
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient cleavage of tertiary amide bonds via radical–polar crossover using a copper(II) bromide/Selectfluor hybrid system†
Zhe
Wang
 a,
Akira
Matsumoto
a and
Keiji
Maruoka
*ab
a,
Akira
Matsumoto
a and
Keiji
Maruoka
*ab
aGraduate School of Pharmaceutical Sciences, Kyoto University Sakyo, Kyoto 606-8501, Japan. E-mail: maruoka.keiji.4w@kyoto-u.ac.jp
bSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China
First published on 14th October 2020
Abstract
A novel approach for the efficient cleavage of the amide bonds in tertiary amides is reported. Based on the selective radical abstraction of a benzylic hydrogen atom by a CuBr2/Selectfluor hybrid system followed by a selective cleavage of an N–C bond, an acyl fluoride intermediate is formed. This intermediate may then be derivatized in a one-pot fashion. The reaction proceeds under mild conditions and exhibits a broad substrate scope with respect to the tertiary amide moiety as well as to nitrogen, oxygen, and carbon nucleophiles for the subsequent derivatization. Mechanistic studies suggest that the present reaction proceeds via a radical–polar crossover process that involves benzylic carbon radicals generated by the selective radical abstraction of a benzylic hydrogen atom by the CuBr2/Selectfluor hybrid system. Furthermore, a synthetic application of this method for the selective cleavage of peptides is described.
Introduction
Organic amide groups are ubiquitous in nature, particularly in peptides and proteins.1 They are also widely used in the chemical and pharmaceutical industry for manufacturing of fine chemicals, polyamide plastics, pesticides, and medicines.2–4 Due to the resonance stabilization of amide groups, the amide N–C bond is relatively stable and unreactive. Accordingly, the selective cleavage of such unreactive amide N–C bonds is highly challenging in organic synthesis.4–6 Indeed, various approaches for the catalytic transamidation of secondary amides have already been developed.7–10 For example, transition-metal-catalyzed or transition-metal-free transamidations of secondary amides, where the amide nitrogen atom is pre-activated with Boc, Ts, or Ms protecting groups, have been developed (Fig. 1a).9,10 In marked contrast to the transamidation of pre-activated secondary amides, the transamidation of tertiary amides is very difficult to achieve and remains under development.11–14 Recently, a promising study on this subject has been published, where the use of an excess of a lithium amide11 or an excess of a manganese (Mn) reagent12 (Fig. 1b) results in the successful transamidation of tertiary amides. Additionally, a Lewis-acid-mediated or Lewis-acid-catalyzed transamidation of tertiary amides has been reported, albeit that the substrate scope is limited in these reactions.13 Although the transamidation of amides is undoubtedly a useful chemical transformation, the synthetic utility of these methods are caveated by the difficulties