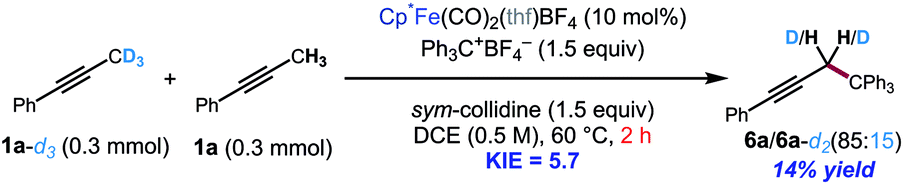DOI:
10.1039/D0SC05091A
(Edge Article)
Chem. Sci., 2020,
11, 12316-12322
Iron-catalyzed α-C–H functionalization of π-bonds: cross-dehydrogenative coupling and mechanistic insights†
Received
14th September 2020
, Accepted 16th October 2020
First published on 16th October 2020
Abstract
The deprotonation of propargylic C–H bonds for subsequent functionalization typically requires stoichiometric metal alkyl or amide reagents. In addition to the undesirable generation of stoichiometric metallic waste, these conditions limit the functional group compatibility and versatility of this functionalization strategy and often result in regioisomeric mixtures. In this article, we report the use of dicarbonyl cyclopentadienyliron(II) complexes for the generation of propargylic anion equivalents toward the direct electrophilic functionalization of propargylic C–H bonds under mild, catalytic conditions. This technology was applied to the direct conversion of C–H bonds to C–C bonds for the synthesis of several functionalized scaffolds through a one-pot cross dehydrogenative coupling reaction with tetrahydroisoquinoline and related privileged heterocyclic scaffolds. A series of NMR studies and deuterium-labelling experiments indicated that the deprotonation of the propargylic C–H bond was the rate-determining step when a Cp*Fe(CO)2-based catalyst system was employed.
Introduction
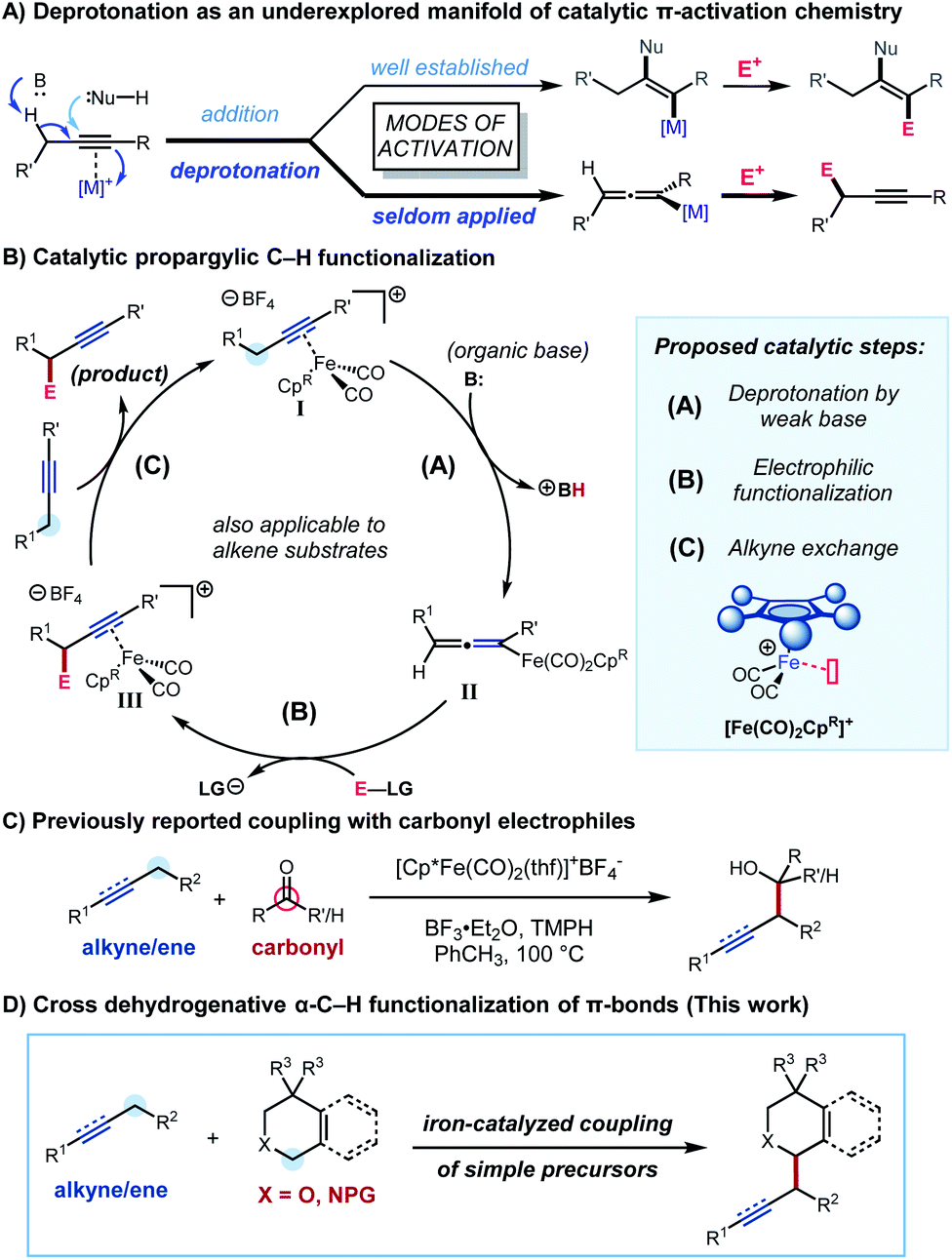
Alkenes and alkynes are synthetically accessible and versatile starting materials for the preparation of stereochemically and functionally complex targets. In addition to a host of catalytic and stoichiometric transformations for hydrofunctionalization and difunctionalization of the C–C multiple bond,1 the allylic and propargylic positions of alkenes and alkynes represent sites of increased chemical reactivity that serve as attractive targets for C–H functionalization processes.2
While several general approaches exist for allylic C–H functionalization, including addition of nucleophiles to electrophilic palladium complexes,3 Kharasch–Sosnovsky-type oxygenation catalysed by copper,4 and direct C–H insertions by rhodium carbenoids and nitrenoids,5 most of these technologies extend poorly to functionalization at the propargylic position of alkynes. As a result, there is a striking dearth of catalytic C–H functionalization processes applicable to the propargylic position compared to those applicable to allylic functionalization. To date, only a small collection of reactions have been developed for propargylic C–H functionalization, primarily through strategies that exploit radical intermediates6 or sigmatropic rearrangement reactions.7 Despite the development of innovative approaches, these processes generally require the presence of directing groups on one or both substrates or are subject to challenges with respect to regioselectivity associated with intermolecular radical processes. Among the handful of methods available for functionalization of propargylic C–H bonds, ones that result in the formation of C–C bonds are particularly rare.6a,e,7a,b
We were interested in developing a general strategy for α-functionalization that would apply to both alkenes and alkynes and specifically address the challenge of forging new C–C bonds. We considered employing π-activation as an underexplored tactic for C–H functionalization by using metal π-coordination to increase the acidity of neighbouring C−H bonds (Scheme 1A). The increased acidity is expected to enable the mild deprotonative cleavage of the C–H bond and subsequent functionalization at the allylic or propargylic site under functional group tolerant conditions. This strategy has been explored using stoichiometric transition metal complexes,8 and Zhang and co-workers have recently reported a similar approach in which bifunctional Au complexes were used for an intramolecular deprotonation at the propargylic position.9 We focused our attention on dicarbonylcyclopentadienyliron complexes,10 whose stoichiometric allylic C–H functionalization chemistry was first investigated by Rosenblum and co-workers,10b as inexpensive and readily accessible scaffolds for catalytic propargylic and allylic C–H functionalization chemistry. We proposed a novel catalytic cycle (Scheme 1B) involving deprotonation of an alkyne–iron (or alkene–iron) π-complex I (step A), electrophilic functionalization of the resultant allenyliron (or allyliron) intermediate II (step B), and exchange of iron-complexed product III with starting alkyne (or alkene) to close the catalytic cycle (step C). In the initial system, we reported the coupling of alkynes and alkenes with carbonyl derivatives to form homopropargylic and homoallylic alcohols (Scheme 1C).11 The hypothesized catalytic cycle was supported by the isolation and reactivity of the proposed allenyliron intermediate.
