
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excitonic Au4Ru2(PPh3)2(SC2H4Ph)8 cluster for light-driven dinitrogen fixation†
Yongnan
Sun‡
a,
Wei
Pei‡
b,
Mingcai
Xie
 a,
Shun
Xu
a,
Si
Zhou
*b,
Jijun
Zhao
b,
Kang
Xiao
c and
Yan
Zhu
*a
a,
Shun
Xu
a,
Si
Zhou
*b,
Jijun
Zhao
b,
Kang
Xiao
c and
Yan
Zhu
*a
aKey Lab of Mesoscopic Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhuyan@nju.edu.cn
bKey Laboratory of Materials Modification by Laser, Ion and Electron Beams, Dalian University of Technology, Dalian 116024, China. E-mail: sizhou@dlut.edu.cn
cSchool of Materials Science and Engineering, Nanjing University of Posts and Telecommunications, Nanjing 210023, China
First published on 27th January 2020
Abstract
The surface plasmon resonance of metal nanoparticles has been widely used to improve photochemical transformations by plasmon-induced charge transfer. However, it remains elusive for the molecular-like metal clusters with non-metallic or excitonic behavior to enable light harvesting including electron/hole pair production and separation. Here we report a paradigm for solar energy conversion on an atomically precise Au4Ru2 cluster supported on TiO2 with oxygen vacancies, in which the electron–hole pairs can be directly generated from the excited Au4Ru2 cluster and the TiO2 support, and the photogenerated electrons can transfer to the Ru atoms. Importantly, the Ru atoms in the Au4Ru2 cluster are capable of injecting the electrons into adsorbed N2 to activate N2 molecules. The cooperative effect in the supported Au4Ru2 catalyst efficiently boosts the photocatalytic activity for N2 fixation in comparison with homogold (Aun) clusters.
Introduction
Atomically precise metal clusters with exact formulas, molecular purity, and total structures have gathered momentum in recent years, owing to their unique physical and chemical properties.1–6 The metal clusters in the quantum size regime possess discrete electron energy levels and show non-metallic or excitonic behaviours,7 which are totally different from the larger metallic-state nanoparticles exhibiting a distinct surface plasmon resonance.8,9 Significant advances in chemical synthesis of the clusters provide an exciting opportunity to unveil previously unknown or inaccessible insights into the applications in optics, catalysis, and biochemistry.10,11 Especially the cluster-based heterogeneous catalysts have exhibited new catalytic properties in many chemical reactions compared to the plasmonic metal nanoparticles.12,13 Furthermore, the precise relation of the properties with atomic-level structures not only reveals the origin of metal catalysis, but also promotes the exploration of important chemical processes with these clusters as well-defined, highly efficient catalysts.14,15One important chemical process is the reduction of dinitrogen to ammonia, which is an essential chemical and energy carrier.16 However, high temperature and pressure are necessary to drive the reaction of N2 with H2 to NH3, due to the strong nonpolar triple bond in N2 and its high activation barrier.17 Many efforts have been made to develop less energy-consuming alternatives that can overcome the kinetic limitation of NH3 production.18–21 Inspired by nitrogenase enzymes that can fix nitrogen under ambient conditions, nanostructured metal catalysts are springing up to enable N2 fixation with the help of photosynthesis.22–25 Despite the important advances in homogeneous complex systems, construction of heterogeneous metal sites for N2 fixation is currently still challenging. Considering that atomically precise metal clusters can bridge the gap between homogeneous and heterogeneous catalysts, we speculate whether metal clusters with excitonic behaviour can convert N2 into ammonia under mild conditions, that is, whether these clusters are capable of generating hot electrons driven by solar light, ensuring the charge separation, and then donating electrons into N2. If this scenario is feasible, it can not only unravel the mystery of non-plasmon-induced solar energy conversion but also offer fundamental insights into exact, heterogeneous metal sites to govern the N2 transformation at the unprecedented level of atomic precision.
Since no example of N2 conversion on atomically precise metal clusters has been documented, a series of ligand-protected Aun (n = gold atom number) clusters with different atomic structures were first screened to catalyse the N2 conversion under light irradiation. As shown in Fig. 1, these Aun clusters failed to give a convincing activity for photocatalytic reaction of N2 fixation, mainly because the N2 molecule cannot coordinate to clusters and be activated on the gold sites of the Aun clusters, according to our density functional theory (DFT) calculations (inset of Fig. 1). We next turned our attention to the bimetal clusters. Since Ru is recognized as a suitable candidate for N2 fixation,24 we sought to explore the wet chemical synthesis of atomically precise Au–Ru clusters.
