
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl urea substituted fatty acids: a new class of protonophoric mitochondrial uncoupler that utilises a synthetic anion transporter†
Tristan
Rawling
 *a,
Hugo
MacDermott-Opeskin
b,
Ariane
Roseblade
a,
Curtis
Pazderka
a,
Callum
Clarke
a,
Kirsi
Bourget
c,
Xin
Wu
d,
William
Lewis
d,
Benjamin
Noble
be,
Philip A.
Gale
d,
Megan L.
O'Mara
b,
Charles
Cranfield
f and
Michael
Murray
c
*a,
Hugo
MacDermott-Opeskin
b,
Ariane
Roseblade
a,
Curtis
Pazderka
a,
Callum
Clarke
a,
Kirsi
Bourget
c,
Xin
Wu
d,
William
Lewis
d,
Benjamin
Noble
be,
Philip A.
Gale
d,
Megan L.
O'Mara
b,
Charles
Cranfield
f and
Michael
Murray
c
aSchool of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Sydney, NSW 2007, Australia. E-mail: Tristan.Rawling@uts.edu.a; Tel: +61-2-9514-7956
bResearch School of Chemistry, College of Science, The Australian National University, Canberra, ACT 0200, Australia
cDiscipline of Pharmacology, School of Medical Sciences, University of Sydney, Sydney, NSW 2006, Australia
dSchool of Chemistry, University of Sydney, Sydney, NSW 2006, Australia
eSchool of Engineering, College of Science, Engineering and Health, RMIT University, Melbourne, VIC 3001, Australia
fSchool of Life Sciences, Faculty of Science, University of Technology Sydney, Sydney, NSW 2007, Australia
First published on 19th August 2020
Abstract
Respiring mitochondria establish a proton gradient across the mitochondrial inner membrane (MIM) that is used to generate ATP. Protein-independent mitochondrial uncouplers collapse the proton gradient and disrupt ATP production by shuttling protons back across the MIM in a protonophoric cycle. Continued cycling relies on the formation of MIM-permeable anionic species that can return to the intermembrane space after deprotonation in the mitochondrial matrix. Previously described protonophores contain acidic groups that are part of delocalised π-systems that provide large surfaces for charge delocalisation and facilitate anion permeation across the MIM. Here we present a new class of protonophoric uncoupler based on aryl-urea substituted fatty acids in which an acidic group and a π-system are separated by a long alkyl chain. The aryl-urea group in these molecules acts as a synthetic anion receptor that forms intermolecular hydrogen bonds with the fatty acid carboxylate after deprotonation. Dispersal of the negative charge across the aryl-urea system produces lipophilic dimeric complexes that can permeate the MIM and facilitate repeated cycling. Substitution of the aryl-urea group with lipophilic electron withdrawing groups is critical to complex lipophilicity and uncoupling activity. The aryl-urea substituted fatty acids represent the first biological example of mitochondrial uncoupling mediated by the interaction of a fatty acid and an anion receptor moiety, via self-assembly.
Introduction
The mitochondrion supports cellular ATP production by oxidative phosphorylation (OxPhos, Fig. 1). The organelle contains two distinct membranes: an outer membrane that is widely permeable to solutes and a relatively impermeable mitochondrial inner membrane (MIM) that encloses the mitochondrial matrix. Nutrients such as fatty acids and pyruvate are converted in the mitochondrial matrix to the high-energy electron carriers NADH and FADH2. These carriers donate electrons to the electron transport chain (ETC), a series of enzyme complexes embedded in the MIM that drive OxPhos. As electrons pass through the ETC complexes protons are pumped out of the matrix and into the inter-membrane space to produce an electrochemical gradient across the MIM. The passage of protons back into the matrix along the electrochemical gradient is utilized by ATP-synthase to produce ATP, thus coupling energy metabolism to electron transport.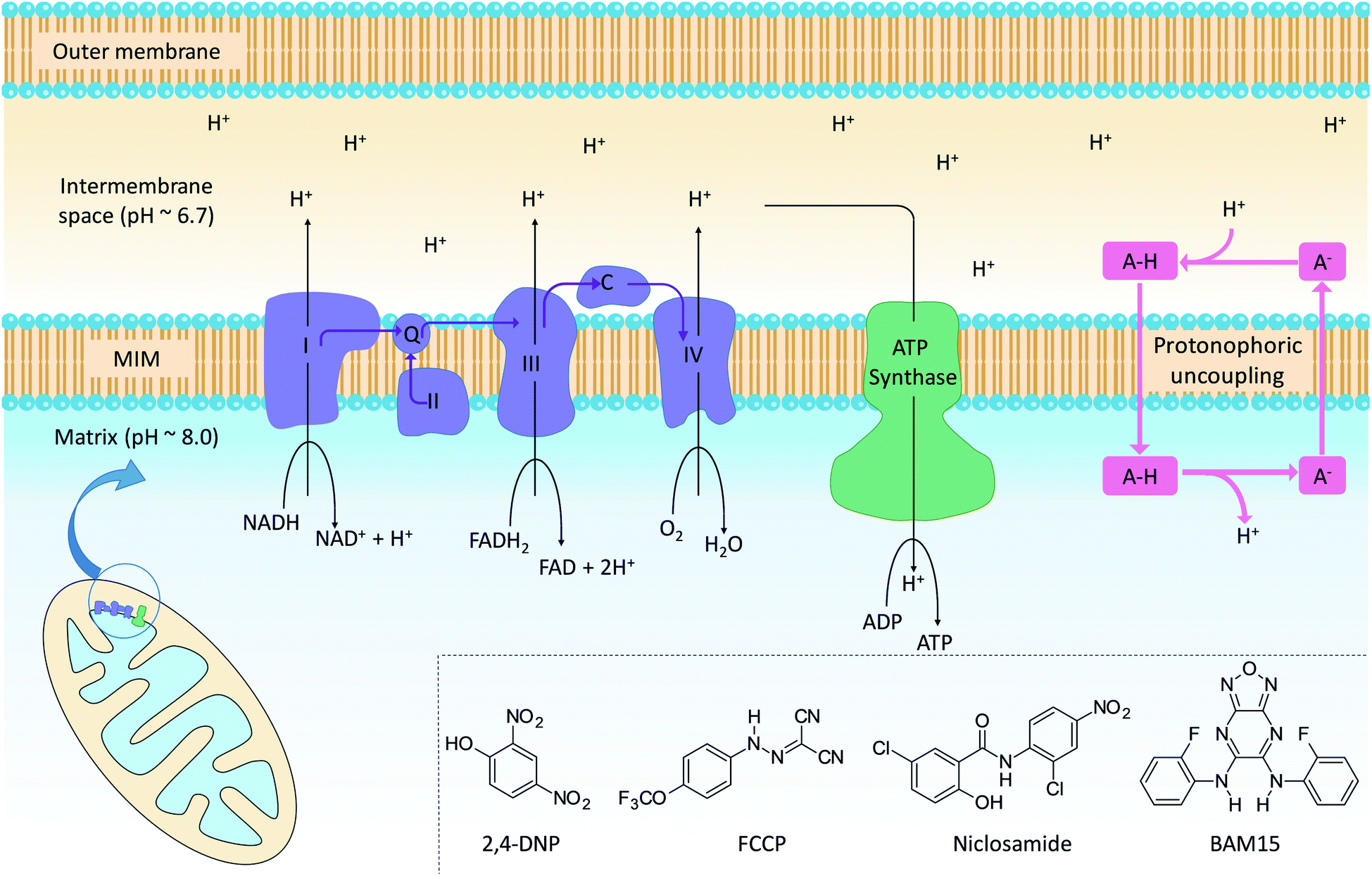 | ||
| Fig. 1 Mitochondrial electron transport, ATP production and protonophoric uncoupling. Left: the citric acid cycle produces NADH and FADH2, which are high energy electron sources for complexes I–IV (light blue). Cytochrome c (C) and coenzyme Q (Q) shuttle electrons between ETC complexes. In parallel, the ETC pumps protons into the intermembrane space to establish an electrochemical gradient across the MIM. Protons flow back across the MIM via ATP synthase (green) to catalyse the formation of ATP. Right: protonophoric uncouplers (A–H) bypass ATP synthase and transport protons from the intermembrane space to the matrix. If sufficiently lipophilic, the conjugate anions (A−) are returned to the intermembrane space for further uncoupling cycles. Insert: chemical structures of representative protonophoric uncouplers. | ||