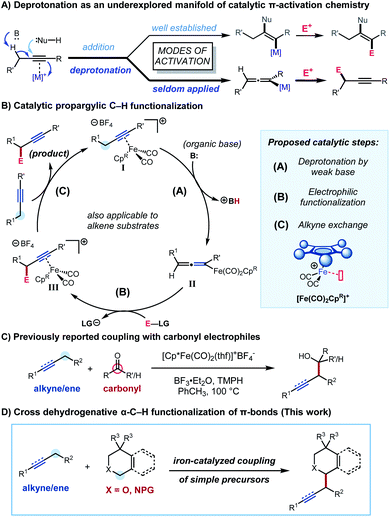 |
| | Scheme 1 Complexation-assisted deprotonation as an underexplored mode of propargylic activation. | |
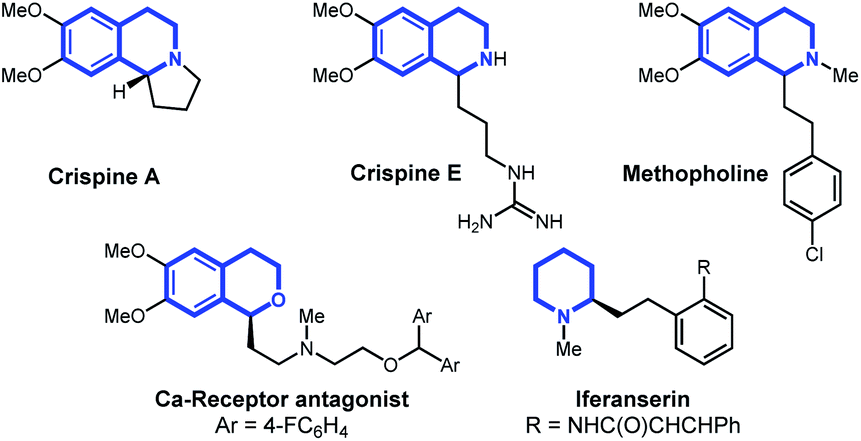
In this article, we report the development of a one-pot formal cross dehydrogenative coupling12 of alkynes and alkenes with tetrahydroisoquinolines and related heterocyclic starting materials using iron-catalyzed α-C–H functionalization (Scheme 1D). Through the coupling of two fragments without prefunctionalization of either component, cross dehydrogenative strategies allow for the reaction of simple, readily available starting materials for the construction of complex structures. Consequently, our approach rapidly affords 1-alkylated tetrahydroisoquinoline and related products which serve as versatile building blocks for a number of natural products and bioactive compounds (Fig. 1).13–15 Notably, this strategy allowed for the clean formation of propargylic functionalization product without formation of the allenyl isomer, a side product that was previously observed when other organometallic species were used.16
 |
| | Fig. 1 Natural products and bioactive compounds containing tetrahydroisoquinoline, piperidine, or dihydroisochroman ring systems. | |
To better understand the catalytic system, we conducted studies to elucidate the behaviour of the catalyst and mechanism of the transformation. It was found that the hindered and electron-rich pentamethylcyclopentadienyl (Cp*)-based catalyst provided superior results for the current process. We investigated the impact of the substituted Cp ligand on the three proposed steps of the catalytic cycle to gain insight into the ligand effect. Kinetic isotope and isotopic labelling experiments, combined with other mechanistic data, allowed us to identify the likely turnover-limiting step of the catalytic cycle. Finally, we investigated the regioselectivity for functionalization of unsymmetrically substituted dialkylacetylenes.