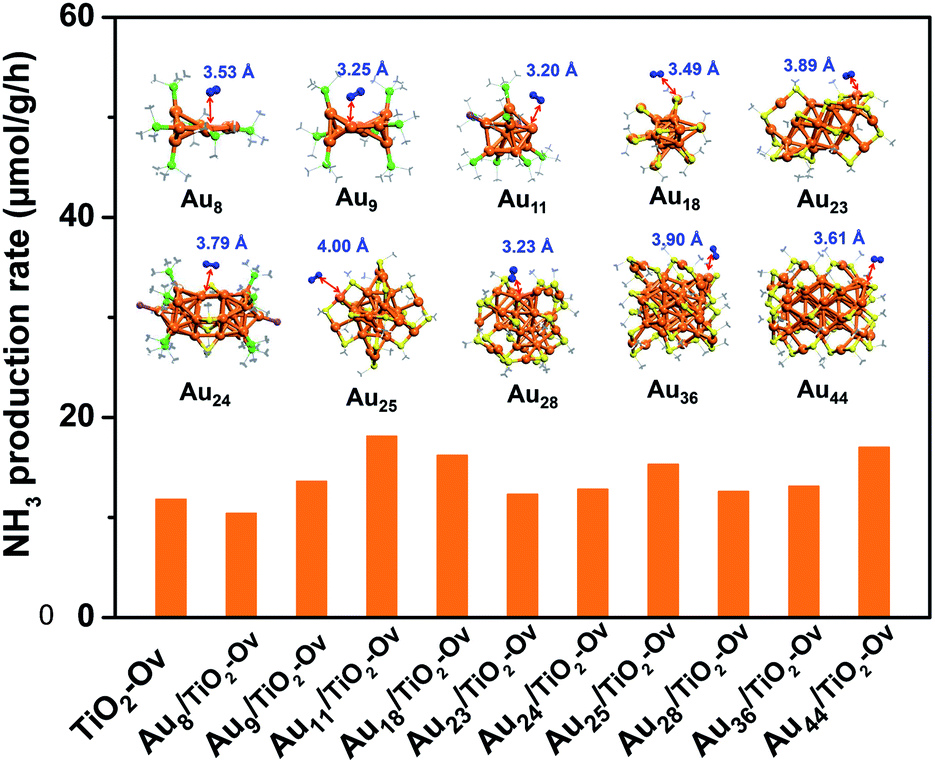 | ||
| Fig. 1 The photocatalytic activity of the ligand-protected Aun clusters supported on TiO2-Ov for N2 reduction. The inset shows the atomic structures of the Aun clusters, which can only physisorb N2 molecule with distance between N2 and Au sites larger than 3.2 Å. The C, N, S, Au, P and Cl are shown in gray, blue, yellow, orange, green and violet colors, respectively. H atoms are omitted for clarity. | ||
In this work, we successfully synthesized a new Au4Ru2 cluster protected by thiolate and triphenylphosphine ligands (namely, Au4Ru2(PPh3)2(SC2H4Ph)8) and solved its crystal structure. Excitingly, the Au4Ru2 cluster supported on TiO2 with oxygen vacancies (hereafter denoted as TiO2-Ov) exhibited a drastic increase in the photocatalytic activity for N2 reduction compared to the supported Aun catalysts. Furthermore, we explicitly demonstrated the cooperative mechanism within the Au4Ru2/TiO2-Ov catalyst for achieving light-driven N2 fixation.
Results and discussion
The crystal structure of the Au4Ru2(PPh3)2(SC2H4Ph)8 cluster is shown in Fig. 2A. This cluster resembles a distorted hexahedron, in which four Au atoms are located at the midpoints of four side edges, two Ru atoms reside on the centres of the top and the bottom planes, and eight S atoms are fixed at the vertexes. The two apex Ru atoms are coordinated by two PPh3 with the average Ru–P bond length of 2.204 Å. Four S atoms binding to a Ru atom are within the same plane as indicated by the average S–Ru–S angle of 90°. The average S–Au–S angle is 172.9°, where S–Au bond distances are 2.310 and 2.318 Å, respectively. The Au–Au distances fall in a very narrow range of 3.045–3.144 Å, which are shorter than the sum of van der Waals radii of two Au atoms (3.32 Å), suggesting the presence of d10–d10 metallophilic contact within the Au4Ru2 cluster.26 Electrospray ionization mass spectrometry (ESI-MS) further confirmed the formula of the cluster, where the m/z 2745 peak was assigned to [Au4Ru2(PPh3)2(SC2H4Ph)8 + Cs]+ adduct supported by the agreement between experimental and simulated isotopic patterns (Fig. 2B). Thermogravimetric analysis further confirmed that the Au4Ru2 cluster was highly pure (Fig. S1†).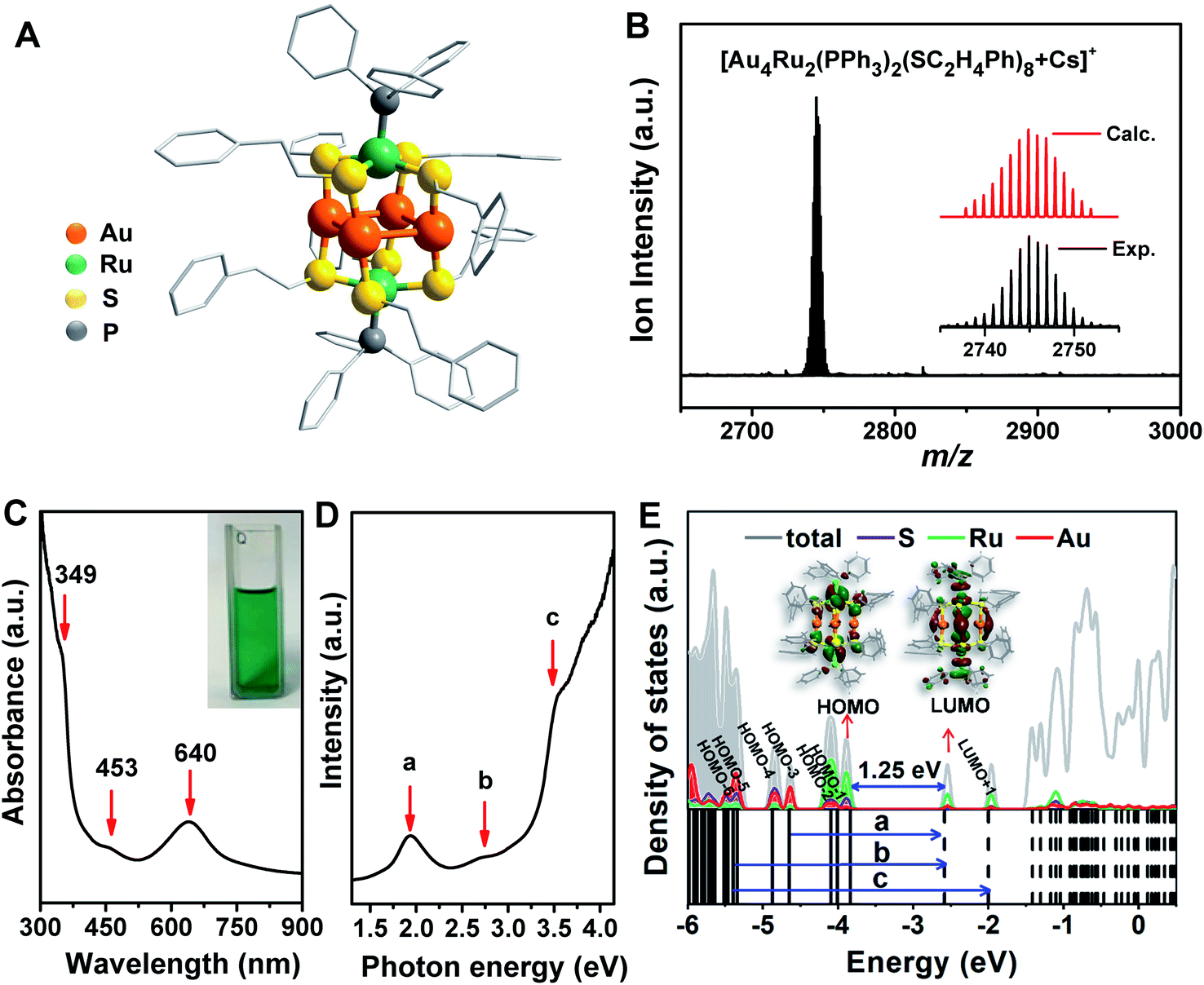 | ||
| Fig. 2 (A) Atomic structure (H atoms are omitted for clarity) and (B) ESI-MS profile of Au4Ru2(PPh3)2(SC2H4Ph)8. (C) UV-vis spectrum of Au4Ru2(PPh3)2(SC2H4Ph)8. (D) UV-vis spectrum plotted on the photon energy scale. (E) Kohn–Sham orbitals (bottom panel) and density of states (top panel) of Au4Ru2(PPh3)2(SC2H4Ph)8 from DFT calculations and the corresponding charge density distributions of HOMO and LUMO (insets). | ||