Mitochondrial uncouplers are compounds that induce proton leak across the MIM and into the matrix, leading to futile cycles of nutrient oxidation without ATP production. One of earliest known uncouplers, 2,4-dinitrophenol (2,4-DNP, Fig. 1), was clinically used as a weight loss drug in the 1930s until it was withdrawn due to unacceptable toxicities. Despite safety concerns, there has been a resurgence of interest in uncouplers because of their potential uses in metabolic diseases such as obesity and non-insulin-dependent diabetes, neurodegenerative diseases and cancer.1
Protonophoric mitochondrial uncouplers are lipophilic weak acids (pKa ∼ 4–8) that shuttle protons across the MIM in a protein-independent cycle. The protonated uncoupler (HA in Fig. 1) first diffuses across the MIM and carries a proton from the intermembrane space to the matrix.1,2 Proton release in the alkaline matrix generates the protonophore anion (A− in Fig. 1), and further uncoupling cycles can only occur if the anions passively diffuse across the MIM and return to the intermembrane space.2 The acidic groups in protonophores are therefore part of delocalised π-systems that spread negative charge over large surfaces and promote anion lipophilicity and protonophoric cycling (Fig. 1).3 In contrast, fatty acids such as palmitic acid cannot complete the cycle because the anion, which has negative charge localized on the carboxylate group, is too polar to permeate the MIM. Instead, fatty acid-mediated uncoupling proceeds by a protein-dependant mechanism in which the anion is actively transported across the MIM by transport proteins such as adenine nucleotide translocase (ANT).4,5
Synthetic anion transporters are small-molecule organic compounds that facilitate the transport of anionic species such as Cl− and HCO3− across lipid bilayers.6,7 Non-channel forming anion transporters contain anion recognition motifs (e.g. urea, thiourea, and sulphonamide groups) that bind their guest anion through non-covalent interactions to form supramolecular complexes on one side of the membrane.8 Delocalisation of the anionic charge within these assemblies allows them to diffuse across the bilayer and translocate the anion. Synthetic anion transporters have received significant research attention due to their potential therapeutic applications,9,10 for example in treating cystic fibrosis11 and as anticancer agents where disruption of intracellular anion concentrations can induce cell death.12
In this paper we reveal the mitochondrial actions of a fatty acid substituted with a urea-based anion transporter motif at the molecular scale. These compounds (termed aryl-ureas) induce mitochondrial dysfunction in breast cancer cells,13 and here we demonstrate that they act as mitochondrial uncouplers. Protein-independent, protonophoric actions of the aryl-ureas is established using a new assay that utilises tethered lipid bilayers in conjunction with electrical impedance spectroscopy. The rate limiting step in the protonophoric cycle, translocation of the deprotonated uncoupler across the MIM, proceeds via self-assembly into dimeric complexes formed by hydrogen bonding between the carboxylate and urea anion binding groups. Aryl-substitution with electron withdrawing and lipophilic groups promotes delocalisation of the carboxylate charge across the aryl urea system and provides membrane permeability to the complexes. By providing the flexibility to separate the acidic moiety from the conjugated π system, these findings open up new possibilities for the design of novel mitochondrial uncouplers that incorporate anion binding motifs.
Results and discussion
Compound library design and synthesis
Aryl ureas capable of disrupting energy production in breast cancer cells possess strong electron withdrawing substituents (σtotal > 0.66) in the meta- and para-positions of the aryl ring.13,14 In this study a series of aryl-ureas was prepared bearing electron withdrawing aryl-substituents of varying polarity, determined from the hydrophobicity constants (πtotal, see Table 1), to assess the influence of substituent lipophilicity on activity. The compounds were synthesised in 2 steps using N,N-carbonyldiimidazole (CDI) chemistry to form the substituted aryl urea moieties (see Scheme S1 in ESI†).|