Results and discussion
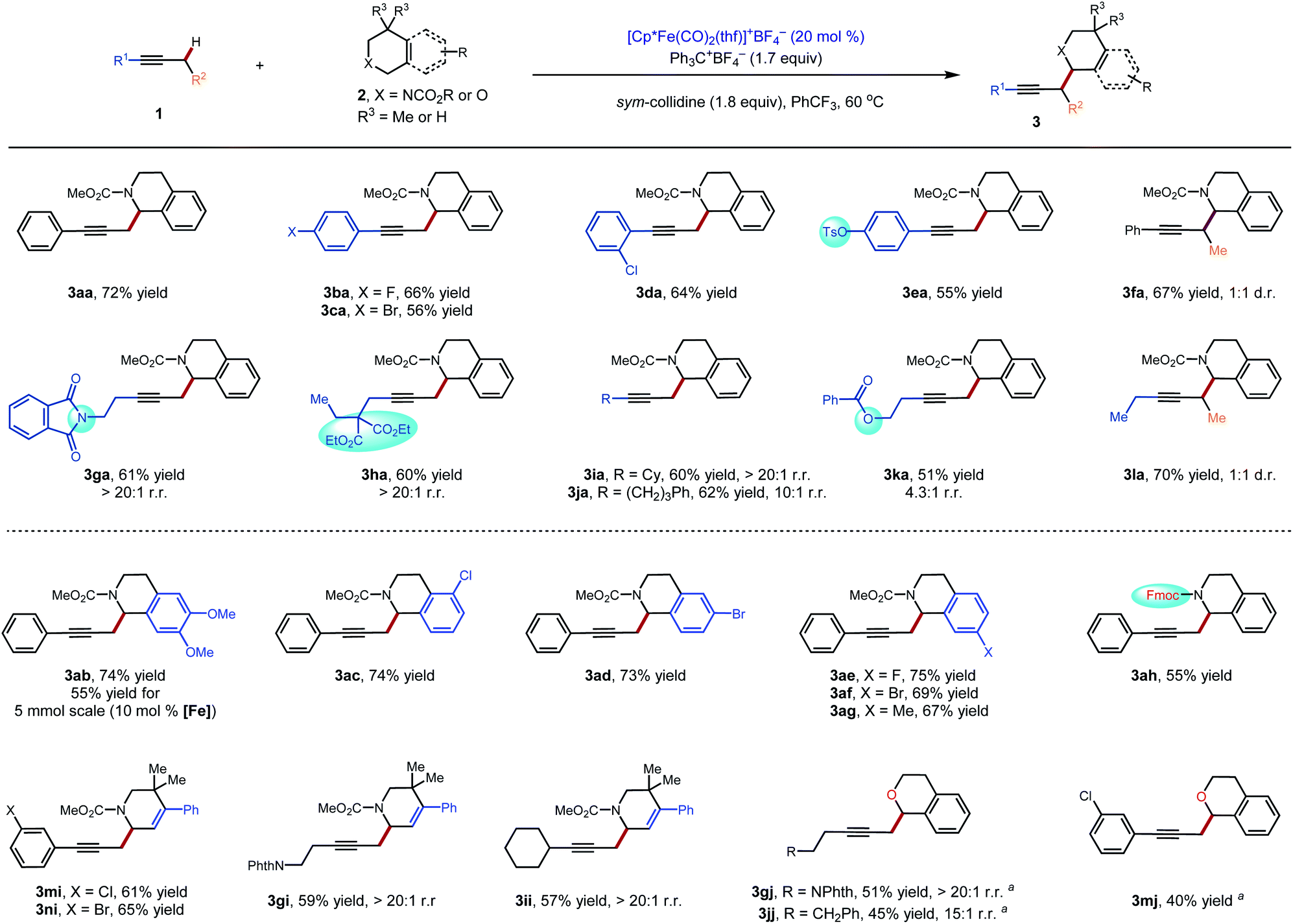
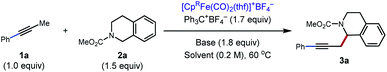
Initially, our investigation began with the reaction of 1-phenyl-1-propyne 1a with N–CO2Me tetrahydroisoquinolines (THIQs) 2a as model coupling partners (Table 1). For generation of the tetrahydroisoquinolinium coupling partner, we chose Ph3C+BF4− as the hydride acceptor, based on earlier investigations of its use,13g,15c likely compatibility with our catalytic system, and commercial availability of the tetrafluoroborate salt. A solution of Ph3C+BF4− and 2a was prestirred for 3 h to generate the electrophile in situ. At the outset of optimization studies, we employed 2,2,6,6-tetramethylpiperidine (TMPH) as the base, as it was previously successful for the coupling of carbonyl derivatives with alkynes. Gratifyingly the coupling reaction took place to deliver the desired product 3a in 31% NMR yield using [Cp*Fe(CO)2(thf)]+BF4− (30 mol%) as the catalyst (entry 1). The strongly hindered amine bases PMP (1,2,2,6,6-pentamethylpiperidine) and i-Pr2NEt, as well as the parent pyridine, were ineffective (entries 2–4), whereas 2,6-lutidine was similar to TMPH (entry 5). The NMR yield could be improved to 62% by using the related sym-collidine as the base (Table 1, entry 6). The supporting ligand of the catalyst was briefly surveyed, and CpR = pentamethylcyclopentadienyl (Cp*) proved superior to less-substituted ligands that were examined (entries 6–9). Finally, when the solvent was switched to PhCF3, the catalyst loading could be decreased to 20 mol% while still delivering 3a in 77% NMR yield (72% isolated yield) (entry 12).
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
CpR |
Base |
Solvent |
Yieldb (%) |
|
All reactions were carried out with 1a (0.1 mmol), 2a (1.5 equiv.), Ph3C+BF4− (1.7 equiv.), base (1.8 equiv.) and 30 mol% of iron catalyst in solvent (0.5 mL).
NMR yield.
20 mol% of iron catalyst.
Isolated yield.
10 mol% iron catalyst. DCE = 1,2-dichloroethane. PMP = 1,2,2,6,6-pentamethylpiperidine. sym-Collidine = 2,4,6-collidine. Cp* = pentamethylcyclopentadienyl. CpR1 = 1,3-(t-Bu)2cyclopentadienyl, CpR2 = tetramethylcyclopentadienyl.
|
| 1 |
Cp* |
TMPH |
DCE |
31 |
| 2 |
Cp* |
PMP |
DCE |
Trace |
| 3 |
Cp* |
i-Pr2NEt |
DCE |
Trace |
| 4 |
Cp* |
Pyridine |
DCE |
0 |
| 5 |
Cp* |
2,6-Lutidine |
DCE |
34 |
| 6 |
Cp* |
sym-Collidine |
DCE |
62 |
| 7 |
Cp |
sym-Collidine |
DCE |
10 |
| 8 |
CpR1 |
sym-Collidine |
DCE |
14 |
| 9 |
CpR2 |
sym-Collidine |
DCE |
20 |
| 10 |
Cp* |
sym-Collidine |
CHCl3 |
60 |
| 11 |
Cp* |
sym-Collidine |
PhCl |
70 |
| 12 |
Cp* |
sym-Collidine |
PhCF3 |
83 |
| 13c |
Cp* |
sym-Collidine |
PhCF3 |
77 (72d) |
| 14e |
Cp* |
sym-Collidine |
PhCF3 |
56 |
These conditions were used to examine the generality of the catalytic system with respect to the alkyne substrate and the electrophile used (Scheme 2). A number of functional groups, including aryl halides (3ba–3da), a sulfonate ester (3ea), a phthalimide (3ga) and carboxylic esters (3ha, 3ka) were tolerated. Substrates with two possible sites of functionalization were then explored (3ga–3ka). For substrates bearing electronically similar alkyl groups, a pronounced steric effect was observed. For instance, methyl groups were functionalized in preference to larger primary alkyl groups with good to excellent regioselectivity (4.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to >20:1 r.r.). Several carbamate and amide N-protecting groups were also suitable nitrogen substituents (see the ESI†). In particular, the tolerance for the alkylamine-sensitive fluorenylmethoxycarbonyl (Fmoc) protecting group demonstrates the mildness of the current reaction conditions (3ah). Finally, the current method was also suitable for gram-scale synthesis. Even at a reduced catalyst loading of 10 mol%, 3ab could be prepared in a synthetically useful isolated yield of 55% (1.01 g).