UV-vis absorption spectrum of the Au4Ru2 cluster shows two prominent peaks at 349 and 640 nm and one weak peak at 453 nm (Fig. 2C), corresponding to excitation energies of 3.55, 1.94 and 2.74 eV (Fig. 2D), respectively. Accordingly, the optical gap is determined to be 1.33 eV based on the photon-energy scale spectrum, which is basically consistent with the computed gap of 1.25 eV between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) from our DFT calculations (Fig. 2E). As revealed by the computed electronic density of states (DOS) in Fig. 2E, the emergence of discrete electronic states and a moderate HOMO–LUMO gap for the Au4Ru2 cluster indicates single electron excitations, that is an exciton.7 Importantly, the projected DOS shows that the low-lying unoccupied states (LUMO and LUMO+1) are mostly localized on the Ru atoms, suggesting that the excited carriers in the Au4Ru2 cluster will be on the Ru atoms, which may act as reaction centers and utilize the excess electrons for N2 activation.
With the newly synthesized cluster, we explored the proposed light-driven N2 fixation using Au4Ru2 as a heterogeneous catalyst. As shown in Fig. 3A, the Au4Ru2/TiO2-Ov catalyst gave rise to an ammonia production rate of 44.5 μmol g−1 h−1 under full spectrum illumination, which exhibited over 3-fold increase in photocatalytic activity compared to the Aun/TiO2-Ov and pure TiO2-Ov catalysts. As much, the Au4Ru2/TiO2-Ov catalyst resulted in a 4-time higher activity than TiO2-Ov in visible light-driven N2 reduction (Fig. 3A), suggesting a strong synergistic effect between Au4Ru2 and TiO2-Ov. Time-dependent photocatalytic ammonia production over the Au4Ru2 catalysts revealed that, not only the ammonia concentration increased linearly with the irradiation time in the visible light region (Fig. 3B), but also the Au4Ru2 loaded on the TiO2 support without abundant oxygen vacancies gave a much lower activity driven by either UV-vis or visible light (Fig. 3A and B). In fact, both TiO2-Ov and TiO2 substrates were in anatase phase (Fig. S2A†). The difference in the two TiO2 samples was that the former contained oxygen vacancies, but the latter not, which was confirmed by electron paramagnetic resonance (EPR). TiO2-Ov showed a characteristic EPR signal at approximately g = 1.998, suggesting the presence of oxygen vacancies,27 whereas no EPR signal was observed on the other TiO2 sample (Fig. S2B†). It can be conjectured that the abundant oxygen vacancies in TiO2 facilitate the photochemical reaction of N2 reduction.17
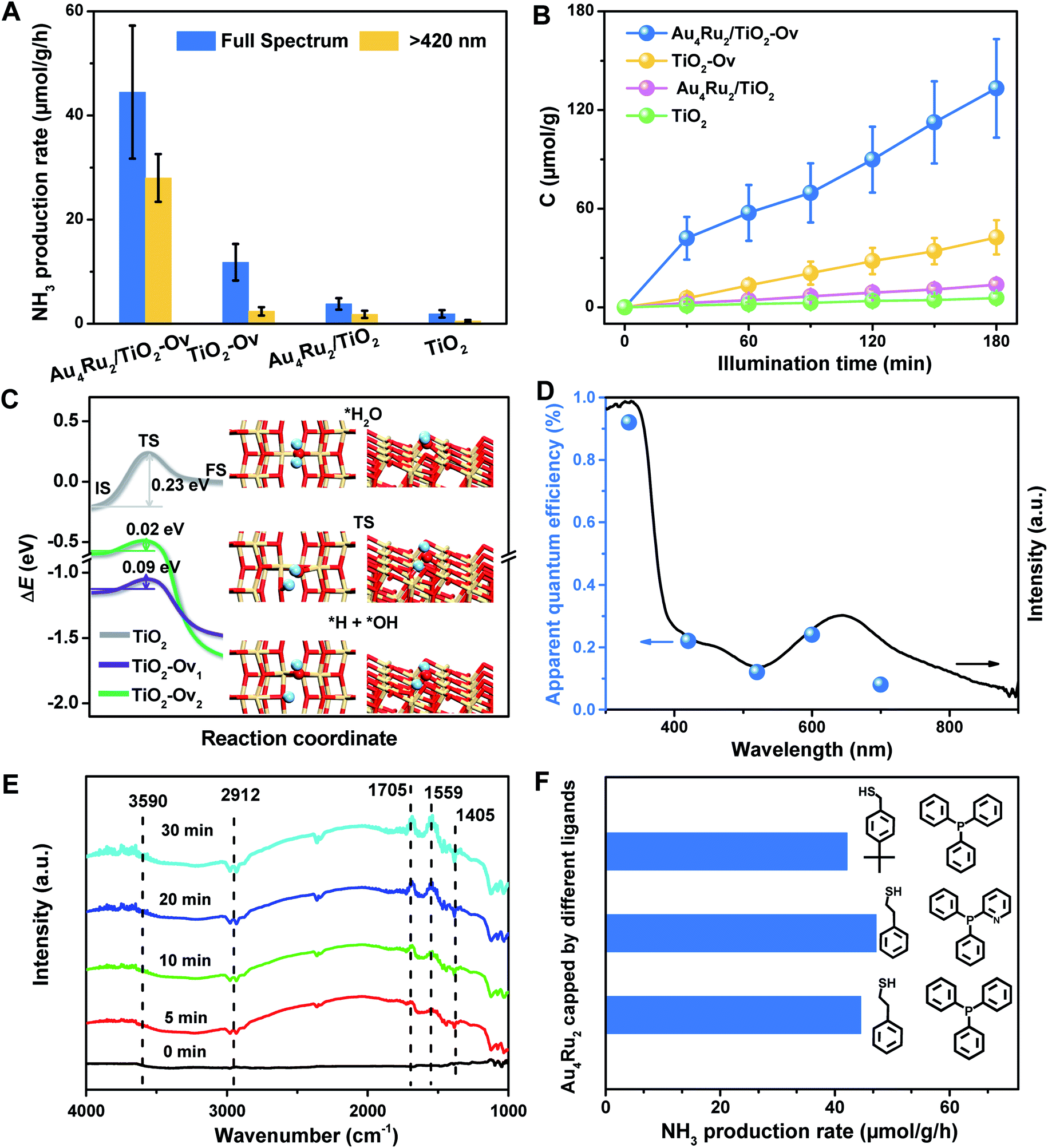 | ||
