of partial differential equations that simulate the elementary steps. Numerous review articles3,17,48,49 have focused on the various aspects of MTO conversion such as mechanistic understanding, spectroscopic characterization, and process technology; however, none has focused on a “multi-scale” quantitative mechanistic understanding of the formation of “primary olefins” from oxygenates. By combining: atomistic simulations (using density functional theory calculations, hybrid quantum mechanics/molecular mechanics and molecular dynamics calculations), macroscale simulations (obtained from physical data-driven ordinary and partial differential equations), and experiments conducted on temporal analysis of products (TAP) reactor systems (sub-vacuum or vacuum pressures) and conventional flow reactors (at atmospheric and higher pressures), we provide herein a quantitative “multiscale” perspective on the formation of the first C–C bond and primary olefins during methanol-to-olefin conversion over zeolite and zeo-type catalysts.
1.2. Our work
Reactions on porous solid materials can be broken down into several sequentiual stages: (1) external diffusion of reactants from the bulk phase (reactant stream) to the catalyst particle (pore mouth), (2) pore diffusion of reactants to the catalyst surface, (3) adsorption onto the catalyst surface, (4) surface reactions, (5) desorption of product species, (6) pore diffusion of product species from the catalyst surface to the pore mouth exterior, (7) external diffusion from the pore mouth exterior to the bulk phase (reactant stream). In this perspective, we consider the implications of the catalyst structure for the kinetics of the induction period and the resulting product distribution. Subsequently, we consider the adsorption, desorption, mobility, and surface chemistries of early species involved in the induction period and the surface/pore chemistry (and mechanisms) through which the first C–C bond is formed.Information on adsorption, desorption, mobility, and surface reactions obtained from vacuum or sub-vacuum TAP reactor experiments, and density functional theory calculations, are consolidated with atmospheric fixed bed reactor studies combined with physical models using partial differential equations that describe the temporal and spatial evolution of species. We review the accompanying spectroscopic evidence for intermediate species during the formation of primary olefins from oxygenates. We also consider the formation of early degradation species during the initial stages of the MTO reaction, and other masking effects on the species (as a result of fast methylation rates, catalyst degradation and presence of steam) involved in the formation of the first C–C bond and primary olefins. New insights on the formation of the first C–C bond are revealed through this multiscale quantitative perspective.
2. Structure: zeolite and zeo-type catalysts
Zeolites and zeo-type structures are important heterogeneous catalysts for MTH conversion due to their pore architecture, thermal stability, and their ability to donate protons, via Brønsted acid sites and accept electrons via Lewis acid sites.50 The framework structure of zeolites, consisting of corner-sharing silicate and alumina tetrahedra, leads to various topologies, whilst the presence of aluminium imparts a negative charge on the framework that has to be balanced by positively charged extra-framework cations.50,51 For acidic zeolites, the bridging hydroxyl protons act as catalytic active sites. The general formula of a zeolite is: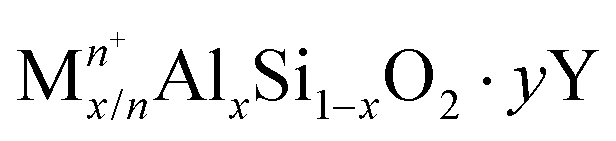 | (1) |
Two widely studied microporous materials for MTH conversion are H-ZSM-5 and H-SAPO-34. Since the discovery of methanol conversion over H-ZSM-5 catalysts in 1977,1,2 other porous materials such as H-ZSM-22 or H-ZSM-23 have been used for hydrocarbon production. Zeo-type catalysts, such as H-SAPO-34, are substituted alumino-phosphates, where the direct replacement of isolated phosphorus atoms by silicon atoms results in a negatively charged framework and an acid site upon calcination.50 SAPO-34 has a three-dimensional (3D) cage structure with small window sizes of 3.8 Å × 3.8 Å (Fig. 5a). The generation of light olefins during the steady-state MTO conversion is rationalised by the small pore windows allowing for only the exit of hydrocarbons with small sizes.52,53 In 2010, MTO conversion was commercialised in China with H-SAPO-34 catalysts in a turbulent fluidised bed reactor.18
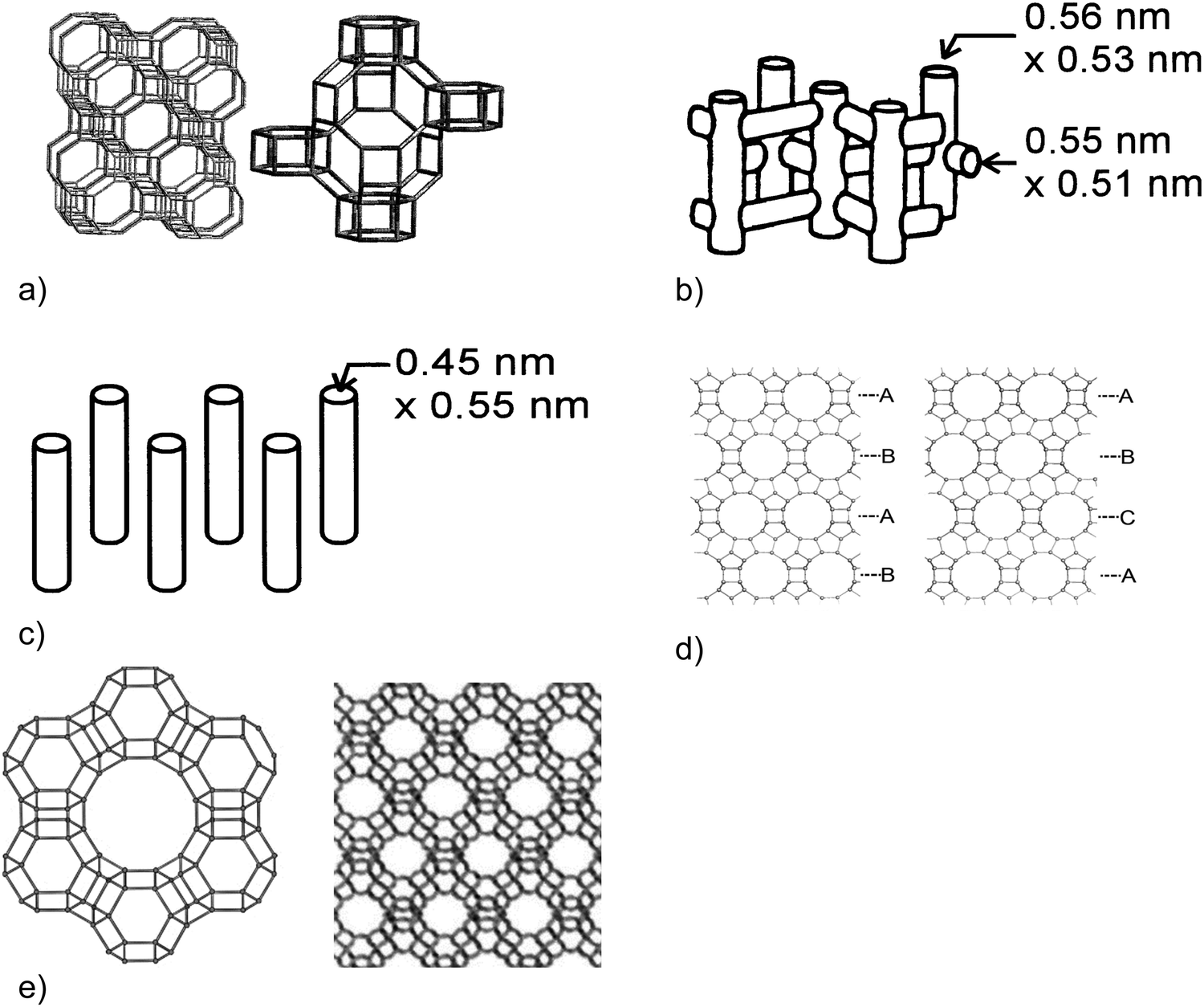 | ||
| Fig. 5 Channel and pore structures of (a) SAPO-34 (CHA) pore structure – [left] extended structure emphasising 6MR windows of sizes of ca. 0.38 nm and [right] expanded structure emphasising a cage of ca. 1 nm × 0.7 nm,54 (b) ZSM-5 (MFI) – the channel structure in ZSM-5,55 (c) ZSM-22 (TON) – the channel structure of ZSM-22,55 (d) beta pore structure (BEA framework). [left] Polymorph A emphasising an AB stacking sequence. [right] Polymorph B emphasising an ABC stacking sequence,56 and (e) H–Y (FAU) catalysts – Faujasite supercage formed of SOD cages [left] and pore structure [right]. Image obtained from ref. 57. | ||
Typical gasoline hydrocarbons (C5+ aliphatics and aromatics) have been produced over H-ZSM-5 catalysts in an MTG process.58,59 The ZSM-5 catalysts have a 3D pore structure with 10-membered ring (10MR) pore windows of size 5.1 × 5.5 Å and 5.3 × 5.6 Å (Fig. 5b). The basic building units of ZSM-5 are 5–5–1 units that link together to form chains. These basic building units link to give silicalite sheets, which contain 10MR openings and these can be connected across inversion centres.50 Under operating conditions of high temperatures (723 K) and low (sub-atmospheric) pressures, the product selectivity over H-ZSM-5 catalysts is tuned towards propylene.27,60 Aromatics can also be generated in an MTA process over Zn-ZSM-5 catalysts.61,62
At 643 K, C5+ aliphatics can be generated over H-ZSM-22 catalysts, which have a one-dimensional (1D) pore structure with 10MR pore windows of size 5.7 × 4.6 Å (Fig. 5c). The product distribution is due to the narrow pore sizes, and the alkenes residing at the pore windows allowing for olefin homologation reactions.63–67 The silicalite sheets of H-ZSM-22 are connected either by translation or by mirror planes (“unit cell twinning”).
The wide-pore H-Beta catalyst, which has a 3D pore structure with 12-membered ring (12MR) pore windows of size 6.6 × 6.7 and 5.6 × 5.6 Å (Fig. 5d), is also an active catalyst for the formation of aromatics, C4 and C5+ aliphatics.68,69 The intergrowth of the A- and B-polymorphs of the Beta framework is made by silicalite sheets stacking layer to layer with one of four possible translations, typically with a high degree of disorder. An additional 3D large pore zeolite, commonly used in fluid catalytic cracking,70 is zeolite H–Y, which has a Faujasite (FAU) framework. The FAU supercage is formed of 10 sodalite cages connected through hexagonal prisms, rendering an inner cavity with a 12 Å diameter (Fig. 5e). The pore, represented by a 12MR, has a relatively large diameter of 7.4 × 7.4 × 7.4 Å.50
2.1. Implications of catalyst structure on the induction period
From sigmoidal observations obtained by “end-of-pipe” measurements in a packed bed reactor, zeolite catalysts are understood to convert methanol to hydrocarbons in three different phases. At the early phase of the reaction, the catalyst is conditioned until full mechanisms are developed. Then, steady state is achieved (at the plateau of the sigmoidal curve). During this period, it is widely established that a hydrocarbon pool mechanism dominates and regulates product distribution. Regimes may be properly defined as “quasi-induction-period”, “quasi-transition-regime” and “quasi-steady-state” as species slowly accumulate during these phases, and they could could block the active sites and prevent the reaction from beginning, depending on the process conditions.19 The final phase involves catalyst deactivation, where site blocking species control the rate of conversion of initial feed (Fig. 3).Most studies on the induction period have focused on three catalysts: H-Y, H-ZSM-5, and H-SAPO-34. The initial stage in the MTH and MTO processes are characterised by an induction period, in which the methanol conversion rate is low.71,72 Several factors are suggested to influence the induction period, such as the presence of steam and a high reactant contact time.73 Water competes with methanol and dimethyl ether to adsorb on the zeolite active sites, and determines the reverse reaction from methoxy species to methanol, limiting the reaction progress; however, water can also stabilize certain carbo-cationic intermediates that are crucial to the MTH process.74,75
Induction periods can also be defined with respect to steady-state investigations (i.e., after a TOS greater than 7 min) conducted by varying contact time. High contact times and temperatures favour the formation of the “hydrocarbon pool reaction centres” and drastically reduce the steady-state induction period. Depending on the type of zeolite used, the induction period can have different profiles under the same reaction conditions; for example, H-SAPO-34 is able to limit the diffusion of hydrocarbons because of its small pores, and accelerate the formation of the “hydrocarbon pool” species (permissible with respect to its pore architecture), whereas the process is more prolonged over H-ZSM-5 catalysts.72 The induction period reduces with an increase in temperature and pressure.76 Consequently, experiments conducted at low temperatures in a low pressure reactor, such as the temporal analysis of products (TAP) reactor, serve to decipher the elementary steps of the induction period of MTO conversion.26,27,34,35,76
Ethylene and/or propylene are reported to oligomerise and form the “hydrocarbon pool reaction centres”, as signalled by the increase in the rate of methanol conversion. The necessary major step that ends the induction period is the formation of the first C–C bond compound leading to ethylene and/or propylene.
We note that there is limited information currently on the effect of catalyst structure on the induction period, either at atmospheric or low pressure conditions. Nonetheless, differentiating between the early intermediates that lead to the first C–C bond and any masking effects is necessary (due to rapid methylation or deactivating species on the catalyst surface).
2.2. Implications of catalyst structure on product distribution
During steady-state MTH conversion under atmospheric pressure conditions, it is widely accepted that a “hydrocarbon pool” mechanism takes places over zeolite catalysts, where an olefin cycle and an aromatic cycle operate (Fig. 6). There are numerous reviews and articles7,8,77–79 on the steady-state hydrocarbon pool mechanism and therefore we will only briefly describe the details here.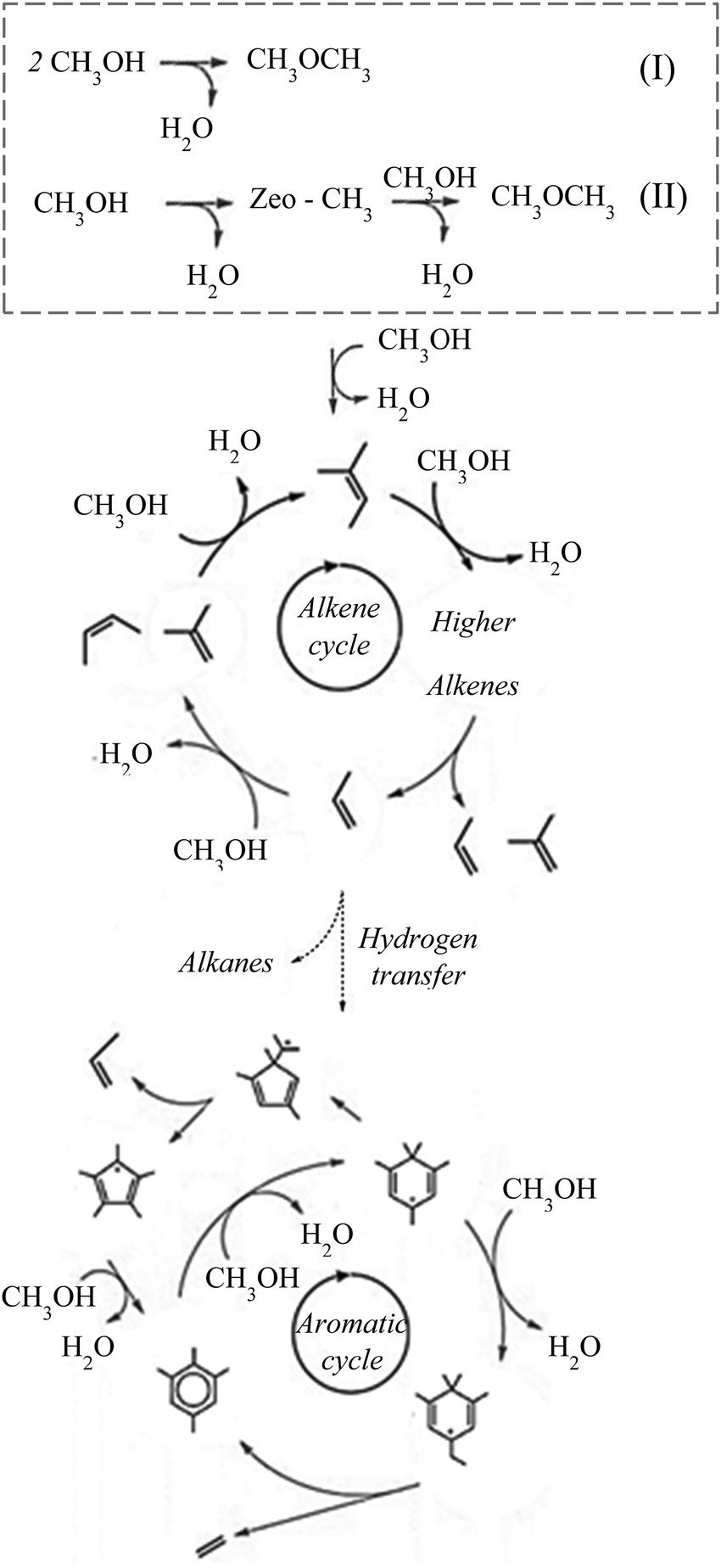 | ||
| Fig. 6 Illustration of the dual cycle mechanism of the MTH process. The initial stage is highlighted in a dashed box illustrating the two main methanol conversion routes (top): direct formation (I) and indirect formation, via methoxy (II), of dimethyl ether, alkene cycle (middle), aromatic cycle (bottom). | ||
In the olefin cycle, primary olefins are methylated or oligomerised in to higher homologues that subsequently undergo catalytic cracking to complete the reaction cycle. It is important to note here that these are not catalytic cycles, where the effects of adsorption, desorption, dissociation and diffusion are decoupled,80 but instead the olefin and aromatic cycles are given as reaction cycles only. Higher olefin (C6+) homologues will undergo hydrogen transfer and cyclisation reactions to form the first aromatic building block, which themselves undergo methylation to higher homologues and subsequently undergo dealkylation to give lower aromatics and small olefins. The small olefins re-propagate the alkene cycle, continuing the reaction.
Other types of zeolite and zeo-type catalysts (e.g., H-SAPO-34, H-ZSM-22, H-Beta, H-Y) are used for MTH conversion4 are suitable for investigating the effect of pore structure on the reaction mechanism. Evidently, the dominant cycle and nature of the hydrocarbon pool depends on zeolite or zeo-type topology and process conditions.
Over H-ZSM-5 catalysts, lower methylbenzenes (toluene, xylene and trimethylbenzenes) are the main hydrocarbon pool species, whilst higher methylbenzenes (tetra-, penta-, and hexamethylbenzenes) are the main hydrocarbon pool species over H-Beta catalysts, both at atmospheric conditions. Over H-SAPO-34 zeo-type catalysts, hexa- and hepta-methylbenzenes have been observed as the main hydrocarbon pool species.77,81,82 On H-ZSM-22 catalysts, no aromatics have been observed in the gas phase at low contact times during MTH conversion, as these process conditions (673 K, atmospheric pressures) limit side reactions.63–65 At higher contact times, however, trimethylbenzenes, ethylbenzene and ethyl toluene, as well as other aromatics, are occluded in the pores of the H-ZSM-22 catalyst, showing an operative “hydrocarbon pool” mechanism. On all the aforementioned catalysts, there is limited knowledge of the most abundant reactive intermediate in the “hydrocarbon pool”, and the variation of the pool with the catalyst pore architecture and process conditions. Knowledge of the hydrocarbon pool specie could simplify e.g., the computational modelling of the MTO process.
Both reaction cycles in the “hydrocarbon pool” mechanism either work distinctively or in concert to control product distribution. For example, the aromatic cycle controls product distribution over H-SAPO-34, and the olefin cycle controls product distribution over H-ZSM-22.4 For H-SAPO-34, pore cages are much wider than pore windows, such that the larger hexa- and hepta-methylbenzenes do not easily move out of the pores but dealkylate/crack to give gaseous light olefins through the aromatic cycle; in contrast, for H-ZSM-22, pore windows and intersections are small and prevent entry of large aromatic species, leading to olefin release through the olefin cycle.
The control of product distribution over H-ZSM-5 and H-Beta catalysts is tuneable as both catalysts have pore windows and intersections that are wide and accessible to olefins and aromatics. The tunability of both cycles in these catalysts allow investigations into kinetic effects, as well as steric hindrance, during MTO conversion.
For H-ZSM-5 catalysts reacting in the steady-state phase, the lack of quantitative information presents a major challenge for understanding the relative propagation of both cycles, and knowledge of kinetic factors that govern product distribution. H-Y provides a framework with very large pores but, due to its weak Brønsted acid sites, its methanol conversion rate is four times lower than H-ZSM-5, producing primarily C1–3 hydrocarbons rather than any aromatic species.
For the MTH process, the high reactivity of zeolite catalysts has been observed to facilitate formation of undesirable side products. In some part, side reactions due to rapid methylation by methanol and/or dimethyl ether can be suppressed by reducing the acidic character of the zeolite. In particular, weakening the strength or concentration of the acid sites as well as “directing” the reactions towards the desired products, by varying the pore size, results in suppression of side reactions. Due to the molecular size of zeolite pores, and the large number of topologies available, the structure of the hydrocarbons can be “moulded” to a specific type through careful framework choices.
Product shape selectivity is evidenced by a comprehensive study at 673 K and 80% methanol conversion, which highlights that 1D large pore zeolites, such as ZSM-22 (10MR TON) and ZSM-23 (10MR MTT), can be used to obtain linear C5+ aliphatics without formation of any aromatic products. For aromatics, 10MR and 12MR zeolites, such as H/Na-ZSM-5 (10MR MFI), 1D H-Mordenite (12MR MOR) and H-Beta (12MR BEA) are more appropriate; the product selectivity for these zeolites is ∼60% C1–C5 aliphatics, 10–30% C6+ aliphatics and 10–20% aromatics.52,83
Bleken et al.84 noted that the acid strength can also determine the functional lifetime of the zeolite catalysts. The pores get blocked by excess coke determined by the high reactivity of the acid sites. On comparing the activity of two CHA topology-materials (the silico-alumino-phosphate H-SAPO-34, and H-SSZ-13 of similar crystal size and acid site densities but different acid strength), a H-SSZ-13 with high acid strength yielded the most coke and deactivated fastest. Similar results were observed by Olsbye et al.6 for two zeolite catalysts with similar topologies but different acid site densities; the more acidic ITQ-13 and IM-5 had higher conversion rates (for butane/butene and C6+ aliphatic/aromatic) than the less acidic ZSM-22 and ZSM-23, but also deactivated faster.