:1 to >20:1 r.r.). Several carbamate and amide N-protecting groups were also suitable nitrogen substituents (see the ESI†). In particular, the tolerance for the alkylamine-sensitive fluorenylmethoxycarbonyl (Fmoc) protecting group demonstrates the mildness of the current reaction conditions (3ah). Finally, the current method was also suitable for gram-scale synthesis. Even at a reduced catalyst loading of 10 mol%, 3ab could be prepared in a synthetically useful isolated yield of 55% (1.01 g).
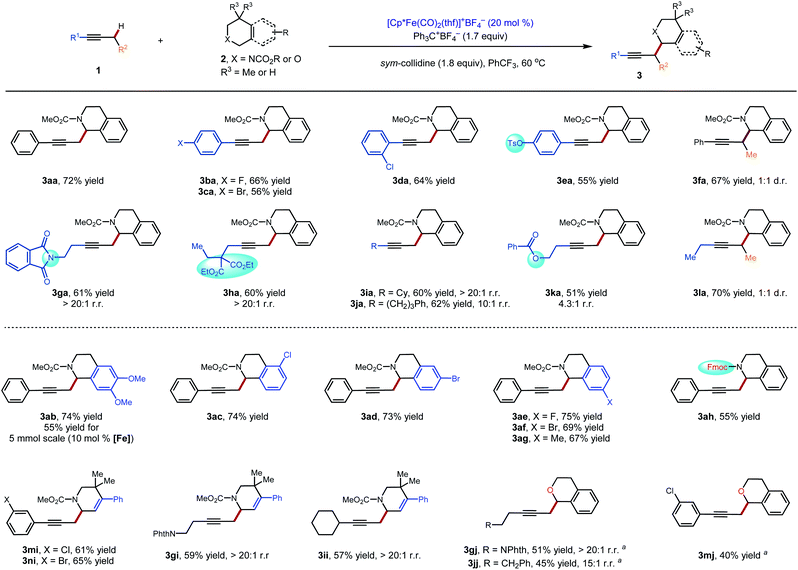 |
| | Scheme 2 Substrate scope for the catalytic C–H propargylic functionalization to form alkylation products 3: standard conditions: 1 (0.30 mmol), 2 (1.5 equiv.), Ph3C+BF4− (1.7 equiv.), sym-collidine (1.8 equiv.), [Cp*Fe(CO)2(thf)]+BF4− (20 mol%), PhCF3 (0.2 M), 60 °C, 48 h. aCHCl3 (0.2 M) was used as the solvent. NPhth = phthalimide. Fmoc = fluorenylmethyloxycarbonyl. | |
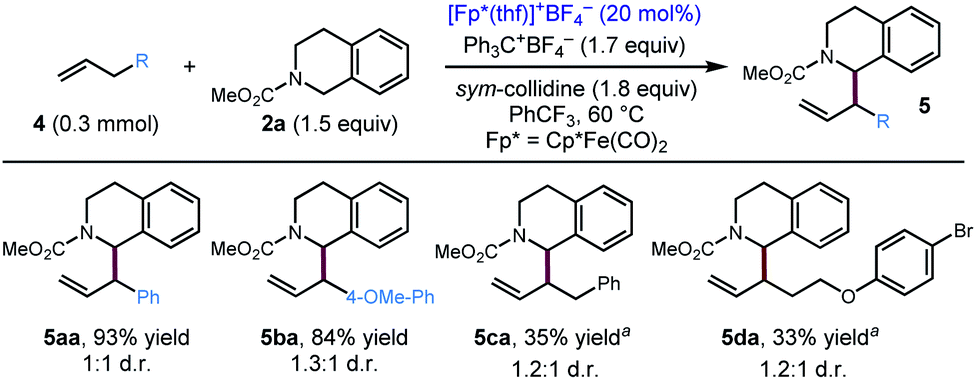
After exploring the scope of the alkyne component, we wondered whether our optimized conditions would be applicable to olefin substrates. To date, there is only one example of a transition metal catalysed reaction, a Rh/Ir dual catalytic photoredox system, reported to deliver 1-allylic tetrahydroisoquinolines with moderate to high branched/linear selectivity.17 Applying our optimized conditions to terminal olefins, we were pleased to find that our conditions resulted in allylic functionalization of these substrates to furnish the 1-allylic tetrahydroisoquinolines products with exclusive (>20:1) branched selectivity (Scheme 3). Notably, even unactivated olefins (4c, 4d) delivered the coupling product in modest yield, while substrates with additional electronic activation (4a, 4b) provided good to excellent yields of the coupling product.
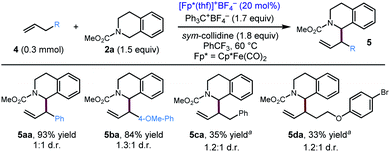 |
| | Scheme 3 Substrate scope for the coupling of olefins with 2a. Standard conditions: 4 (0.3 mmol), 2a (1.5 equiv.), Ph3C+BF4− (1.7 equiv.), sym-collidine (1.8 equiv.), [Cp*Fe(CO)2(thf)]+BF4− (20 mol%), PhCF3 (0.2 M), 60 °C, 48 h. aCpR1Fe(CO)2(thf)BF4 (20 mol%) was used as the iron catalyst, PhCF3 (0.3 M). CpR1 = 1,3-(t-Bu)2cyclopentadienyl. | |
Mechanistic studies
A mechanistic investigation was undertaken to gain a better understanding of the ligand effects involved, including the superior performance of catalysts based on Cp* relative to Cp, as well as the relative rates of the steps of the catalytic cycle. Due to the formation of rotameric mixtures and the complications presented by the in situ generation of the tetrahydroisoquinolinium electrophiles, we elected to perform some of these investigations using Ph3C+BF4− as a single component electrophile whose products are readily analysed by NMR spectroscopy.