| Fig. 3 (A) The photocatalytic performances of the Au4Ru2/TiO2-Ov, Au4Ru2/TiO2, TiO2-Ov and TiO2 catalysts for N2 reduction. (B) Time-dependent photocatalytic ammonia production over the Au4Ru2/TiO2-Ov, Au4Ru2/TiO2, TiO2-Ov and TiO2 catalysts. (C) Reaction energy diagrams of water dissociation on the anatase TiO2(101) surface without defect (TiO2) and with an oxygen vacancy per supercell on surface (TiO2-Ov1) and subsurface (TiO2-Ov2), respectively. ΔE is relative to the energy of each substrate plus a free water molecule. Insets are the structures of water adsorption (top) and dissociation into *H and *OH species adsorbed on TiO2-Ov1 (bottom), and the transition state (middle). The H, O and Ti are shown in cyan, red and yellow. (D) UV-DRS and quantum efficiency of Au4Ru2/TiO2-Ov for NH4+ evolution by photocatalytic N2 reduction under monochromatic light of different wavelengths. (E) In situ FTIR spectra of the photocatalytic N2 reduction over Au4Ru2/TiO2-Ov during full spectrum illumination in the presence of N2 and water. (F) Performances of the Au4Ru2 clusters protected by different ligands for photocatalytic N2 reduction. | ||
DFT calculations demonstrated that TiO2 with oxygen vacancies can efficiently promote the photolysis of water to produce hydrogen as the proton source of ammonia (Fig. S3†). The anatase TiO2(101) surfaces with an oxygen vacancy on the surface (TiO2-Ov1) and subsurface (TiO2-Ov2), have low kinetic barriers (ΔEa) of 0.09 and 0.02 eV for water dissociation, respectively, compared to 0.23 eV for the perfect TiO2(101) surface (Fig. 3C). Moreover, they all provide moderate binding strength with H atoms (binding energy ΔEH = 0.14–0.43 eV relative to the energy of H2 molecule), which is beneficial for protons transfer from TiO2 to Au4Ru2. Photocatalytic N2 fixation on the Au4Ru2/TiO2-Ov catalyst in CH3CN solvent did not produce ammonia, again corroborating the origin of protons in ammonia from water splitting. For comparison, the Au4Ru2 cluster supported on SiO2 gave a low ammonia production rate of 2.4 μmol g−1 h−1, implying the key role of TiO2-Ov in water splitting.
Furthermore, the action spectrum for NH3 formation on the Au4Ru2/TiO2-Ov catalyst was determined under monochromatic light irradiation at wavelengths of 334, 420, 520, 600, and 700 nm. The trend of apparent quantum efficiencies (AQEs) well matched that of the optical absorption spectrum of the Au4Ru/TiO2-Ov (Fig. 3D). This proved that the photocatalytic N2 fixation originated from the light absorption by the Au4Ru2 cluster. In addition, the catalytic activity decreased slightly with multiple cycles (Fig. S4†), mainly due to the partial detachment of Au4Ru2 from TiO2-Ov (∼8 wt% metal loss detected by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis). The diffuse reflectance optical spectra (DRS) of the Au4Ru2/TiO2-Ov sample did not significantly change after the reaction (Fig. S5A and B†) and transmission electron microscopy (TEM) studies showed that the spent catalyst had no obvious aggregation (Fig. S5C and D†), suggesting that the Au4Ru2 cluster was robust throughout the reaction.