3. Influence of aluminium distribution and the nature of acid sites on primary olefin formation
Distribution of aluminium in zeolites varies with Si/Al ratio.85,86 Dědeček and co-workers observed that Al atoms are distributed in the pore channels and intersections as single atoms or pairs. With a higher Al content, zeolites have a higher predominance of Al pairs that sit at pore intersections, and single Al atoms mostly sit on pore channels and windows, though the distribution depends also on the hydrothermal synthesis methodology.Adsorption of probe molecules such as pyridine, carbon monoxide, and other amines, can be used to investigate the location of Al atoms.87,88 H-ZSM-5 is known to have bridging hydroxyl groups, silanol groups, and coordinated unsaturated Al3+ ions present in its structure. The bridging hydroxyl groups are located within the internal zeolite channels, while the unsaturated Al3+ are located on the external zeolite surface as extra-framework cations. The bridging hydroxyl groups act as Brønsted acid sites that donate protons, while the weakly acidic coordinated Al3+ ions, and the extra-framework Al atoms, act as Lewis acid sites that accept electrons. Extra-framework material is composed of small particles mostly containing Al cations complexed by OH groups but sometimes also involves silicate species interacting with framework walls, either in the cavities or on the external surface. The detection of octahedral Al ions by 27Al NMR is evidence of extra-framework material,89as Al ions in Brønsted acid positions most commonly have tetrahedral coordination.90
Protonic zeolites can have high acid strength because the Al–O bond is weak. Interaction of protonic zeolites with a base strengthens the bond, subsequently allowing for cation solvation. The 3D solvation occurs within the zeolite cages, by van der Waals interactions with the walls of the pore channels. When the strength of the Brønsted acid site is higher, the conjugate base can stabilise a more negative charge.90
The internal acidic hydroxyl groups of H-ZSM-5 interact with alkanes, alkenes, and aromatics at low temperatures through hydrogen bonding. The hydrogen bonds follow the strength trend: olefins > aromatics > paraffins.87,91 Olefins undergo an important vibrational perturbation upon H-bonding, showing that the interaction involves C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-type orbitals. For aromatics, π-type electronic clouds are involved but partial steric hindrance weakens this interaction. The interaction with the localized π-type orbitals of olefins (bond order = 2) is stronger than that of the delocalised π-type orbitals of aromatics (bond order = 1.5). The strength of interaction increases with the number of carbon atoms of the aromatic i.e., p-xylene > toluene > benzene. The same occurs for olefins where the strength of interaction decreases in the following order: butenes > propylene > ethylene. Furthermore, Busca and co-workers91 showed that hydrogen bonds with alkanes involve C–C σ-type orbitals predominantly, and the strength of this interaction increases with carbon chain length.
C π-type orbitals. For aromatics, π-type electronic clouds are involved but partial steric hindrance weakens this interaction. The interaction with the localized π-type orbitals of olefins (bond order = 2) is stronger than that of the delocalised π-type orbitals of aromatics (bond order = 1.5). The strength of interaction increases with the number of carbon atoms of the aromatic i.e., p-xylene > toluene > benzene. The same occurs for olefins where the strength of interaction decreases in the following order: butenes > propylene > ethylene. Furthermore, Busca and co-workers91 showed that hydrogen bonds with alkanes involve C–C σ-type orbitals predominantly, and the strength of this interaction increases with carbon chain length.
The type of Al distribution can influence not only the olefin and aromatic cycles but also the initial stages of the MTH process.92 Experimental kinetic analysis comparing the dehydration rate of methanol in H-CHA and H-ZSM-5 determined that paired protons are an order of magnitude more reactive than isolated ones. Furthermore, the indirect reaction pathway of methanol (via methoxy) to dimethyl ether is inhibited when employing isolated active sites, whereas the production of methoxy species is the primary dehydration pathway in the case of paired Al sites. Meta-dynamics simulations highlighted that the activation barrier to methoxylate H-ZSM-5 is lowered by ∼40 kJ mol−1 in presence of paired protons, as opposed to a single acid site, validating the crucial role of Al distribution in the MTH process. Though steam de-alumination occurs above 700 K, water molecules break the Al–O bonds releasing Al(OH)3 out of the framework, thus producing different types of condensed and non-condensed extra-framework aluminium.93 Although still debated, the condensed species can be characterised as an Al2O3 phase, mostly formed at the zeolite surface. Non-condensed extra-framework aluminium defects can be represented by mono-nuclear species such as Al(OH)2+, Al(OH)2+, AlOOH or other proposed clusters of multi-nuclear species such as [Al3O4H3]4+.
At lower temperatures (383 K) and hydration conditions, additional types of defects can be encountered in the form of framework associated aluminium. These species are aluminium partially dislodged from the framework, having one or more –OH or –OH2 groups coordinated to the cation. Both extra-framework and framework-associated Al can be found in H-ZSM-5.94
The impact of extra-framework and framework-associated Al on adsorption and catalysis is not fully understood. Previous studies reported that extra-framework Al can lower the activation barrier for propene cracking or isomerization of glucose to fructose. There is a synergetic effect from the two types of acid sites when considering hydrogen transfer processes involving methanol, which leads to an enhanced selectivity for small alkanes and aromatics.95 Specifically, when both methanol and olefins adsorb on the Lewis acid sites (LAS), a proton is transferred from methanol to the olefin, resulting in the formation of an alkane and formaldehyde. Afterwards, formaldehyde is adsorbed on Brønsted acid sites (BAS) and reacts with another olefin to form dienes, which themselves oligomerise and cyclise to produce aromatics (Fig. 7).
 | ||
| Fig. 7 Reprinted from ref. 94. Copyright 2016 American Chemical Society. BAS is Brønsted acid sites. LAS is Lewis acid sites. | ||
The presence of LAS was found not to enhance the acidity or the activity of BAS in the previously mentioned experiments involving methanol; the limited influence of the LAS in this instance could be due to the distance between the two types of sites. Computational studies96 show that dimethyl ether adsorbed on BAS separated by two T-sites from any LAS would enhance the reactivity of dimethyl ether, and mediate an energetically favourable C–C bond formation of ethylene via methane and Al–OCH2 intermediates. Specifically, dimethyl ether adsorbed on BAS could convert into a methanol adsorbed on BAS and Al–OCH3 (LAS-CH3) with a Gibbs free energy activation (ΔGact) of 26.7 kJ mol−1 at 573 K. Afterwards, the LAS-CH3 donates a hydride to methanol to form methane (ΔGact = 29.8 kJ mol−1) and a formaldehyde like species, Al–OCH2, which combines with methane to form ethylene (ΔGact = 30.2 kJ mol−1). Since controlling the position of the acid sites is challenging, investigations are needed to clearly identify the role of LAS on methanol activation.
4. Challenges in MTO conversion
As detailed in the previous sections, there is a considerable understanding on the structure and composition of the zeolite environment, in terms of topology, acidity and their effects on the induction period and production distribution; however, significant knowledge is still lacking on the reaction mechanisms that occur inside the zeolite pores, and the ability to control the corresponding kinetics of the overall process.Although the hydrocarbon pool mechanism that occurs during atmospheric steady-state MTH conversion is widely accepted, the formation of primary species during the early stages has been extensively debated. The dehydration reaction, when two methanol form dimethyl ether and water, is predicted to be the fastest reaction during steady-state MTH conversion.97 In understanding the early stages of the reaction, three key challenges exist: (i) identifying the key oxygenate, (ii) validating the first and/or major olefin, and (iii) deciphering the pathway that leads from the key oxygenate to the first olefin. The first challenge deals with identifying the source of methylating agent and the key oxygenate (methanol or dimethyl ether) during MTH conversion. Methanol, dimethyl ether, and water compete initially for active sites on the zeolite catalyst. Although methanol and dimethyl ether can methylate olefins or aromatics (Fig. 6), the source of the methylating agent has only been identified recently.34
The second challenge deals with identifying reaction intermediates that lead to the first C–C bond and primary olefins, as well as the conditions that determine their reaction rates. Spectroscopic studies identify the first C–C bond by monitoring species occluded in the zeolite either during the reaction (operando) or after the reaction (ex situ) while kinetic studies observe the primary gaseous olefin(s) through a gas chromatograph. Much debate has centred on whether ethylene and/or propylene are the primary products98–100 though, in the earlier kinetic studies spanning the 1980s and 1990s, the structure of the zeolite catalyst was not considered. The studies did not describe whether experiments were conducted under intrinsic kinetic conditions, mass transport or heat transport limitations; such considerations are necessary as the release of ethylene has recently been mechanistically linked to the formation and propagation of the aromatic cycle11 as shown in Fig. 6.
Under steady-state atmospheric conditions, heavy aromatics can clog zeolite pores, which makes the release of large hydrocarbons severely constrained. Small molecules, such as ethylene, can be produced under such mass transport limitations. If such transport limitations do not exist, paring and side chain mechanisms (Fig. 8) regulate the formation of olefins, albeit after the “hydrocarbon pool” is established.
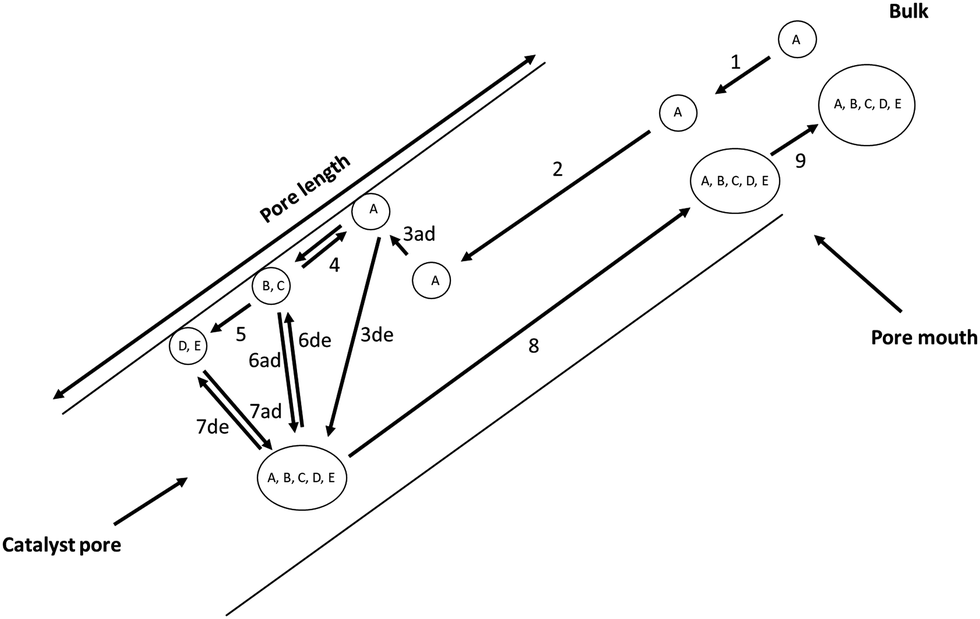 | ||
| Fig. 8 An overview of interrelated processes occurring during the conversion of methanol to olefins over zeolite catalysts. A, B, C, D, and E represent methanol, dimethyl ether, water, ethylene, and propylene respectively. Step 1 is external mass transfer of methanol from bulk to the pore mouth, step 2 is the internal mass transfer, 3ad and 3de represent adsorption and desorption of methanol, step 4 is the equilibration reaction yielding dimethyl ether and water. Step 5 is the formation of primary olefins from the equilibration mixture, steps 6ad and 6de, and 7ad and 7de represent the adsorption and desorption of the dimethyl ether and water as well as ethylene and propylene, respectively. Step 8 is the internal mass transfer of educt, intermediates and products to the pore mouth and step 9 is the external mass transfer of educt, intermediates, and products to the bulk. The pore length regulates the rates and diffusion of the species in the aforementioned processes, leading to the specific catalytic activity for MTO conversion. The elementary steps involving adsorption, desorption and reaction are studied through transient kinetic methods (TPD, TPSR, Step-response) and quasi-elastic neutron scattering studies for mobility. N.B. The formation of surface methoxy species and/or adsorbed species constituting the hydrocarbon pool are not represented here. | ||
Recent synchrotron operando micro-spectroscopic studies with a temporal resolution of 0.25 s provide evidence for the formation of the first alkenes directly from methoxy groups formed at the acid sites. These alkenes either desorb or oligomerise and cyclize to form dimethylcyclopentenyl cations, which are the first major components that precede aromatic hydrocarbons and lead to indirect alkene formation.101 The indirect formation of alkenes has been described as “secondary alkene formation” following transient experiments conducted in a temporal analysis of products (TAP) reactor.26
The last major challenge during MTH conversion relates to the exact pathway between the key reactant and the primary olefin(s). More than 20 mechanisms have been proposed; however, there exists no consensus. The species with the first C–C bond may be formed and occluded in the pores of the zeolite before the gaseous primary olefin(s) is released. For the numerous mechanisms, spectroscopic studies and quantitative determination of surface coverages are required to validate the intermediates formed. Shortly after the start of the millennium, two major routes were suggested: (1) a direct mechanism, where olefins are generated directly from the methanol and/or dimethyl ether feed, and (2) an indirect mechanism, where impurities in the methanol or dimethyl ether feed (such as ethanol and acetone) are precursors to the hydrocarbon pool, which subsequently lead to primary olefin formation. Recent evidence supports the direct formation of olefins that serve as initial precursors for the formation of aromatics (and other cyclopentenyl species) before the initialisation of the hydrocarbon pool.35
In addition to the three major challenges, the possibility that the zeolite could be altered during the induction period should be considered. This modification, particularly during MTH conversion under industrial conditions, where large quantities of steam are generated for extended periods at high temperatures, could alter the acidic properties of the zeolite leading to formation of extra-framework Al and enhanced Brønsted acid sites.102 Enhanced Brønsted acid sites are formed following intense activation conditions during zeolite steaming, and result in the generation of a high number of carbenium ions. These extra-framework Al and enhanced Brønsted acid sites could change product distribution, and the propagation of competing mechanisms during the induction period and at steady-state. During laboratory experiments, efforts are made to reduce the reaction time of the zeolite catalyst to prevent such acid site modification. The presence of steam, template removal conditions, activation temperature, pressure, and the heating rate (resulting in gradients) during activation determine the precondition of the zeolite catalyst before the onset of the MTO reaction.
In summary, in addition to adsorption, desorption and surface reactions, diffusion of species in the porous solid is important and should be considered. The evolution and modification of active sites as molecules adsorb, desorb, diffuse, and react makes the quantitative description of MTH conversion very challenging. Steam generated during the initial reaction can alter the dynamics of the induction period; for instance, steam alters the reaction rate and deactivation rate of H-ZSM-5 catalysts during the MTO or dimethyl ether-to-olefin (DTO) conversion reaction.74,103 Not only is the nature of the hydrocarbon pool unclear, but also understanding is needed of the hydrocarbon pool mechanisms, such as proton transfer, hydride transfer, rearrangement, reformation, and co-ordination of the active sites influence the mechanisms occurring in the dual cycle.
Experiments should be conducted under intrinsic conditions (without masking transport limitations, rapid methylation, or deactivating conditions) to understand the behaviour of the zeolite catalyst under each isolated phenomenon and for each species. Thereafter, a microkinetic model describing catalyst behaviour (adsorption, desorption and reaction of species) can be constructed, and co-joined with a diffusion model to emulate industrial catalysts. The microkinetic model and diffusion model can then be placed in an energy, momentum, and mass balance of a reactor model, which can be combined with a fluid dynamics model, and scaled to optimum industrial conditions and used in the design of process plants for MTH conversion.
There is currently no proof of a hydrocarbon pool mechanism operating under steady-state vacuum conditions. The induction period is marked by a regime where oxygenates undergo equilibration reactions and convert to primary olefins, and the transition regime encompasses the chemistry that conditions the catalyst from an initial state to a working steady-state where the hydrocarbon pool dominates.26 A mechanistic approach towards understanding the product distributions obtained during MTH conversion can only come from validating the reaction mechanisms that take place and obtaining accurate kinetic parameters that describe the process.4,104–109 Ultimately, understanding the initial transient stage and steady-state process can give further clarification of the relation between catalyst structure and product distribution, and lead to the design of superior catalysts. An understanding of the kinetic parameters that govern the MTH process can be used to tailor and optimise maximum throughput, selectivity, and reactor design.
In view of these challenges, a useful strategy is to thoroughly investigate the adsorption, desorption, surface reaction and mobility of early species (Fig. 8).
4.1. Adsorption and desorption during the induction period of methanol conversion
During the MTH process over H-ZSM-5 catalysts, methanol initially undergoes a rapid equilibration reaction leading to the formation of dimethyl ether and water. Readily available oxygenates (methanol and dimethyl ether) compete for active sites.97 Several pieces of theoretical work have considered the adsorption energy of initial species over H-ZSM-5 catalysts. Density functional theory calculations of the adsorption of one methanol molecule onto an active site give adsorption energies in the range of 75–126 kJ mol−1.38,110–112 Blaszkowski and van Santen showed that the simultaneous adsorption and activation of two methanol molecules is the preferred pathway for the formation of dimethyl ether and water when excluding surface methoxy group formation; however, surface methoxy groups have been readily observed with stopped flow NMR studies over H-ZSM-5 catalysts.113 These surface methoxy groups can be formed after the adsorption of methanol or dimethyl ether. The presence of surface methoxy groups, which could validate computational studies, can be linked to the dissociative adsorption behaviour of oxygenates, respectively and the presence of single or paired Al atoms. Previously, methanol and dimethyl ether were lumped in kinetic modelling studies,4,114,115 which excluded consideration of both species having different interactions with the active sites of the H-ZSM-5 catalysts, and avoided the mechanistic descriptions necessary for a microkinetic model.H-ZSM-5 catalysts with Si/Al ratios of 25 and 36 exhibit three types of adsorption site (“low”, “medium”, and “high” temperature sites), while there is no difference between medium and high temperature sites at a higher Si/Al ratio of 135 (Table 1). Molecular adsorption on the low temperature site and dissociative adsorption on the medium and high temperature sites match closely between experiment and a microkinetic model.34 The activation energy for dimethyl ether desorption was systematically higher than that of methanol i.e., 121 vs. 112 kJ mol−1 on strongest binding sites.34 Adsorption stoichiometry shows that methanol and dimethyl ether form clusters at the binding sites.
| Catalyst | Si/Al ratios | E des,experimental(methanol) | ΔHads,DFT(methanol) loading (mpuc) = 1 |
E
des,experimental(dimethyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ether) ether)
|
ΔHads,DFT(dimethylether) loading (mpuc) = 1 |
|---|---|---|---|---|---|
| a Energies were obtained only for medium temperature site. E des – activation energies of desorption (kJ mol−1).mpuc – molecules per unit cell.The activation energy of desorption is the sum of the heat of adsorption and the activation energy of adsorption: Edes = −ΔHads + Eads.122Citations are given in brackets.Level of theory:38,39 cluster: QM/MM TZVP, DFT, B-97-D, Hill and Sauer forcefield;123,124 PBC: PAW, DFT, PBE-D2 corrected with MP2;116 PBC: PAW, DFT, PBE-D;117 PBC: QM/QM: GPAW, MP2; DFT, PBE-D;119 cluster: ONIOM(DFT, B3LYP/dgtzvp:HF/dgtzvp) for energy and ONIOM(DFT, B3LYP/dgtzvp:MNDO) for geometry optimisation;121 cluster: DZVP, DFT, BLYP. | |||||
| ZSM-5 | 25 | 92.0–112.034 | — | 103.6–121.034 | — |
| 36 | 93.1–115.034 | — | 103.5–119.234 | — | |
| 135a | 94.434 | 100–12638 | 11034 | 107–13239 | |
| 94–116116 | |||||
| 115117 | |||||
| 100120 | |||||
| H-Y | — | 96–10638 | — | 10539 | |
| 89–104116 | |||||
| SAPO-34 | 64118 | 86119 | 45120 | — | |
| 80120 | |||||
| ZSM-22 | — | 107116 | — | — | |
| AlOH–(SiOH) | — | 75121 | — | — | |
The dimethyl-methanol-to-olefin turbulent fluidised bed reactor runs uses a methanol feed of 5.5 × 106 kg day−1, a catalyst inventory of 45000 kg of H-SAPO-34 catalyst, with a space-velocity of maximum 2 m s−1, temperature of 723 K.18 At these temperatures, and under close to vacuum conditions, dissociative adsorption of methanol and dimethyl ether dominates in a temporaral analysis of products (TAP) reactor. Kinetic experiments conducted in the TAP reactor on a H-ZSM-5 crystal, at vacuum conditions, could be considered close to the density functional theory calculations and complement this work; however, in the TAP reactor, and other atmospheric reactors, it is important to consider the influence of limiting conditions such as heat, mass, and momentum transport.6,123,125 The accuracy of density functional theory calculations allow for consistent predictions that could be used in the catalyst screening. In Table 1, the activation energies obtained for the medium and high temperatures sites are given for macroscale studies, while density functional theory values are provided for adsorption enthalpies.
Theoretical QM/MM simulations – with the quantum mechanical (QM) region modelled using the density functional theory (DFT) method with B97-D exchange correlation functional, and the Hill and Sauer forcefield for the molecular mechanical (MM) region38 – and periodic DFT simulations with the PBE-D exchange correlation functional117 agree with experimental results, suggesting adsorption energies of 92–115 kJ mol−1.34 Theoretical modelling of specific H-ZSM-5 active sites in the channels, with the representative topologies of the main zeolite intersection (T12), zig-zag channel (T4) and straight channel (T1), show that the T12 and T4 sites have the greatest adsorption energy, with a difference of ∼10 kJ mol−1 compared to T1 for a single methanol adsorption. Adsorption of dimethyl ether is ∼15 kJ mol−1 stronger than methanol, and a slightly contrasting trend is observed; specifically, the intersectional T12 site has a considerable stabilising effect towards dimethyl ether over the channel sites, with ∼25 kJ mol−1 difference. In addition, dimethyl ether was observed to spontaneously deprotonate T12, in contrast to single methanol adsorption. Molecular dynamics simulations with both dimethyl ether and methanol adsorbed on the same acid site show that the adsorbate cluster was very stable, regardless of whether it was bonded through methanol or dimethyl ether. Additional meta-dynamics analysis determined that an activation energy of 27 kJ mol−1 is necessary for the two adsorbates to switch places on the acid site.