associated with the generation of the reactive acyl derivatives required for further synthetic transformations.15 Therefore, the development of an alternative approach that can generate such reactive acyl derivatives under mild conditions would be highly desirable. In this context, we herein report an efficient strategy for the generation of reactive acyl fluorides16 from inert tertiary amides via a novel radical–polar crossover approach (Fig. 1c). | ||
| Fig. 1 Transamidation of secondary and tertiary amides. | ||
Results and discussion
Our strategy is based on the selective radical abstraction of a benzylic hydrogen atom using a Cu(II)/Selectfluor hybrid system,17–19 which induces the selective cleavage of an amide N–C bond and enables the formation of the acyl fluoride. We explored the reaction using N-(p-methoxybenzyl)-N-phenyl-3-phenylpropanamide (1a), as a model compound for the desired reactivity (Table 1). After optimizing a range of reaction conditions, we found that the reaction of amide 1a with Selectfluor (1.5 equiv.) in the presence of a catalytic amount of CuBr2 (2 mol%) in MeCN at 80 °C for 1 h gave rise to the corresponding acyl fluoride 2a in 82% NMR yield and the one-pot transamidation product, N-benzyl-3-phenylpropionamide, was isolated in 86% yield (entry 1). As demonstrated in entry 2, the copper salt is essential to produce acyl fluoride 2a. When the reaction was carried out under atmospheric conditions, the yield decreased (entry 3). Other copper(II) and copper(I) salts were screened and exhibited a comparable efficiency (entries 4–6). In consideration of costs and availability, CuBr2 was selected for the subsequent experiments in this study. Relative to ligand-free systems, a CuBr2(2,2′-bipyridine) complex afforded a lower yield of 2a (entry 7 vs. entry 1). This result suggests that a ligand-free system may be more advantageous for our strategy and more widely applicable. Next, we examined various electrophilic fluorine sources. Among these, Selectfluor afforded the highest chemical yield of 2a. When N-fluorobenzenesulfonimide (NFSI) was used, the yield of 2a was merely moderate (entry 8). Switching the counter anion of Selectfluor from [BF4]− to [PF6]− also afforded a lower yield of 2a (entry 9). Other factors such as different metal salts, other fluorine sources, the Selectfluor loading, as well as solvent and temperature effects, were examined and the results are shown in ESI Table S1.†|
|
|||
|---|---|---|---|
| Entry | Variation from the standard conditions | Conversionb (%) | Yield of 2ac (%) |
| a Reaction conditions: 1a (0.20 mmol), Selectfluor (0.30 mmol), CuBr2 (4 μmol, 2.0 mol% of Cu), and MeCN (1.0 mL) at 80 °C for 1 h. b The conversion of 1a was determined by a 1H NMR analysis of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard. c The yield of 2a was determined by a 19F NMR analysis of the crude reaction mixture using 4,4′-difluorobenzophenone as an internal standard. d The isolated yield of the one-pot transamidation product, N-benzyl-3-phenylpropionamide, is shown in parentheses (for details, see ESI). e 2 mol% of 2,2′-bipyridine (bpy) was used. f NFSI: N-fluorobenzenesulfonimide. g Selectfluor·PF6: 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(hexafluorophosphate). | |||