|
||||
|---|---|---|---|---|
| Compound | R | σ total | π total | JC-1 IC50 (μM) |
| a Hammett substituent constants were taken from published values.15σp was used for ortho substituents. b Inactive: no change in the JC-1 red: green ratio when tested at 50 μM. | ||||
| 1 |
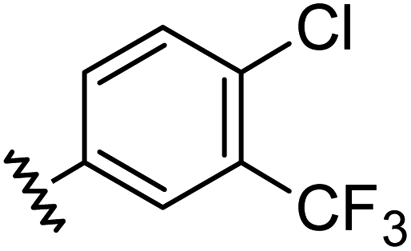
|
0.66 | 1.56 | 4.5 ± 1.1 |
| 2 |
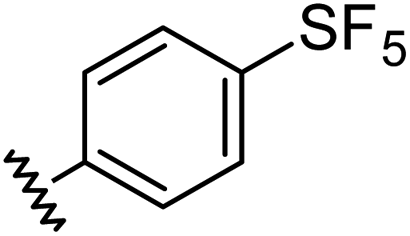
|
0.68 | 1.23 | 14 ± 1 |
| 3 |
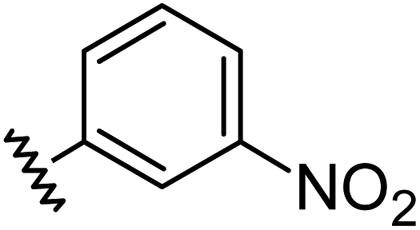
|
0.71 | −0.28 | inactiveb |
| 4 |
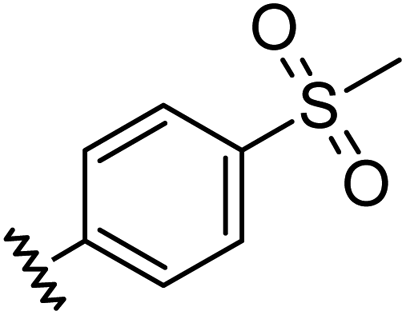
|
0.72 | −1.63 | inactiveb |
| 5 |

|
0.78 | −0.28 | inactiveb |
| 6 |
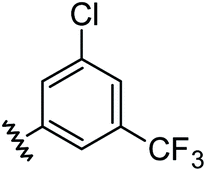
|
0.80 | 1.56 | 2.9 ± 1.1 |
| 7 |
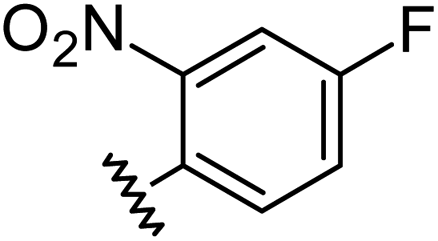
|
0.84 | −0.14 | inactiveb |
| 8 |
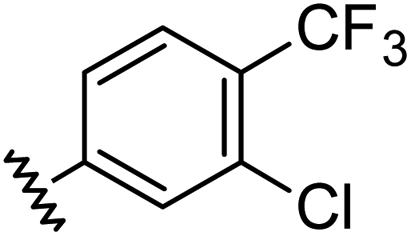
|
0.91 | 1.56 | 7.2 ± 1.2 |
| 9 |
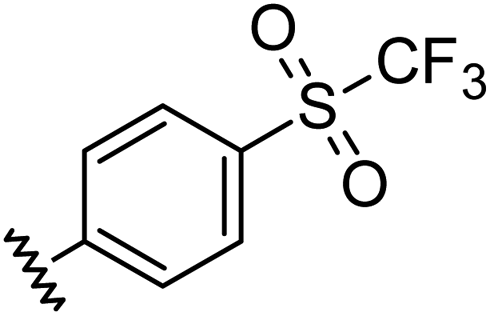
|
0.96 | 0.55 | 6.4 ± 1.2 |
| 10 |

|
0.97 | 1.76 | 7.6 ± 1.2 |
| 11 |
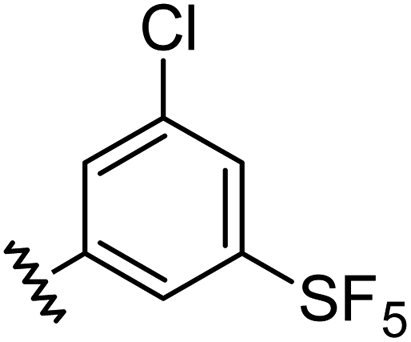
|
0.98 | 1.94 | 6.0 ± 1.0 |
| 12 |
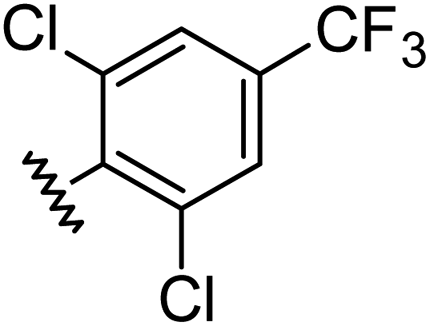
|
1.00 | 2.30 | inactiveb |
| 13 |
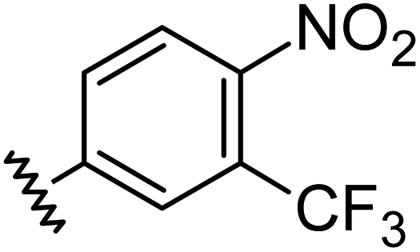
|
1.21 | 0.60 | 7.6 ± 1.2 |
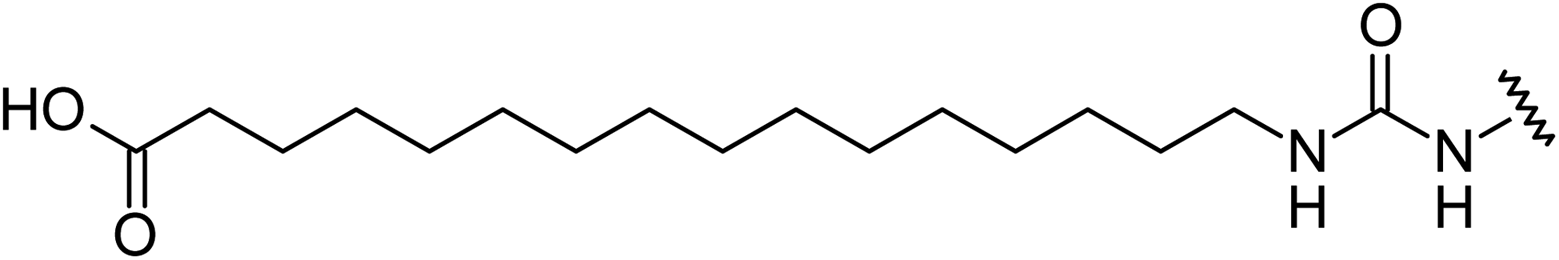
Mitochondrial actions of aryl ureas
We evaluated the capacity of the aryl-ureas to depolarize the MIM in MDA-MB-231 cells using the JC-1 assay. JC-1 is a redox active dye that forms aggregates that fluoresce red in energized mitochondria with high membrane potential. Mitochondrial uncouplers dissipate the membrane potential so that JC-1 remains in its monomeric form and fluoresces green. We determined JC-1 IC50s for aryl-ureas as the concentrations required to shift the red![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :green fluorescence ratio by 50% (Table 1). The active analogues 1, 2, 6, 8, 9, 10, 11, and 13 shifted the JC-1 red:green fluorescence ratio with IC50s between 3–14 μM. These analogues also markedly impaired ATP production at concentrations above their JC-1 IC50 values (10 and 40 μM, Fig. 2a). In contrast, the ortho-substituted analogues 7 and 12 did not alter the JC-1 red:green fluorescence ratio or modulate ATP production. These findings are consistent with our previous finding that only aryl-ureas with strongly electron-withdrawing groups in the meta- and para-positions promoted mitochondrial dysfunction.13,14
:green fluorescence ratio by 50% (Table 1). The active analogues 1, 2, 6, 8, 9, 10, 11, and 13 shifted the JC-1 red:green fluorescence ratio with IC50s between 3–14 μM. These analogues also markedly impaired ATP production at concentrations above their JC-1 IC50 values (10 and 40 μM, Fig. 2a). In contrast, the ortho-substituted analogues 7 and 12 did not alter the JC-1 red:green fluorescence ratio or modulate ATP production. These findings are consistent with our previous finding that only aryl-ureas with strongly electron-withdrawing groups in the meta- and para-positions promoted mitochondrial dysfunction.13,14
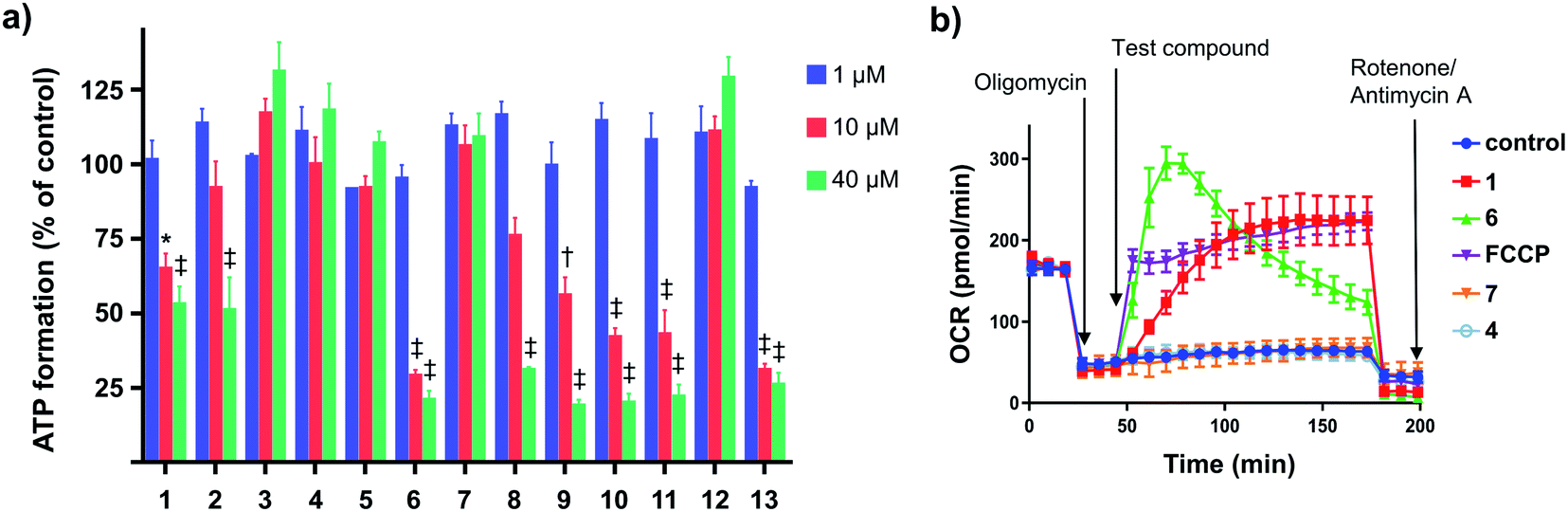 | ||
| Fig. 2 Effects of aryl-ureas 1–13 of mitochondrial function in MDA-MB-231 cells. (a) ATP formation by MDA-MB-231 cells after 24 hours treatments with 1–13. (b) Oxygen consumption rates (OCR) in MDA-MB-231 cells after sequential addition of the ATP-synthase inhibitor oligomycin (1 μM; arrow (a)), protonophore FCCP (1 μM) or aryl-ureas (20 μM; arrow (b)), and then the ETC complex inhibitors rotenone/antimycin A (1 μM). Data represents the mean ± SEM of 3 independent experiments. Different from DMSO treated control: (‡) P < 0.0001, (†) P < 0.001, (*) P < 0.05. | ||
Analogues 3, 4, and 5 were also inactive. These analogues possess the hydrophilic substituents (negative πtotal values), indicating that substituent lipophilicity is also critical to activity. This is further illustrated by comparison of 9 and 4. These compounds have similar electronic and steric properties, but differ markedly in lipophilicity. The more lipophilic 9 effectively depolarized the mitochondrion and impaired ATP production, while 4 was inactive.