4.2. Surface chemistry occurring during methanol conversion
The chemistry of steady-state MTH conversion have been briefly described by Ilias and Bhan.4 The process includes olefin methylation, olefin cracking, hydrogen transfer, cyclisation, aromatic methylation, and aromatic dealkylation. Different reaction phenomena can occur at higher temperatures, when methanol transformation is tuned towards olefin formation, such as paraffin cracking and hydrogen transfer between paraffins and olefins.As various mechanisms can account for the same kinetic data, the verification of reaction mechanisms through spectroscopy is very important (Table 2). Recently, Oyama et al.126 developed a method for scrutinizing adsorbed intermediates by in situ spectroscopy through analysis of coverage transients (ACT).
| Mechanism | Spectroscopic method | Accuracy/resolution | Nature of spectroscopic study |
|---|---|---|---|
| a Experiments were not conducted under ultra-high vacuum (UHV) conditions, technique included here for completion purposes. | |||
| (A) Oxonium ylide | N/A127 | — | — |
| (B) Carbene | Synchrotron infrared micro-spectroscopy101 | 0.25 s | Qualitative |
| (C) Methane-formaldehyde | Temperature programmed desorptiona128,129 |
— | Quantitative |
| (D) Carbocation | — | — | — |
| (E) Radical | Electron paramagnetic resonance130,131 | Qualitative | |
| (F) Methoxy methyl | Infrared132 | — | Qualitative |
| (G) Carbon monoxide | Infrared40 | 4 cm−1 | Qualitative |
Dimethyl ether and water are initially formed at high rates of methanol reaction over H-ZSM-5.3,6,141 The dehydration reaction, when two methanol molecules form dimethyl ether and water, is the fastest reaction during MTH conversion, and methanol and dimethyl ether are present simultaneously at less than 100% methanol conversion.97 By using an in situ stopped flow cross-polarisation magic-angle spinning nuclear magnetic resonance (CP-MAS NMR) spectroscopy method, Hunger and co-workers observed surface methoxy species and proposed a route to form dimethyl ether via these surface methoxy species: an adsorbed methanol molecule converts to a methoxy species, which subsequently reacts with another methanol molecule to form dimethyl ether.142,143 The surface methoxy species are proposed to act as precursors for carbene and ylide intermediates, which are involved in direct methanol conversion at temperatures ≥ 523 K, because the C–H bonds of the surface methoxy groups are weak and hydrogen can be readily abstracted by the basic oxygen atoms of the framework.101,144,145 The surface methoxy groups react with water to form methanol, and with methanol to form dimethyl ether.113,145 At exceptionally low temperatures, signals corresponding to Brønsted acid sites, methoxy groups, adsorbed methanol, and dimethyl ether have been obtained from 1H MAS NMR and 13C MAS dimethyl ether studies. Surface methoxy groups are readily observed during MTH conversion and are also important in the mechanisms of direct olefin formation from methanol.52,113,145 For the mechanism involving surface methoxy intermediates, the rate limiting step is the dehydration step involving the first methanol molecule;146 here, the methoxonium ion that was formally proposed to be an intermediate was found to be a transition state.
In contrast, Blaszkowski and van Santen147 concluded that a concerted, single step mechanism of two methanol molecules is highly favoured over the mechanism involving a surface methoxy group as an intermediate, when using cluster DFT calculations. They obtained an activation barrier for dimethyl ether formation of 145 kJ mol−1 when two methanol molecules adsorbed simultaneously on a Brønsted acid site.146 Extended cluster models produce lower activation barriers of 132 kJ mol−1,97 whilst non-local periodic DFT calculations over chabazite give activation barriers of 119 kJ mol−1 for the same mechanism.148
4.2.3.1. Indirect mechanism. The indirect mechanism was originally proposed by Dahl and Kolboe over H-SAPO-34 catalysts, and was further described as the formation of scaffolds, which are composed of well-defined organic species (polymethylbenzenes) on one or more inorganic acid sites that preserves the organic components and regulates selectivity through steric constraints.149 Earlier research showed that the hydrocarbon pool operates through aromatic methylation and dealkylation chemistry. These reactions lead to the release of light olefins during MTH conversion over H-SAPO-34 catalysts.
Two different dealkylation reactions have been proposed for the indirect mechanism, namely: (1) a side chain methylation mechanism162–165 and, (2) a paring mechanism166 (Fig. 9); however, the dominant mechanism during MTH conversion for the aromatic dealkylation is still debated.167 Both mechanisms (Fig. 8) are initiated by the germinal methylation of a methylbenzene, and the activation energy for gem-methylation decreases as the number of methyl groups on the benzene ring increases.168 In the side chain methylation mechanism, an exocyclic double bond forms after elimination of a methyl hydrogen; this double bond can undergo side chain methylation, which further cracks to form ethylene or propylene.4,33,169 In contrast, during the paring mechanism, the gem-methylation of a methylbenzene results in ring contraction, leading to the formation and release of propylene4,169 or isobutene.151 Meta-dynamics and static calculations for the methylation of alkenes with methanol, dimethyl ether, and methoxy species showed that the reaction rates are similar between each species employed.44 For instance, a high methanol loading can lower the reaction barrier by ∼30 kJ mol−1 when methylating benzene and can lead to the spontaneous methoxylation of the H-ZSM-5 at 300 K.170–172
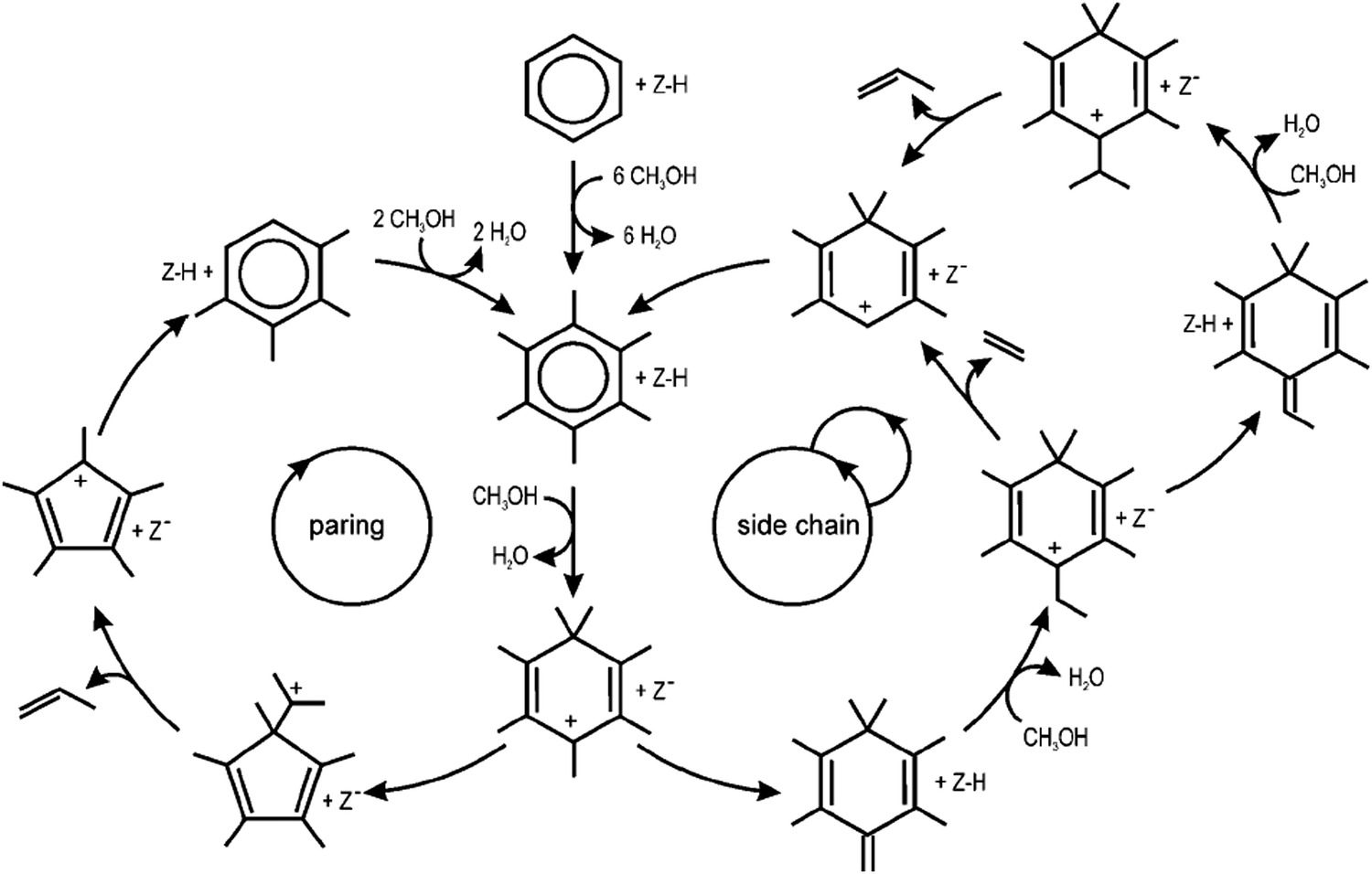 | ||
| Fig. 9 Schematic representation of the paring and side-chain reaction concepts in MTO catalysis. Reprinted from ref. 169. Copyright 2009. Wiley-VcH Verlag GmBH & Co. | ||
Haw and co-workers reported the formation of methylbenzenes during MTH conversion over H-SAPO-34 catalysts after four seconds of reaction time, as studied using 13C NMR spectroscopy at 673 K. Polymethylbenzenes were observed as trapped in the zeolite cages, and were assumed to form from impurities in the methanol feed, trace impurities in the carrier gas, and incomplete combustion of organics on the catalysts.150 Surface methoxy groups were observed and their concentration decreased during the reaction, indicating a role as reactive intermediates. The surface methoxy groups were shown to be reactive at lower temperatures (433–523 K) than the starting temperature of steady-state (573 K) in the MTO process. Specifically, at temperatures above 433 K, methoxy groups are prone to methylate aromatics, whereas above 523 K, methoxy groups alone can form hydrocarbons.145 If any organic impurities were present on solid acid catalysts, they should react with the surface methoxy groups at low temperatures, but this work did not show any reactivity of surface methoxy groups at temperatures ≤ 473 K, thereby raising doubts on whether the impurity route leads to aromatics that can then produce olefins through a dealkylation reaction. Moreover, the quantity of impurities (1 ppm ethanol and 11 ppm organic compounds) may be too small for the initial formation of polymethylbenzenes.
Haw and co-workers151 also combined theoretical studies, using extended cluster models, with experimental clues of the existence of a 5-membered ring cation (1,3-dimethylcyclopentadienyl)173 and a 6-membered ring cation (1,1,2,4,6-pentamethylbenzenium)174 to show the co-conversion of toluene with methanol to give isobutene through a paring mechanism over H-ZSM-5 catalysts. The paring mechanism used to explain the co-conversion of toluene with methanol for the initial carbon–carbon formation presents a major challenge for macroscale kinetic modelling, as the pre-existence of toluene in the methanol feed is difficult to realise.
Polymethylbenzenes formed over H-SAPO-34 catalysts, and alkyl substituted cyclopentyl carbenium ions formed over H-ZSM-5 catalysts, have been observed at relatively low contact times during MTH conversion.149,173 Polymethylbenzenes form olefins, observed at low contact times and conversions, through the aromatic dealkylation step. Conversely, using in situ13C NMR spectroscopy and gas chromatography, Goguen et al.161 observed ethylene traces during MTH conversion on H-ZSM-5 catalysts at reaction times of one second. Also, Kaeding and Butter observed ethylene formation at low conversions.98 Ethylene also forms alkyl substituted cyclopentyl carbenium ions at a reaction time of four seconds.173,175 These cyclopentenyl carbenium ions are intermediates for toluene formation (another postulated hydrocarbon pool species) over H-ZSM-5 catalysts. Derouane and co-workers99 observed ethylene at low conversions, using GC coupled with 13C NMR data, and concluded that the high reactivity of ethylene with Brønsted acid sites readily leads to carbenium ions.
In summary, olefins and aromatics have been observed as occuring after an initial C–C bond is formed from oxygenates (methanol, dimethyl ether). Where aromatics are formed initially from impurities in the feed, olefins form indirectly through an aromatic dealkylation chemistry; on the contrary, olefins can form directly from the methanol feed without accompanying aromatics in the initial stages.
4.2.3.2. Direct mechanisms. There is currently no consensus on the direct mechanism that leads to the formation of the first C–C bond. The direct formation of olefins from oxygenates was initially proposed through various mechanisms,3 which are identified by their key intermediate. Among the intermediates considered are the oxonium ylide, carbene, methane-formaldehyde, surface methoxy groups, and carbon monoxide. IR and 13C MAS NMR investigations of the conversion of surface methoxy species over acidic zeolites indicate that the initial C–C bond forms via the direct mechanism.143,145,176,177 Isolated surface methoxy groups can be rigidly bound on the catalyst surface and have a high thermal stability.113,159 Using IR and MAS NMR spectroscopy, surface methoxy species are characterised as present and directly coordinated to the aluminosilicate framework prior to the onset of hydrocarbon formation.142,143,178–180 Surface methoxy groups show high thermal stability below 200 °C, after which they start to form hydrocarbons themselves. Other direct mechanisms are described below.
A. Oxonium ylide mechanism. Mole and co-workers proposed the formation of oxonium-ylide species during MTH conversion at 573 K. Recent studies by Lesthaeghe and co-workers using extended cluster (46 T-sites) DFT modelling showed that the primary formation of ethylene over H-ZSM-5 catalysts could occur through two routes, starting from methanol or dimethyl ether (Scheme 1).
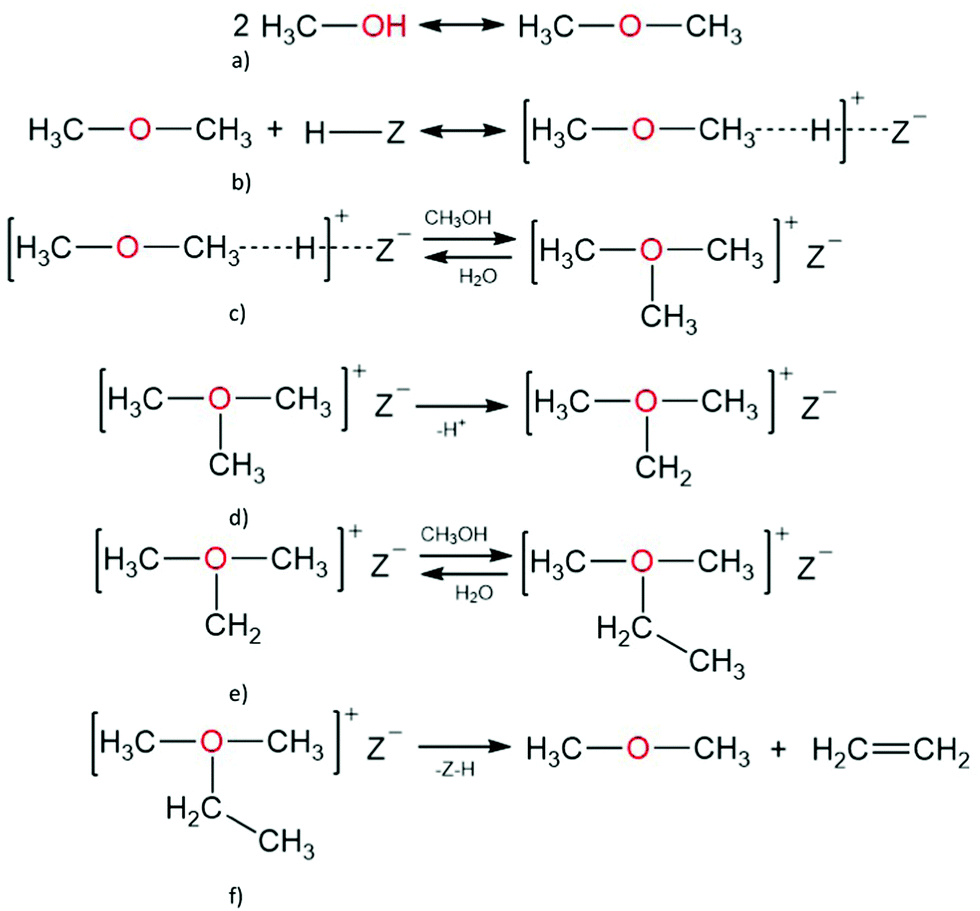 | ||
| Scheme 1 Ethene formation via the oxonium ylide mechanism, starting from (a) methanol and (b) dimethyl ether, followed by (c) dimethyl ether activation to form (d) trimethyl oxonium, which is (e) deprotonated and (f) converted to dimethyl ethyl ether resulting in (g) ethene and dimethyl ether. | ||
Following the equilibration of methanol, dimethyl ether, and water, dimethyl oxonium ions form directly from adsorbed dimethyl ether. Dimethyl oxonium ion can be deprotonated to obtain methyl oxonium methylide, which further undergoes a Stevens-type intramolecular rearrangement leading to ethanol and the first C–C bond. Alternatively, trimethyl oxonium ion can be formed from the co-reaction of dimethyl ether and methanol, co-reaction of two adsorbed dimethyl ether molecules, or reaction of a framework bound methoxy species with dimethyl ether.153 Using periodic DFT, trimethyl oxonium ion has been shown to be stabilised by the zeolite framework, having an adsorption energy of 70 kJ mol−1, and the dimethyl oxonium ion can also react with dimethyl ether to yield trimethyl oxonium ion, with a reaction energy (Er) of 22 kJ mol−1, and and activation energy of 132 kJ mol−1.154 A trimethyl oxonium ion can further undergo proton abstraction to yield dimethyl oxonium methylide, which can undergo a Stevens-type intramolecular rearrangement to form the first carbon–carbon bond, giving rise to methyl ethyl ether, or an intermolecular methylation to form ethyldimethyl oxonium ion; ethyldimethyl oxonium ion can then undergo β-elimination to form ethylene and dimethyl ether.
Using extended cluster models based on the ONIOM method, and using DFT, Lesthaeghe and co-workers showed that the formation of trimethyl oxonium and ethyldimethyl oxonium ions is stable on H-ZSM-5 catalysts; however, due to steric constraints, proton abstraction from trimethyl oxonium ion was deemed improbable.155 Tajima et al.181 and Lesthaeghe et al.155 showed, using cluster and extended cluster models, that the deprotonation steps from an dimethyl oxonium ion to a methyl oxonium methylide, and from an trimethyl oxonium ion to dimethyl oxonium methylide, are infeasible because both methyl oxonium methylide and dimethyl oxonium methylide can not be stabilised on the zeolite framework;148,154,155 periodic DFT calculations, considering the whole unit cell of the zeolite, could provide results that help draw conclusions on the stability of the species given, and the feasibility of the direct route. Using extended cluster models, and applying DFT, Lesthaeghe and co-workers concluded that the deprotonation steps from a dimethyl oxonium ion to methyl oxonium methylide, and from a trimethyl oxonium ion to dimethyl oxonium methylide, are non-existent;148,154,155 and that the formation of trimethyl oxonium and ethyldimethyl oxonium ions are stable on H-ZSM-5 catalysts. These studies148,155 show ethyldimethyl oxonium and trimethyl oxonium ions are feasible species in the reaction mechanism; however, because of the difficulty to stabilise the product states, their direct interconversion (by insertion of a carbene group) has not been investigated either experimentally or theoretically.155
B. Methoxy – carbene mechanism. The conversion of pure methoxy species directly to olefins and other hydrocarbons has been observed using 13C MAS NMR over H-Y, H-ZSM-5 and H-SAPO-34 catalysts at temperatures between 473–673 K.145 Methoxy groups were reported to aid formation of a wide range of hydrocarbons (predominantly paraffins, alongside olefins, aromatics), in addition to carbon monoxide and carbon dioxide, when working at 523 K.144,145 In order for methoxy species to react and form higher order hydrocarbons, the main reactants have to be in the vicinity of each other. Considering that only one methanol can convert on one acid site, the possibility of the C1 species migrating next to each other was considered. Theoretical QM/MM and metadynamics calculations show that it is energetically too expensive for methyl or carbene to migrate away from the active site, so the conversion of methanol outside of the active site was investigated; the investigations concluded that the activation barrier for methylation on a Si–O–Si bridge in H-ZSM-5 is three times larger than on the Al–O–Si site.182
As previously mentioned, the conversion of methoxy can initially produce carbon monoxide, carbon dioxide and iso-paraffins. Since carbon monoxide and carbon dioxide are formed, a viable route from methoxy would be via dehydrogenation and oxidation of a methyl group, leading to carbon monoxide and hydrogen, with the most viable intermediate being carbene.
Carbene was suggested as the dominant intermediate in the methanol to gasoline reaction following a decrease of iso-butane to n-butane ratio.183 There are two proposed routes for the carbene mechanism (Scheme 2). The first route involves the formation of carbenes via surface methoxy groups, and has been studied theoretically considering a methanol and dimethyl ether molecule, and from two methanol molecules.121 The conjugate base oxygen of the methylated acid site is the basic site for the deprotonation of the surface methoxy species that leads to carbene formation.121 In the second route, carbenes could be formed from α-elimination of water from methanol molecules. Here, the formation of carbene species is based on the proximity of two acid sites (one in conjugate base form), and the carbenes can undergo subsequent oligomerisation to form olefins, or by sp3 insertion of the carbene into the methanol or dimethyl ether in a free carbene mechanism.1,3,184
 | ||
| Scheme 2 Ethene formation via methoxy-carbene mechanism, starting from (a) methanol and (b) dimethyl ether to (c) methoxy converted to carbene, finally coupling with another carbene to form ethene. | ||
In addition to being highly endothermic, both routes do not account for the methyl ethyl ether that is observed in the conversion of methanol over H-ZSM-5 catalysts.99 The conjugate base of the zeolite is not strongly basic enough to abstract hydrogen from methanol.100 Using extended cluster approximations, the activation energy for carbene formation is calculated as 215–232 kJ mol−1.121,154 The formation of carbenes involves high activation barriers of similar magnitude to the transformation of the carbene itself,121 which suggests that ethylene formation via carbene polymerisation is unlikely.121,185
Hutchings and co-workers found the carbene proposal to be inconsistent with their observations of decreased methane/alkene ratios as conversion increased with contact times.100,185 Mihail and co-workers suggested that high methane concentrations observed at low conversions is due to the reaction of carbene with hydrogen, with hydrogen proposed to be available from the water gas shift reaction or methanol decomposition. Hutchings and co-workers checked for hydrogen formation through the water gas shift reaction over Na-ZSM-5 catalysts at temperatures between 623–673 K and found no evidence of this moiety. Here, methanol decomposition was studied over Na-ZSM-5 to avoid the complication of hydrocarbon formation, and no evidence was found for methanol decomposition at 523 K for a wide range of methanol feed rates; methanol decomposition was only observed to a limited extent at higher temperatures (623–673 K). These experiments demonstrate that methanol decomposition and water gas shift reaction are not viable sources of hydrogen for the formation of methane at low temperatures. Nonetheless, Kondo and co-workers,186 using IR spectroscopy, provided evidence for carbene-like intermediates on reaction between methoxy species and light alkenes.