Deprotonation step
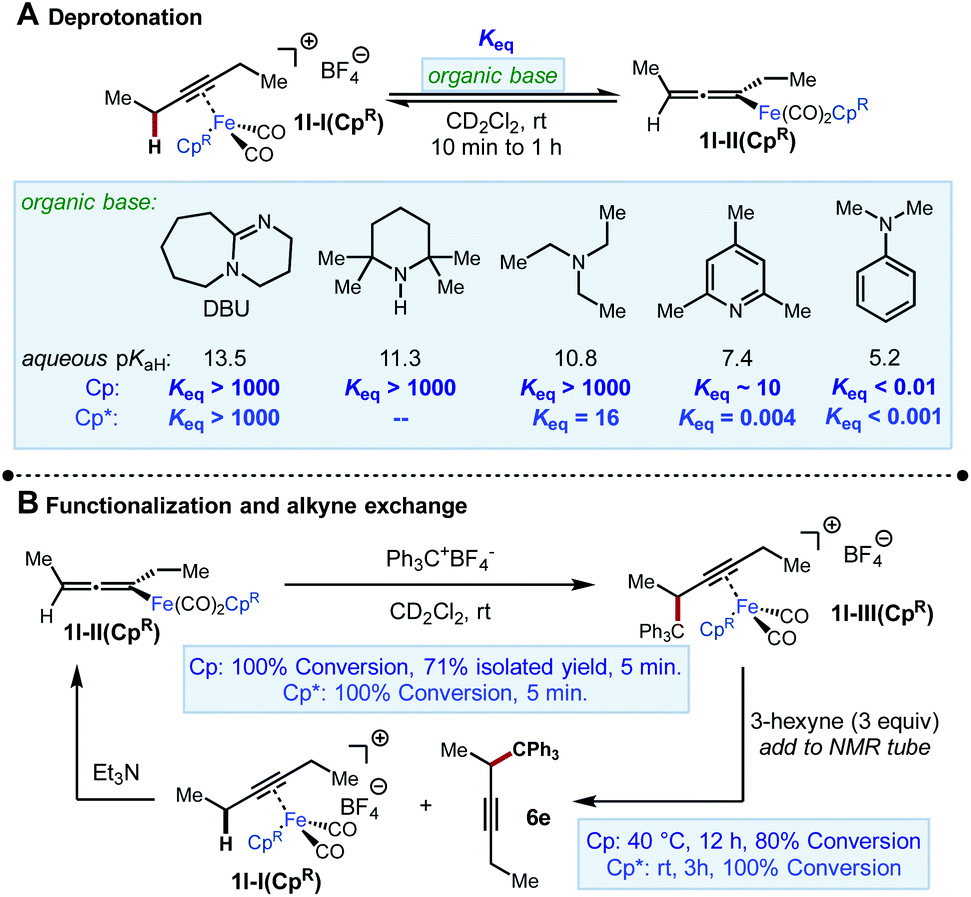
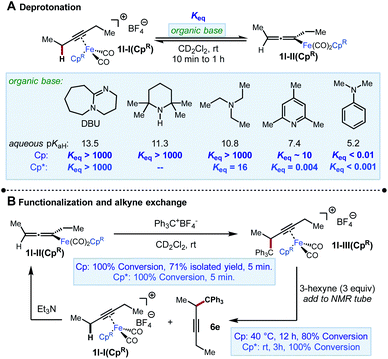
We began our investigation by exploring the stoichiometric reactivity of the previously characterized Fp-alkyne complex 1l-I(Cp) ([CpFe(CO)2(3-hexyne)]+[BF4]−) (Scheme 4). Fp-alkyne complex 1l-I(Cp) was treated with a series of bases to determine upper and lower bounds for its acidity. Exposure of 1l-I(Cp) to a range of amines (10 ≤ aq. pKaH ≤ 14) resulted in complete conversion to previously known σ-allenyliron complex 1l-II(Cp). When deprotonation was attempted with 2,4,6-collidine, an unexpected and yet unidentified product was produced that precluded accurate determination of Keq. However, when a base of similar strength, N-methylmorpholine (pKaH 7.6), was used in its place, an equilibrium constant Keq = 30 was measured. Taken together, these data imply an approximate range of 6–7 for the pKa of 1l-I(Cp) (Scheme 4A).18
 |
| | Scheme 4 Stoichiometric NMR experiments using [CpFe(CO)2(3-hexyne)]+BF4− and [Cp*Fe(CO)2(3-hexyne)]+BF4− . | |
On the other hand, when we performed the same analysis using 1l-I(Cp*), we found that this complex was more difficult to deprotonate, both kinetically and thermodynamically. For instance, while complete deprotonation of 1l-I(Cp) was achieved in <5 min using Et3N, the deprotonation of 1l-I(Cp*) took approximately 1 h to approach a constant composition corresponding to an equilibrium constant Keq = 16, as measured by 1H NMR spectroscopy. Moreover, deprotonation by 2,4,6-collidine was found to reach an unfavourable position of equilibrium (Keq = 0.004) over the same timescale of about 1 h. The equilibrium constant for the deprotonation of 1l-I(Cp*) by TMPH could not be determined, as equilibrium was not reached even after 24 h, by which time the NMR sample had started to undergo decomposition. Taken together, we estimate 1l-I(Cp*) have an approximate pKa range of 9–10 (i.e., about 3 pKa units less acidic than 1l-I(Cp)). The difference in thermodynamic acidity between these species is comparable to the differences in acidities between Cp- and Cp*-ligated transition metal hydride complexes (2 to 5 pKa units).19
Electrophilic functionalization step
Next, the reaction of 1l-II(Cp) with the electrophile Ph3C+BF4− was studied (Scheme 4B). This reaction was found to give 1l-III(Cp) cleanly and rapidly (100% conversion, >90% NMR yield, 71% isolated yield in 5 min). A similar rapid reaction was observed when 1l-II(Cp*) was subjected to the same conditions. However, in this case, the iron-bound functionalization product 1l-III(Cp*) could only be observed by NMR, and a portion of 1l-III(Cp*) was found to undergo decoordination in 5 minutes at room temperature to release the organic product 6e (Fig. 2). The same stoichiometric functionalization experiments were repeated using the N-methoxycarbonyl tetrahydroisoquinolinium as the electrophile. It was found that 1l-II(Cp) and 1l-II(Cp*) were consumed within 10 min and 5 min, respectively (see the ESI†). These experiments indicate that regardless of electrophile and ligand, the reaction of allenyliron complexes II with the electrophilic reagent takes place rapidly.