Impaired ATP production and mitochondrial membrane depolarization are consistent with uncoupling. To confirm that the active aryl ureas are uncouplers we used the Seahorse Mito Stress test, which measures cellular oxygen consumption rate (OCR) in the presence of the ATP synthase inhibitor oligomycin. Addition of uncouplers under these conditions increases OCR. When added to MDA-MB-231 cells treated with oligomycin all of the active aryl-ureas, as well as the known protonophore FCCP, increased OCR approximately 5-fold over control (Fig. 2b and S1 in ESI†). In contrast, the inactive analogues did not increase OCR (Fig. 2b and S1†). Taken together, these data show that aryl-ureas with lipophilic and electron withdrawing substituents in the meta and para positions are mitochondrial uncouplers that depolarise the MIM and impair ATP production.
Mechanism of uncoupling
We next sought to define the mechanism of aryl-urea mediated uncoupling. The recoupling agent 6-ketocholesterol (6-KCH) is commonly used in cell-based assays to distinguish protein-independent uncouplers from those that rely on transport proteins embedded in the MIM to complete the uncoupling cycle.16 6-KCH is suggested to incorporate into the MIM and selectively inhibit anion permeation, which is the rate limiting step in protonophoric cycling. In JC-1 assays, pre- or co-treatment of MDA-MB-231 cells with 6-KCH failed to prevent 6 mediated mitochondrial depolarisation (data not shown). These findings however do not rule out a protonophoric mechanism for 6 because 6-KCH mediated-recoupling only affects protonophores of certain structural classes.17 Indeed, unambiguous identification of a protein-dependant mechanism involves complex and time-consuming cell-based assays.18 We therefore sought to develop a new assay to rapidly and unambiguously identify compounds with protonophoric activity.Lipid bilayers tethered to a thin film gold electrode, when used in conjunction with electrical impedance spectroscopy, permit a measure of ionic membrane conductance over prolonged periods.19 We hypothesized that this system could be used to identify protonophores because their addition to the lipid membrane should increase conductance by transporting protons across the bilayer, while protein-dependant uncouplers should have no effect. 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipid bilayers were used in the assays because phosphatidylcholine is the major lipid component of the MIM.20 Consistent with our hypothesis, addition of active aryl-ureas 1 and 6, as well as the known protonophore FCCP, increased bilayer conductance compared to a control (Fig. 3a), consistent with increased ionic transport across the bilayer. In contrast, the inactive aryl urea 7 and the protein-dependant uncoupler palmitic acid failed to effect conductance. To confirm that changes in conductance were due to proton transport, and not to other metal cations, experiments were conducted over an expanded pH range on a background of a constant concentration of 100 mM NaCl (Fig. 3b). At lower pH values (3–5) higher proton concentrations increased membrane conductance in the presence of active aryl-ureas 1 and 6, but not inactive analogue 7. Decreased membrane conductance at pH 6 has been previously observed in studies using this model lipid bilayer platform, and is attributed to tighter packing of the lipids in the bilayer due to increased hydrogen bonding at the lipid interface.19,21 Thus the changes in membrane conductance observed over the pH range are consistent with the ability of 1 and 6 to transport protons across the lipid bilayer. To the best of our knowledge, this is the first example of tethered lipid bilayers in conjunction with electrical impedance spectroscopy to identify protonophoric agents capable of directly modulating membrane proton transport.
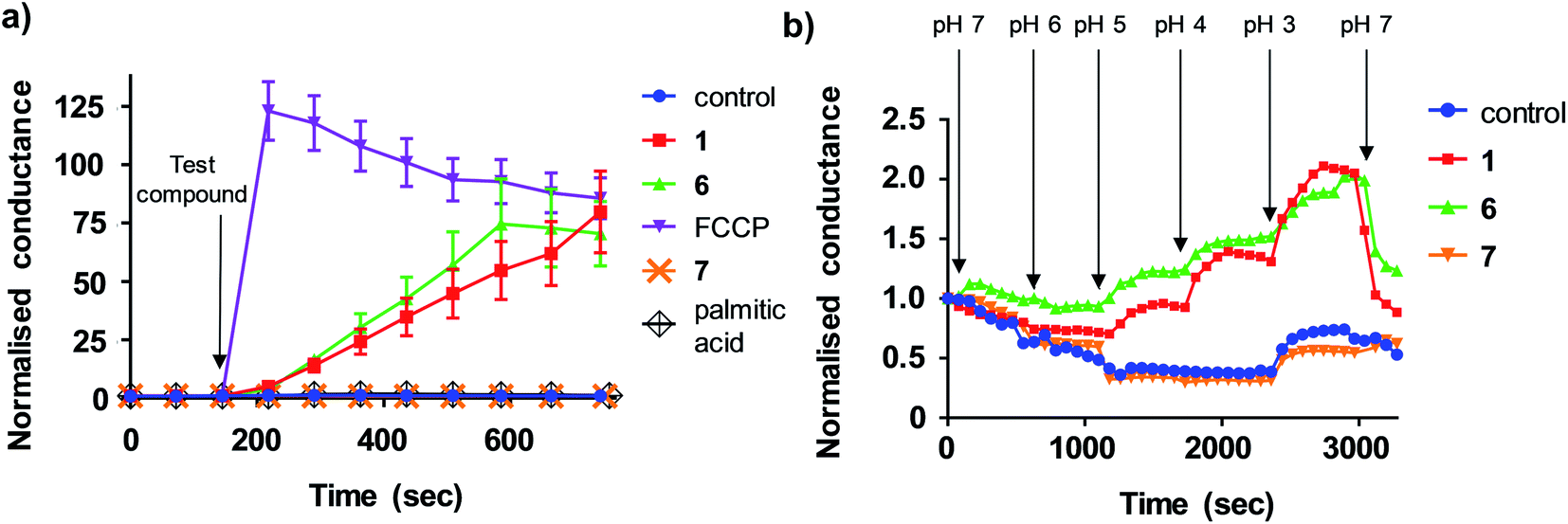 | ||
| Fig. 3 Aryl-ureas and the protonophore FCCP modulate proton conductance in tethered lipid bilayer membranes. (a) Addition of active aryl ureas 1 and 6 (10 μM) and FCCP (0.5 μM) increased conductance across a DOPC lipid bilayer tethered to a gold electrode, as measured by electrical impedance spectroscopy. The inactive aryl urea 7 (10 μM) and protein-dependant uncoupler palmitic acid failed to increase bilayer conductance. Data normalised to baseline control. (b) Sequential reduction of membrane pH leads to an increase in relative membrane conductivity for active aryl ureas 1 and 6 (10 μM). For the inactive aryl urea 7 (10 μM) there was no relative conductance increase. Data normalised to conductance of aryl-urea treated membranes at pH = 7. Data represents the mean ± SEM of 3 independent experiments. | ||
To further characterise the uncoupling mechanism, we sought to identify the acidic functional groups in the aryl-ureas that facilitate protonophoric cycling. The carboxylic acid groups lack connection to a delocalised π system and would not be expected to generate a MIM-permeable anion after deprotonation. The urea protons adjacent to the aryl ring are part of the delocalised aryl urea system and are potentially acidic due to the strongly electron withdrawing aryl substituents that are required for activity. However, high-level ab initio calculations indicate that pKa values for these protons (∼11–14, see Table S1 in ESI†) are too high to dissociate in the matrix. To evaluate the potential roles of the carboxylate and urea moieties in uncoupling, we prepared esterified (6-Me) and N,N-dimethylated urea (6-NNMe) analogues of 6 (Fig. 4a and ESI†) and assessed their protonophoric activity.