C. Methane-formaldehyde mechanism. Methane and formaldehyde have been observed in similar amounts above 630 K and at low conversions,128 with less formaldehyde detected with increasing temperature and methanol conversion.12,128 Theoretically, there are multiple routes towards methane and formaldehyde formation, starting from: two methanol molecules, surface methoxy species and methanol, or a trimethyl oxonium ion adsorbed with methanol (Scheme 3).153,154,181 Once formed, the adsorbed methane and formaldehyde molecules could react to form ethanol, which subsequently undergoes a dehydration step to form ethylene; however, extended cluster models have shown this direct route to be improbable.154 The work suggested that the conversion of methane and formaldehyde proceeds too slowly for the mechanism to be suitable. However, recent studies, using periodic DFT calculations, suggest that the methane-formaldehyde mechanism could be possible with methane formation having an activation energy of 149.6 kJ mol−1 and ethanol formation to be 124.5 kJ mol−1.132 The mechanism may not be faster than ethylene formation, which occurs from ethanol impurities in the methanol feed through the indirect mechanism, due to the smaller number of steps.
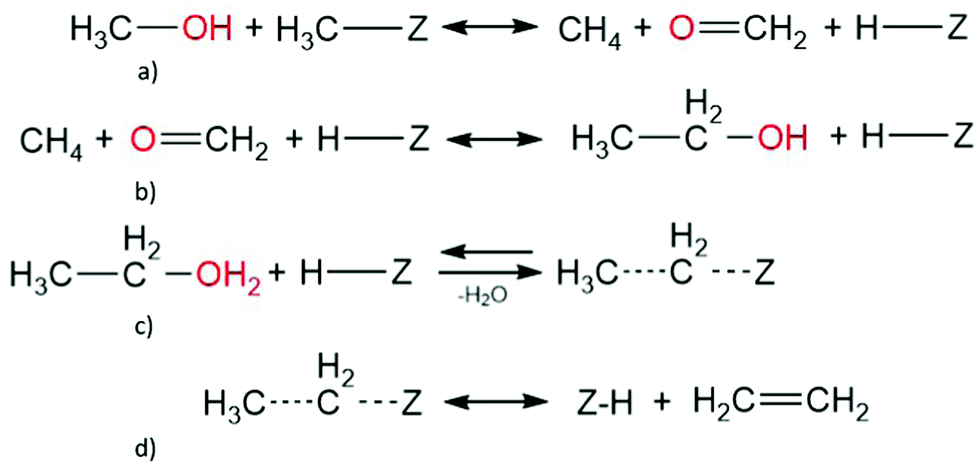 | ||
| Scheme 3 Ethene formation via methane-formaldehyde mechanism, with (a) methanol and methoxy form methane and formaldehyde, that (b) further convert into ethanol which (c) forms ethoxy and eventually (d) ethene. | ||
D. Carbocation mechanism. A carbocation mechanism was originally proposed by Olah and co-workers, and modified by Kagi, and involves methanol protonation in an acidic medium resulting in carbenium ion formation. The carbenium species react on vacant zeolite sites to form methyl ethyl ether, which can react with methyl carbenium ions supplied by protonated dimethyl ether. The resultant methyl isopropyl ether can react with a methyl carbenium ion or eliminate methanol to form propylene. Another carbocation route involves the formation of carbenium ions that react with methanol to produce higher ethers, which then decompose to give olefins;99,143 however, because the scission of the C–H bond in TMO has an activation barrier that exceeds 200 kJ mol−1,187 this route was deemed unfeasible.99,143
E. Radical mechanism. Free radicals were observed using electron spin and electron paramagnetic resonance during the conversion of dimethyl ether to hydrocarbons at temperatures above 344 K. Radicals were associated with solid-state defects in the H-ZSM-5 catalyst.130 A mechanism was proposed by Clarke et al.130 for the formation of the initial C–C bond that involves either direct coupling of radical species (methoxymethyl) or C–H bond insertion by methylene, generated by methoxy radical scission. These mechanisms were disputed by Hunter et al.185,188 who observed no effect of adding 1–3% NO, which is a known radical scavenger, during methanol/dimethyl ether conversion over Na-ZSM-5 at 523 K. Na-ZSM-5 was chosen because it is essentially inactive towards dimethyl ether under these conditions.
Chang et al.131 studied the effect of NO concentration (0, 450 and 5000 ppm) on zeolites with SiO2/Al2O3 ratios of 1800 and 70 at 723 K and 1 bar in a packed bed reactor. With lower acidity, addition of 5000 ppm NO completely suppressed hydrocarbon formation within 30 min. At 450 ppm, NO suppression required an induction period of 2 h. Similar behaviour was observed at higher acidity, albeit with a longer methanol breakthrough. They concluded that the catalyst was poisoned by a process involving radicals.131 The results of Chang et al.131 are in agreement with Clarke et al.130 but in contrast to Hunter et al.185,188
F. Methoxymethyl-ethoxy mechanism. Fan and co-workers132 provided spectroscopic and theoretical evidence for the direct mechanism for initial C–C bond formed from methanol over H-SAPO-34. They identified a methoxymethyl cation (CH3OCH2+) using IR spectroscopy as the primary reaction intermediate and, by using DFT calculations, they concluded that methoxymethyl is stable in the CHA cage of SAPO-34. CH3OCH2+ forms from the reaction of dimethyl ether and surface methoxy groups, with a relatively high activation energy of 135 kJ mol−1. Despite the slightly demanding formation path, CH3OCH2+ has been experimentally (spectroscopy) identified, and the following reaction paths are less energetically demanding: CH3OCH2+ combines with another dimethyl ether or methanol molecule to give 1,2-dimethoxyethane or 2-methoxyethanol, respectively (Scheme 4), with activation energies of 95 and 102 kJ mol−1, respectively. The latter components are methylated by methoxy species and decompose in a series of reactions to give propylene as the primary olefin.
 | ||
| Scheme 4 Ethene formation via the methoxymethyl/ethoxy mechanism, where (a) dimethyl ether polarises methoxy, which leads to (b) the production of methanol and ethoxy; (c) ethyl methyl ether and (d) ethanol convert to zeolite bound ethoxy that (e) produces ethene. | ||
Chowdhury et al.137 used solid-state NMR and UV/Vis diffuse reflectance spectroscopy to provide evidence for intermediates such as surface acetate, methyl acetate and dimethoxymethane during MTO conversion over H-SAPO-34. Fan and co-workers189 also showed a pathway involving the formation of CH3OCH2+. These studies highlight a new direction in MTO chemistry involving the role of secondary oxygenates. The evidence (DFT and spectroscopy) suggests that primary oxygenates (methanol, dimethyl ether) lead to secondary oxygenates (1,2-dimethoxyethane, dimethoxymethane, 2-methoxyethanol), which subsequently lead to primary olefins (ethylene and/or propylene). However, no steady-state isotopic kinetic evidence exists for the formation of 1,2-dimethoxyethane or 2-methoxyethanol, either on H-SAPO-34 or H-ZSM-5 catalysts.
In situ 13C solid-state nuclear magnetic resonance analysis coupled with periodic simulations indicated that the chemical shift at 69 ppm, detected when reacting methanol in H-ZSM-5 at 573 K, corresponded to an activated C–H bond on dimethyl ether, which could have been polarized by the interaction with a surface methyl species.190 The observation that ZeO–CH3 interactes with an activated methyl (–CH2–H) of dimethyl ether, while the main product is ethene, led to the conclusion that a methoxymethyl analogue (CH3–O–CH2–H) could be the main intermediate capable of explaining the transition from a C1 to C2 species.190
Further investigations on a continuous flow of 13C methanol inserted in H-SSZ-13 at 493 K provided the same NMR signal at 68.5 ppm, and additional ex situ analysis at room temperature using 13C CP/MAS NMR and 2D 13C–13C CORD spin diffusion MAS NMR captured in real time the signals corresponding to activated dimethyl ether (68.5 ppm) and surface ethoxy species (70.5 and 14.2 ppm), which were only present for the first 20 min of the reaction.191 In order to determine the energetics required in the reaction cycle (Scheme 4) involving activated dimethyl ether, ab initio metadynamics simulations have been employed to model the initial stages leading to ethene via methoxymethyl analogue and surface ethoxy species in H-SSZ-13.191 Thus, the free energy barrier for the C–C bond formation from dimethyl ether and surface methyl species was shown to be 142 kJ mol−1. Similarly, additional reactions of other hydrocarbon pools species that lead to ethene via surface ethoxy species were investigated, with ethanol and methyl ethyl ether converting to surface ethoxy species with a free energy barrier of 74 kJ mol−1 and 99 kJ mol−1, respectively. Finally, the deprotonation of surface ethoxy species to ethene was calculated to have a barrier of 103 kJ mol−1, which indicated an overall feasible reaction cycle.
G. Carbon monoxide mechanism. The carbonylation of methoxy groups by carbon monoxide was originally proposed by Jackson and Bertsch192 in 1990, and has regained prominence40,137 as carbon monoxide was recently shown to form slowly during the induction period, and reacts with surface methoxy species to form methyl acetate and acetic acid intermediates.40,192 Liu et al.40 postulated that these intermediates undergo a series of acid-catalysed reactions such as acetylation, decarboxylation, aldol condensation, and cracking to obtain surface bound hydrocarbons, the hydrocarbon pool and the first olefin (Scheme 5). The carbon monoxide mechanism is increasingly accepted as DFT calculations show that the carbonylation step has an activation energy as low as 80 kJ mol−1 and an exothermic reaction energy of 93 kJ mol−1,40 in contrast to the results highlighted previously, where acid catalysed steps involve prohibitively high activation energies. The intermediates observed here are similar to those involved with the indirect mechanism involving acetone impurities leading to primary C–C bond formation.193
 | ||
| Scheme 5 First C–C bond formation in MTH through coupling between nucleophilic and electrophilic carbon atoms. Adsorption of methanol or dimethyl ether on a zeolite Brønsted acid site forms a surface methoxy group. The methoxy group undergoes nucleophilic attack by carbon monoxide, forming a surface acetyl group which contains the first C–C bond. The acetyl group dissociates into methyl acetate and acetic acid in the gas phase. Energy barrier of the carbonylation step is 80 kJ mol−1. Reprinted from ref. 40 Copyright 2016. Wiley-VcH. GmbH & Co. | ||
As mentioned above, over H-SAPO-34 zeo-type catalysts, Chowdhury et al.137,138 provided evidence of the formation of surface acetate and methyl acetate, as well as dimethoxymethane, during the MTO process using a combination of solid-state NMR, UV/Vis diffuse reflectance, and mass spectroscopy. Chowdhury et al.137,138 further investigated the conversion of methyl acetate to olefinic and aromatic hydrocarbons using the same combination spectroscopic methods, and the combined pieces of work provide spectroscopic proof of the carbon monoxide carbonylation mechanism leading to methyl acetate, and later to olefins and aromatics. A major challenge now lies in bridging observed kinetics with spectroscopic studies. Furthermore, the pathway over the H-ZSM-5 catalyst may be different from H-SAPO-34, due to their different pore architectures. Following the work of Dědeček and co-workers,194 the siting of single Al atoms and Al pairs are different for pore channels, intersections, and cages; therefore, due to the different architectures of H-ZSM-5 and H-SAPO-34, the siting and varied distribution of Al species could affect the MTH chemistry differently. Additional studies195 were conducted with a two bed reactor, where carbon dioxide initially reacted in the first phase (Fe2O3/KO2) of the reactor to produce mainly carbon monoxide and methanol, and these were then inserted in to different types of zeolites (H-MOR and H-ZSM-5), though with similar MTH chemistry observed.
There are many detailed reviews and articles on the spectroscopic investigations into intermediates involved in each mechanism,40,48,130–132 and Table 2 gives information about the spectroscopic methods used to determine the intermediates in the key mechanisms mentioned and Table 3 provides a summary of the rate limiting activation barriers for each proposed mechanism above. The accuracy and/or resolution of these methods and the quantitative measurements of surface coverage are also provided.
| Pathway | First C–C bond | Primary Olefin | Barrier of key/bottleneck step in pathway obtained through experiments, kJ mol−1 | Condition under which primary C–C bond is formed (intrinsic, mass transport, heat transport) | Macrokinetics or microkinetics | DFT calculated rate limiting barrier in the pathway, kJ mol−1 | Cluster size | Catalyst |
|---|---|---|---|---|---|---|---|---|
| a Rate-parameters were calculated at 623 K. b Rate-parameters were calculated at 720 K. c 1st step = methane and formaldehyde formation. d 2nd step = formation of ethanol. e Cluster approximation and spin-restricted, gradient-corrected density functional theory (DFT). f Surface acetyl groups formed through the carbonylation of methoxy groups on zeolite acid sites. g Light olefin formation from dimethyl ether. h Light olefin formation from methanol. Route II TMO deprotonation resulting in the formation of DOMY (dimethyl oxonium methylide) and MEE (methyl ethyl ether) through Stevens’ rearrangement. bs: conjugate Lewis basic sites. | ||||||||
| Methane-formaldehyde | Ethanol | Ethylene | — | — | — | 149.6 (1st step)a,c | Periodic132 | H-SAPO-34 |
| 124.5 (2nd step)b,d | 5T153 | H-ZSM-5 | ||||||
| Methane-formaldehyde196 | Ethanol | Ethylene | — | — | — | — | — | H-ZSM-5 |
| Carbon monoxide/ketene | Surface acetyl groupf | Ethylene | — | — | — | 80 | Cluster – 74 atoms embedded in a field of 1481 force field particles40 | H-ZSM-5 |
| Radical131 | Direct coupling of methoxymethyl radicals or C–H bond insertion by methylene, generated by methoxy radical scission | Ethylene | — | — | — | — | — | H-ZSM-5 |
| Methoxymethyl cation | 1,2-Dimethoxyethane or 2-methoxy-ethanol | Ethylene, propylene | — | — | — | 135.1 (1st step)a,c | Periodic132 | H-SAPO-34 |
| Tri-methyl oxonium ylide (TMO) | MEE | Ethylene | — | — | — | 112–139 (route II) | 5T–46T155 | H-ZSM-5 |
| Carbene185 | Carbene leading to ethylene | Ethylene | — | — | — | 215–232 | Clustere,121 | H-ZSM-5 |
| Comparison of experiments and macroscale modelling results | ||||||||
| Direct107 | Through methane-formaldehyde route | Ethylene | 121 ± 18107 | Intrinsic | Single event microkinetics | — | — | H-ZSM-5 (Si/Al = 200) |
| Direct104,105 | TMO | Ethylene | 137.8bs | Intrinsic | Single event microkinetics | — | — | H-ZSM-5 (Si/Al = 200)105 |
| Direct198 | — | Ethylene | (11.3 ± 0.6) | — | Macrokinetics | — | — | H-SAPO-34198 |
| Propylene | (9.7 ± 0.75) | — | Macrokinetics | — | — | H-SAPO-34198 | ||
| Direct199 | — | Light olefins | (129 ± 48)g | Macrokinetics (lumped model) | H-ZSM-5 (Si/Al = 15) | |||
| (267 ± 33)h | ||||||||
| Direct199 | — | Light olefins | (87.9 ± 7.3)g | Macrokinetics (lumped model) | H-ZSM-5 (Si/Al = 140) | |||
| (270 ± 25)h | ||||||||
| Carbocation197 | MEE | Propylene | 160143 | — | Macrokinetics | — | — | H-ZSM-5 (Si/Al = 18) |
| Direct103 | — | Ethylene, propylene and butene from dimethyl ether | (42 ± 1) | Intrinsic | Macrokinetics | — | — | H-ZSM-5 (Si/Al = 140)103 |
| Direct35 | Methoxy methyl | Propylene | ∼200 | Intrinsic | Microkinetics using a temporal analysis of products reactor | — | — | H-ZSM-5 (Si/Al = 36) |
4.3. Diffusion and mobility of species during the MTO process
The combination of neutron spectroscopy and computational modelling has a unique role in elucidating both structure and dynamics during operation of catalytic systems. Quasielastic neutron scattering techniques provide key information about hydrocarbon transport in microporous catalysts. Quasielastic neutron scattering can yield values for self-diffusion coefficients, which are useful for isotopic and jump models.200 Using a combination of quasielastic neutron scattering and molecular dynamic simulations, the mobility of initial species was studied in H-ZSM-5 catalysts (Si/Al = 30) and H-Y (Si/Al = 30),41,42,172,201 with the latter considered because it is inactive for the MTH process and thus provides a reference material. O’Malley et al.41,172 showed from quasielastic neutron scattering measurements (Fig. 10) that methanol is immobile (with respect to the timescale of their measurements) in H-ZSM-5 (Si/Al = 30), while the spectral broadening in the H-Y (Si/Al = 30) system is consistent with significant movement. The close fit to the resolution spectra for methanol in H-ZSM-5 (Si/Al = 30) catalysts suggests that virtually all the methanol protons remain static, indicating that the methanol is immobilized due to methoxylation. In the studies of O’Malley et al.,172 it is important to note that the spectra was collected at room temperatures and up to 373 K, and also to note that quasielastic neutron scattering experiments are not conducted under flow conditions, which is in contrast to many of the previously discussed studies. Evidently, during the conversion of methanol to olefins in a flow reactor at high temperatures, methanol is observed at <100% feed conversion in the outlet stream.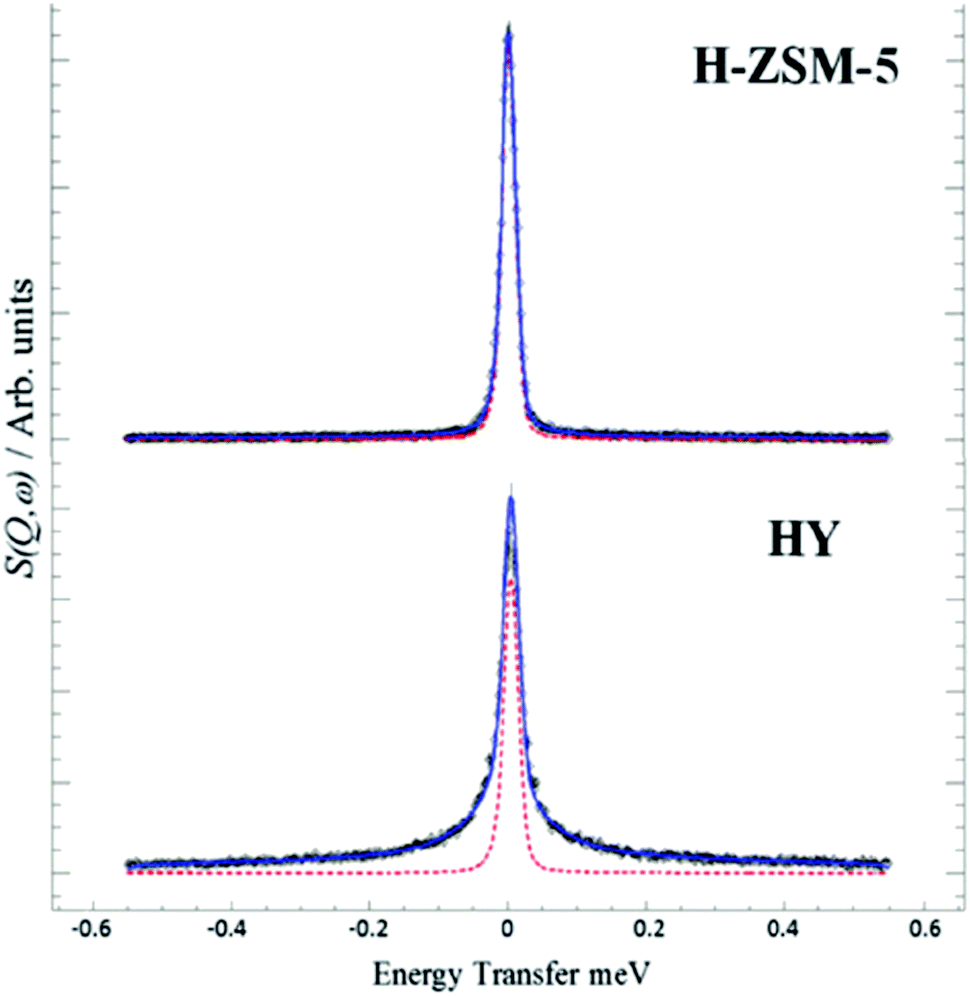 | ||
| Fig. 10 QENS spectra at 298 K of methanol loaded in zeolites H-ZSM-5 and HY at Q = 0.9 Å, (--) represents the resolution data taken at 5 K. Reprinted from ref. 172 with permission of the Royal Society of Chemistry. | ||
Table 4 gives the barriers for diffusion and mobility of methanol and dimethyl ether at various loadings in several catalysts, themselves with differing chemical compositions. Such quantitative analysis can only be produced when there is a uniform diffusion mechanism across all temperatures of observation for each catalyst. As observed by Catlow and co-workers,200 several mechanisms could be involved depending on catalyst composition, loading and temperature; these include the Chudley and Elliot jump diffusion model,202 Volino–Dianoux model of translational diffusion confined to a sphere,203 isotopic rotation model by Sears.204 At room temperature over H-ZSM-5 (Si/Al = 36) catalysts, isotopic methanol rotation was observed with a rotational diffusional coefficient of 2.6 × 1010 s−1.205 At these conditions, bridging molecular behaviour with data-driven ordinary and partial differential equations that describe the macroscale behaviour of the reactor is challenging. The latter (macroscale investigations) allows for a surface diffusion/mobility coefficient (m2 s−1) while the former provides rotational rates (s−1). The rotational diffusion coefficient is most likely integrated into the adsorption constant of the specie on the active sites of the H-ZSM-5 catalysts. The mobility behaviour contrasts qualitatively with H-ZSM-5 (Si/Al = 135) catalysts, where diffusion was observed as confined in a sphere matching the 5.5 Å channel width. The formation of water during the equilibration period in H-ZSM-5 catalysts could either retard or promote the motion of methanol, dimethyl ether, methoxy species, and other intermediates formed during the induction period of MTO conversion.