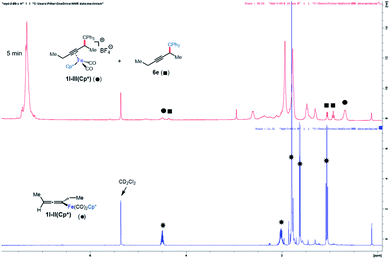 |
| | Fig. 2 NMR study of functionalization with Cp* as the supporting ligand. | |
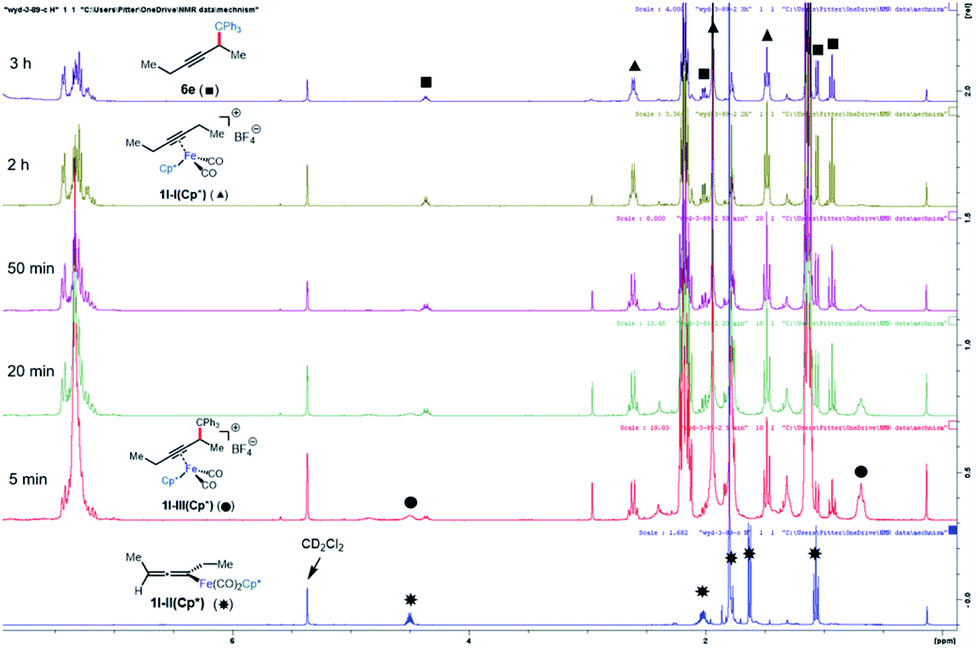
Alkyne exchange step
This system was selected for further NMR study of the alkyne exchange step. Consistent with the stability of 1l-III(Cp), the addition of 3-hexyne (3 equiv.) to a solution of this complex at room temperature resulted in no observable alkyne exchange over 1 h. However, heating a solution of 1l-III(Cp) and 3-hexyne (3 equiv.) at 40 °C for 12 h resulted in the release of functionalization product 6e in 80% conversion by NMR, and the regeneration of 1l-I(Cp) was observed.10b In stark contrast to the sluggish alkyne exchange for 1l-III(Cp), a solution of in situ generated 1l-III(Cp*) and 3-hexyne (3 equiv.) was found to undergo complete alkyne exchange over 3 h at room temperature (Fig. 3). The faster exchange by the Cp*-based catalyst, combined with the release of the organic product (6e) even in the absence of added alkyne (Fig. 2), suggests that exchange may proceed through a dissociative mechanism, which is facilitated by the sterically hindered and electron-donating ligand.
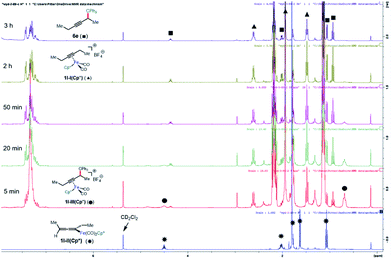 |
| | Fig. 3 NMR study of functionalization and alkyne exchange with Cp* as the supporting ligand. | |
Overall implications
Taken as a whole, these results suggest that while exchange of the alkyne is challenging in the case of catalysts derived from Cp and may be turnover-limiting, the alkyne exchange takes place with much greater facility in the case of catalysts derived from Cp*. This difference may play a role in the superior performance of the Cp*-derived catalyst. On the other hand, the deprotonation step is more challenging in both a thermodynamic and kinetic sense for the Cp*-derived catalyst, and our observations suggest that this step may be turnover-limiting for these electron-rich complexes. To obtain some addition evidence of this, kinetic isotope effect and deuterium labelling experiments were performed.
Kinetic isotope effect and labelling studies
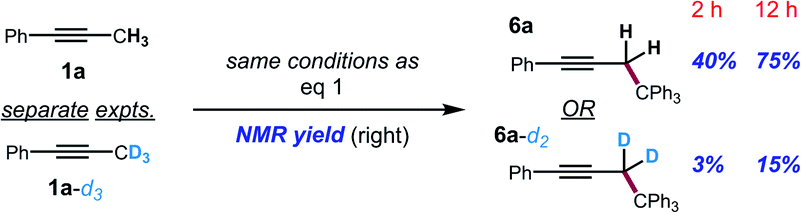
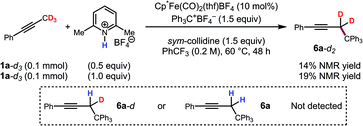
The kinetic isotope effect was studied using 1a and its methyl-deuterated isotopologue (1a-d3). An experiment by intermolecular competition yielded a kH/kD of 5.7 (eqn (1)). Moreover, reactions of 1a and 1a-d3 conducted in parallel indicated significantly slower formation of functionalization product for the deuterated substrate, although precise measurement of the rate constants was hampered by formation of side products in the case of 1a-d3 (eqn (2)). Nevertheless, the results of these experiments strongly suggest that deprotonation is in fact the turnover-limiting step of the catalytic cycle.
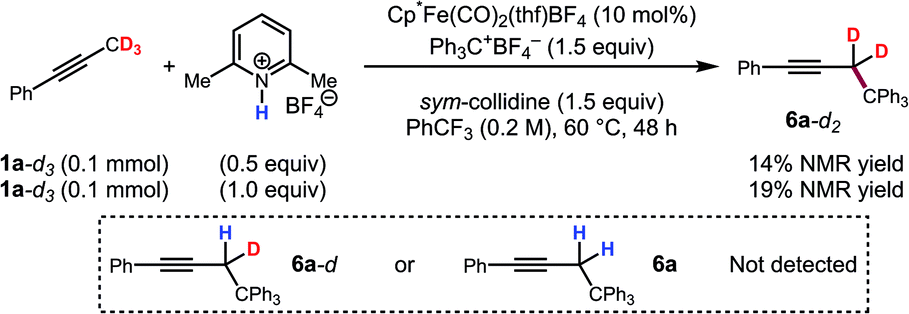
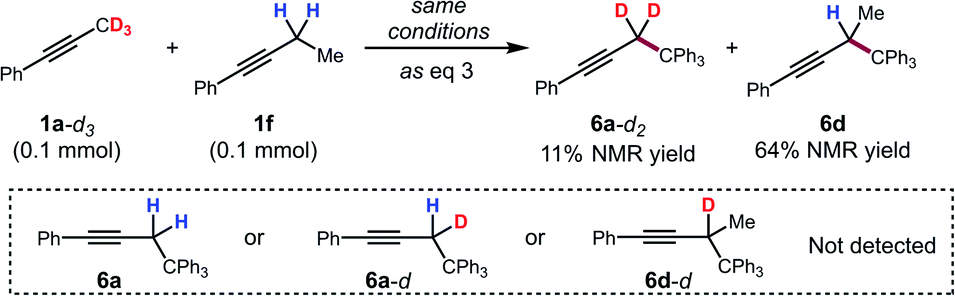
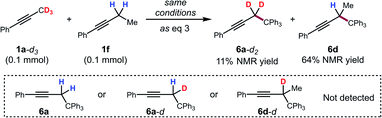
To further support this conclusion, isotope wash-in experiments were performed in which a proton source, collidinium tetrafluoroborate, was added to catalytic tritylation reactions of 1a-d3. When the product was isolated, only 6a-d2 was observed. In particular, there was no evidence of the incorporation of protium at the propargylic position (eqn (3)). Finally, an isotope crossover experiment was conducted with alkynes 1a-d3 and 1f in one pot to deliver the 6a-d2 and 6d, respectively. In line with expectations, no isotope crossover products were detected (eqn (4)). In summary, these results are consistent with rapid electrophilic functionalization that renders the deprotonation step irreversible.