To directly visualize the N2 conversion on the Au4Ru2/TiO2-Ov catalyst, in situ infrared Fourier transform (DRIFT) spectroscopy was utilized to monitor the time-dependent change of the functional nitrogenous intermediates on the surface of Au4Ru2/TiO2-Ov. No signal change was observed in the DRIFT spectra within 30 min of incident light exposure in the absence of water (Fig. S6†), suggesting that the H atoms in ammonia indeed came from water. After water was introduced into the reaction cell, several absorption peaks appeared gradually with the irradiation time (Fig. 3E). The broad band at 3590 cm−1 is assigned to the ν(N–H) stretching vibration, and the two absorption bands at 1705 and 1559 cm−1 are attributed to the σ(N–H) bending vibration.25,28 Besides, the bands at 1405 and 2912 cm−1 assigned to the NH4+ deforming vibration became stronger gradually with the irradiation time.29,30 The result validated that the Au4Ru2/TiO2-Ov catalyst can convert N2 into ammonia under the light irradiation.
Considering that the Au4Ru2 cluster contained thiolate and PPh3 ligands, it is natural to ask whether the ligands can affect the catalytic conversion of N2. To address this, the comparison experiments were conducted, where a series of Au4Ru2 clusters protected by different ligands were prepared (Fig. S7†). As shown in Fig. 3F, the Au4Ru2 clusters with different ligands showed no drastic difference in the photocatalytic performance for N2 reduction, manifesting that the catalytic reaction was mainly determined by metal sites, rather than the ligands. Furthermore, when all the ligands in Au4Ru2 were removed via the thermal treatment, the Au4Ru2 clusters crashed into large nanoparticles and hence lost the activity for N2 fixation (Fig. S8†).
We now discuss the mechanism that Au4Ru2/TiO2-Ov can achieve an extraordinary activity for photocatalytic N2 reduction, but Aun/TiO2-Ov and TiO2-Ov cannot. Mott–Schottky (M–S) plots were first collected to provide the flat band potentials of Au4Ru2 and TiO2-Ov. The obtained tangent positive slopes indicated that both Au4Ru2 and TiO2-Ov were likely n-type semiconductors (Fig. 4A). The flat band potentials of Au4Ru2 and TiO2-Ov versus the saturated Ag/AgCl were −0.18 and −0.38 V, respectively. Based on the UV-DRS and M–S measurements, the band alignments of Au4Ru2 and TiO2-Ov were shown in Fig. 4B. From the thermodynamic point of view, photogenerated electron carriers can transfer from TiO2-Ov to Au4Ru2, while the Au4Ru2 cluster was more capable of light-driven N2 reduction than the TiO2-Ov substrate.
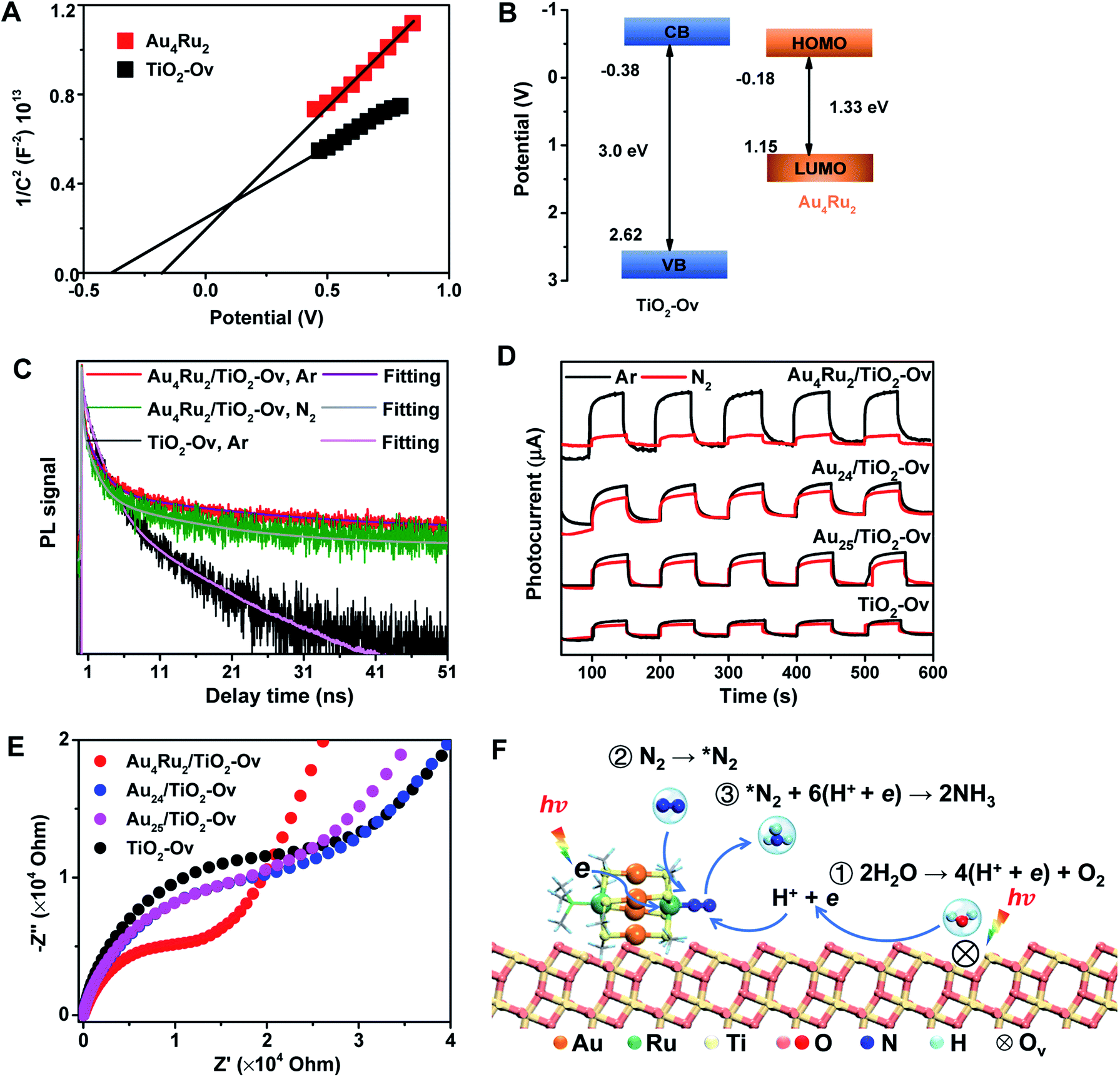 | ||
| Fig. 4 (A) Mott–Schottky plots of Au4Ru2 and TiO2-Ov (potential V vs. Ag/AgCl at pH 6.4). (B) Schematic diagram of band alignment (potential V vs. Ag/AgCl at pH 6.4). VB means valence band, and CB denotes conduction band. (C) Time-resolved PL spectra of the Au4Ru2/TiO2-Ov and TiO2-Ov under Ar and N2 atmospheres, respectively. (D) Photocurrent responses of Au4Ru2/TiO2-Ov, Au24/TiO2-Ov, Au25/TiO2-Ov and TiO2-Ov under Ar and N2 atmospheres, respectively. (E) Electrochemical impedance spectra of Au4Ru2/TiO2-Ov, Au24/TiO2-Ov, Au25/TiO2-Ov and TiO2-Ov in the presence of 300 W xenon lamp. (F) Proposed mechanism for the photocatalytic N2 reduction on the Au4Ru2/TiO2-Ov catalyst. | ||