| Specie | Catalyst | Si/Al ratio | Loading (mpuc)a | T (K) | QENS (Ds, m2 s−1) × 10−10 | QENS (Ed, kJ mol−1) | MD (Ds, m2 s−1) × 10−9 |
|---|---|---|---|---|---|---|---|
| a mpuc = molecules per unit cell. b Change in diffusion mechanisms across temperatures. | |||||||
| Methanol | H-Y41 | 30 | 6 | 400 | 4.9 ± 1.2 | 8.8 | 3.21 |
| H-Beta42 | 31 | 4 | 373 | — | 6.8 | 8.99 | |
| H-ZSM-5205 | 36 | 22 | 373 | 8.98 ± 0.3 | —b | — | |
| 135 | 373 | 8.87 ± 0.43 | 0.58 | — | |||
| Dimethyl ether | H-ZSM-5205 | 36 | 14 | 373 | 9.89 ± 0.36 | 0.96 | — |
| 135 | 373 | 10.80 ± 0.25 | 1.33 | — | |||
As previously mentioned in Section 4.2.3.1, olefins can be formed either via the olefins mechanism involving methylation and oligomerisation of small olefins, followed by cracking of larger ones, or the aromatic route in which polymethylated aromatic species undergo a series of skeletal rearrangement steps to produce alkenes. To identify the role of pore size and aromatic methylated species on olefins diffusion and the dual-cycle mechanism, the mobility was analysed inside two 1D zeolites (H-ZSM-12 and H-ZSM-22) with a pore size difference of 0.3 Å, where H-ZSM-12 has larger pores. The forcefield MD simulations highlighted that the diffusion of methanol, ethene, propene and 1-butene are ∼2.5 times faster in the larger pore zeolite.206 The inclusion of methoxide and ethoxide species inside the zeolites pores identified that ethene has a higher contact time with both surface oxi-species in the narrower pore zeolite, indicating that the methylation and dimerization reactions are more feasible, and thus the MTO process could be more favoured, in H-ZSM-22 than in H-ZSM-12. Previous experimental and theoretical analysis suggested that polymethylate species, in particular the pentamethyl benzenium, are key intermediates to the formation of olefins via a skeletal rearrangement.174,207 Therefore, polymethylated benzene species (PMBs), specifically p-xylene and 1,2,4-trimethylbenzene, were considered as co-adsorbates. At 673 K, ethene diffusion is an order of magnitude lower than when the reaction is performed in the absence of any neighbouring species inside H-ZSM-12, and then completely stops when a pentamethylated benzene was included. Additional MD analysis showed that the trimethylbenzene blocks the pores of H-ZSM-22, whereas tetramethylbenzene blocks the larger by 0.3 Å pores of H-ZSM-12,206 which further suggests that H-ZSM-12 is better suited for olefin formation via the aromatic cycle.
To determine the energetic conditions influencing the methylation rate of PMBs, the theoretical ONIOM (ωB97XD/6-31(d,p):MNDO) modelling technique was employed to analyse the vicinal methylation, i.e. the methylation of C neighboring a methylated C on benzene, and geminal methylation, i.e., methylation of a C already containing a methyl group, in H-ZSM-12 and H-ZSM-22.208 The geminal methylation activation barriers for H-ZSM-12 for xylene, trimethylbenzene, and tetramethylbenzene were shown to be 175, 165 and 158 kJ mol−1, respectively, while for vicinal methylation, 135, 134 and 169 kJ mol−1 were calculated for the same respective PMBs. The contrasting trends in activation energies, which depends on the type of methylation, is attributed to the variation in charge distribution and basicity with increase in number of methyl groups on the aromatic ring, as proven by Mulliken charge analysis. For H-ZSM-22, the activation barriers for geminal methylation were 142, 137 and 208 kJ mol−1 for xylene, trimethylbenzene, and tetramethylbenzene, respectively, while 105, 131 and 156 kJ mol−1 were obtained for vicinal methylation of xylene, trimethylbenzene and tetramethylbenzene, respectively. Although the activation barriers over H-ZSM-22 are more favorable than over H-ZSM-12, the barrier to the more reactive geminal pentamethyl benzene is higher in H-ZSM-22, which could be to the higher stress imposed on the transition state by the narrower pore. Analysis of propene formation in the two zeolites via the alkene cycle, through butene and pentene cracking, was further investigated; whilst the rate determining step for the formation of olefins via the aromatic cycle is expected to be at least 158 kJ mol−1 in H-ZSM-12, the highest energies for butene and pentene cracking are 141 and 152 kJ mol−1, respectively, highlighting that the olefins cycle is dominant over the aromatic cycle in H-ZSM-12.208 For H-ZSM-22, the highest energy barriers for butene and pentene cracking are 174 and 162 kJ mol−1, respectively, which leads to the conclusion that it is energetically more feasible to produce olefins with H-ZSM-12 than H-ZSM-22 through both the aromatic and olefinic cycles. The differences in reaction energetics and diffusion capability of the two zeolites were considered experimentally and shown to have a significant impact on catalytic activity. GC/MS analysis showed that the insertion of the methanol at 400 °C in H-ZSM-22 only produced traces of olefins,209 which were attributed to the external and pore mouth acid sites catalysis.210–212 In the case of H-ZSM-12, methanol was completely converted and produced a large amount of olefins and paraffins, highlighting the crucial role of pore size on zeolite catalysis.209
While it is important to study the isolated diffusion of species (oxygenates, olefins) in the zeolite pores, future investigations should continue reducing the gap between experiment and theory, with more realistic models where the diffusion of species is analyzed under equilibration conditions (i.e., in the presence of methanol, dimethyl ether, water and surface methoxy species) and/or also in the presence of primary olefins (Fig. 8). Furthermore, the effect of hydronium ions,213,214 methanol,34,215 and DME clusters34 on the diffusion of other individual species in the pores should be quantitatively evaluated (Fig. 8). Typically, a thorough description, through the application of species balance with Maxwell–Stefan equations, should highlight the competitive nature of reaction and diffusion of species in the zeolite pores during the formation of primary olefins. To uncover the effect of acidity and adsorbate loading on the transport mechanism behind the diffusion of paraffins and olefins, the dynamic properties of ethene, ethane, propene and propane inserted in H-SAPO-34 have been investigated using Pulsed-field gradient (PFG) NMR analysis together with ab initio MD (AIMD) and ab initio umbrella sampling (AI-US) modelling techniques.216 The umbrella sampling technique was employed to determine the free energy profile of diffusion through the 8 membered ring (8MR) windows of the H-SAPO-34 zeolite, having 1–3 adsorbates and 0–2 Brønsted acid sites per unit cell. The increase in adsorbate loading from one to three molecules per cage led to a slightly enhanced self-diffusion rate of ethene, while ethane mobility decreased. Furthermore, alkene diffusion was enhanced with increasing acid site density, whereas alkane diffusion remained constant, regardless of the temperature, in the range of 300 K to 600 K. At 600 K, in the presence of two acid sites per unit cell, the barrier for ethene and propene diffusion is lowered by ∼25 kJ mol−1 compared to diffusion within a zeolite with no acid sites. 1H PFG NMR analysis at room temperature provided further evidence that the diffusion coefficient (D) of ethene is directly proportional to Brønsted acid site (BAS) density, as D ≈ 10−11 m2 s−1 at BAS = 0.65 mmol g−1 goes to D ≈ 2 × 10−11 m2 s−1 at BAS = 1.13 mmol g−1.216 The improved diffusion profile of alkenes at higher BAS density is attributed to the polarization induced by the BAS on the adsorbed olefins creating an electrostatic field in the proximity of the 8MR, which leads to a flatter free energy surface that facilitates the transition from one cage to another. Supportive evidence has been found in the formation of alkene π-H complexes, detected at 3307 cm−1 by FT-IR measurements.216
To further determine the effect of product distribution on olefins diffusion, additional computational analysis replicating the conditions closer to experimental work has been conducted by employing a series of techniques such as force-field MD and MTD, together with AIMD and AI-US, to model ethene and propene mobility in H-SAPO-34, considering the transfer from one CHA cage to another cage via an 8MR window.217 Several simulations determining the mobility of propene next to or with additional propene molecules, from one cage to another was modelled. For the transition of propene from a cage having three propene molecules to an empty cage, the diffusion barrier is lowered by 20 kJ mol−1, whereas when two propene molecules were added per cage, the diffusion barrier decreases by 15 kJ mol−1. The diffusion of propene in the presence of different hydrocarbon pool species was also carried out, specifically the transition of propene was not possible from a cage with four methanol molecules to a cage containing hexamethyl benzene (HMB) and three methanol molecules, which led to the conclusion that any propene formed would be expelled immediately. However, when HMB was substituted by toluene, a local minimum for propene was found inside the cage, although with a high barrier ∼150 kJ mol−1, highlighting the significant role that methyl groups and methylation rate play.217 By controlling the number of methyl groups on a benzene ring, the diffusion of small olefins can be varied and, in turn, the kinetics and lifetime of the zeolite catalyst can be influenced.
5. Masking effects on the first C–C bond formation: influence of early degradation species and fast methylation chemistries (autocatalysis)
Under atmospheric conditions,19 the catalyst lifetime of a H-ZSM-5 catalysts is about 70 times longer than other zeolites when used for MTO conversion. The MTH reaction over a H-ZSM-5 catalyst bed is accompanied by coke formation, which leads to catalyst deactivation; this occurs through blocking the access to the active sites, either by deposition of carbonaceous compounds directly on the active sites and in the micropore channels of the zeolite (internal coke), or by blocking the entrance to the micropores (external coke), thereby preventing the diffusion of methanol molecules into the zeolite structure.218–220 The catalyst lifetime, as depicted by the breakthrough of methanol, depends on the deactivation rate, the catalyst activity, and the contact time or space velocity.220Janssen et al.220,221 considered two models for catalyst deactivation. One model assumed MTO conversion as a first-order reaction with a deactivation rate proportional to the conversion. The proportionality implies that the formation of coke deposits follow methanol conversion; however, the build-up of hydrocarbon fragments in the zeolite channels may take some time, and, during the initial phase, the deactivation may not take place or is slower compared to the methanol conversion. The product distribution may be different on a working catalyst than on a fresh catalyst, even if the methanol conversion is the same.221 Such selective deactivation at atmospheric conditions has been observed for H-SAPO-34 catalysts.158 Selective deactivation was also observed in a temporal analysis of products (TAP) reactor,26,27 where deactivation was slow during the induction period. Janssen considered a second model where the deactivation rate is not proportional to methanol conversion.221 Müller et al.15 compared the MTH reaction over H-ZSM-5 in plug-flow and back-mixed reactors and hypothesized that the evolution of coke species depends on the position of the catalyst particle. In the early stages of the MTH reaction, the H-ZSM-5 zeolite deactivated fast by blocking of individual Brønsted acid sites due to oxygen-containing surface species; these species were further transformed to aromatic compounds and deactivation proceeded by forming large coke species that blocked the catalyst pores. Deactivation kinetics were not affected by the formation of condensed aromatic species.
Large primary or secondary products may lead to catalyst deactivation mainly due to blocking of the zeolite pores and preventing the reactants from accessing the active sites or the intermediates necessary to methylate. Depending on the temperature regime, important intermediates, such as derivatives from formaldehyde, ethyl-trimethyl-benzene, and iso-propyl-dimethylbenzene, can hinder diffusion due to their large size through the catalytic channels.222,223 Ethyl-trimethyl-benzene and isopropyl-dimethyl-benzene were found to remain inert at medium temperatures (573 K), while alkene formation was observed when the temperature was increased to 613 K.223 By continuing to increase the temperature, after a series of methylation steps and skeletal rearrangements, the methylated monocyclic arenes could form other “coke species”, such as polymethylated naphthalene and anthracene, that may block the zeolite pores.
Catalyst deactivation is not only determined by the products obtained in the later stages, but also by those formed at the very beginning. Disproportionation of methanol to formaldehyde influences the deactivation rate because formaldehyde could react with aromatic molecules, resulting in the formation of polycondensed aromatics. The conversion of methanol to methane or carbon dioxide would also be accounted as a carbon loss, since both by-products are considered inert in common industrial conditions and cannot contribute to the formation of any desired hydrocarbons. Substituting methanol with dimethyl ether was attempted in order to limit the production of the previously mentioned side-products.26,27,35,49 The use of dimethyl ether leads, however, to a higher ratio of aromatic to aliphatic compounds, and also a lower water content, which can be used to stabilise different intermediates, in addition to regenerating the necessary acid sites for methylation of the aromatic species, which can lead to an increased deactivation rate.103
Nørskov and co-workers224 provided additional evidence, through adsorption energies, that polycylic arenes can exist within the framework of H-ZSM-5 during MTH conversion, thereby hindering the passage of reactants and products particularly at the adsorption sites. They propose, buttressing the conclusions of Mores et al.,225 that the graphitic layer observed on deactivated H-ZSM-5 crystals is formed by polycyclic arenes growing in straight channels and joining on the outer surface.
The initial precursors have been decoupled from the deactivating species26,27,35 using a temporal analysis of products (TAP) reactor operating under vacuum conditions, allowing for full elucidation of the species that lead to the formation of primary olefins, even with dimethyl ether as a feed. Although the degradation challenges can be regressed under the right conditions, methylation effects can mask the formation of the first C–C bond. On formation of the ethylene and/or propylene, methylation occurs in a first order process with respect to the olefin and zero order with respect to the oxygenate i.e., methanol or dimethyl ether,30,226,227 with lower barriers for propylene compared to ethylene. Consequently, deciphering the primary olefin becomes challenging. Fast methylation occurs due to the autocatalytic nature of the MTO conversion process and, as soon as the first olefin is formed, methylation and/or cracking processes are initiated that can mask the first olefin.
6. Towards a quantitative mechanistic understanding of the formation of primary olefins
Given the primary reactant (methanol and/or dimethyl ether) and the primary olefin(s), such as ethylene and/or propylene, various mechanisms (direct or indirect) have been investigated. Density functional theory calculations have shown the improbability of many direct pathways due to activation energies exceeding 200 kJ mol−1 and the instability of intermediates such as oxonium ylides.153–155 Nonetheless, there is recent spectroscopic evidence for three main pathways for the formation of the first C–C bond: the carbon monoxide mechanism,13,40,137,138 methoxymethyl mechanism49,132 and the carbene mechanism.101 Omojola et al.27,35 showed recently that simulations of the methoxymethyl mechanism gives the closest agreement to transient experimental data obtained from the temporal analysis of products (TAP) reactor, corroborating the direct mechanism of primary olefin formation.Kinetic modelling is an indispensable tool that could be used to discriminate between direct and indirect mechanisms of primary olefin formation, but quantitative determination of the most abundant surface/reactive specie hydrocarbon pool molecules under atmospheric and vacuum conditions is necessary to simplify the complex reaction network.
The indirect olefin formation route was suggested by Haw and co-workers150 due to the possible presence of impurities in the feed and artefacts in the zeolites. Primary olefin formation occurs via aromatic dealkylation chemistry. A reaction mechanism, hitherto absent in the archived literature, is needed to account for the “impurity pathway”. Studies by Hunger and co-workers145,176,177 refuted the impurities hypothesis. Understanding the induction period, where the first C–C bond and the primary olefins are formed, is challenging due to fast methylation rates via methanol, dimethyl ether, or methoxy species, that could mask the observation of the true primary olefins,30,226 and also through the formation of degrading species. The presence of steam plays an important role in regulating this phenomenon.214,228 The influence of intermediates, such as carbon monoxide, carbon dioxide, aromatics, and dienes, on the induction period can be quantified only if the reactions corresponding to these initial precursors are included in the reaction network; however, there are many precursors that could play a role and complete elucidation of the reaction network is yet unattainable. Development of a full network could be facilitated through a top-down approach229 or by automated reaction network generators.230,231 In the absence of the latter, “heuristic” models, such as S-shaped logistic curves,26 have been used to quantify the influence of intermediates.
An understanding of the relationship between the strength and structure of the acid site and the activation of sorbates remains unclear. Density functional theory calculations38,232,233 that consider the adsorption of different types of compounds, such as carbon monoxide, methanol, dimethyl ether, and pyridine, have not been conclusive on what characteristics determine the overall bonding capacity of the zeolite. Several observables, such as acid site deprotonation energy, vibrational frequencies, number of hydrogen bonds formed, charge, and dipole moment of the adsorbate, have proven to describe a non-linear direct correlation for guest–host bonding; however, a clear understanding that can be applied on a large range of compounds and zeolite structures has not been identified yet due to the complex nature of the interactions that can occur between the Brønsted acid sites and also with the surrounding siliceous framework.
Experimental studies34 in a temporal analysis of products (TAP) reactor on the desorption of methanol and dimethyl ether over fresh and working H-ZSM-5 catalysts of differing Si/Al ratios (with minimum re-adsorption masking effects) show that binding over oxygenates occurs over ensembles of active sites (non-uniform Brønsted acid sites). The non-uniformity can be attributed to different coordination and local environment present in the H-ZSM-5 crystal. Three ensembles of active sites were obtained over H-ZSM-5 (Si/Al = 25) and H-ZSM-5 (Si/Al = 36), and two ensembles of active sites were obtained over H-ZSM-5 (Si/Al = 136) catalysts, as identified from combined temperature programmed desorption (TPD) experiments and microkinetic modelling studies. The activation energies of desorption of dimethyl ether over the H-ZSM-5 catalysts were systematically higher than methanol over the corresponding sites, and increased from low temperature activated sites to high temperature activated sites. In modelling desorption profiles, only a small percentage of the total Brønsted active sites were required for binding. A further study with combined temperature programmed surface reaction (TPSR) experiments and microkinetic modelling35 was conducted over H-ZSM-5 (Si/Al = 36) and H-ZSM-5 (Si/Al = 135); only a minority of the active sites were responsible for adsorption, desorption, and surface reactions in the zeolite catalysts, although there are differences in the number of ensembles of Brønsted active sites responsible in the TPD and TPSR studies.35
Theoretical studies confirm that H-ZSM-5 catalysts have Brønsted acid sites with a difference of ∼20 kJ mol−1 in adsorption strength for both methanol and dimethyl ether.39 Previous QM/MM studies indicate that the co-adsorption of the second methanol on the acid sites will lower the average adsorption energy per methanol molecule by ∼15 kJ mol−1. A key aspect to consider when accounting for adsorption ensembles are adsorbate clusters connected through hydrogen bonds.34 Adsorption of two or more methanol on the Brønsted acid site lead to the spontaneous deprotonation of the active site, which facilitate the formation of protonated methanol clusters stable at 300 K and 673 K.43 At room temperature, the methanol cluster was composed of five adsorbates, whereas at 673 K, only three adsorbates manage to stay connected for longer periods of the simulation. The behaviour is also dependent on the ensemble of active sites onto which the adsorbates are bonded;34 molecular dynamic simulations also indicate that acid site distribution is important since two close acid sites can form a hydrogen bonding network from one another via methanol adsorbates that may block a pore and thus influence desorption rates.
In addition, the Si/Al ratio can play a major role on crystallite size. An increase in the Si/Al ratio leads to larger crystallite particles.34 The size of zeolite crystallite (and particle) influence mass transfer234 and methanol conversion. Large particle sizes were shown to have an accumulation of “coke” at the edges of the particle235,236 leading to the reactant being prohibited from accessing the active sites in the centre of the zeolite. Further investigations conducted on H-ZSM-5236 show that the position of the acid sites within the zeolite channels can significantly alter the conversion rate. Active sites located predominantly in larger intersection cavities (∼9 Å diameter) had a similar product distribution to those expected from the aromatic based cycle (enriched in ethylene and aromatics), whereas acid sites placed in smaller (∼5 Å diameter) sinusoidal or straight channels had more products of the olefin-based cycle (enriched in propene and higher olefins). Moreover, QENS studies by Omojola et al.205 showed that dimethyl ether is predominantly resident in the pore intersection of ZSM-5 facilitating its higher mobility. Methanol, on the other hand, is primarily located in the pore channels. Placing this mobility/diffusion information in the context of acid-site location above provides proof of why the use of dimethyl ether as a feed leads to the formation of more aromatics in comparison to methanol.