| 1 | None | >99 | 82 (86)d |
| 2 | Without CuBr2 | 56 | <1 |
| 3 | Atmospheric conditions | >99 | 71 |
| 4 | Cu(OAc)2 | >99 | 78 |
| 5 | CuBr | >99 | 82 |
| 6 | CuI | >99 | 81 |
| 7 | CuBr2/bpye | 93 | 73 |
| 8 | NFSIf (1.5 equiv.) | 71 | 53 |
| 9 | Selectfluor·PF6g (1.5 equiv.) | >99 | 74 |
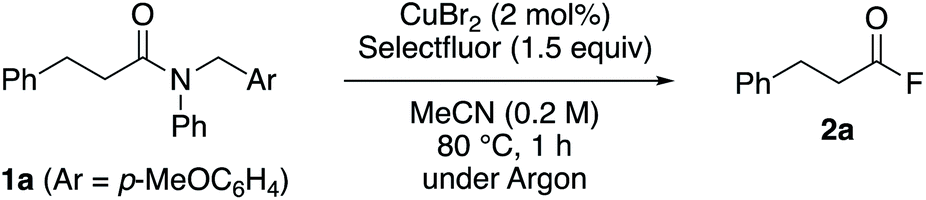
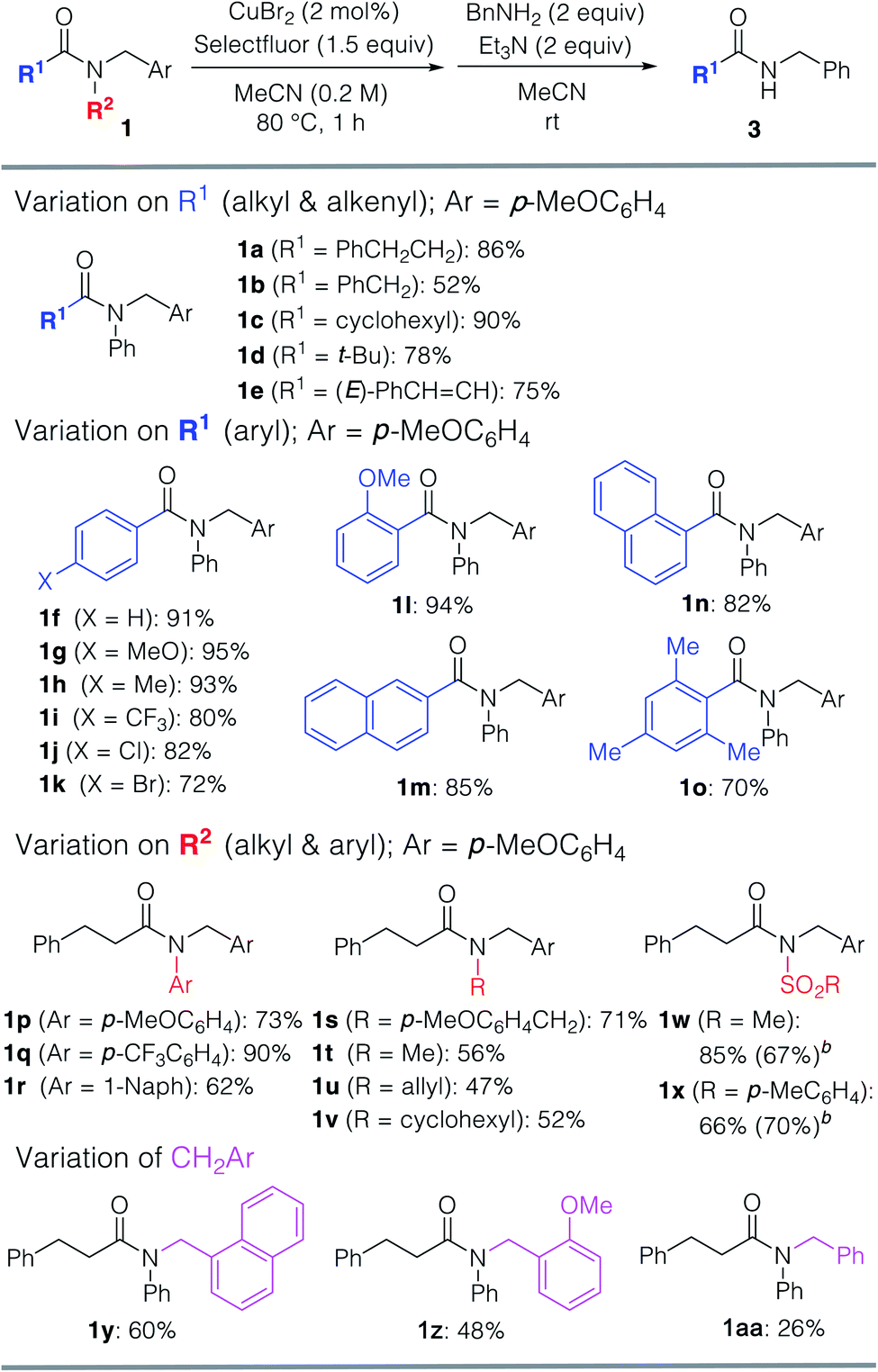
With the optimized reaction conditions in hand, we examined the scope of the reaction with respect to other tertiary amides (Table 2). The efficiency of this transformation was determined using the yield of the product of the one-pot transamidation with benzylamine. Firstly, we examined the reactivity of prim-, sec-, and tert-alkanoyl amides 1a–d, and found that our approach afforded the corresponding transamidation products in moderate to high yields. Notably, sterically hindered pivalamide 1d could be transformed under the applied conditions. Furthermore, α,β-unsaturated carboxamide 1e also underwent a smooth transamidation. Moreover, we discovered that a broad scope of benzamides with electron-donating (1g, 1h, 1l, 1o) or -withdrawing (1i–k) motifs furnished the corresponding transamidation products in high yield. For example, para-bromo and ortho-substituted benzamides 1k, 1l, and 1n are well-tolerated, albeit that these represent challenging substrates for transition-metal-catalyzed methods.9 Moreover, the transamidation of sterically hindered 2,4,6-trimethylbenzamide 1o was also possible using our method.
| a Reaction conditions: 1a (0.20 mmol), Selectfluor (0.30 mmol), CuBr2 (4 μmol, 2.0 mol% of Cu), and MeCN (1.0 mL) at 80 °C for 1 h under argon. To isolate the product as N-benzylamides, the one-pot transamidation was carried out for 3–24 h. For details, see ESI. b Isolated yield of ethyl 3-phenylpropionate. |
|---|
|
Then, we explored which amide N-aryl substituents 1p–r are tolerated under the optimized conditions. Replacement of the phenyl group with an electron-rich arene motif in 1p did not improve the yield of the transamidation. However, when electron-deficient arene 1q was used, the reaction proceeded with a satisfactory yield. Interestingly, transamidation of much less activated N,N-dialkyl amides 1s–v resulted in the formation of the desired products in moderate to good yields. Pre-activated sulfone amides 1w and 1x were not only converted into the corresponding amides, but also into esters via the versatile acyl fluoride intermediates. Notably, the N–C amide bond cleavage could be triggered when using the CH2Ar motif (Ar = p-MeOC6H4), and also when using other CH2Ar motifs. Substrates bearing 1-naphthyl (1y) and o-MeOC6H4 (1z) CH2Ar moieties afforded transamidation products in lower yields.
Furthermore, various amines and alcohols were screened as nucleophiles for the one-pot transformation of inert tertiary amide 1a (Table 3). Prim-, sec-, and even bulky tert-alkylamines furnished the corresponding transamidation products 3a and 3p–s in high yield. Despite being a weak nucleophile, aniline can also be employed in our one-pot transamidation (3t). The synthetically versatile Weinreb-amide 3v was also obtained in high yield. Moreover, our strategy allows the facile synthesis of a reversible inhibitor of monoamine oxidase A, moclobemide (3w). By taking advantage of the reactive acyl fluoride intermediate, one-pot transesterifications20,21 (4a–c) were accomplished in a highly efficient manner. In addition, thiophene can be used as a carbon nucleophile for the one-pot transformation of inert tertiary amide 1a to the corresponding ketone 5 (Scheme 1). This result demonstrates that our strategy is highly versatile on account of the synthetic utility of the acyl fluoride intermediates.22
| a Reaction conditions: 1a (0.20 mmol), Selectfluor (0.30 mmol), CuBr2 (4 μmol, 2.0 mol% of Cu), and MeCN (1.0 mL) at 80 °C for 1 h under argon. The one-pot transamidation or transesterification was carried out at room temperature for 3–24 h. For details, see ESI. |
|---|

|
 | ||
| Scheme 1 Synthesis of unsymmetrical ketone 5 from tertiary amide 1a. | ||
We discovered that our approach is, in principle, also applicable to the selective cleavage and reconstruction of peptides.23,24 Our initial findings on this subject are illustrated in Scheme 2. When N-phthaloyl-N-(p-methoxybenzyl)-Gly-Gly-OMe (6) was treated with the CuBr2/Selectfluor hybrid system, followed by addition of L-proline methyl ester, dipeptide 7 was obtained in moderate yield (Scheme 2a). Moreover, when dipeptide dl-8 from N-Phth-Ala-Gly-OMe was treated with CuBr2/Selectfluor, the intermediary acyl fluoride 9 (81% NMR yield) was subsequently treated with an β-amino acid methyl ester to afford dipeptide 10 in good yield (Scheme 2b). Treatment of N-p-methoxybenzyl derivatives 11 and 13 from N-Phth-L-Ala-L-Ala-OMe and N-Phth-D-Ala-L-Ala-OMe with the CuBr2/Selectfluor system and subsequent addition of L-Valine methyl ester furnished diastereomerically enriched dipeptides 12 and 14, respectively. Only minor epimerization was observed and both products were obtained in good yield (Scheme 2c and d).
 | ||
| Scheme 2 Selective cleavage of dipeptide bonds in 6, 8, 11, and 13 gave new dipeptides 7, 10, 12, and 14, respectively, via the corresponding acyl fluorides. For details, see ESI.† (a) CuBr2 (2 mol%), Selectfluor (2 equiv.), MeCN (0.2 M), 80 °C, 1.5 h. (b) L-Pro-OMe·HCl (2 equiv.), N,N-diisopropylethylamine (DIPEA) (4 equiv.), THF, rt. (c) H2N(CH2)2CO2Me·HCl (2 equiv.), DIPEA (4 equiv.), THF, rt. (d) L-Val-OMe·HCl (2 equiv.), DIPEA (4 equiv.), THF, rt. | ||
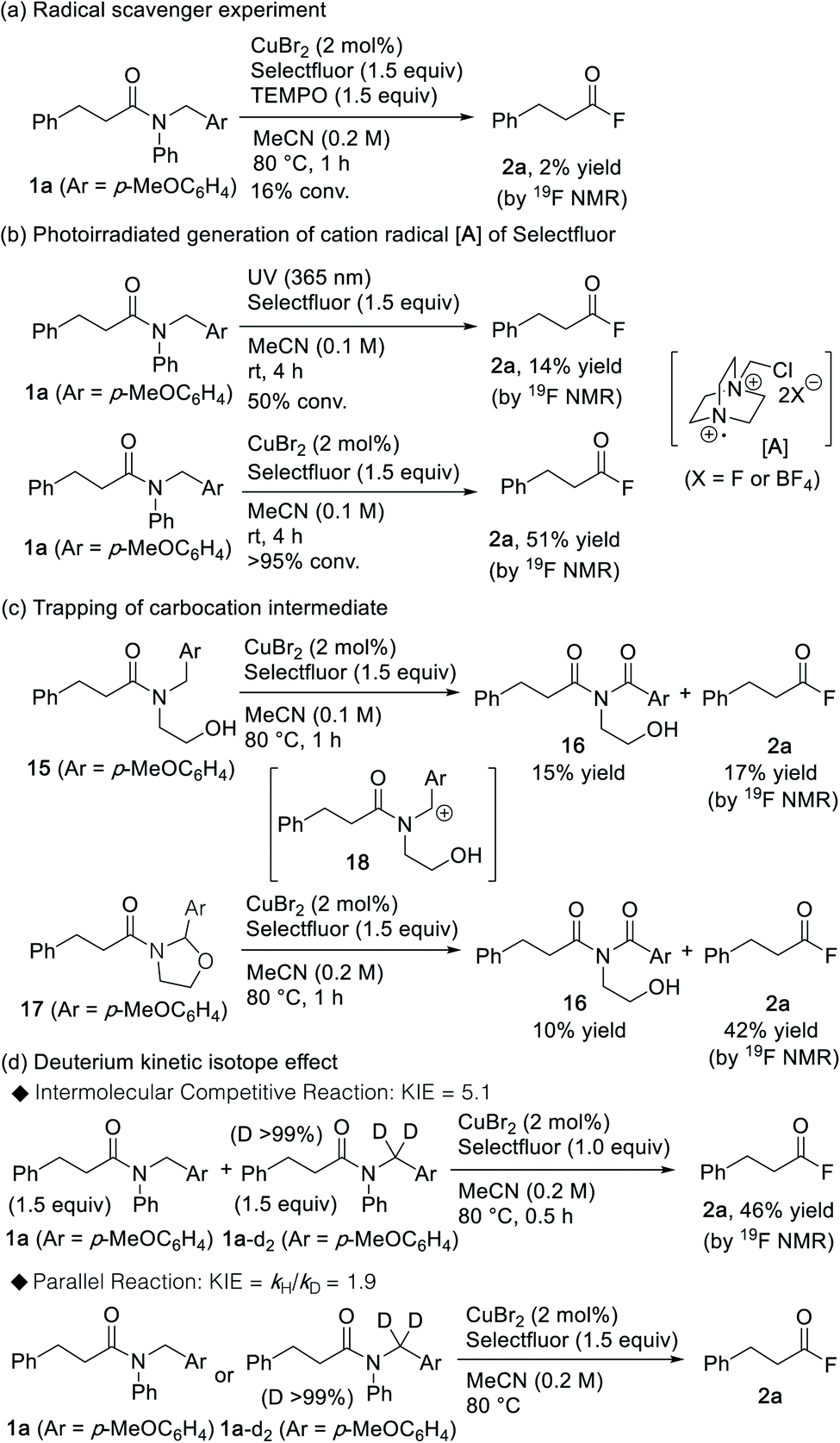
Several experiments were conducted in order to gain insight into the underlying mechanistic details of this promising cleavage reaction of amide bonds. As depicted in Scheme 3a, the reaction was fully inhibited in the presence of TEMPO, thereby indicating that a radical-promoted process occurs on the catalytic pathway. It should be noted here that Selectfluor can be activated by photoirradiation, where the generated cation radical A is responsible for the hydrogen atom abstraction (HAA) during the C–H activation of various hydrocarbons.25,26 Interestingly, acyl fluoride 2a was also obtained when the reaction was triggered via photoirradiation (Scheme 3b). However, when compared to the photoirradiation approach, the CuBr2/Selectfluor system showed higher efficiency, which suggests that the role of the copper species may not be that of an initiator, but that of a catalyst involved in the amide-cleavage process. Moreover, when N-(2-hydroxyethyl) amide 15 was treated with the CuBr2/Selectfluor system, imide2716 was obtained in addition to acyl fluoride 2a (Scheme 3c). Under similar conditions, cyclic N-acyl-N,O-acetal 17 afforded imide 16 (Scheme 3c). This result implies that cyclic N-acyl-N,O-acetal 17 may be formed via the intermediary carbocation 18 during the reaction between amide 15 and CuBr2/Selectfluor. Next, we studied the deuterium kinetic isotope effect (KIE) for the generation of the acyl fluoride from substrates 1a and 1a-d2. KIEs of 5.1 and 1.9 were observed for the intermolecular competition reaction and the parallel reaction, respectively (Scheme 3d). This result indicates that C–H bond cleavage is likely to be involved in the rate-determining step of the reaction.28
 | ||
| Scheme 3 Control experiments conducted to elucidate the reaction mechanism. | ||
Based on these experimental findings, a feasible catalytic cycle is proposed for the CuBr2/Selectfluor-promoted amide cleavage in Fig. 2. We propose that the formation of a nitrogen-centered cation radical A from Selectfluor is initially triggered in the presence of a Cu(II) salt via a single electron transfer (SET) mechanism.29,30 A subsequent HAA process could then lead to the formation of benzylic radical species B which is readily oxidized to give a carbocation and acyliminium species C. This ultimately leads to the formation of an acyl fluoride (RCOF) and imine (ArCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR′), most likely via an ionic pathway.31 Although the acyl fluoride formation ascribed to an acyl radical D with Selectfluor cannot be ruled out in our system, with the high yields of one-pot transamidation of 1c and 1d (see Table 2), it would be reasonable to understand that the acyl radical is unlikely mainly responsible for the acyl fluoride formation, since the competitive decarbonylation of acyl radicals,32 like pivaloyl radical, probably give much lower yield of transamidation.
NR′), most likely via an ionic pathway.31 Although the acyl fluoride formation ascribed to an acyl radical D with Selectfluor cannot be ruled out in our system, with the high yields of one-pot transamidation of 1c and 1d (see Table 2), it would be reasonable to understand that the acyl radical is unlikely mainly responsible for the acyl fluoride formation, since the competitive decarbonylation of acyl radicals,32 like pivaloyl radical, probably give much lower yield of transamidation.
 | ||
| Fig. 2 Proposed catalytic cycle for the radical–polar crossover cleavage of tertiary amides. | ||
In summary, we have realized a novel approach for the efficient cleavage of tertiary amides using a CuBr2/Selectfluor hybrid system. The resulting acyl fluoride intermediate can be subsequently derivatized in a one-pot fashion with various nitrogen, oxygen, and carbon nucleophiles. Mechanistic studies suggest that the present reaction proceeds via a radical–polar crossover process that involves benzylic carbon radicals generated by the selective radical abstraction of a benzylic hydrogen atom by the CuBr2/Selectfluor hybrid system. At this point, we expect that our approach may be useful for the late-stage functionalization of multi-functionalized peptides. For example, cyclic peptides such as cyclosporine33 could, via the selective benzylation and subsequent selective peptide-bond cleavage offered by our approach, be easily derivatized to furnish various types of ring-opened peptides. The exploration of these research avenues is currently in progress in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support via JSPS KAKENHI Grant Numbers JP26220803, and JP17H06450 (Hybrid Catalysis).Notes and references
- A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
- (a) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS; (b) A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS; (c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- K. Marchildon, Macromol. React. Eng., 2011, 5, 22–54 CrossRef CAS.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, Wiley-VCH, Weinheim, 2003 Search PubMed.
- M. E. Gonzalez-Rosende, E. Castillo, J. Lasri and J. Sepúlveda-Arques, Prog. React. Kinet. Mech., 2004, 29, 311–332 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS.
- For reviews and references therein, see: (a) G. Li and M. Szostak, Chem. Rec., 2020, 20, 649–659 CrossRef CAS; (b) P. Acosta-Guzmán, A. Mateus-Gómez and D. Gamba-Sánchez, Molecules, 2018, 23, 2382 CrossRef; (c) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS; (d) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS; (e) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS.
- Selected examples of Lewis-acid-mediated or Lewis-acid-catalyzed transamidation of secondary amides: (a) D. A. Kissounko, J. M. Hoerter, I. A. Guzei, Q. Cui, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 1776–1783 CrossRef CAS; (b) J. M. Hoerter, K. M. Otte, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 5177–5183 CrossRef CAS.
- Selected examples of transition-metal-catalyzed transamidation of secondary amides: for Ni-catalyzed transamidation: (a) S. Yu, T. Shin, M. Zhang, Y. Xia, H. Kim and S. Lee, Org. Lett., 2018, 20, 7563–7566 CrossRef CAS; (b) J. E. Dander, E. L. Baker and N. K. Garg, Chem. Sci., 2017, 8, 6433–6438 RSC; (c) C. W. Cheung, M. L. Ploeger and X. Hu, ACS Catal., 2017, 7, 7092–7096 CrossRef CAS; (d) E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 11554 CrossRef; For Pd-catalyzed transamidation: (e) T. Zhou, G. Li, S. P. Nolan and M. Szostak, Org. Lett., 2019, 21, 3304–3309 CrossRef CAS; (f) G. Meng, P. Lei and M. Szostak, Org. Lett., 2017, 19, 2158–2161 CrossRef CAS.
- Selected examples of transition-metal-free transamidation of secondary amides: (a) J. Chen, Y. Xia and S. Lee, Org. Lett., 2020, 22, 3504–3508 CrossRef CAS; (b) A. Mishra, S. Singh and V. Srivastava, Asian J. Org. Chem., 2018, 7, 1600–1604 CrossRef CAS; (c) Y. Liu, S. Shi, M. Achtenhagen, R. Liu and M. Szostak, Org. Lett., 2017, 19, 1614–1617 CrossRef CAS; (d) T. B. Nguyen, J. Sorres, M. Q. Tran, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2012, 14, 3202–3205 CrossRef CAS.
- (a) M. Fairley, L. J. Bole, F. F. Mulks, L. Main, A. R. Kennedy, C. T. O'Hara, J. García-Alvarez and E. Hevia, Chem. Sci., 2020, 11, 6500–6509 RSC; (b) G. Li, C.-L. Ji, X. Hong and M. Szostak, J. Am. Chem. Soc., 2019, 141, 11161–11172 CrossRef CAS; (c) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165 CrossRef.
- C. W. Cheung, J.-A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789–6792 CrossRef CAS.
- Lewis-acid-mediated or Lewis-acid-catalyzed transamidation of tertiary amides: (a) H. Sheng, R. Zeng, W. Wang, S. Luo, Y. Feng, J. Liu, W. Chen, M. Zhu and Q. Guo, Adv. Synth. Catal., 2017, 359, 302–313 CrossRef CAS; (b) N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS; (c) J. M. Hoerter, K. M. Otte, S. H. Gellman, Q. Cui and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 647–654 CrossRef CAS; (d) E. Bon, D. C. H. Bigg and G. Bertrand, J. Org. Chem., 1994, 59, 4035–4036 CrossRef CAS.
- Transamidation of N,N-dimethylformamide (DMF) and N-acyl-glutarimides: (a) T. Ghosh, S. Jana and J. Dash, Org. Lett., 2019, 21, 6690–6694 CrossRef CAS; (b) J. Yin, J. Zhang, C. Cai, G.-J. Deng and H. Gong, Org. Lett., 2019, 21, 387–392 CrossRef CAS; (c) Y. Liu, M. Achtenhagen, R. Liu and M. Szostak, Org. Biomol. Chem., 2018, 16, 1322–1329 RSC.
- Only a few reports on the in situ formation of acyl fluoride intermediate from preactivated secondary amide bond cleavage are known: (a) Z. Luo, H. Wu, Y. Li, Y. Chen, J. Nie, S. Lu, Y. Zhu and Z. Zeng, Adv. Synth. Catal., 2019, 361, 4117–4125 CrossRef CAS; (b) Y. Li, H. Wu and Z. Zeng, Eur. J. Org. Chem., 2019, 4357–4361 CrossRef CAS; (c) W. Guo, J. Huang, H. Wu, T. Liu, Z. Luo, J. Jian and Z. Zeng, Org. Chem. Front., 2018, 5, 2950–2954 RSC; (d) H. Wu, W. Guo, S. Daniel, Y. Li, C. Liu and Z. Zeng, Chem.–Eur. J., 2018, 24, 3444–3447 CrossRef CAS.
- Recent reviews and highlight on the high synthetic versertility of acyl fluorides: (a) Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS; (b) Z. Wang, X. Wang and Y. Nishihara, Chem.–Asian J., 2020, 15, 1234–1247 CrossRef CAS; (c) N. Blanchard and V. Bizet, Angew. Chem., Int. Ed., 2019, 58, 6814–6817 CrossRef CAS; (d) G. Prabhu, N. Narendra, Basavaprabhu, V. Panduranga and V. V. Sureshbabu, RSC Adv., 2015, 5, 48331–48362 RSC.
- Selected reviews on the benzylic C–H functionalization: (a) R. Yazaki and T. Ohshima, Tetrahedron Lett., 2019, 60, 151225 CrossRef; (b) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS; (c) R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC.
- Selected examples of Cu-catalyzed selective functionalization of benzylic C–H bond with electrophilic fluorinating agents: (a) S.-E. Suh, S.-J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS; (b) H. Hu, S.-J. Chen, M. Mandal, S. M. Pratik, J. A. Buss, S. W. Krska, C. J. Cramer and S. S. Stahl, Nat. Catal., 2020, 3, 358–367 CrossRef CAS; (c) W. Zhang, L. Wu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2019, 58, 6425–6429 CrossRef CAS; (d) H. Xiao, Z. Liu, H. Shen, B. Zhang, L. Zhu and C. Li, Chem, 2019, 5, 940–949 CrossRef CAS; (e) W. Zhang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 7709–7712 CrossRef CAS; (f) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS; (g) Z. Ni, Q. Zhang, T. Xiong, Y. Zheng, Y. Li, H. Zhang, J. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2012, 51, 1244–1247 CrossRef CAS.
- Recent reviews on Selectfluor: (a) K. Yang, M. Song, A. I. M. Ali, S. M. Mudassir and H. Ge, Chem.–Asian J., 2020, 15, 729–741 CrossRef CAS; (b) S. Stavber, Molecules, 2011, 16, 6432–6464 CrossRef CAS; (c) P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS.
- For a review, see: K. Mashima, Y. Nishii and H. Nagae, Chem. Rec., 2020, 20, 332–343 CrossRef CAS and references therein.
- Selected examples of transesterification of tertiary amides: (a) H. Nagae, T. Hirai, D. Kato, S. Soma, S. Akebi and K. Mashima, Chem. Sci., 2019, 10, 2860–2868 RSC; (b) T. Toyano, M. N. Rashed, Y. Morita, T. Kamachi, S. M. A. H. Siddiki, M. A. Ali, A. S. Touchy, K. Kon, Z. Maeno, K. Yoshizawa and K. Shimizu, ChemCatChem, 2019, 11, 449–456 CrossRef; (c) D. Ye, Z. Liu, H. Chen, J. L. Sessler and C. Lei, Org. Lett., 2019, 21, 6888–6892 CrossRef CAS; (d) G. Li, P. Lei and M. Szostak, Org. Lett., 2018, 20, 5622–5625 CrossRef CAS; (e) Y. Bourne-Branchu, C. Gosmini and G. Danoun, Chem.–Eur. J., 2017, 23, 10043–10047 CrossRef CAS; (f) L. Hie, E. L. Baker, S. M. Anthony, J.-N. Desrosiers, C. Senanayake and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS; (g) L. Hie, N. F. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS.
- K. V. Raghavendra Rao and Y. Vallée, Tetrahedron, 2016, 72, 4442–4447 CrossRef CAS.
- Review: S. Mahesh, K.-C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef.
- Peptide cleavage promoted by Cu/N-oxyl radical system: Y. Seki, K. Tanabe, D. Sasaki, Y. Sohma, K. Oisaki and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 6501–6505 CrossRef CAS.
- Recent review: F. J. Aguilar Troyano, K. Merkens and A. Gómez-Suárez, Asian J. Org. Chem., 2020, 9, 992–1007 CrossRef CAS.
- Recent reports on the nitrogen-centered cation radical generation from Selectfluor under photoirradiation condition: (a) F. J. Aguilar Troyano, F. Ballaschk, M. Jaschinski, Y. Özkaya and A. Gómez-Suárez, Chem.–Eur. J., 2019, 25, 14054–14058 CrossRef CAS; (b) X.-A. Liang, L. Niu, S. Wang, J. Liu and A. Lei, Org. Lett., 2019, 21, 2441–2444 CrossRef CAS; (c) H. Zhao and J. Jin, Org. Lett., 2019, 21, 6179–6184 CrossRef CAS; (d) L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef.
- (a) Review: J. Sperry, Synthesis, 2011, 3569–3580 CrossRef CAS; (b) Z. Jin, B. Xu and G. B. Hammond, Tetrahedron Lett., 2011, 52, 1956–1959 CrossRef CAS . In this publication, the authors reported that trace water in the reaction acts as the oxygen source for imide formation.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS.
- Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef CAS.
- S. Sathyamoorthi, Y.-H. Lai, R. M. Bain and R. N. Zare, J. Org. Chem., 2018, 83, 5681–5687 CrossRef CAS.
- R. Wagner, B. Wiedel, W. Günther, H. Görls and E. Anders, Eur. J. Org. Chem., 1999, 2383–2390 CrossRef CAS . In this work, acyl fluoride is favorably obtained in high yield from in situ formed 1-acyl-4-benzylpyridinium tetrafluoroborate.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2069 CrossRef CAS.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05137c |
| This journal is © The Royal Society of Chemistry 2020 |