The charge carrier kinetics of the Au4Ru2 cluster with N2, including separation, transfer and recombination, was investigated by room temperature steady-state and time-resolved photoluminescence (PL) spectroscopy. As shown in Fig. S9,† when the Ar atmosphere was changed to the N2 atmosphere, the steady-state PL spectrum of Au4Ru2/TiO2-Ov was significantly quenched, which was related to the non-radiative transfer of the photoexcited electrons from Au4Ru2 to the π* antibonding orbitals of N2 adsorbed on the cluster.30 The time-resolved PL spectroscopy studies (Fig. 4C) showed that the average decay time (τ = 14.40 ns) of Au4Ru2/TiO2-Ov was longer than that of TiO2-Ov (τ = 5.26 ns). The prolonged lifetime of the photogenerated electrons illustrated that Au4Ru2 supported on TiO2-Ov can reduce the charge recombination, thereby possess highly effective separation of electron–hole pairs.31
We argued that the high efficiency of N2 fixation on the Au4Ru2/TiO2-Ov catalyst was acquired by a synergic effect between Au4Ru2 and TiO2-Ov. To confirm this, transient photocurrent responses were conducted on the Au4Ru2/TiO2-Ov, TiO2-Ov, and two reference systems (Au24/TiO2-Ov and Au25/TiO2-Ov) under the Ar and N2 atmospheres with light (Fig. 4D), respectively. Compared to TiO2-Ov, the photocurrents of Au24/TiO2-Ov and Au25/TiO2-Ov were enhanced under the Ar atmosphere, suggesting that the Aun clusters also can serve as trapping sites for the photogenerated electrons.30 However, the transient photocurrent responses of TiO2-Ov, Au24/TiO2 and Au25/TiO2-Ov samples under N2 atmosphere were similar to those under Ar atmosphere, indicating that the interfacial electron transfer in the three samples was not interfered by surrounding N2. It partially accounted for the catalytic performance of the Aun/TiO2-Ov catalysts shown in Fig. 1 that the Aun/TiO2-Ov catalysts did not significantly enhance the photoactivity of N2 reduction in comparison with the TiO2-Ov. Notably, the photocurrent of the Au4Ru2/TiO2-Ov sample under the N2 atmosphere was only a quarter of that under the Ar atmosphere (Fig. 4D), in which the quenching of the other three-quarters of photocurrent was possibly due to the electrons consumed by the adsorbed N2 molecules.
Moreover, electrochemical impedance spectroscopy (EIS) was measured to investigate the interfacial charge-transfer properties of the above four samples under illumination. As shown in Fig. 4E, the semicircular diameters of Au24/TiO2-Ov and Au25/TiO2-Ov measured under light irradiation were slightly smaller than that of TiO2-Ov, indicating that the Aun clusters had an inherent ability of electron transport, but this ability was not extraordinary. Notably, the impedance of Au4Ru2/TiO2-Ov was the smallest among the four samples, providing a solid evidence that there existed a fast transfer of the interfacial charges between Au4Ru2 and TiO2-Ov.32 The charge transfer resistance on the Au4Ru2/TiO2-Ov sample without illumination was also investigated (Fig. S10†). It was found that the impedance of Au4Ru2/TiO2-Ov in the absence of light was much higher than that in the presence of light. Therefore, these observations supplied a clue that the Aun clusters were able to generate the electrons under the light irradiation, but lacked the ability to activate N2, and thus the Ru atoms in Au4Ru2 should be crucial for N2 binding and activation. To further elucidate the critical contribution of the Ru atoms in the Au4Ru2 cluster to the activation of the inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, the two Ru atoms of Au4Ru2 were replaced by the two Pd atoms (Fig. S11A†), that is, Au4Pd2. No increase in the photocatalytic reduction of N2 on the Au4Pd2/TiO2-Ov was observed when compared to the Aun/TiO2-Ov (Fig. S11B†). The result definitely confirmed that the Ru atoms in the Au4Ru2 cluster indeed can provide unique reaction sites for the NN cleavage by strong coordination.