| | 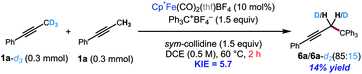 | (1) |
| | 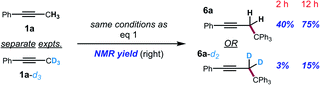 | (2) |
| |  | (3) |
| |  | (4) |
Regioselectivity
Given the evidence in support of deprotonation as an irreversible step, we conducted studies of the regioselectivity of functionalization for unsymmetrically substituted alkyne starting materials. The observed selectivities provided insights into the relative kinetic acidities of the two possible sites of deprotonation (Scheme 5). A methyl group and an ethyl group could be distinguished with useful levels of selectivity (1p, 3.7:1 r.r.). The selectivity could be increased when the hindered substituents were introduced at either the α, β or γ positions (1q, 1g, 1j, 8.1:1 to >20:1 r.r.). However, electronic effects also impact regioselectivity, and an anion-stabilizing group (benzyl) was functionalized in preference to an unhindered group (methyl), overriding the steric preference (1s, 1:11 r.r.). Interestingly, even an ethyl group and a 6-chlorohexyl group could be distinguished with useful levels of selectivity (1t, 3.8:1 r.r.).
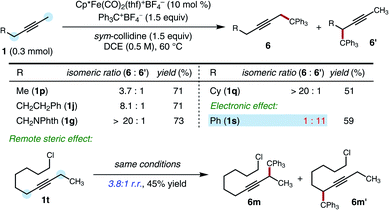 |
| | Scheme 5 Regioselectivity studies. Standard conditions: 1 (0.30 mmol), Ph3C+BF4− (1.5 equiv.), sym-collidine (1.5 equiv.), [Cp*Fe(CO)2(thf)]+BF4− (10 mol%), DCE (0.5 M), 60 °C, 48 h. NPhth = phthalimide. | |
Conclusions
In summary, we report the development of a two-step, one-pot cross dehydrogenative coupling of alkynes and alkenes with tetrahydroisoquinolines and related heterocyclic starting materials using iron-catalyzed α-C–H functionalization. This strategy allowed for the clean formation of propargylic functionalization product without formation of the allenyl isomer as a side product. Mechanistic studies using stoichiometric NMR experiments, kinetic isotope effect studies, and deuterium-labelling experiments revealed the deprotonation of α-position of alkynes to be the rate-determining step when a Cp*-based iron catalyst was employed. Based on these insights, further efforts to improve catalyst performance are ongoing and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge startup support from the University of Pittsburgh. We thank Professors Scott Nelson and Xinyu Liu for the sharing of laboratory resources with the Wang group. We thank Austin Durham for proofreading of the manuscript. We would like to acknowledge Professors Dean Toste (Berkeley) and Paul Floreancig (Pitt) for helpful discussions.
Notes and references
- Selected books and reviews on the functionalization of alkynes and alkenes by π activation:
(a)
Modern Alkyne Chemistry, ed. C.-J. Li and B. M. Trost, Wiley, Weinheim, 2015 Search PubMed
 ;
(b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS ;
(c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef ;
(d) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS ;
(e) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS ;
(f) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS ;
(g) D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677 RSC ;
(h) C. Lin and L. Shen, ChemCatChem, 2019, 11, 961 CAS .
;
(b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS ;
(c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef ;
(d) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS ;
(e) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS ;
(f) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS ;
(g) D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677 RSC ;
(h) C. Lin and L. Shen, ChemCatChem, 2019, 11, 961 CAS .
- For C–H functionalization:
(a) L. Ilies, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 7672 CAS ;
(b) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030 CrossRef CAS ;
(c) M. Sekine, L. Ilies and E. Nakamura, Org. Lett., 2013, 15, 714 CrossRef CAS ;
(d) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400 RSC ;
(e) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS ;
(f) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS ;
(g) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS ;
(h) W. N. Oloo and L. Que, Jr., Acc. Chem. Res., 2015, 48, 2612 CrossRef CAS ;
(i) J. R. Griffin, C. I. Wendell, J. A. Garwin and M. C. White, J. Am. Chem. Soc., 2017, 139, 13624 CrossRef CAS ;
(j) S. Tnabe, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 12374 CrossRef .
-
(a) G. Liu and Y. Wu, Top. Curr. Chem., 2010, 292, 195 CrossRef CAS ;
(b) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863 CrossRef CAS ;
(c) P.-S. Wang, M.-L. Shen, T.-C. Wang, H.-C. Lin and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 16032 CrossRef CAS . Report of differential outcome of allylic and propargylic systems:
(d) T. Jiang, X. Quan, C. Zhu, P. G. Andersson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 5824 CrossRef CAS ;
(e) T. M. Locascio and J. A. Tunge, Chem.–Eur. J., 2016, 22, 18140 CrossRef CAS .
-
(a)
L. P. Jacques, S. Gunther and B. Carsten, Transition Metals for Organic Synthesis, ed. M. Beller, and C. Bolm, 2004, vol. 2, p. 256 Search PubMed . Sporadic reports of application to propargylic systems:
(b) H. Kropf, R. Schröder and R. Fölsing, Synthesis, 1977, 894 CrossRef CAS ;
(c) J. S. Clark, K. F. Tolhurst, M. Taylor and S. Swallow, Tetrahedron Lett., 1998, 39, 4913 CrossRef CAS ;
(d) L. X. Alvarez, M. L. Christ and A. B. Sorokin, Appl. Catal., A, 2007, 325, 303 CrossRef CAS ;
(e) C. C. Black and A. E. V. Gorden, Tetrahedron Lett., 2018, 59, 803 CrossRef CAS .
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem.
Rev., 2010, 110, 704 CrossRef CAS .
-
(a) D. Cheng and W. Bao, J. Org. Chem., 2008, 73, 6881 CrossRef CAS ;
(b) T. Wang, W. Zhou, H. Yin, J.-A. Ma and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 10823 CrossRef CAS ;
(c) R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14, 280 CrossRef CAS ;
(d) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS ;
(e) D. Cheng, X. Zhou, X. Xu and J. Yan, RSC Adv., 2016, 6, 52459 RSC ;
(f) M. Ju, E. E. Zerull, J. M. Roberts, M. Huang, I. A. Guzei and J. M. Schomaker, J. Am. Chem. Soc., 2020, 142, 12930 CrossRef CAS .