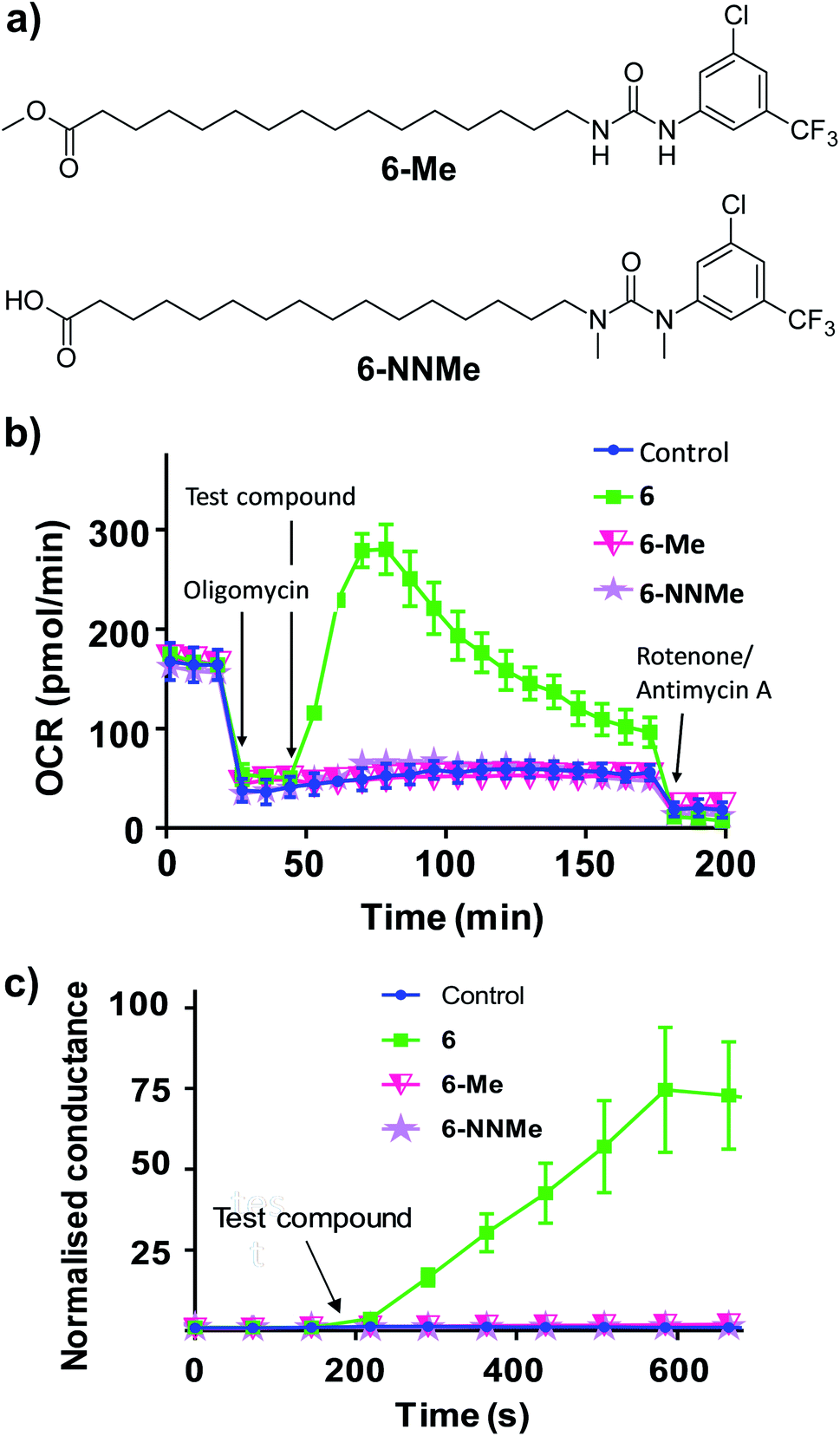 | ||
| Fig. 4 Methyl ester (6-Me) and N,N-dimethylated (6-NNMe) analogues of aryl-urea 6 lack uncoupling and protonophoric activity. (a) Chemical structure of 6 analogues. (b) OCR in MDA-MB-231 cells treated sequentially with the ATP-synthase inhibitor oligomycin (1 μM), test compound (20 μM), and then ETC complex inhibitors rotenone/antimycin A (1 μM), as indicated. (c) Effects of 6, 6-Me and 6-NNMe (10 μM) on the conductance of DOPC tethered lipid bilayers measured by impedance spectroscopy. Data represents the mean ± SEM of 3 independent experiments. | ||
As shown in Fig. 4b, removal of either the carboxylic acid group (6-Me, 20 μM) or the urea NHs (6-NNMe, 20 μM) produced analogues that were devoid of uncoupling activity in Seahorse Mito Stress tests. Similarly, neither compound (10 μM) increased proton conductance in the tethered lipid bilayer model (Fig. 4c).
The inactivity of 6-Me and 6-NNMe indicate that both functional groups are essential for uncoupling activity and that interaction of these groups occurs during the protonophoric cycle. This led us to postulate that the urea group functions as a synthetic anion transporter to facilitate diffusion of a deprotonated species across the MIM. Indeed, urea derivatives are known anion receptors that form strong interactions with carboxylate groups comprised of two parallel NH⋯O hydrogen bonds.22 Through-bond propagation of electron density delocalizes the carboxylate negative charge across the aryl-urea system, and the resulting complexes are stable in non-polar solvents (reflecting the membrane environment).23,24 The structure–activity relationship of the aryl-ureas is also consistent with that of anion transporters, where inclusion of electron withdrawing25 and lipophilic26 aryl substituents promotes transporter activity.9,27 Further, it has been shown that thioureas act as fatty acid anion membrane transporters by a mechanism that relies on the formation of hydrogen-bonded membrane–permeable complexes.28 To understand how urea/carboxylate interaction could facilitate aryl urea-mediated uncoupling the formation and nature of the membrane permeable anionic species formed by the aryl ureas was evaluated using Density Functional Theory (DFT).
Binding free energies were calculated for the complexation of model (tail-truncated) aryl ureas with propanoate at the M06-2X/6-31+G(d) level of theory (see Computational methodology).29 Complexation in both water and in n-pentadecane (chosen to mimic the low dielectric environment of a lipid membrane) was investigated using the SMD continuum solvation model.30 In water, binding free energies ranged from ∼−1 to 8 kJ mol−1 over the full compound set (Table S1†), indicating that carboxylate complexation by aryl ureas is thermo-neutral to slightly unfavourable in an aqueous environment. In contrast, binding free energies in n-pentadecane were highly favourable: ranging from −117 to −95 kJ mol−1 for the active analogues and −99 to −79 kJ mol−1 for the inactive analogues (Table S1†). The highly favourable complexation of carboxylates by aryl ureas in non-aqueous solvent, combined with the thermo-neutral to slightly unfavourable binding in water, indicates that the thermodynamic stability of these complexes is highly dependent on their local dielectric environment. Specifically, these complexes would be stable to dissociation within the lipid membrane but not at the water interface. This mechanism is consistent with the reversible transport of carboxylate moieties by aryl ureas required for uncoupling.