Previous quantitative mechanistic understanding of the three regimes of MTO conversion has focused primarily on Brønsted acid sites.103 Lewis acid sites have been shown to influence the catalyst lifetime while Brønsted acid sites in H-ZSM-5 catalysts impact the tailored generation of propylene,237 and so further quantitative mechanistic studies should include both types of acid sites due to the origin of activating and deactivating species during atmospheric studies, and site-specific behaviour of H-ZSM-5 catalysts observed through integrated adsorption, desorption, and surface reaction studies.35
Another challenge in understanding the first C–C bond and formation of primary olefins is the identification of key species that limit methanol activity. Hydrocarbon pool species could create pore-blockages and limit the transport of olefins to the exterior of the zeolite or zeotype crystal (intra-diffusion limitations). Conversely, inter-particle diffusion limitations could occur through the transfer of oxygenates (methanol, dimethyl ether) to the active sites, which could limit olefin formation. Ethylene formation from the dual cycle may be controlled by the formation of bulky hydrocarbons in the aromatic cycle. The bulky hydrocarbons may allow only little molecules, such as ethylene and methane, to escape the zeolite porous matrix in view of intra-diffusion limitations; however, ethylene is not too reactive during MTO conversion and could be formed from the cracking of larger olefins.226 Regulation of the process conditions that affect thermal and catalytic cracking could regulate both ethylene and propylene formation. Such studies dealing with the intra-grain diffusion limitations may be challenging under atmospheric conditions, as aromatics re-adsorb under atmospheric pressure conditions.238 Reducing the pressure mitigates the possibility for re-adsorption, such that aromatics flow out of the pores easily and the coking possibility is reduced. Recently, Omojola and van Veen showed that aromatics are easily removed from the surface of the H-ZSM-5 catalyst compared to dimethyl ether.239 The temporal analysis of products (TAP) reactor serves a unique benefit in this regard as the re-adsorption of such species is reduced allowing identification of the first C–C bond and primary olefin formation. Moreover, the TAP reactor uses Knudsen diffusion (under vacuum conditions) as a yardstick for characterizing catalytic reactions, where the influence of gas–gas interactions within the zeolite pore is reduced compared to atmospheric conditions. Consequently, external mass transfer limitations are reduced in the temporal analysis of products due to the low density of gas molecules in the thin boundary layer surrounding the catalyst particle. During step response, temperature-programmed surface reaction (TPSR), and temperature-programmed desorption (TPD) studies, experiments are carried in the temporal analysis of products (TAP) reactor at an inlet pressure of close to 1000 Pa. While Knudsen diffusion is widely used as a yard-stick during pulse experiments, the diffusion regime under flow conditions for other transient and non-conventional TAP experiments (TPD, TPSR and step response) has not been investigated.
7. Outlook
The conversion of methanol to hydrocarbons was commercialised again in 2010 with the construction of turbulent fluidised bed reactors, facilitating the clean conversion of solid fuels into olefins. The recent industrial maturity has left some fundamental challenges in MTO conversion chemistry unsolved. Several research groups have tried to decipher the challenge. Recently, Deng and co-workers48 provided a solid-state nuclear magnetic resonance perspective on the formation of the first C–C bond; Olsbye and co-workers49 provided a mechanistic perspective on the formation of the first C–C bond and the masking effects of degradation species; Weckhuysen, Gascon and co-workers242 provided a mechanistic perspective on the first C–C bond and the promotional effect of zeolite topology and acidity on the alkene cycle; and Van Speybroeck and co-workers240 gave a detailed molecular perspective of the formation of the first C–C bond during MTO conversion. Our quantitative multiscale perspective of the mechanisms and kinetics involved in the formation of the first C–C bond decouples the adsorption, desorption, reaction, and mobility of species on both the molecular and macroscale level, and provides new insights that could guide rational catalyst design.The adsorption enthalpies (1 molecule per unit cell) and activation energies of desorption for methanol and dimethyl ether range between 92–126 and 103–132 kJ mol−1, respectively, over H-ZSM-5 of different Si/Al ratios. For H–Y catalysts, adsorption enthalpies of 89–106 and between 105 kJ mol−1 were obtained for methanol and dimethyl ether, respectively. For H-SAPO-34, adsorption enthalpies between 64–86 kJ mol−1 and of 45 kJ mol−1 were obtained for methanol and dimethyl ether, respectively. For H-ZSM-22 and AlOH–(SiOH), adsorption enthalpies of methanol of 107 and 75 kJ mol−1 were obtained, respectively.
Although increases in the resolution of spectroscopic methods have been reported, for instance using a synchrotron light source with a resolution down to 0.25 s, there remains a large opportunity to obtain definite quantification of the lifetime, mobility and reactivity of adspecies during MTO conversion. A comparison of the barriers involved in the different proposed surface chemistries of MTO conversion shows that low activation energies exist for the carbon monoxide mechanism (80 kJ mol−1) over H-ZSM-5 catalysts, while carbene formation is energetically demanding with barriers up to 232 kJ mol−1, calculated using density functional theory. Conversely, barriers range from 42 kJ mol−1 up until 200 kJ mol−1 using data-driven simulation of differential–algebraic equations that represent the elementary steps of the formation of the first C–C bond and primary olefins. Relatively lower energetics are observed over H-SAPO-34 compared to H-ZSM-5, justifying its current industrial application.
Low barriers to methanol and dimethyl ether diffusion in H-ZSM-5, H-β, and H-Y catalysts have been reported, with values ranging between 0.96 to 8.8 kJ mol−1. Although dimethyl ether is a larger molecular than methanol, it shows higher diffusion coefficients in H-ZSM-5 catalysts at low temperatures.205
The adsorption and reaction barriers have been identified as competitive with each other in this work, and could each limit the activity of zeolite catalysts for MTO conversion. The adsorption and reaction barriers have been studied separately by density functional theory calculations and data-driven simulations of partial differential equations that represent the elementary steps on the catalyst surface. These data-driven simulations represent static microkinetic models that do not consider the dynamic nature of active sites. Ryder, Chakraborty and Bell241 conducted density functional theory calculations of proton mobility in zeolites and, in the absence of water, the acidic proton “hops” between two of the four O atoms surrounding an aluminium centre with an activation barrier of 117.2 kJ mol−1. The barrier is further reduced to 100 kJ mol−1 in the presence of adsorbed water; in the absence of adsorbed water, during proton transfer, the O atoms stretch closer together in order to stabilize the transition state, which is identified by a 13.4° decrease in O–Al–O bond angle. Inclusion of proton mobility in the density functional theory-assisted microkinetic models could lead to a more realistic representation of the industrial MTO process, given that acidic protons in zeolites are known to be mobile at elevated temperatures.
The current perspective presents the cumulative effort of the MTO catalysis community in understanding the formation of the first C–C bond and primary olefins over the last 44 years. There is, however, still a lack of systematic studies that link site distribution (Al pairs, single Al atoms, and isolated Al atoms) with zeolite structure, duration of the induction period, and the ensuing product distribution. Such studies could guide the rational design of catalysts, by relating synthesis procedures, which regulate site distribution with zeolite/zeotype structure, and kinetics and mechanisms occurring during MTO conversion. Additionally, the impact of proton mobility should be studied for each site-distribution and catalyst structure, and linked with the corresponding duration of the induction period and ensuing product distribution. Density functional theory assisted microkinetic models can also help provide a more realistic representation of the industrial MTO process. Finally, although there are detailed studies for H-ZSM-5 or H-SAPO-34 catalysts (of varying composition) towards the adsorption and reaction energies involved in the first C–C bond formation, there are currently few systematic studies providing scaling-relations between adsorption and reaction energies during MTO conversion.
Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
Toyin Omojola acknowledges financial support from the Petroleum Technology Development Fund (PTDF/ED/PHD/OO/766/15) for his PhD work conducted at the University of Bath and the University of Warwick and is grateful to the Royal Society of Chemistry, Society of Chemical Industry, Armourers and Brasiers Gauntlet Trust, University of Warwick, and the Alumni Travel Fund of the University of Bath for additional financial support. Stefan Adrian F. Nastase wishes to thank the School of Chemistry, Cardiff University for a PhD studentship. Andrew J. Logsdail acknowledges funding by the UKRI Future Leaders Fellowship program (MR/T018372/1). We are grateful to Dr Dmitry Lukyanov and Professor Richard Catlow for fruitful scientific discussions.References
- C. D. Chang and A. J. Silvestri, The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts, J. Catal., 1977, 47(2), 249–259 CrossRef CAS.
- C. D. Chang, W. H. Lang and R. L. Smith, The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts. II. Pressure effects, J Catal., 1979, 56(2), 169–173 CrossRef CAS.
- M. Stöcker, Methanol-to-hydrocarbons: Catalytic materials and their behavior, Microporous Mesoporous Mater., 1999, 29(1–2), 3–48 CrossRef.
- S. Ilias and A. Bhan, Mechanism of the catalytic conversion of methanol to hydrocarbons, ACS Catal., 2013, 3, 18–31 CrossRef CAS.
- J. L. White, Methanol-to-hydrocarbon chemistry: The carbon pool (r)evolution, Catal.: Sci. Technol., 2011, 1(9), 1630–1635 RSC.
- U. Olsbye, S. Svelle, M. Bjrgen, P. Beato, T. V. W. Janssens and F. Joensen, et al., Conversion of methanol to hydrocarbons: How zeolite cavity and pore size controls product selectivity, Angew. Chem., Int. Ed., 2012, 51(24), 5810–5831 CrossRef CAS PubMed.
- I. M. Dahl and S. Kolboe, On the reaction mechanism for hydrocarbon formation from methanol over SAPO-34. I. Isotopic labeling studies of the co-reaction of ethene and methanol, J. Catal., 1994, 149(2), 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, On the reaction mechanism for hydrocarbon formation from methanol over SAPO-34: 2. Isotopic labeling studies of the Co-reaction of propene and methanol, J. Catal., 1996, 161(1), 304–309 CrossRef CAS.
- S. Kolboe and I. M. Dahl, Methanol conversion to hydrocarbons. Use of isotopes for mechanism studies, in Stud Surf Sci Catal. 94, ed. H. K. Beyer, H. G. Karge, I. Kiricsi and J. B. Nagy, Elsevier, 1995, pp. 427–434 Search PubMed.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud and S. Kolboe, et al., Conversion of methanol into hydrocarbons over zeolite H-ZSM-5: Ethene formation is mechanistically separated from the formation of higher alkenes, J. Am. Chem. Soc., 2006, 128(46), 14770–14771 CrossRef CAS PubMed.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe and F. Bonino, et al., Conversion of methanol to hydrocarbons over zeolite H-ZSM-5: On the origin of the olefinic species, J. Catal., 2007, 249(2), 195–207 CrossRef.
- O. Dewaele, V. L. Geers, G. F. Froment and G. B. Marin, The conversion of methanol to olefins: A transient kinetic study, Chem. Eng. Sci., 1999, 54(20), 4385–4395 CrossRef CAS.
- P. N. Plessow, A. Smith, S. Tischer and F. Studt, Identification of the reaction sequence of the MTO initiation mechanism using ab initio-based kinetics, J. Am. Chem. Soc., 2019, 141(14), 5908–5915 CrossRef CAS PubMed.
- D. Prinz and L. Riekert, Formation of ethene and propene from methanol on zeolite ZSM-5. I. Investigation of rate and selectivity in a batch reactor, Appl. Catal., 1988, 37(C), 139–154 CrossRef CAS.
- S. Müller, Y. Liu, M. Vishnuvarthan, X. Sun, A. C. Van Veen and G. L. Haller, et al., Coke formation and deactivation pathways on H-ZSM-5 in the conversion of methanol to olefins, J. Catal., 2015, 325, 48–59 CrossRef.
- S. Ilias and A. Bhan, Tuning the selectivity of methanol-to-hydrocarbons conversion on H-ZSM-5 by co-processing olefin or aromatic compounds, J. Catal., 2012, 290, 186–192 CrossRef CAS.
- F. J. Keil, Methanol-to-hydrocarbons: Process technology, Microporous Mesoporous Mater., 1999, 29(1), 49–66 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, Methanol to olefins (MTO): From fundamentals to commercialization, ACS Catal., 2015, 5(3), 1922–1938 CrossRef CAS.
- H. Schulz, “Coking” of zeolites during methanol conversion: Basic reactions of the MTO-, MTP- and MTG processes, Catal. Today, 2010, 154(3), 183–194 CrossRef CAS.
- G. B. Marin and G. S. Yablonsky, Kinetics of Chemical Reactions: Decoding Complexity Weinheim, Wiley-VCH, Germany, 2011, pp. 1–428 Search PubMed.
- P. Del Campo, W. A. Slawinski, R. Henry, M. W. Erichsen, S. Svelle and P. Beato, et al., Time- and space-resolved high energy operando X-ray diffraction for monitoring the methanol to hydrocarbons reaction over H-ZSM-22 zeolite catalyst in different conditions, Surf. Sci., 2016, 648, 141–149 CrossRef CAS.
- D. S. Wragg, M. G. O'Brien, F. L. Bleken, M. Di Michiel, U. Olsbye and H. Fjellvãg, Watching the methanol-to-olefin process with time- and space-resolved high-energy operando X-ray diffraction, Angew. Chem., Int. Ed., 2012, 51(32), 7956–7959 CrossRef CAS PubMed.
- B. E. Langner, Reactions of methanol on zeolites with different pore structures, Appl. Catal., 1982, 2(4), 289–302 CrossRef CAS.
- L. Qi, Y. Wei, L. Xu and Z. Liu, Reaction behaviors and kinetics during induction period of methanol conversion on HZSM-5 zeolite, ACS Catal., 2015, 5(7), 3973–3982 CrossRef CAS.
- L. Qi, J. Li, Y. Wei, L. Xu and Z. Liu, Role of naphthalene during the induction period of methanol conversion on HZSM-5 zeolite., Catal.: Sci. Technol., 2016, 6(11), 3737–3744 RSC.
- T. Omojola, D. B. Lukyanov, N. Cherkasov, V. L. Zholobenko and A. C. van Veen, Influence of precursors on the induction period and transition regime of dimethyl ether conversion to hydrocarbons over ZSM-5 catalysts, Ind. Eng. Chem. Res., 2019, 58(36), 16479–16488 CrossRef CAS.
- T. Omojola, D. B. Lukyanov and A. C. van Veen, Transient kinetic studies and microkinetic modeling of primary olefin formation from dimethyl ether over ZSM-5 catalysts, Int. J. Chem. Kinet., 2019, 51(7), 528–537 CrossRef CAS.
- L. Bentea, M. A. Watzky and R. G. Finke, Sigmoidal nucleation and growth curves across nature fit by the Finke–Watzky model of slow continuous nucleation and autocatalytic growth: Explicit formulas for the lag and growth times plus other key insights, J. Phys. Chem. C, 2017, 121(9), 5302–5312 CrossRef CAS.
- Y. Zhou, L. Qi, Y. Wei and Z. Liu, Comparative investigation of the MTH induction reaction over HZSM-5 and HSAPO-34 catalysts, Mol. Catal., 2017, 433, 20–27 CrossRef CAS.
- S. Svelle, P. O. Rønning, U. Olsbye and S. Kolboe, Kinetic studies of zeolite-catalyzed methylation reactions. Part 2. Co-reaction of [12C]propene or [12C]n-butene and [13C]methanol, J Catal., 2005, 234(2), 385–400 CrossRef CAS.
- N. A. Bhore, M. T. Klein and K. B. Bischoff, The Delplot technique: A new method for reaction pathway analysis, Ind. Eng. Chem. Res., 1990, 29(2), 313–316 CrossRef CAS.
- N. A. Bhore, M. T. Klein and K. B. Bischoff, Species rank in reaction pathways: Application of Delplot analysis., Chem. Eng. Sci., 1990, 45(8), 2109–2116 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, The Mechanism of methanol to hydrocarbon catalysis, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed.
- T. Omojola, N. Cherkasov, A. I. McNab, D. B. Lukyanov, J. A. Anderson and E. V. Rebrov, et al., Mechanistic insights into the desorption of methanol and dimethyl ether over ZSM-5 catalysts, Catal. Lett., 2018, 148(1), 474–488 CrossRef CAS.
- T. Omojola and A. C. van Veen, Mechanistic insights into the conversion of dimethyl ether over ZSM-5 catalysts: A combined temperature-programmed surface reaction and microkinetic modelling study, Chem. Eng. Sci., 2021, 239, 116620 CrossRef CAS.
- A. Rasmuson, B. Andersson, L. Olsson and R. Andersson, Mathematical Modeling in Chemical Engineering, Cambridge University Press, USA, 2014 Search PubMed.
- B. Zijlstra, R. J. P. Broos, W. Chen, I. A. W. Filot and E. J. M. Hensen, First-principles based microkinetic modeling of transient kinetics of CO hydrogenation on cobalt catalysts, Catal. Today, 2020, 342, 131–141 CrossRef CAS.
- S. A. F. Nastase, A. J. O'Malley, C. R. A. Catlow and A. J. Logsdail, Computational QM/MM investigation of the adsorption of MTH active species in H-Y and H-ZSM-5, Phys. Chem. Chem. Phys., 2019, 21(5), 2639–2650 RSC.
- S. A. F. Nastase, C. R. A. Catlow and A. J. Logsdail, QM/MM study of the stability of dimethyl ether in zeolites H-ZSM-5 and H-Y, Phys. Chem. Chem. Phys., 2021, 23, 2088–2096 RSC.
- Y. Liu, S. Müller, D. Berger, J. Jelic, K. Reuter and M. Tonigold, et al., Formation mechanism of the first carbon–carbon bond and the first olefin in the methanol conversion into hydrocarbons, Angew. Chem., Int. Ed., 2016, 55(19), 5723–5726 CrossRef CAS PubMed.
- A. J. O'Malley, V. García Sakai, I. P. Silverwood, N. Dimitratos, S. F. Parker and C. R. A. Catlow, Methanol diffusion in zeolite HY: A combined quasielastic neutron scattering and molecular dynamics simulation study, Phys. Chem. Chem. Phys., 2016, 18(26), 17294–17302 RSC.
- C. H. Botchway, R. Tia, E. Adei, A. J. O'Malley, N. Y. Dzade and C. Hernandez-Tamargo, et al., Influence of topology and Brønsted acid site presence on methanol diffusion in zeolites beta and MFI, Catalysts, 2020, 10(11), 1342 CrossRef CAS.
- S. A. F. Nastase, P. Cnudde, L. Vanduyfhuys, K. De Wispelaere, V. Van Speybroeck and C. R. A. Catlow, et al., Mechanistic insight into the framework methylation of H-ZSM-5 for varying methanol loading and Si/Al ratio using first principles molecular dynamics simulations, ACS Catal., 2020, 10(15), 8904–8915 CrossRef CAS PubMed.
- J. Van der Mynsbrugge, S. L. C. Moors, K. De Wispelaere and V. Van Speybroeck, Insight into the formation and reactivity of framework-bound methoxide species in H-ZSM-5 from static and dynamic molecular simulations, ChemCatChem, 2014, 6(7), 1906–1918 CrossRef CAS.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Advances in theory and their application within the field of zeolite chemistry., Chem. Soc. Rev., 2015, 44(20), 7044–7111 RSC.
- C. R. A. Catlow, R. van Santen and B. Smit, Computer Modelling of Microporous Materials, 2004 Search PubMed.
- C. R. A. Catlow, V. van Speybroeck and R. van Santen, Modelling and Simulation in the Science of Micro- and Meso-Porous Materials, Elsevier, 2017 Search PubMed.
- C. Wang, J. Xu and F. Deng, Mechanism of methanol-to-hydrocarbon reaction over zeolites: A solid-state NMR perspective, ChemCatChem, 2020, 12, 965 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen and J. F. Li, et al., The formation and degradation of active species during methanol conversion over protonated zeotype catalysts, Chem. Soc. Rev., 2015, 44(20), 7155–7176 RSC.
- P. A. Wright, Microporous framework solids, RSC, Cambridge, 2008 Search PubMed.
- P. Payra and P. K. Dutta, Zeolites: A primer, in Handbook of Zeolite Science and Technology United States of America, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker, Inc., 2003 Search PubMed.
- W. Wang, Y. Jiang and M. Hunger, Mechanistic investigations of the methanol-to-olefin (MTO) process on acidic zeolite catalysts by in situ solid-state NMR spectroscopy, Catal. Today, 2006, 113(1–2), 102–114 CrossRef CAS.
- J. Q. Chen, A. Bozzano, B. Glover, T. Fuglerud and S. Kvisle, Recent advancements in ethylene and propylene production using the UOP/hydro MTO process, Catal. Today, 2005, 106(1–4), 103–107 CrossRef CAS.
- J. F. Haw and W. Song, inventorsShip in a bottle catalysts, USA, 2006 Search PubMed.
- J. Weitkamp, Zeolites and catalysis., Solid State Ionics, 2000, 131(1–2), 175–188 CrossRef CAS.
- M. Tong, D. Zhang, W. Fan, J. Xu, L. Zhu and W. Guo, et al., Synthesis of chiral polymorph A-enriched zeolite Beta with an extremely concentrated fluoride route., Sci. Rep., 2015, 5, 11521 CrossRef PubMed.
- C. Baerlocher and L. B. McCusker, Database of zeolite structures, http://www.iza-structure.org/databases/.
- J. Cobb, New Zealand Synfuel: The Story of the World's First Natural Gas to Gasoline Plant, ed. G. Connell, Cobb/Horwood Publications, Auckland, New Zealand, 1995 Search PubMed.
- T. Omojola, N. Cherkasov, E. V. Rebrov, D. B. Lukyanov and S. P. Perera, Zeolite minilith: A unique structured catalyst for the methanol to gasoline process, Chem. Eng. Process., 2018, 131, 137–143 CrossRef CAS.
- H. Koempel and W. Liebner, Lurgi's methanol to propylene (MTP®) report on a successful commercialisation, Stud. Surf. Sci. Catal., 2007, 261–267 CrossRef.
- T. Wang, X. Tang, X. Huang, W. Qian, Y. Cui and X. Hui, et al., Conversion of methanol to aromatics in fluidized bed reactor., Catal. Today, 2014, 233, 8–13 CrossRef CAS.
- J. G. Zhang, W. Z. Qian, X. P. Tang, K. Shen, T. Wang and X. F. Huang, et al., Influence of catalyst acidity on dealkylation, isomerization and alkylation in MTA process, Wuli Huaxue Xuebao/Acta Phys. – Chim. Sin., 2013, 29(6), 1281–1288 CAS.
- Z. M. Cui, Q. Liu, W. G. Song and L. J. Wan, Insights into the mechanism of methanol-to-olefin conversion at zeolites with systematically selected framework structures, Angew. Chem., Int. Ed., 2006, 45(39), 6512–6515 CrossRef CAS PubMed.
- S. Teketel, S. Svelle, K. P. Lillerud and U. Olsbye, Shape-selective conversion of methanol to hydrocarbons over 10-ring unidirectional-channel acidic H-ZSM-22, ChemCatChem, 2009, 1(1), 78–81 CrossRef CAS.
- Z. M. Cui, Q. Liu, Z. Ma, S. W. Bian and W. G. Song, Direct observation of olefin homologations on zeolite ZSM-22 and its implications to methanol to olefin conversion, J. Catal., 2008, 258(1), 83–86 CrossRef CAS.
- J. Li, Y. Wei, G. Liu, Y. Qi, P. Tian and B. Li, et al., Comparative study of MTO conversion over SAPO-34, H-ZSM-5 and H-ZSM-22: Correlating catalytic performance and reaction mechanism to zeolite topology, Catal. Today, 2011, 171(1), 221–228 CrossRef CAS.
- S. Teketel, W. Skistad, S. Benard, U. Olsbye, K. P. Lillerud and P. Beato, et al., Shape selectivity in the conversion of methanol to hydrocarbons: The catalytic performance of one-dimensional 10-ring zeolites: ZSM-22, ZSM-23, ZSM-48, and EU-1, ACS Catal., 2012, 2(1), 26–37 CrossRef CAS.
- M. Bjørgen, S. Akyalcin, U. Olsbye, S. Benard, S. Kolboe and S. Svelle, Methanol to hydrocarbons over large cavity zeolites: Toward a unified description of catalyst deactivation and the reaction mechanism., J. Catal., 2010, 275(1), 170–180 CrossRef.
- O. Mikkelsen and S. Kolboe, The conversion of methanol to hydrocarbons over zeolite H-beta, Microporous Mesoporous Mater., 1999, 29(1–2), 173–184 CrossRef CAS.
- C. H. L. Tempelman, X. Zhu, K. Gudun, B. Mezari, B. Shen and E. J. M. Hensen, Texture, acidity and fluid catalytic cracking performance of hierarchical faujasite zeolite prepared by an amphiphilic organosilane, Fuel Process. Technol., 2015, 139, 248–258 CrossRef CAS.
- Y. Wei, D. Zhang, F. Chang and Z. Liu, Direct observation of induction period of MTO process with consecutive pulse reaction system, Catal. Commun., 2007, 8(12), 2248–2252 CrossRef CAS.
- Y. Zhou, L. Qi, Y. Wei, C. Yuan, M. Zhang and Z. Liu, Methanol-to-olefin induction reaction over SAPO-34, Chinese J. Catal., 2016, 37(9), 1496–1501 CrossRef CAS.
- A. Hwang, M. Kumar, J. D. Rimer and A. Bhan, Implications of methanol disproportionation on catalyst lifetime for methanol-to-olefins conversion by HSSZ-13, J. Catal., 2017, 346, 154–160 CrossRef CAS.
- K. De Wispelaere, C. S. Wondergem, B. Ensing, K. Hemelsoet, E. J. Meijer and B. M. Weckhuysen, et al., Insight into the effect of water on the methanol-to-olefins conversion in H-SAPO-34 from molecular simulations and in situ microspectroscopy, ACS Catal., 2016, 6(3), 1991–2002 CrossRef CAS.
- S. Bailleul, I. Yarulina, A. E. J. Hoffman, A. Dokania, E. Abou-Hamad and A. D. Chowdhury, et al., A supramolecular view on the cooperative role of Brønsted and Lewis acid sites in zeolites for methanol conversion, J. Am. Chem. Soc., 2019, 141(37), 14823–14842 CrossRef CAS PubMed.
- T. Omojola, Experimental and Kinetic Modelling Studies of Methanol Conversion to Hydrocarbons over Zeolite Catalysts, PhD Thesis, University of Bath, 2019 Search PubMed.
- M. Bjørgen, F. Joensen, K. P. Lillerud, U. Olsbye and S. Svelle, The mechanisms of ethene and propene formation from methanol over high silica H-ZSM-5 and H-beta, Catal. Today, 2009, 142(1–2), 90–97 CrossRef.
- I. M. Dahl and S. Kolboe, On the reaction mechanism for propene formation in the MTO reaction over SAPO-34, Catal. Lett., 1993, 20(3–4), 329–336 CrossRef CAS.
- C. M. Wang, Y. D. Wang, Y. J. Du, G. Yang and Z. K. Xie, Computational insights into the reaction mechanism of methanol-to-olefins conversion in H-ZSM-5: Nature of hydrocarbon pool, Catal.: Sci. Technol., 2016, 6(9), 3279–3288 RSC.
- R. van Santen and M. Neurock, Molecular Heterogeneous Catalysis: A Conceptual and Computational Approach, Wiley-VCH Verlag GmbH, 2006 Search PubMed.
- S. Svelle, U. Olsbye, F. Joensen and M. Bjørgen, Conversion of methanol to alkenes over medium- and large-pore acidic zeolites: Steric manipulation of the reaction intermediates governs the ethene/propene product selectivity, J. Phys. Chem. C, 2007, 111(49), 17981–17984 CrossRef CAS.
- S. Teketel, U. Olsbye, K. P. Lillerud, P. Beato and S. Svelle, Selectivity control through fundamental mechanistic insight in the conversion of methanol to hydrocarbons over zeolites, Microporous Mesoporous Mater., 2010, 136(1–3), 33–41 CrossRef CAS.
- D. Rojo-Gama, S. Etemadi, E. Kirby, K. P. Lillerud, P. Beato, S. Svellea and U. Olsbye, Time- and space-resolved study of the methanol to hydrocarbons (MTH) reaction – influence of zeolite topology on axial deactivation patterns, Faraday Discuss., 2017, 197, 421–446 RSC.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle and K.-P. Lillerud, et al., The effect of acid strength on the conversion of methanol to olefins over acidic microporous catalysts with the CHA topology, Top. Catal., 2009, 52(3), 218–228 CrossRef CAS.
- J. Dedecek, V. Balgová, V. Pashkova, P. Klein and B. Wichterlová, Synthesis of ZSM-5 zeolites with defined distribution of Al atoms in the framework and multinuclear MAS NMR analysis of the control of Al distribution, Chem. Mater., 2012, 24(16), 3231–3239 CrossRef CAS.
- W. O. Haag, R. M. Lago and P. B. Weisz, The active site of acidic aluminosilicate catalysts, Nature, 1984, 309(5969), 589–591 CrossRef CAS.
- M. Trombetta, T. Armaroli, A. D. Gutièrrez Alejandre, J. Ramirez Solis and G. Busca, An FT-IR study of the internal and external surfaces of HZSM5 zeolite, Appl. Catal., A, 2000, 192(1), 125–136 CrossRef CAS.
- T. Armaroli, L. J. Simon, M. Digne, T. Montanari, M. Bevilacqua and V. Valtchev, et al., Effects of crystal size and Si/Al ratio on the surface properties of H-ZSM-5 zeolites, Appl. Catal., A, 2006, 306, 78–84 CrossRef CAS.
- T. Armaroli, M. Trombetta, A. Gutièrrez Alejandre, J. R. Solis and G. Busca, FTIR study of the interaction of some branched aliphatic molecules with the external and internal sites of H-ZSM5 zeolite, Phys. Chem. Chem. Phys., 2000, 2(14), 3341–3348 RSC.
- T. K. Phung and G. Busca, On the Lewis acidity of protonic zeolites, Appl. Catal., A, 2015, 504, 151–157 CrossRef CAS.
- M. Trombetta, A. Gutièrrez Alejandre, J. Ramirez Solis and G. Busca, An FT-IR study of the reactivity of hydrocarbons on the acid sites of HZSM5 zeolite, Appl. Catal., A, 2000, 198(1-2), 81–93 CrossRef CAS.
- J. R. Di Iorio, C. T. Nimlos and R. Gounder, Introducing catalytic diversity into single-site chabazite zeolites of fixed composition via synthetic control of active site proximity, ACS Catal., 2017, 7(10), 6663–6674 CrossRef CAS.
- D. L. Bhering, A. Ramírez-Solís and C. J. A. Mota, A density functional theory based approach to extraframework aluminum species in zeolites, J. Phys. Chem. B, 2003, 107(18), 4342–4347 CrossRef CAS.
- S. Müller, Y. Liu, F. M. Kirchberger, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Hydrogen transfer pathways during zeolite catalyzed methanol conversion to hydrocarbons, J. Am. Chem. Soc., 2016, 138(49), 15994–16003 CrossRef PubMed.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, Nature and catalytic role of extraframework aluminum in Faujasite zeolite: A theoretical perspective, ACS Catal., 2015, 5(11), 7024–7033 CrossRef CAS.
- Y. Chu, X. Yi, C. Li, X. Sun and A. Zheng, Brønsted/Lewis acid sites synergistically promote the initial C–C bond formation in the MTO reaction, Chem. Sci., 2018, 9(31), 6470–6479 RSC.
- S. Svelle, S. Kolboe, O. Swang and U. Olsbye, Methylation of alkenes and methylbenzenes by dimethyl ether or methanol on acidic zeolites, J. Phys. Chem. B, 2005, 109(26), 12874–12878 CrossRef CAS PubMed.
- W. W. Kaeding and S. A. Butter, Production of chemicals from methanol. I. Low molecular weight olefins, J. Catal., 1980, 61(1), 155–164 CrossRef CAS.
- E. G. Derouane, J. B. Nagy, P. Dejaifve, J. H. C. van Hooff, B. P. Spekman and J. C. Védrine, et al., Elucidation of the mechanism of conversion of methanol and ethanol to hydrocarbons on a new type of synthetic zeolite, J. Catal., 1978, 53(1), 40–55 CrossRef CAS.
- G. J. Hutchings, F. Gottschalk, M. V. M. Hall and R. Hunter, Hydrocarbon formation from methylating agents over the zeolite catalyst ZSM-5. Comments on the mechanism of carbon–carbon bond and methane formation, J. Chem. Soc., Faraday Trans. 1, 1987, 83(3), 571–583 RSC.
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley and G. Cinque, et al., Elementary steps in the formation of hydrocarbons from surface methoxy groups in HZSM-5 seen by synchrotron infrared microspectroscopy, ACS Catal., 2019, 9(7), 6564–6570 CrossRef CAS.
- D. B. Lukyanov, On the extremely high ability of enhanced activity sites in HZSM-5 to generate carbenium ions, Zeolites, 1994, 14(3), 233–234 CrossRef.
- P. Pérez-Uriarte, A. Ateka, A. T. Aguayo, A. G. Gayubo and J. Bilbao, Kinetic model for the reaction of DME to olefins over a HZSM-5 zeolite catalyst, Chem. Eng. J., 2016, 302, 801–810 CrossRef.
- T. Y. Park and G. F. Froment, Kinetic modeling of the methanol to olefins process. 1. Model formulation, Ind. Eng. Chem. Res., 2001, 40(20), 4172–4186 CrossRef CAS.
- T. Y. Park and G. F. Froment, Kinetic modeling of the methanol to olefins process. 2. Experimental results, model discrimination, and parameter estimation, Ind. Eng. Chem. Res., 2001, 40(20), 4187–4196 CrossRef CAS.
- R. Mihail, S. Straja, G. Maria, G. Musca and G. Pop, A kinetic model for methanol conversion to hydrocarbons, Chem. Eng. Sci., 1983, 38(9), 1581–1591 CrossRef CAS.
- P. Kumar, J. W. Thybaut, S. Svelle, U. Olsbye and G. B. Marin, Single-event microkinetics for methanol to olefins on H-ZSM-5, Ind. Eng. Chem. Res., 2013, 52(4), 1491–1507 CrossRef CAS.
- P. Kumar, J. W. Thybaut, S. Teketel, S. Svelle, P. Beato and U. Olsbye, et al., Single-Event MicroKinetics (SEMK) for methanol to hydrocarbons (MTH) on H-ZSM-23, Catal. Today, 2013, 215, 224–232 CrossRef CAS.
- S. M. Alwahabi and G. F. Froment, Single event kinetic modeling of the methanol-to-olefins process on SAPO-34, Ind. Eng. Chem. Res., 2004, 43(17), 5098–5111 CrossRef CAS.
- F. Haase and J. Sauer, Ab initio molecular dynamics simulation of methanol interacting with acidic zeolites of different framework structure, Microporous Mesoporous Mater., 2000, 35-36, 379–385 CrossRef CAS.
- S. P. Greatbanks, I. H. Hillier, N. A. Burton and P. Sherwood, Adsorption of water and methanol on zeolite Brønsted acid sites: An ab initio, embedded cluster study including electron correlation, J. Chem. Phys., 1996, 105(9), 3770–3776 CrossRef CAS.
- A. J. O'Malley, A. J. Logsdail, A. A. Sokol and C. R. A. Catlow, Modelling metal centres, acid sites and reaction mechanisms in microporous catalysts, Faraday Discuss., 2016, 188, 235–255 RSC.
- W. Wang, M. Seiler and M. Hunger, Role of surface methoxy species in the conversion of methanol to dimethyl ether on acidic zeolites investigated by in situ stopped-flow MAS NMR spectroscopy, J. Phys. Chem. B, 2001, 105(50), 12553–12558 CrossRef CAS.
- C. D. Chang, A kinetic model for methanol conversion to hydrocarbons, Chem. Eng. Sci., 1980, 35(3), 619–622 CrossRef CAS.
- N. Y. Chen and W. J. Reagan, Evidence of autocatalysis in methanol to hydrocarbon reactions over zeolite catalysts, J. Catal., 1979, 59(1), 123–129 CrossRef CAS.
- C. M. Nguyen, M.-F. Reyniers and G. B. Marin, Adsorption thermodynamics of C1–C4 alcohols in H-FAU, H-MOR, H-ZSM-5, and H-ZSM-22, J. Catal., 2015, 322, 91–103 CrossRef CAS.
- S. Svelle, C. Tuma, X. Rozanska, T. Kerber and J. Sauer, Quantum chemical modeling of zeolite-catalyzed methylation reactions: Toward chemical accuracy for barriers, J. Am. Chem. Soc., 2009, 131(2), 816–825 CrossRef CAS PubMed.
- G. Pop, G. Bozga, R. Ganea and N. Natu, Methanol conversion to dimethyl ether over H-SAPO-34 catalyst, Ind. Eng. Chem. Res., 2009, 48(15), 7065–7071 CrossRef CAS.
- K. Hemelsoet, A. Ghysels, D. Mores, K. De Wispelaere, V. Van Speybroeck and B. M. Weckhuysen, et al., Experimental and theoretical IR study of methanol and ethanol conversion over H-SAPO-34, Catal. Today, 2011, 177(1), 12–24 CrossRef CAS.
- Y. Kobayashi, Y. Li, Y. Wang and D. Wang, Adsorption isotherms of methanol and dimethyl ether on SAPO-34 measured together with differential adsorption heat measurement, Chin. J. Catal., 2013, 34(12), 2192–2199 CrossRef CAS.
- P. E. Sinclair and C. R. A. Catlow, Generation of carbenes during methanol conversion over Brønsted acidic aluminosilicates. A computational study, J. Phys. Chem. B, 1997, 101(3), 295–298 CrossRef CAS.
- C. Aharoni and F. C. Tompkins, Kinetics of adsorption and desorption and the elovich equation, in Advances in Catalysis. 21, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1970, pp. 1–49 Search PubMed.
- G. Piccini, M. Alessio and J. Sauer, Ab initio study of methanol and ethanol adsorption on Brønsted sites in zeolite H-MFI, Phys. Chem. Chem. Phys., 2018, 20(30), 19964–19970 RSC.
- J.-R. Hill and J. Sauer, Molecular mechanics potential for silica and zeolite catalysts based on ab initio calculations. 2. Aluminosilicates, J. Phys. Chem., 1995, 99(23), 9536–9550 CrossRef CAS.
- S. Etemadi and U. Olsbye, Catalytic investigations of zeolite based methanol to hydrocarbons catalysts, University of Oslo, Olsbye.
- S. Ted Oyama, G.-N. Yun, S.-J. Ahn, K. K. Bando, A. Takagaki and R. Kikuchi, How to scrutinize adsorbed intermediates observed by in situ spectroscopy: Analysis of coverage transients (ACT), J. Catal., 2021, 394, 273–283 CrossRef CAS.
- G. A. Olah, H. Doggweiler, J. D. Felberg, S. Frohlich and M. J. Grdina, et al. , J. Am. Chem. Soc., 1984, 106(7), 2143–2149 CrossRef CAS.
- J. Nováková, L. Kubelková, K. Habersberger and Z. Dolejšek, Catalytic activity of dealuminated Y and HZSM-5 zeolites measured by the temperature-programmed desorption of small amounts of preadsorbed methanol and by the low-pressure flow reaction of methanol, J. Chem. Soc., Faraday Trans. 1, 1984, 80(6), 1457–1465 RSC.
- J. Nováková, L. Kubelková and Z. Dolejšek, Intermediates in methanol and ethanol transformation on HZSM-5 zeolite studied by chemical trapping and deuterium labeling, J. Mol. Catal., 1988, 45(3), 365–372 CrossRef.
- J. K. A. Clarke, R. Darcy, B. F. Hegarty, E. O'Donoghue, V. Amir-Ebrahimi and J. R. Rooney, Free Radicals in Dimethyl Ether on H-ZSM-5 Zeolite. A Novel Dimension of Heterogeneous Catalysis, J. Chem. Soc., Chem. Commun., 1986, 5, 425–426 RSC.
- C. D. Chang, S. D. Hellring and J. A. Pearson, On the existence and role of free radicals in methanol conversion to hydrocarbons over HZSM-5. I. Inhibition by NO, J. Catal., 1989, 115(1), 282–285 CrossRef CAS.
- J. Li, Z. Wei, Y. Chen, B. Jing, Y. He and M. Dong, et al., A route to form initial hydrocarbon pool species in methanol conversion to olefins over zeolites, J. Catal., 2014, 317(0), 277–283 CrossRef CAS.
- P. Cheung, A. Bhan, G. J. Sunley, D. J. Law and E. Iglesia, Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites, J. Catal., 2007, 245(1), 110–123 CrossRef CAS.
- P. F. Gay and M. W. Travers, On the thermal decomposition of dimethyl ether, Trans. Faraday Soc., 1937, 33, 756–770 RSC.
- C. T. W. Chu and C. D. Chang, Methanol conversion to olefins over ZSM-5. II. Olefin distribution, J Catal., 1984, 86(2), 297–300 CrossRef CAS.
- G. J. Hutchings, F. Gottschalk and R. Hunter, Comments on “kinetic model for methanol conversion to olefins” with respect to methane formation at low conversion, Ind. Eng. Chem. Res., 1987, 26(3), 635–637 CrossRef CAS.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri and S. A. Al-Thabaiti, et al., Initial carbon–carbon bond formation during the early stages of the methanol-to-olefin process proven by zeolite-trapped acetate and methyl acetate, Angew. Chem., Int. Ed., 2016, 55(51), 15840–15845 CrossRef CAS PubMed.
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Bridging the gap between the direct and hydrocarbon pool mechanisms of the methanol-to-hydrocarbons process, Angew. Chem., Int. Ed., 2018, 57, 8095 CrossRef CAS PubMed.
- H. Pines, Acid-Catalyzed Reactions The Chemistry of Catalytic Hydrocarbon Conversions, Academic Press, UK, 1981 Search PubMed.
- B. C. Gates, J. R. Katzer and G. C. A. Schuit, Cracking. Chemistry of Catalytic Processes, McGraw-Hill, USA, 1979 Search PubMed.
- S. A. Butter, W. W. Kaeding and A. T. Jurewicz, inventors. U.S. 1975.
- T. R. Forester and R. F. Howe, In situ FTIR studies of methanol and dimethyl ether in ZSM-5, J. Am. Chem. Soc., 1987, 109(17), 5076–5082 CrossRef CAS.
- Y. Ono and T. Mori, Mechanism of methanol conversion into hydrocarbons over ZSM-5 zeolite, J. Chem. Soc., Faraday Trans. 1, 1981, 77(9), 2209–2221 RSC.
- Y. Jiang, M. Hunger and W. Wang, On the reactivity of surface methoxy species in acidic zeolites, J. Am. Chem. Soc., 2006, 128(35), 11679–11692 CrossRef CAS PubMed.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, Evidence for an Initiation of the methanol-to-olefin process by reactive surface methoxy groups on acidic zeolite catalysts, J. Am. Chem. Soc., 2003, 125(49), 15260–15267 CrossRef CAS PubMed.
- S. R. Blaszkowski and R. A. Van Santen, The mechanism of dimethyl ether formation from methanol catalyzed by zeolitic protons, J. Am. Chem. Soc., 1996, 118(21), 5152–5153 CrossRef CAS.
- S. R. Blaszkowski and R. A. Van Santen, Theoretical study of the mechanism of surface methoxy and dimethyl ether formation from methanol catalyzed by zeolitic protons, J. Phys. Chem. B, 1997, 101(13), 2292–2305 CrossRef CAS.
- R. Shah, J. D. Gale and M. C. Payne, In situ study of reactive intermediates of methanol in zeolites from first principles calculations, J. Phys. Chem. B, 1997, 101(24), 4787–4797 CrossRef CAS.
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, Methylbenzenes are the organic reaction centers for methanol-to-olefin catalysis on HSAPO-34, J. Am. Chem. Soc., 2000, 122(43), 10726–10727 CrossRef CAS.
- W. Song, D. M. Marcus, H. Fu, J. O. Ehresmann and J. F. Haw, An oft-studied reaction that may never have been: Direct catalytic conversion of methanol or dimethyl ether to hydrocarbons on the solid acids HZSM-5 or HSAPO-34, J. Am. Chem. Soc., 2002, 124(15), 3844–3845 CrossRef CAS PubMed.
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman and V. Van Speybroeck, et al., A complete catalytic cycle for supramolecular methanol-to-olefins conversion by linking theory with experiment, Angew. Chem., Int. Ed., 2008, 47(28), 5179–5182 CrossRef CAS PubMed.
- D. M. Marcus, K. A. McLachlan, M. A. Wildman, J. O. Ehresmann, P. W. Kletnieks and J. F. Haw, Experimental evidence from H/D exchange studies for the failure of direct C–C coupling mechanisms in the methanol-to-olefin process catalyzed by HSAPO-34, Angew. Chem., Int. Ed., 2006, 45(19), 3133–3136 CrossRef CAS PubMed.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, Understanding the failure of direct C–C coupling in the zeolite-catalyzed methanol-to-olefin process, Angew. Chem., Int. Ed., 2006, 45(11), 1714–1719 CrossRef CAS PubMed.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, The rise and fall of direct mechanisms in methanol-to-olefin catalysis: An overview of theoretical contributions, Ind. Eng. Chem. Res., 2007, 46(26), 8832–8838 CrossRef CAS.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, What role do oxonium ions and oxonium ylides play in the ZSM-5 catalysed methanol-to-olefin process?, Chem. Phys. Lett., 2006, 417(4–6), 309–315 CrossRef CAS.
- W. Dai, C. Wang, M. Dyballa, G. Wu, N. Guan and L. Li, et al., Understanding the early stages of the methanol-to-olefin conversion on H-SAPO-34, ACS Catal., 2015, 5(1), 317–326 CrossRef CAS.
- W. Song, J. B. Nicholas and J. F. Haw, A persistent carbenium ion on the methanol-to-olefin catalyst HSAPO-34: Acetone shows the way, J. Phys. Chem. B, 2001, 105(19), 4317–4323 CrossRef CAS.
- H. Fu, W. Song and J. F. Haw, Polycyclic aromatics formation in HSAPO-34 during methanol-to-olefin catalysis: Ex situ characterization after cryogenic grinding, Catal. Lett., 2001, 76(1-2), 89–94 CrossRef CAS.
- W. Wang and M. Hunger, Reactivity of surface alkoxy species on acidic zeolite catalysts, Acc. Chem. Res., 2008, 41(8), 895–904 CrossRef CAS PubMed.
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Direct production of propene from methoxy species and dimethyl ether over H-ZSM-5, J. Phys. Chem. C, 2012, 116(45), 24091–24097 CrossRef CAS.
- P. W. Goguen, T. Xu, D. H. Barich, T. W. Skloss, W. Song and Z. Wang, et al., Pulse-quench catalytic reactor studies reveal a carbon-pool mechanism in methanol-to-gasoline chemistry on zeolite HZSM-5, J. Am. Chem. Soc., 1998, 120(11), 2650–2651 CrossRef CAS.
- T. Mole, G. Bett and D. Seddon, Conversion of methanol to hydrocarbons over ZSM-5 zeolite: An examination of the role of aromatic hydrocarbons using 13carbon- and deuterium-labeled feeds, J. Catal., 1983, 84(2), 435–445 CrossRef CAS.
- T. Mole, J. A. Whiteside and D. Seddon, Aromatic co-catalysis of methanol conversion over zeolite catalysts, J. Catal., 1983, 82(2), 261–266 CrossRef CAS.
- B. Arstad, J. B. Nicholas and J. F. Haw, Theoretical study of the methylbenzene side-chain hydrocarbon pool mechanism in methanol to olefin catalysis, J. Am. Chem. Soc., 2004, 126(9), 2991–3001 CrossRef CAS PubMed.
- A. Sassi, M. A. Wildman, H. J. Ahn, P. Prasad, J. B. Nicholas and J. F. Haw, Methylbenzene chemistry on zeolite HBeta: Multiple insights into methanol-to-olefin catalysis, J. Phys. Chem. B, 2002, 106(9), 2294–2303 CrossRef CAS.
- R. F. Sullivan, R. P. Sieg, G. E. Langlois and C. J. Egan, A new reaction that occurs in hydrocracking of certain aromatic hydrocarbons, J. Am. Chem. Soc., 1961, 83(5), 1156–1160 CrossRef CAS.
- M. Bjørgen, U. Olsbye, S. Svelle and S. Kolboe, Conversion of methanol to hydrocarbons: The reactions of the heptamethylbenzenium cation over zeolite H-beta, Catal. Lett., 2004, 93(1–2), 37–40 CrossRef.
- D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Zeolite shape-selectivity in the gem-methylation of aromatic hydrocarbons, Angew. Chem., Int. Ed., 2007, 46(8), 1311–1314 CrossRef CAS PubMed.
- D. Lesthaeghe, A. Horré, M. Waroquier, G. B. Marin and V. Van Speybroeck, Theoretical insights on methylbenzene side-chain growth in zsm-5 zeolites for methanol-to-olefin conversion, Chem. – Eur. J., 2009, 15(41), 10803–10808 CrossRef CAS PubMed.
- S. K. Matam, S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, Methanol loading dependent methoxylation in zeolite H-ZSM-5, Chem. Sci., 2020, 11(26), 6805–6814 RSC.
- S. K. Matam, R. F. Howe, A. Thetford and C. R. A. Catlow, Room temperature methoxylation in zeolite H-ZSM-5: An operando DRIFTS/mass spectrometric study, Chem. Commun., 2018, 54(91), 12875–12878 RSC.
- A. O'Malley, S. F. Parker, A. Chutia, M. R. Farrow, I. P. Silverwood and V. Garcia-Sakai, et al., Room temperature methoxylation in zeolites: insight into a key step of the methanol-to-hydrocarbons process, Chem. Commun., 2016, 52, 2897–2900 RSC.
- J. F. Haw, J. B. Nicholas, W. Song, F. Deng, Z. Wang and T. Xu, et al., Roles for cyclopentenyl cations in the synthesis of hydrocarbons from methanol on zeolite catalyst HZSM-5, J. Am. Chem. Soc., 2000, 122(19), 4763–4775 CrossRef CAS.
- T. Xu, D. H. Barich, P. W. Goguen, W. Song, Z. Wang and J. B. Nicholas, et al., Synthesis of a benzenium ion in a zeolite with use of a catalytic flow reactor, J. Am. Chem. Soc., 1998, 120(16), 4025–4026 CrossRef CAS.
- F. G. Oliver, E. J. Munson and J. F. Haw, High-temperature in situ magic angle spinning NMR studies of chemical reactions on catalysts, J. Phys. Chem., 1992, 96(20), 8106–8111 CrossRef CAS.
- Y. Jiang, W. Wang, V. R. R. Marthala, J. Huang, B. Sulikowski and M. Hunger, Effect of organic impurities on the hydrocarbon formation via the decomposition of surface methoxy groups on acidic zeolite catalysts, J. Catal., 2006, 238(1), 21–27 CrossRef CAS.
- Y. Jiang, W. Wang, V. R. Reddy Marthala, J. Huang, B. Sulikowski and M. Hunger, Response to comments on the paper: “Effect of organic impurities on the hydrocarbon formation via the decomposition of surface methoxy groups on acidic zeolite catalysts” by Y. Jiang, W. Wang, V.R.R. Marthala, J. Huang, B. Sulikowski, M. Hunger, J. Catal., 2006, 244(1), 134–136 CrossRef CAS.
- V. Bosáček, Formation of surface-bonded methoxy groups in the sorption of methanol and methyl iodide on zeolites studied by 13C MAS NMR spectroscopy, J. Phys. Chem., 1993, 97(41), 10732–10737 CrossRef.
- J. Nováková, L. Kubelková and Z. Dolejšek, Deuterium-labeled methanol in reactions with HZSM-5 zeolite, J. Catal., 1986, 97(1), 277–279 CrossRef.
- E. G. Derouane, J. P. Gilson and J. B. Nagy, Adsorption and conversion of ethylene on H-ZSM-5 zeolite, Zeolites, 1982, 2, 42 CrossRef CAS.
- N. Tajima, T. Tsuneda, F. Toyama and K. Hirao, A new mechanism for the first carbon–carbon bond formation in the MTG process: A theoretical study, J. Am. Chem. Soc., 1998, 120(32), 8222–8229 CrossRef CAS.
- S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, QM/MM study of the reactivity of zeolite bound methoxy and carbene groups, Phys. Chem. Chem. Phys., 2021, 23, 17634–17644 RSC.
- D. V. Dass, R. W. Martin, A. L. Odell and G. W. Quinn, A re-examination of evidence for carbene (CH2:) As an intermediate in the conversion of methanol to gasoline. The effect of added propane, in Stud Surf Sci Catal. 36, ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, Elsevier, 1988, pp. 177–81 Search PubMed.
- E. A. Swabb and B. C. Gates, Diffusion, reaction, and fouling in H-mordenite crystallites. The catalytic dehydration of methanol, Ind. Eng. Chem. Fundam., 1972, 11(4), 540–545 CrossRef CAS.
- R. Hunter and G. J. Hutchings, Hydrocarbon formation from methanol using WO3/Al 2O3 and zeolite H-ZSM-5 catalysts: Further evidence on the reaction mechanism, J. Chem. Soc., Chem. Commun., 1987, 5, 377–379 RSC.
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Evidence for a “carbene-like” intermediate during the reaction of methoxy species with light alkenes on H-ZSM-5, Angew. Chem., Int. Ed., 2011, 50(8), 1853–1856 CrossRef CAS PubMed.
- K. Hemelsoet, J. Van Der Mynsbrugge, K. De Wispelaere, M. Waroquier and V. Van Speybroeck, Unraveling the reaction mechanisms governing methanol-to-olefins catalysis by theory and experiment, ChemPhysChem, 2013, 14(8), 1526–1545 CrossRef CAS PubMed.
- R. Hunter, G. J. Hutchings and W. Pickl, Mechanistic studies on initial C–C bond formation in the zeolite ZSM-5 catalysed methanol conversion reaction: Evidence against a radical pathway, J. Chem. Soc., Chem. Commun., 1987, 11, 843–844 RSC.
- Z. Wei, Y. Y. Chen, J. Li, W. Guo, S. Wang and M. Dong, et al., Stability and Reactivity of Intermediates of Methanol Related Reactions and C–C Bond Formation over H-ZSM-5 Acidic Catalyst: A Computational Analysis, J. Phys. Chem. C, 2016, 120(11), 6075–6087 CrossRef CAS.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li and B. Yu, et al., Direct Mechanism of the First Carbon-Carbon Bond Formation in the Methanol-to-Hydrocarbons Process, Angew. Chem., 2017, 129(31), 9167–9171 CrossRef.
- T. Sun, W. Chen, S. Xu, A. Zheng, X. Wu and S. Zeng, et al., The first carbon–carbon bond formation mechanism in methanol-to-hydrocarbons process over chabazite zeolite, Chem, 2021 DOI:10.1016/j.chempr.2021.05.023.
- J. E. Jackson and F. M. Bertsch, Conversion of methanol to gasoline: A new mechanism for formation of the first carbon–carbon bond, J. Am. Chem. Soc., 1990, 112(25), 9085–9092 CrossRef CAS.
- T. Tago, H. Konno, M. Sakamoto, Y. Nakasaka and T. Masuda, Selective synthesis for light olefins from acetone over ZSM-5 zeolites with nano- and macro-crystal sizes., Appl. Catal., A, 2011, 403(1–2), 183–191 CrossRef CAS.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Siting and distribution of framework aluminium atoms in silicon-rich zeolites and impact on catalysis, Catal. Rev., 2012, 54(2), 135–223 CrossRef.
- A. Ramirez, A. Dutta Chowdhury, A. Dokania, P. Cnudde, M. Caglayan and I. Yarulina, et al., Effect of zeolite topology and reactor configuration on the direct conversion of CO2 to light olefins and aromatics, ACS Catal., 2019, 9(7), 6320–6334 CrossRef CAS.
- L. Kubelková, J. Nováková and P. Jírů, Reaction of small amounts of methanol on Hzsm-5, Hy and modified Y zeolites, in Stud Surf Sci Catal. Volume 18, ed. P. A. Jacobs NIJPJVBK and G. Schulz-Ekloff, Elsevier, 1984, pp. 217–24 Search PubMed.
- D. Kagi, In re: Mechanism of conversion of methanol over ZSM-5 catalyst, J Catal., 1981, 69(1), 242–243 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. E. S. del Campo, A. M. Tarrio and J. Bilbao, Kinetic modeling of methanol transformation into olefins on a SAPO-34 catalyst, Ind. Eng. Chem. Res., 2000, 39(2), 292–300 CrossRef CAS.
- T. Cordero-Lanzac, A. T. Aguayo and J. Bilbao, Reactor–regenerator system for the dimethyl ether-to-olefins process over HZSM-5 catalysts: Conceptual development and analysis of the process variables, Ind. Eng. Chem. Res., 2020, 59(33), 14689–14702 CrossRef CAS.
- A. J. O'Malley, S. F. Parker and C. R. A. Catlow, Neutron spectroscopy as a tool in catalytic science, Chem. Commun., 2017, 53(90), 12164–12176 RSC.
- S. K. Matam, A. J. O’Malley, C. R. A. Catlow, A. Suwardiyanto, P. Collier and A. P. Hawkins, et al., The effects of MTG catalysis on methanol mobility in ZSM-5, Catal. Sci. Technol., 2018, 8(13), 3304–3312 RSC.
- C. T. Chudley and R. J. Elliott, Neutron scattering from a liquid on a jump diffusion model, Proc. Phys. Soc., 1961, 77(2), 353–361 CrossRef.
- F. Volino and A. J. Dianoux, Neutron incoherent scattering law for diffusion in a potential of spherical symmetry: General formalism and application to diffusion inside a sphere, Mol. Phys., 1980, 41(2), 271–279 CrossRef CAS.
- V. F. Sears, Theory of cold neutron scattering by homonuclear diatomic liquids: 1. Free rotation, Can. J. Phys., 1966, 44(6), 1279–1297 CrossRef CAS.
- T. Omojola, I. P. Silverwood and A. J. O'Malley, Molecular behaviour of methanol and dimethyl ether in H-ZSM-5 catalysts as a function of Si/Al ratio: A quasielastic neutron scattering study, Catal. Sci. Technol., 2020, 10(13), 4305–4320 RSC.
- Z. Liu, Y. Chu, X. Tang, L. Huang, G. Li and X. Yi, et al., Diffusion dependence of the dual-cycle mechanism for MTO reaction inside ZSM-12 and ZSM-22 zeolites, J. Phys. Chem. C, 2017, 121(41), 22872–22882 CrossRef CAS.
- D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Zeolite shape-selectivity in thegem-methylation of aromatic hydrocarbons, Angew. Chem., Int. Ed., 2007, 46(8), 1311–1314 CrossRef CAS PubMed.
- Y. Chu, X. Sun, X. Yi, L. Ding, A. Zheng and F. Deng, Slight channel difference influences the reaction pathway of methanol-to-olefins conversion over acidic H-ZSM-22 and H-ZSM-12 zeolites, Catal. Sci. Technol., 2015, 5(7), 3507–3517 RSC.
- Q. Wang, Z.-M. Cui, C.-Y. Cao and W.-G. Song, 0.3 Å makes the difference: Dramatic changes in methanol-to-olefin activities between H-ZSM-12 and H-ZSM-22 zeolites, J. Phys. Chem. C, 2011, 115(50), 24987–24992 CrossRef CAS.
- S. Teketel, U. Olsbye, K.-P. Lillerud, P. Beato and S. Svelle, Selectivity control through fundamental mechanistic insight in the conversion of methanol to hydrocarbons over zeolites, Microporous Mesoporous Mater., 2010, 136(1-3), 33–41 CrossRef CAS.
- J. Li, Y. Wei, Y. Qi, P. Tian, B. Li and Y. He, et al., Conversion of methanol over H-ZSM-22: The reaction mechanism and deactivation, Catal. Today, 2011, 164(1), 288–292 CrossRef CAS.
- R. A. Ocakoglu, J. F. M. Denayer, G. B. Marin, J. A. Martens and G. V. Baron, Tracer chromatographic study of pore and pore mouth adsorption of linear and monobranched alkanes on ZSM-22 zeolite, J. Phys. Chem. B, 2003, 107(1), 398–406 CrossRef CAS.
- E. Grifoni, G. Piccini, J. A. Lercher, V. A. Glezakou, R. Rousseau and M. Parrinello, Confinement effects and acid strength in zeolites, Nat. Commun., 2021, 12(1), 2630 CrossRef CAS PubMed.
- S. Eckstein, P. H. Hintermeier, R. Zhao, E. Baráth, H. Shi and Y. Liu, et al., Influence of hydronium ions in zeolites on sorption, Angew. Chem., Int. Ed., 2019, 58(11), 3450–3455 CrossRef CAS PubMed.
- G. Mirth, J. A. Lercher, M. W. Anderson and J. Klinowski, Adsorption complexes of methanol on zeolite ZSM-5, J. Chem. Soc., Faraday Trans., 1990, 86(17), 3039–3044 RSC.
- P. Cnudde, E. A. Redekop, W. Dai, N. G. Porcaro, M. Waroquier and S. Bordiga, et al., Experimental and theoretical evidence for the promotional effect of acid sites on the diffusion of alkenes through small-pore zeolites, Angew. Chem., Int. Ed., 2021, 60(18), 10016–10022 CrossRef CAS PubMed.
- P. Cnudde, R. Demuynck, S. Vandenbrande, M. Waroquier, G. Sastre and V. V. Speybroeck, Light olefin diffusion during the MTO process on H-SAPO-34: A complex interplay of molecular factors, J. Am. Chem. Soc., 2020, 142(13), 6007–6017 CrossRef CAS PubMed.
- D. M. Bibby, R. F. Howe and G. D. McLellan, Coke formation in high-silica zeolites, Appl. Catal., A, 1992, 93(1), 1–34 CrossRef CAS.
- M. Guisnet and P. Magnoux, Organic chemistry of coke formation, Appl. Catal., A, 2001, 212(1), 83–96 CrossRef CAS.
- T. V. W. Janssens, A new approach to the modeling of deactivation in the conversion of methanol on zeolite catalysts, J. Catal., 2009, 264(2), 130–137 CrossRef CAS.
- T. V. W. Janssens, S. Svelle and U. Olsbye, Kinetic modeling of deactivation profiles in the methanol-to-hydrocarbons (MTH) reaction: A combined autocatalytic-hydrocarbon pool approach, J. Catal., 2013, 308, 122–130 CrossRef CAS.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle and K. P. Lillerud, et al., The effect of acid strength on the conversion of methanol to olefins over acidic microporous catalysts with the CHA topology, Top. Catal., 2009, 52(3), 218–228 CrossRef CAS.
- H. Schulz, “Coking” of zeolites during methanol conversion: Basic reactions of the MTO-, MTP- and MTG processes, Catal. Today, 2010, 154(3-4), 183–194 CrossRef CAS.
- R. Y. Brogaard, B. M. Weckhuysen and J. K. Nørskov, Guest–host interactions of arenes in H-ZSM-5 and their impact on methanol-to-hydrocarbons deactivation processes, J. Catal., 2013, 300, 235–241 CrossRef CAS.
- D. Mores, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Coke formation during the methanol-to-olefin conversion: In situ microspectroscopy on individual H-ZSM-5 crystals with different brønsted acidity, Chem. – Eur. J., 2011, 17(10), 2874–2884 CrossRef CAS PubMed.
- S. Svelle, P. O. Rønning and S. Kolboe, Kinetic studies of zeolite-catalyzed methylation reactions: 1. Coreaction of [12C]ethene and [13C]methanol, J Catal., 2004, 224(1), 115–123 CrossRef CAS.
- I. M. Hill, S. A. Hashimi and A. Bhan, Kinetics and mechanism of olefin methylation reactions on zeolites, J. Catal., 2012, 285(1), 115–123 CrossRef CAS.
- M. Wang, N. R. Jaegers, M. S. Lee, C. Wan, J. Z. Hu and H. Shi, et al., Genesis and stability of hydronium ions in zeolite channels, J. Am. Chem. Soc., 2019, 141(8), 3444–3455 CrossRef CAS PubMed.
- M.-F. Reyniers and G. B. Marin, Experimental and theoretical methods in kinetic studies of heterogeneously catalyzed reactions, Ann. Rev. Chem. Biomol. Eng., 2014, 5(1), 563–594 CrossRef CAS PubMed.
- S. Vernuccio and L. J. Broadbelt, Discerning complex reaction networks using automated generators, AlChE J., 2019, 65(8), e16663 Search PubMed.
- A. B. Bendtsen, P. Glarborg and K. Dam-Johansen, Visualization methods in analysis of detailed chemical kinetics modelling, Comput. Chem., 2001, 25(2), 161–170 CrossRef CAS PubMed.
- M. John, K. Alexopoulos, M.-F. Reyniers and G. B. Marin, Effect of zeolite confinement on the conversion of 1-butanol to butene isomers: mechanistic insights from DFT based microkinetic modelling, Catal. Sci. Technol., 2017, 7(14), 2978–2997 RSC.
- M. Boronat and A. Corma, What is measured when measuring acidity in zeolites with probe molecules?, ACS Catal., 2019, 9(2), 1539–1548 CrossRef CAS PubMed.
- S. M. Rao, E. Saraçi, R. Gläser and M. O. Coppens, Surface barriers as dominant mechanism to transport limitations in hierarchically structured catalysts – application to the zeolite-catalyzed alkylation of benzene with ethylene, Chem Eng J., 2017, 329, 45–55 CrossRef CAS.
- M. Gao, H. Li, W. Liu, Z. Xu, S. Peng and M. Yang, et al., Imaging spatiotemporal evolution of molecules and active sites in zeolite catalyst during methanol-to-olefins reaction, Nat. Commun., 2020, 11(1), 3641 CrossRef CAS PubMed.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang and S. Wang, et al., Conversion of methanol to olefins over H-ZSM-5 zeolite: Reaction pathway is related to the framework aluminum siting, ACS Catal., 2016, 6(11), 7311–7325 CrossRef CAS.
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma and E. Abou-Hamad, et al., Structure–performance descriptors and the role of Lewis acidity in the methanol-to-propylene process, Nat. Chem., 2018, 10(8), 804–812 CrossRef CAS PubMed.
- A. Jentys, H. Tanaka and J. A. Lercher, Surface processes during sorption of aromatic molecules on medium pore zeolites, J. Phys. Chem. B, 2005, 109(6), 2254–2261 CrossRef CAS PubMed.
- T. Omojola and A. C. van Veen, Competitive adsorption of oxygenates and aromatics during the initial steps of the formation of primary olefins over ZSM-5 catalysts, Catal. Commun., 2020, 140, 106010 CrossRef CAS.
- V. Van Speybroeck, J. Van Der Mynsbrugge, M. Vandichel, K. Hemelsoet, D. Lesthaeghe and A. Ghysels, et al., First principle kinetic studies of zeolite-catalyzed methylation reactions, J. Am. Chem. Soc., 2011, 133(4), 888–899 CrossRef CAS PubMed.
- J. A. Ryder, A. K. Chakraborty and A. T. Bell, Density functional theory study of proton mobility in zeolites: Proton migration and hydrogen exchange in ZSM-5, J. Phys. Chem. B, 2000, 104(30), 6998–7011 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Recent trends and fundamental insights in the methanol-to-hydrocarbons process, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
Footnotes |
| † Current address: Department of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, Dahlem, 14195 Berlin, Germany. |
| ‡ Current address: Department of Chemistry and Engineering, KAUST Catalysis Center, KAUST 4700, Thuwal 23955-6900, Kingdom of Saudi Arabia. |
| This journal is © the Owner Societies 2021 |