-
(a) J. A. Fernandez-Salas, A. J. Eberhart and D. Procter, J. Am. Chem. Soc., 2016, 138, 790 CrossRef CAS ;
(b) G. Hu, J. Xu and P. Li, Org. Chem. Front., 2018, 5, 2167 RSC Application to allylic systems:
(c) L. Bayeh and U. K. Tambar, ACS Catal., 2017, 7, 8533 CrossRef CAS .
- For a similar strategy applied to alkenyl C–H functionalization, see:
(a) J. M. Schomaker, W. C. Boyd, I. C. Stewart, F. D. Toste and R. G. Bergman, J. Am. Chem. Soc., 2008, 130, 3777 CrossRef CAS ;
(b) W. C. Boyd, M. R. Crimmin, L. E. Rosebrugh, J. M. Schomaker, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 16365 CrossRef CAS ;
(c) C. Zhao, F. D. Toste and R. G. Bergman, J. Am. Chem. Soc., 2011, 133, 10787 CrossRef CAS ;
(d) C. Zhao, M. R. Crimmin, F. D. Toste and R. G. Bergman, Acc. Chem. Res., 2014, 47, 517 CrossRef CAS . For the reversed-polarity strategy of stabilizing propargylic cations, see:
(e) K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207 CrossRef CAS .
-
(a) Z. Wang, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2014, 136, 8887 CrossRef CAS ;
(b) T. Li and L. Zhang, J. Am. Chem. Soc., 2018, 140, 17439 CrossRef CAS .
-
(a) M. Rosenblum, Acc. Chem. Res., 1974, 7, 122 CrossRef CAS ;
(b) A. Cutler, D. Ehntholt, P. Lennon, K. Nicholas, D. F. Marten, M. Madhavarao, S. Raghu, A. Rosan and M. Rosenblum, J. Am. Chem. Soc., 1975, 97, 3149 CrossRef CAS ;
(c) A. Cutler, D. Ehntholt, W. P. Giering, P. Lennon, S. Raghu, A. Rosan, M. Rosenblum, J. Tancrede and D. Wells, J. Am. Chem. Soc., 1976, 98, 3495 CrossRef CAS ;
(d) S. Jiang, G. E. Agoston, T. Chen, M.-P. Cabal and E. Turos, Organometallics, 1995, 14, 4697 CrossRef CAS . Previous work on alkyne complexes:
(e) D. L. Reger, C. J. Coleman and P. J. Mcelligott, J. Organomet. Chem., 1979, 171, 73 CrossRef CAS ;
(f) M. Akita, S. Kakuta, S. Sugimoto, M. Terada, M. Tanaka and Y. Moro-oka, Organometallics, 2001, 20, 2736 CrossRef CAS ;
(g) M. Rosenblum and J. C. Watkins, J. Am. Chem. Soc., 1990, 112, 6316 CrossRef CAS ;
(h) M. D. Redlich, M. F. Mayer and M. M. Hossain, Aldrichimica Acta, 2003, 36, 3 CAS ;
(i) D. L. Reger and P. J. McElligott, J. Am. Chem. Soc., 1980, 102, 5923 CrossRef CAS .
-
(a) Y. Wang, J. Zhu, A. C. Durham, H. Lindberg and Y.-M. Wang, J. Am. Chem. Soc., 2019, 141, 19594 CrossRef CAS ;
(b) A. C. Durham, Y. Wang and Y.-M. Wang, Synlett, 2020, 31 DOI:10.1055/s-0040-1707271 .
- For reviews on CDC reactions, see:
(a) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS ;
(b) W. J. Yoo and C. J. Li, Top. Curr. Chem., 2010, 292, 281 CrossRef ;
(c) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS ;
(d) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353 CrossRef CAS ;
(e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS . For examples:
(f) Z. Li and C. J. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS ;
(g) Z. Li and C. J. Li, J. Am. Chem. Soc., 2005, 127, 3672 CrossRef CAS ;
(h) Y. Zhang and C. J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949 CrossRef CAS ;
(i) Y. Zhang and C. J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS ;
(j) Z. Meng, S. Sun, H. Yuan, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 543 CrossRef CAS ;
(k) P.-S. Gao, X.-J. Weng, Z.-H. Wang, C. Zheng, B. Sun, Z.-H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254 CrossRef CAS .
-
(a) L. J. Cass and W. S. Frederik, Am. J. Med. Sci., 1963, 246, 550 CrossRef CAS ;
(b)
M. Shamma and J. L. Moniot, The Isoquinoline Alkaloids, Academic Press, New York and London, 1972 Search PubMed ;
(c) K. T. Wanner, I. Praschak, G. Höfner and H. Beer, Arch. Pharm., 1996, 329, 11 CrossRef CAS ;
(d) L. F. Tietze, N. Rackelmann and I. Müller, Chem.–Eur. J., 2004, 10, 2722 CrossRef CAS ;
(e) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS ;
(f) I. P. Singh and P. Shah, Expert Opin. Ther. Pat., 2016, 27, 17 CrossRef ;
(g) Z. Xie, L. Liu, W. Chen, H. Zheng, Q. Xu, H. Yuan and H. Lou, Angew. Chem., Int. Ed., 2014, 53, 390 Search PubMed .
-
(a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC ;
(b) X. Liu, Z. Meng, C. Li, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 6012 CrossRef CAS ;
(c) G. Wang, Y. Mao and L. Liu, Org. Lett., 2016, 18, 6476 CrossRef CAS .
-
(a) J. Xu, J. Kjer, J. Sendker, V. Wray, H. Guan, R. Edrada, W. E. G. Müller, M. Bayer, W. Lin, J. Wu and P. Proksch, Bioorg. Med. Chem., 2009, 17, 7362 CrossRef CAS ;
(b) M. Braun and W. Kotter, Angew. Chem., Int. Ed., 2004, 43, 514 CrossRef CAS ;
(c) W. Chen, Z. Xie, H. Zheng, H. Lou and L. Liu, Org. Lett., 2014, 16, 5988 CrossRef CAS .
- S. Agarwal, O. Kataeva, U. Schmidt and H.-J. Knölker, RSC Adv., 2013, 3, 1089 RSC .
- J. Zheng and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 3392 CrossRef CAS .
- Measurement of equilibrium constants were conducted in CD2Cl2 because 1a-I(Cp) reacted with standard solvents for pKa measurements, including CD3CN, THF-d8, and (CD3)2SO.
- For a detailed discussion on acidity trends for metal hydride complexes, see:
(a) R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1948 CrossRef CAS ;
(b) R. H. Morris, Chem. Rev., 2016, 116, 8588 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds, experimental procedures, and theoretical studies on mechanisms. CCDC 1909839 and 1909840. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05091a |
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jin
Zhu
,
Rui
Guo
,
Haley
Lindberg
and
Yi-Ming
Wang
,
Jin
Zhu
,
Rui
Guo
,
Haley
Lindberg
and
Yi-Ming
Wang