N triple bond, the two Ru atoms of Au4Ru2 were replaced by the two Pd atoms (Fig. S11A†), that is, Au4Pd2. No increase in the photocatalytic reduction of N2 on the Au4Pd2/TiO2-Ov was observed when compared to the Aun/TiO2-Ov (Fig. S11B†). The result definitely confirmed that the Ru atoms in the Au4Ru2 cluster indeed can provide unique reaction sites for the NN cleavage by strong coordination.
To gain atomistic insight into the photochemical N2 reduction on the Au4Ru2/TiO2-Ov catalyst, we performed DFT calculations to determine the active sites and reaction pathways. Our calculations show that the Au atoms do not have any activity for N2 fixation, but N2 can be adsorbed onto the Ru atom in the side-on or end-on configuration, with adsorption energies of −0.07 eV and −1.33 eV and Ru–N bond length of 2.14 Å and 1.84 Å, respectively (Fig. 5A–D). The N–N bond is elongated to 1.14–1.16 Å compared to 1.13 Å for the gaseous N2 molecule, which manifests that N2 is activated on the Ru site of the Au4Ru2 cluster. Moreover, we examined the structure of Au4Ru2(SCH3)8(P(CH3)3)2 cluster supported on the anatase TiO2(101) surface (Fig. S12†), which exhibits a weak interfacial interaction with a distance of 2.57 Å between the cluster and substrate and a binding energy of only −0.28 eV per Au(Ru) atom. Therefore, the presence of substrate should not affect the adsorption properties of the Au4Ru2 cluster with N2. Hereafter, we considered N2-to-NH3 conversion on the Au4Ru2 cluster without support of the substrate.
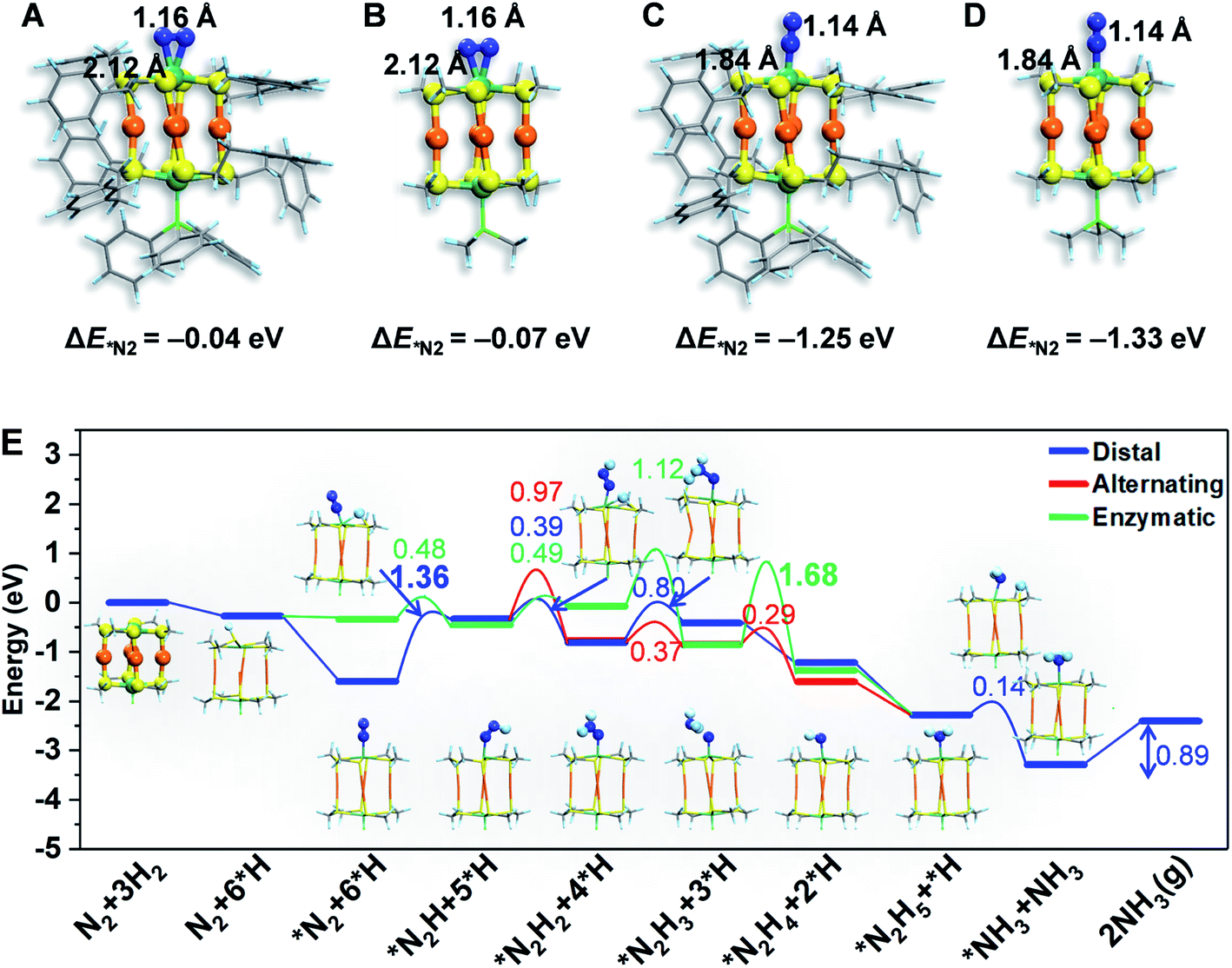 | ||
| Fig. 5 The atomic structures of N2 molecule adsorption via (A and B) end-on and (C and D) side-on patterns on the Au4Ru2(SC2H4Ph)8PPh3 and Au4Ru2(SCH3)8P(CH3)3. The H, C, N, S, Au and Ru are shown in light blue, gray, blue, yellow, orange and blackish green colors, respectively. The adsorption energy of a N2 molecule is shown for each system, revealing that simplifying the ligands of –PPh3 and –SC2H4Ph by –P(CH3)3 and –SCH3, respectively, has a minor effect on the binding properties of the cluster with N2. (E) The colors numbers indicate the kinetic barriers of N2 fixation on the Au4Ru2 cluster, and the barriers of rate-limiting step are bolded. The atomic structures of reaction intermediates are displayed as insets. The H, C, N, S, Au and Ru are shown in light blue, gray, blue, yellow, orange and green colors, respectively. | ||
Ammonia synthesis on the Au4Ru2 cluster can proceed through three pathways, i.e. distal, alternating and enzymatic mechanisms (Fig. 5E).24,33 For the former two paths, the N2 molecule strongly binds with the underlying Ru atom in the end-on configuration. Protonation of the chemisorbed *N2 species to form a *NNH intermediate is endothermic and has a kinetic barrier of 1.36 eV, which is the rate-limiting step of N2-to-NH3 conversion. The following reaction steps are exothermic involving barriers below 0.97 eV or even barrierless. For the enzymatic mechanism, the adsorption strength of *N2 species on Ru is relatively weak. The reaction proceeds almost down-hill in energy. Reaction from *N2 to form a *HN–NH species is favorable with a kinetic barrier of only 0.48 eV. Protonation of *H2N–NH leads to the breaking of N–N bond and generation of two *H2N species, which requires the largest barrier of 1.68 eV during the whole reaction. Finally, desorption of *NH3 has to overcome a moderate energy barrier of 0.89 eV. In brief, our DFT calculations suggest that the synergistic effect of the Au4Ru2/TiO2-Ov catalyst stems from the cooperation between cluster and substrate during the catalytic reaction (Fig. 4F): the Ru atom in the Au4Ru2 cluster serves as the active site for N2 fixation and ammonia synthesis through the distal or alternating pathways; the anatase TiO2(101) substrate plays important roles in water splitting to generate hydrogen protons that transfer to the cluster for the N2-to-NH3 reaction.