Aryl-ureas containing electron-withdrawing substituents ortho to the urea moiety did not disrupt the mitochondrial membrane potential or impair ATP production (Table 1, Fig. 2). To rationalize this, the carboxylate binding affinity and electronic structure of the inactive 12 were compared with the highly active 6. Despite the favourable aryl substitution pattern and lipophilicity (σtotal and πtotal values of 1.0 and 2.30 respectively) for 12, compared to 6 (0.80 and 1.56 respectively), the complexation free energy of 12 and propanoate in n-pentadecane was low compared to that of 6 (−79 kJ mol−1vs. −95 kJ mol−1 respectively). This is due to an unfavourable steric interaction between the carboxylate moiety and the ortho substituent of 12. Additionally, ortho substitution forces the urea group out of plane with the aryl π-system, which hinders charge delocalization in the respective complex. Fig. 5 shows the HOMOs of 6 and 12 complexed with propanoate (Fig. 5a and b respectively). In the case of the 12-propanoate complex greater HOMO density is localized on the propanoate moiety and the distorted π-system prevents constructive HOMO overlap between the urea and aryl moieties (Fig. 5b). In contrast, the HOMO density in the 6-propanoate complex is significantly delocalised (Fig. 5a).
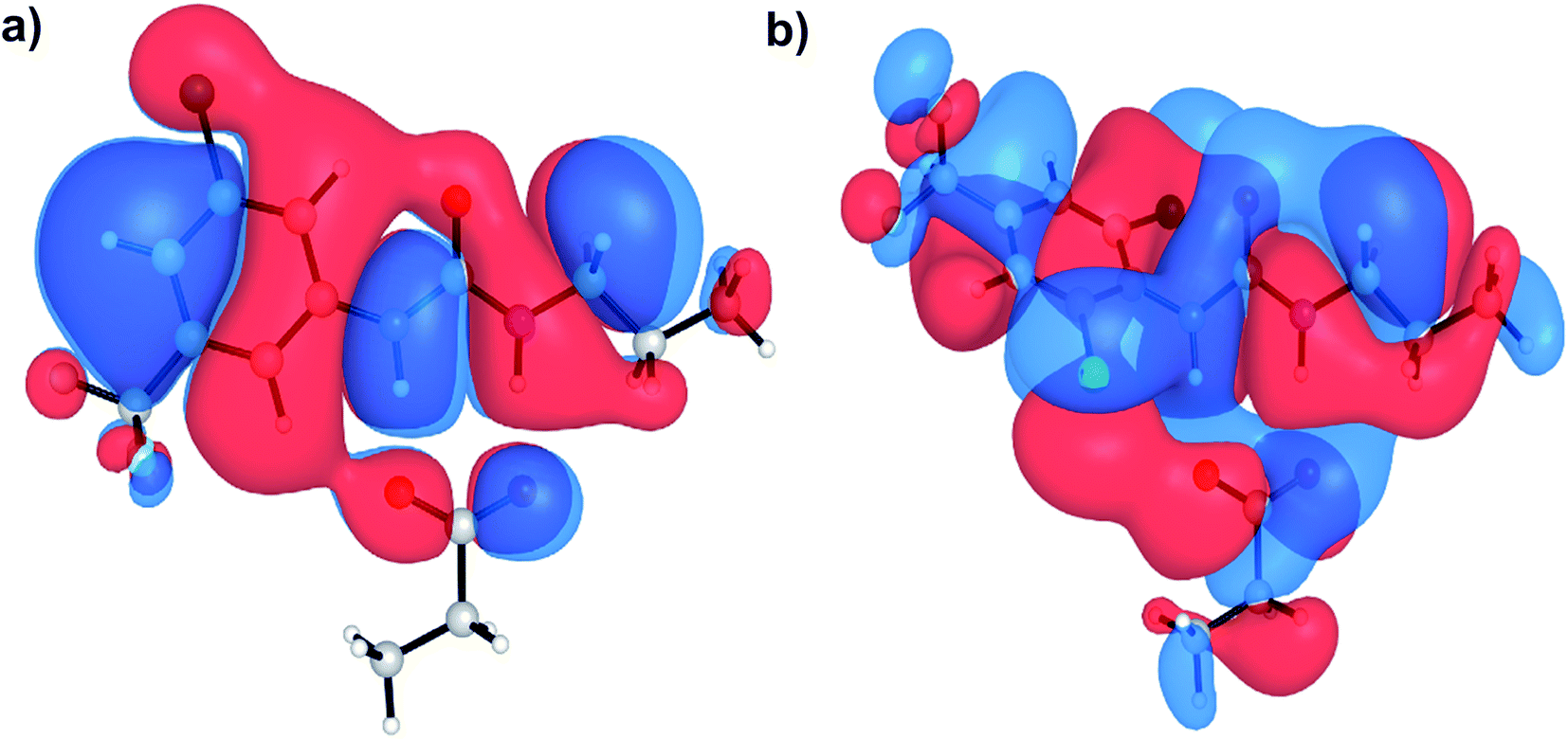 | ||
| Fig. 5 HOMOs of aryl urea model compounds, (a) 6 and (b) 12 in complex with propanoate. Calculations were conducted at the M06-2X/6-31+G(d) level of theory. | ||
Lipophilicity is a major determinant of trans-bilayer ion transport activity and is closely linked to charge delocalization of the ionic species. To assess the lipophilicity of the aryl-urea analogues, free energies for transfer of propanoate-complexed aryl-ureas from water to n-pentadecane were calculated (ΔGSolvcomplex(W–P), see Computational methodology). Impaired anion delocalization results in high free energies of solvent transfer for 12 relative to 6 (99 kJ mol−1 and 80 kJ mol−1 respectively, Table S2†) and consequently much lower expected membrane permeability of the anionic complex. In contrast, the free energy of solvent transfer for isolated 12 (ΔGSolvmonomer(W–P), see Computational methodology) is more favourable than 6 (5 vs. 7 kJ mol−1 respectively, Table S2†). Thus, the unfavourable HOMO localization and π-system orientation of carboxylate complexed aryl-ureas can account for the lack of activity of ortho-substituted analogues.
Interaction of the urea and carboxylate moieties to form anionic complexes was demonstrated with DFT calculations, however whether this interaction occurs via intra- or inter-molecular complexation was unclear. To clarify this, we sought crystal structures of 6 in it's protonated and deprotonated carboxylate from (6-D). 6-D failed to crystallise, however we obtained single crystals of 6 and 6-DMF solvate. In both crystal structures (Fig. 6a, b), 6 formed head-to-tail dimers stabilised by multiple hydrogen bonds between urea NH donors and carboxylic acid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acceptors. The dimerisation observed in the solid-state indicates possible intermolecular association of 6 within the low polarity environment of lipid bilayers.
O acceptors. The dimerisation observed in the solid-state indicates possible intermolecular association of 6 within the low polarity environment of lipid bilayers.
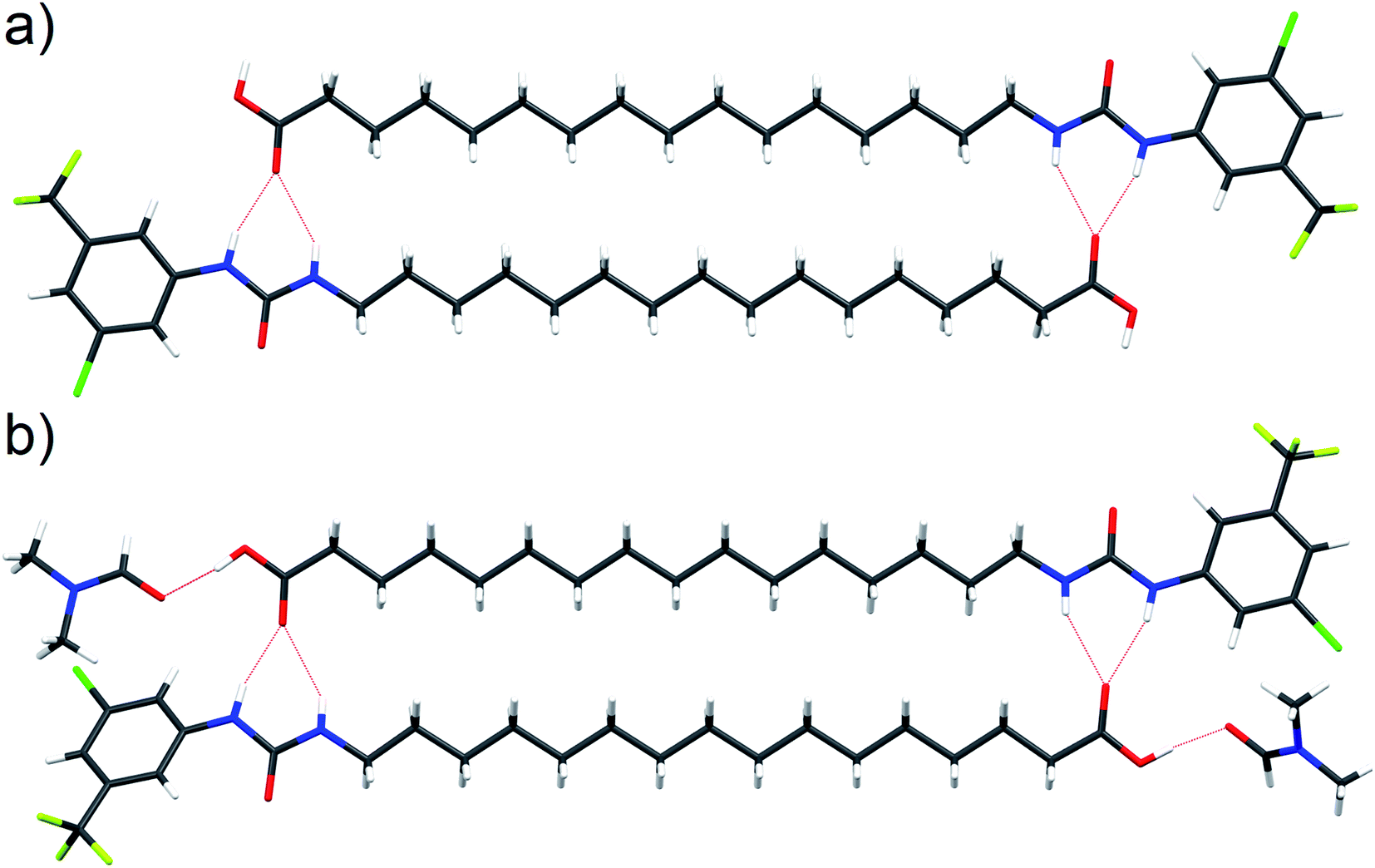 | ||
| Fig. 6 Single crystal structure of 6 ((a), CCDC 1998411†) and 6-DMF solvate ((b), CCDC 1998415†) with hydrogen bonds represented by red dashed lines. | ||
To further investigate the possible formation of membrane permeable dimers in a bilayer environment, we conducted molecular dynamics (MD) simulations of the active analogue 6 interacting with a DOPC bilayer in relevant protonation states (see Computational methodology). 6 in its deprotonated carboxylate form (6-D) and the neutral carboxylic acid form (6) readily integrated into a GROMOS-54A7 DOPC bilayer30,31 in microsecond timescale MD simulations. The carboxylate tail of 6-D was excluded from the bilayer interior and was restricted to the upper leaflet (Fig. S2a and b†), while the neutral 6 readily diffused between the two leaflets over the timescale of the simulation (Fig. S2c and d†). The exclusion of the carboxylate tail of 6-D from the bilayer interior is consistent with the protein-dependent uncoupling mechanisms of isolated fatty acids, where anion transport is transporter mediated.
Intra-molecular complexation of the urea and carboxylate moieties was not observed over the simulation timescale. However, similar to the aggregation of 6 observed in crystal structures (Fig. 6), both 6-D and 6 formed head-to-tail dimeric or multimeric structures in the DOPC bilayer environment (Fig. 7a and b). Dimers and multimers occurred both in simulations containing a single species (e.g. only 6-D or only 6) and in simulations containing both 6-D and 6 (see Computational methodology). Despite the tendency of 6-D to aggregate with 6 or other 6-D molecules in the bilayer, no inter-leaflet translocation of the resultant carboxylate-containing complexes was observed over the simulation timescale. However, it should be noted that there was no charge gradient across the bilayer in our MD simulations; in mitochondria anion translocation across the MIM is driven by the electrochemical gradient. The higher inter-leaflet permeability of 6, and the formation of head-to-tail 6-D−6 complexes suggests that co-transport of the carboxylate moieties with protonated aryl ureas is likely to be preferred over co-transport of two complexed carboxylates. In support of this, the translocation of anionic species complexed with their conjugate acids has previously been proposed for other uncouplers.16,32
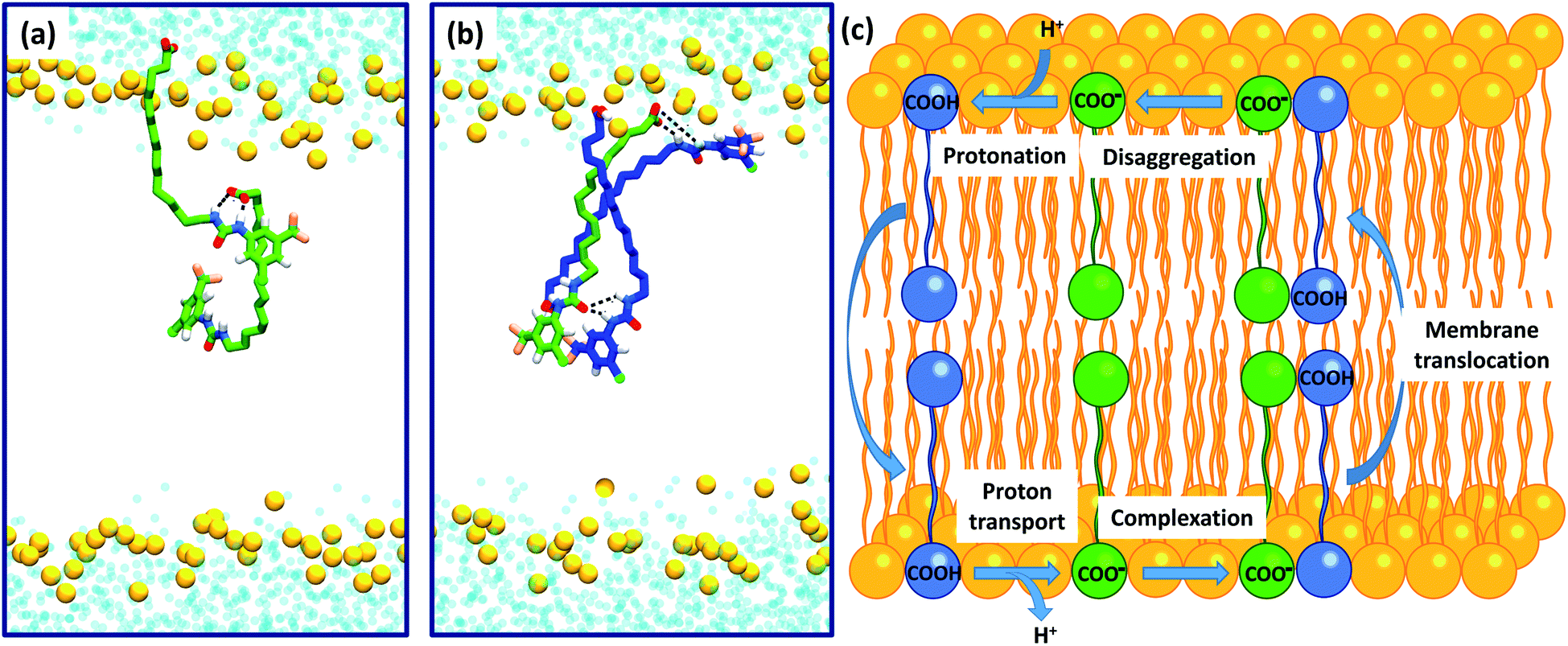 | ||
| Fig. 7 Self-assembly of aryl-ureas into membrane permeable dimeric complexes. Panels (a) and (b) Dimeric and multimeric assemblies of 6-D and 6 formed in a DOPC bilayer. 6-D and 6 are shown in green and blue respectively, while the phosphorous atoms of the phospholipid head groups are shown as orange spheres. Lipid acyl chains and headgroup structures are omitted for clarity and water is shown in light blue. Partial dimerization of 6-D is shown in (a), while formation of a head-to-tail 6-D-(6)2 trimer is shown in (b). (c) Proposed mechanism of uncoupling via dimer formation. | ||
To confirm the identity of the membrane permeable species we performed concentration-dependent 1H NMR studies of 6 in CDCl3 after deprotonation by tetrabutylammonium hydroxide (TBAOH). By increasing the concentration of 6-TBAOH from 50 μM to 5.0 mM, we observed dramatic downfield shifts in the aromatic CH peaks of 6-TBAOH (Fig. S3†), which is consistent with the inter-molecular complexation in CDCl3. The translational diffusion coefficients of 6-TBAOH at 50 μM and 5.0 mM determined by diffusional NMR do not show a significant difference within experimental errors (Table S3†), which rules out the formation of large aggregates. The concentration-dependent chemical shifts could be fitted to a monomer-dimer aggregation model (Fig. S4†), giving a dimerisation equilibrium constant of 5.9 × 103 M−1 for 6-TBAOH in CDCl3. To further support dimer formation we also measured the concentration dependant effects of 6 on bilayer conductance. Uncouplers that form membrane permeable dimers display a quadratic relationship between bilayer conductance and uncoupler concentration,32 and in tethered bilayers treated with 6 conductance was proportional to the square of the concentration of 6 (Fig. S5†). Together with the DFT calculations and MD, these studies show that aryl urea mediated uncoupling proceeds via self-assembly into lipophilic dimers as summarised in Fig. 7c.
Conclusions
In this paper we report the first example of a protonophoric mitochondrial uncoupler in which the acidic group and delocalised π system are separated by an alkyl chain. Uncoupling activity was established in cell-based assays and we developed a simple method using tethered lipid bilayers in conjunction with electrical impedance spectroscopy to show that the aryl-ureas are protein independent protonophores. The rate-limting step in the protonophoric cycle, diffusion of the deprotonated uncoupler across the MIM, proceeds via self-assembly of the aryl-ureas into dimeric complexes. Intermolecular hydrogen bonds between the carboxylate and anion binding urea group delocalizes the negative charge to promote membrane permeability, and overall complex lipophilicity is enhanced by electron withdrawing and lipophilic meta- and para-aryl substituents. The aryl-ureas studied herein represent a new type of mitochondrial uncoupler and their discovery widens the structural diversity of this potentially important class of therapeutic agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the Australian National Health and Medical Research Council (1031686 and 1087248). PAG thanks the Australian Research Council (DP200100453 and DP180100612) for funding. We acknowledge the NMR Facility of Mark Wainwright Analytical Centre at the University of New South Wales for NMR support. We would like to thank Tasya Salian and Natalie Mazurek for their assistance with the synthetic chemistry. We would also like to thank Lily Wang for her assistance with the computational study.References
- E. S. Childress, S. J. Alexopoulos, K. L. Hoehn and W. L. Santos, J. Med. Chem., 2018, 61, 4641–4655 CrossRef CAS PubMed.
- S. Spycher, P. Smejtek, T. I. Netzeva and B. I. Escher, Chem. Res. Toxicol., 2008, 21, 911–927 Search PubMed.
- R. T. Naven, R. Swiss, J. Klug-McLeod, Y. Will and N. Greene, Toxicol. Sci., 2013, 131, 271–278 CrossRef CAS PubMed.
- L. Wojtczak, M. R. Wieckowski and P. Schonfeldt, Arch. Biochem. Biophys., 1998, 357, 76–84 CrossRef CAS PubMed.
- M. Di Paola and M. Lorusso, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1330–1337 CrossRef CAS PubMed.
- P. A. Gale, E. N. W. Howe, X. Wu and M. J. Spooner, Coord. Chem. Rev., 2018, 375, 333–372 CrossRef CAS.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- B. A. McNally, W. M. Leevy and B. D. Smith, Supramol. Chem., 2007, 19, 29–37 CrossRef CAS PubMed.
- X.-H. Yu, X.-Q. Hong, Q.-C. Mao and W.-H. Chen, Eur. J. Med. Chem., 2019, 184, 111782 CrossRef CAS PubMed.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- H. Li, H. Valkenier, L. W. Judd, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurcek, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24–32 CrossRef CAS PubMed.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed.
- T. Rawling, H. Choucair, N. Koolaji, K. Bourget, S. E. Allison, Y.-J. Chen, C. R. Dunstan and M. Murray, J. Med. Chem., 2017, 60, 8661–8666 CrossRef CAS PubMed.
- Y. Al-Zubaidi, C. Pazderka, N. Koolaji, M. K. Rahman, H. Choucair, B. Umashankar, K. Bourget, Y. Chen, T. Rawling and M. Murray, Eur. J. Pharm. Sci., 2019, 129, 87–98 CrossRef CAS PubMed.
- C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS PubMed.
- A. A. Starkov, V. I. Dedukhova and V. P. Skulachev, FEBS Lett., 1994, 355, 305–308 CrossRef CAS PubMed.
- A. A. Starkov, D. A. Bloch, B. V. Chernyak, V. I. Dedukhova, S. E. Mansurova, I. I. Severina, R. A. Simonyan, T. V. Vygodina and V. P. Skulachev, Biochim. Biophys. Acta, Bioenerg., 1997, 1318, 159–172 CrossRef CAS.
- J. L. Figarola, J. Singhal, J. D. Tompkins, G. W. Rogers, C. Warden, D. Horne, A. D. Riggs, S. Awasthi and S. S. Singhal, J. Biol. Chem., 2015, 290, 30321–30341 CrossRef CAS PubMed.
- C. G. Cranfield, T. Berry, S. A. Holt, K. R. Hossain, A. P. Le Brun, S. Carne, H. Al Khamici, H. Coster, S. M. Valenzuela and B. Cornell, Langmuir, 2016, 32, 10725–10734 CrossRef CAS PubMed.
- S. E. Horvath and G. Daum, Prog. Lipid Res., 2013, 52, 590–614 CrossRef CAS PubMed.
- E. Deplazes, D. Poger, B. Cornell and C. G. Cranfield, Phys. Chem. Chem. Phys., 2018, 20, 357–366 RSC.
- V. Blazek Bregovic, N. Basaric and K. Mlinaric-Majerski, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef CAS.
- V. Amendola, M. Bonizzoni, D. Esteban-Gomez, L. Fabbrizzi, M. Licchelli, F. Sancenon and A. Taglietti, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- C.-C. Peng, M.-J. Zhang, X.-X. Sun, X.-J. Cai, Y. Chen and W.-H. Chen, Org. Biomol. Chem., 2016, 14, 8232–8236 RSC.
- X.-H. Yu, X.-Q. Hong and W.-H. Chen, Org. Biomol. Chem., 2019, 17, 1558–1571 RSC.
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Felix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC.
- X. Wu and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 16508–16514 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS PubMed.
- S. McLaughlin, J. Membr. Biol., 1972, 9, 361–372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1998415 and 1998411. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02777d |
| This journal is © The Royal Society of Chemistry 2020 |