Conclusions
In conclusion, we have synthesized an excitonic Au4Ru2 cluster, which enables light harvesting including electron/hole pair production and separation. The experimental studies combined with theoretical calculations demonstrate that the cooperative effect between Au4Ru2 cluster and TiO2 substrate with oxygen vacancies leads to an extraordinary activity for light-driven N2 reduction. The electron–hole pairs can be generated from the excited Au4Ru2 cluster; the heterojunction between Au4Ru2 cluster and the TiO2-Ov substrate also facilitates photocarriers separation; the photoelectrons transfer to the Ru atoms of the cluster; meanwhile, TiO2-Ov induces water splitting to produce hydrogen protons for N2 fixation and conversion on the Ru atoms. Certainly, this work provides deep insights into non-plasmon-induced charge transfer from atomically precise metal clusters and develops a feasible strategy to enable highly efficient solar energy utilization via pursuing heterogeneous catalysts with atomic precision.Conflicts of interest
No conflicts of interest.Acknowledgements
We acknowledge financial supports from National Natural Science Foundation of China (21773109, 91845104).References
- M. Azubel, J. Koivisto, S. Malola, D. Bushnell, G. L. Hura, A. L. Koh, H. Tsunoyama, T. Tsukuda, M. Pettersson, H. Häkkinen and R. D. Kornberg, Science, 2014, 345, 909–912 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS PubMed.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- S. Yuan, C. Xu, J. Li and Q. Wang, Angew. Chem., Int. Ed., 2019, 58, 5967–5970 CrossRef.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- M. Zhou, C. Zeng, Y. Chen, S. Zhao, M. Y. Sfeir, M. Zhu and R. Jin, Nat. Commun., 2016, 7, 13240 CrossRef CAS PubMed.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- R. Huang, Y. Wei, X. Dong, X. Wu, C. Du, S. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS.
- S. Chen, H. Ma, J. W. Padelford, W. Qinchen, W. Yu, S. Wang, M. Zhu and G. Wang, J. Am. Chem. Soc., 2019, 141, 9603–9609 CrossRef CAS PubMed.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS.
- X. Cai, G. Saranya, K. Shen, M. Chen, R. Si, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2019, 58, 9964–9968 CrossRef CAS.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, ACS Catal., 2017, 7, 3713–3720 CrossRef CAS.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- Y. Gong, J. Wu, M. Kitano, J. Wang, T. Ye, J. Li, Y. Kobayashi, K. Kishida, H. Abe, Y. Niwa, H. Yang, T. Tada and H. Hosono, Nat. Catal., 2018, 1, 178–185 CrossRef CAS.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef.
- J. Yang, Y. Guo, R. Jiang, F. Qin, H. Zhang, W. Lu, J. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS.
- L. Xu, J. Wang, X. Zhu, X. Zeng and Z. Chen, Adv. Funct. Mater., 2015, 25, 3033–3042 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Mater., 2019, 1806482 CrossRef PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Z. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS.
- C. Li, T. Wang, Z. J. Zhao, W. Yang, J. F. Li, A. Li, Z. Yang, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 5278–5282 CrossRef CAS.
- J. Xia, J. Di, H. Li, H. Xu, H. Li and S. Guo, Appl. Catal., B, 2016, 181, 260–269 CrossRef CAS.
- J. Liu, X. Ma, Y. Li, Y. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1972938. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06424a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |