
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCsAlB3O6F: a beryllium-free deep-ultraviolet nonlinear optical material with enhanced thermal stability†
Hongkun
Liu
a,
Ying
Wang
 *a,
Bingbing
Zhang
a,
Zhihua
Yang
b and
Shilie
Pan
*b
*a,
Bingbing
Zhang
a,
Zhihua
Yang
b and
Shilie
Pan
*b
aKey Laboratory of Analytical Science and Technology of Hebei Province, College of Chemistry and Environmental Science, Hebei University, 180 East Wusi Road, Baoding 071002, China. E-mail: wangy@hbu.edu.cn
bCAS Key Laboratory of Functional Materials and Devices for Special Environments, Xinjiang Technical Institute of Physics & Chemistry, CAS, Xinjiang Key Laboratory of Electronic Information Materials and Devices, 40-1 South Beijing Road, Urumqi 830011, China. E-mail: slpan@ms.xjb.ac.cn
First published on 5th November 2019
Abstract
The design of new beryllium-free deep-ultraviolet nonlinear optical materials is important but challenging. Here, we describe a new strategy to search for such materials based on rational selection of fundamental structural units. By combining asymmetric AlO3F tetrahedra and π-conjugated B3O6 rings, a new aluminum borate fluoride, CsAlB3O6F was obtained. It exhibits excellent linear and nonlinear optical properties including a high optical transmittance with a cut-off edge shorter than 190 nm, large second harmonic generation intensities (2.0× KH2PO4, KDP), and suitable birefringence for phase-matching under 200 nm. It also has good thermal stability and can be synthesized easily in an open system.
The exploration of new deep-ultraviolet (DUV) nonlinear optical (NLO) materials is intriguing and of great importance because these materials are crucial for the development of all-solid-state DUV lasers.1–3 For NLO materials, the main obstacles to DUV application are 3-fold:4,5 (i) a wide DUV transparency window (wavelength cut-off edge < 200 nm), (ii) high NLO coefficients (>1× commercial KH2PO4, KDP), and (iii) sufficient birefringence to satisfy phase matching conditions in the DUV region. Until now, only KBe2BO3F2 (KBBF) can certainly break through these barriers and generate lasers with wavelengths shorter than 200 nm by direct second harmonic generation (SHG).6 However, KBBF is limited in practical use because of its adverse layered crystal growth habit and use of the highly toxic beryllium component. To find a KBBF replacement, many new NLO crystals have been developed continuously, but have so far been unable to achieve desired NLO properties.7–10
Recently, it was shown that fluorooxoborates and fluorophosphates with mixed O/F anionic groups might be recognized as new sources for discovering DUV NLO materials. For example, Pan's group proposed that the [BOxF4−x] (x = 1, 2, 3) tetrahedra are good units to balance the multiple criteria of DUV NLO materials.11 Accordingly, monofluorophosphates with non-π-conjugated asymmetric [PO3F] units were also paid attention, exhibiting superior optical properties.12,13 Thereafter, numerous fluorooxoborates and fluorophosphates with an unprecedented crystal structure and high performance as potential DUV NLO materials have been reported.14–17 Nevertheless, because these materials are relatively unstable at high temperature in air, one need to develop a suitable crystal growth method under sealed conditions and/or search for a suitable flux (solvent) system at relatively low temperature.14,16 Alternatively, it is possible to achieve a beryllium-free, higher stability DUV NLO crystal by combining other anionic groups, such as [AlO4], [PO4], [SiO4], and [ZnO4].18–21
Enlightened by the successful synthesis of NLO-active fluorooxoborates and fluorophosphates, we proposed that aluminum borate fluorides with [AlOmFn] (m + n = 4, 5, 6, AlOF for short) units might be another choice for exploring new NLO materials. Four specific aspects were considered: (I) aluminum borates are preferred because of the chemical and coordination environment similarity between Al3+ and Be2+ cations.22 (II) similar to fluorooxoborates, the substitution of O2− with larger electronegativity F− can increase the bandgap and optical anisotropy due to the ionicity of the Al–F bond. (III) The rich coordination environment of AlOF groups provides more structural possibilities than those of other mixed-anionic groups (e.g. [BeO3F], [BO3F], [BO2F2], see Fig. S1†). Different from the B or Be atom, the Al atom has empty d orbitals, and it can form sp3, sp3d, and sp3d2 hybrid orbitals when bonding with O/F atoms. Consequently, diverse AlOF groups without anion-site disorder, such as [AlO3F] tetrahedra, [AlO3F2] or [AlO4F] trigonal bipyramids, and [AlO5F], [AlO4F2], [AlO2F4], or [AlOF5] octahedra, have been achieved (Fig. S1†). Replacing oxygen atoms with fluorine in the Al–O polyhedra not only increases the degree of freedom (e.g. cis/trans conformation), but also causes a (local) symmetry breaking and results in increased microscopic susceptibility and optical anisotropy, which are beneficial to build a noncentrosymmetric (NCS) material. (IV) Aluminum borate fluorides without B–F bonds (e.g. BaAlBO3F2,23 Rb3Al3B3O10F,24 and K3Ba3Li2Al4B6O20F20) show good thermal stability, and large crystals could be obtained in air.
By applying the strategy described above, we tried to design a new DUV NLO material according to the blueprints shown in Fig. 1a. From KBBF to fluorooxoborates (e.g. NH4B4O6F,25 ABF), the nontoxic [BO3F] units were selected to replace the [BeO3F] tetrahedra of KBBF while the NLO properties were retained. Meanwhile, benzene-like [B3O6] rings (the same as that in β-BaB2O4, BBO) with a better conjugated π-orbital system were utilized to replace [BO3] triangles (the case in CsB4O6F,26 CBF), which could contribute to the increase of SHG responses compared to KBBF. In this work, more stable [AlO3F] units combined with the [B3O6] rings were chosen as fundamental building units (FBUs) and a new potential DUV NLO material CsAlB3O6F (CABF) has been successfully synthesized. The material can be obtained easily in an open system and it exhibits a short UV cutoff edge below 190 nm with a powder SHG response of 2.0× KDP under 1064 nm incident radiation. The first-principles calculations reveal that CABF possesses moderate SHG coefficients and sufficient birefringence for DUV phase-matching. With the introduction of new AlO3F units, CABF continues to maintain the excellent structural features of both KBBF and β-BBO, and thus exhibits a great potential to be a Be-free DUV material for nonlinear light–matter interactions.
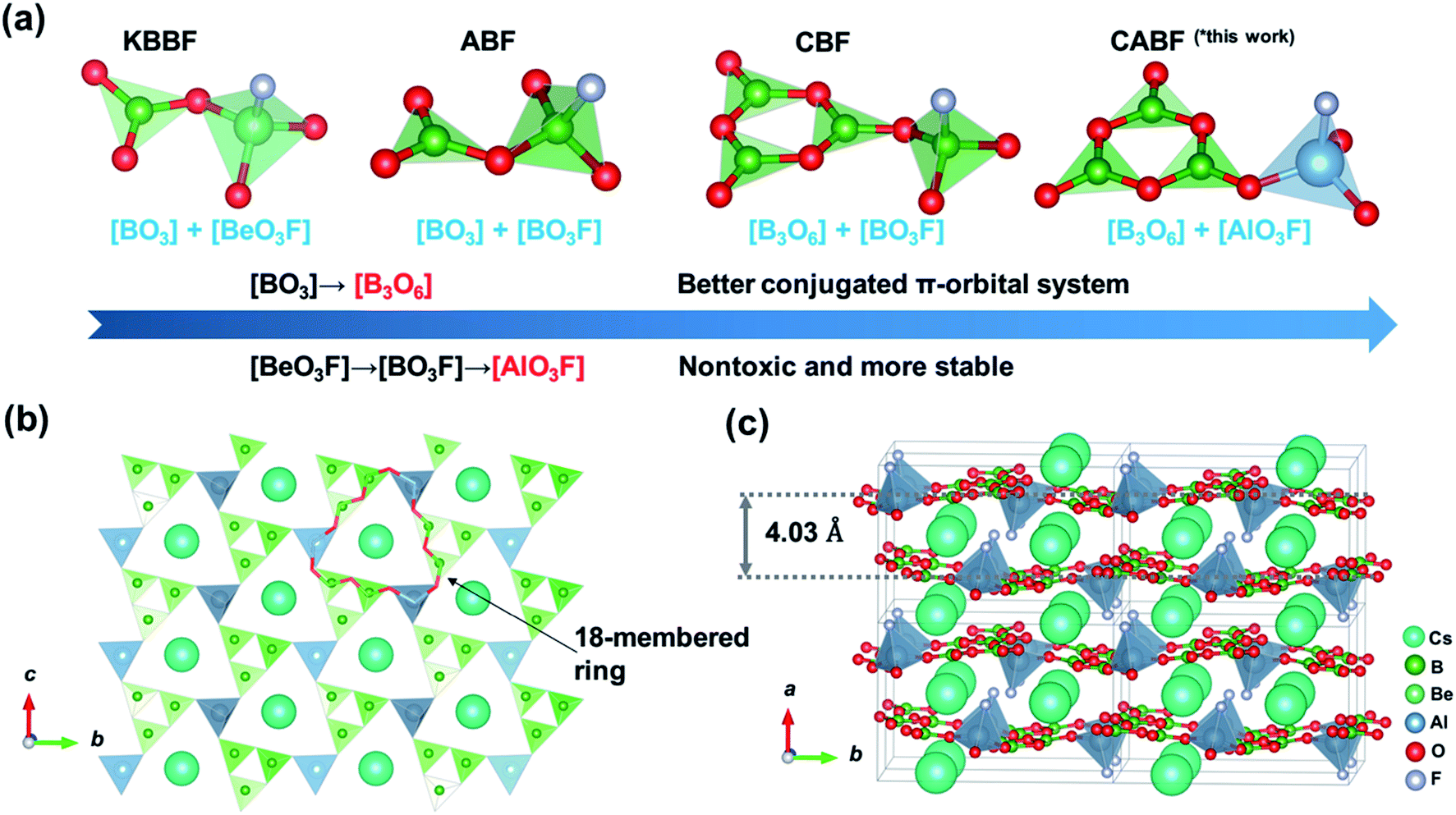 | ||
| Fig. 1 (a) The structural evolution from KBe2BO3F2 to CsAlB3O6F. (b) The 2D [AlB3O6F]∞ layer of CABF. (c) Crystal structure of CABF. | ||
Single crystals of CABF were obtained by the conventional flux method in an open system (see the Experimental details in the ESI†). CABF crystallizes in the NCS orthorhombic polar space group Pna21 (Tables S1–S3†). Its crystal structure is built up from [AlB3O6F]∞ 2D layers and the Cs+ cations reside in the interlayer space (Fig. 1b and c). The FBUs of CABF are the [AlB3O8F] groups composed of one [B3O6] ring and one [AlO3F] tetrahedron. Three crystallographically independent boron atoms are all three-coordinated to oxygen atoms forming the BO3 triangles, which further form a B3O6 ring by sharing their terminal O atoms. The B–O bond distances and the O–B–O angles are in the range of 1.342(10) to 1.395(11) Å and 117.2(7) to 121.9(8)°, respectively. The Al3+ cation is coordinated to three oxygen atoms (Al–O bond lengths: 1.719(6)–1.734(6) Å) and one fluorine atom (Al–F bond length equals 1.675(5) Å) to form a distorted AlO3F tetrahedron. In the bc-plane, the [AlO3F] tetrahedra bond with discrete [B3O6] rings to create [AlB3O6F]∞ layers that contain 18-membered rings (Fig. 1b). In one layer, the apical F atoms of [AlO3F] tetrahedra point upward and downward regularly. The Cs+ cations are twelve-coordinated, forming the [CsO11F] polyhedra with a Cs–F distance of 3.292(7) Å and Cs–O distances ranging from 3.256(5) to 3.585(6) Å (Fig. S2†). Because the Cs+ cations reside in the tunnels created by the 18-membered rings and adjacent [AlB3O6F]∞ layers, the interlayer distance in CABF is 4.03 Å. Compared to the distance of two adjacent layers in KBBF (6.25 Å), CABF should exhibit stronger interlayer interactions. Theoretical calculation based on the density functional theory (DFT) also confirms that CABF shows a larger interlayer binding energy than KBBF (0.178 vs. 0.0314 eV Å−2 per layer). These results indicate that CABF might possess a better crystal growth habit without layering. The bond valence sum calculation27 (Table S2†) and IR spectrum (Fig. S3†) indicate that all atoms of CABF have the expected oxidation states and coordination environments.
Polycrystalline CABF was synthesized by a stoichiometric solid-state reaction of CsF, Al2O3 and H3BO3 at 500 °C, and the phase purity was confirmed by powder X-ray diffraction (PXRD) Rietveld refinement (Fig. S4†). To check the stability of CABF, thermal measurements and further PXRD analysis were performed on its polycrystalline samples. Based on the re-crystallization experiment, the CABF sample after melting was almost the same as the original one; however, owing to the loss of volatile fluorines, a small amount of Cs2Al2(B3O6)2O28 was obtained (Fig. S6a†). Thermogravimetric (TG) analysis shows that CABF has nearly no weight loss until 900 °C, and the differential scanning calorimetry (DSC) data show two sharp endothermic peaks around 505 and 702 °C on the heating curve (Fig. 2a). In addition, variable temperature PXRD measurements were performed (Fig. S6b†), demonstrating a possible phase transition of CABF near 500 °C (consistent with the first peak on the DSC curve). Therefore, an excessive amount of fluorides should be used as the flux for crystal growth of CABF. Different from many fluorooxoborates (e.g. CaB5O7F3 and SrB5O7F3 (ref. 29–31)), CABF can survive at relatively high temperature in an open system after replacing BO3F with AlO3F units, which is an important advantage for large crystal growth.
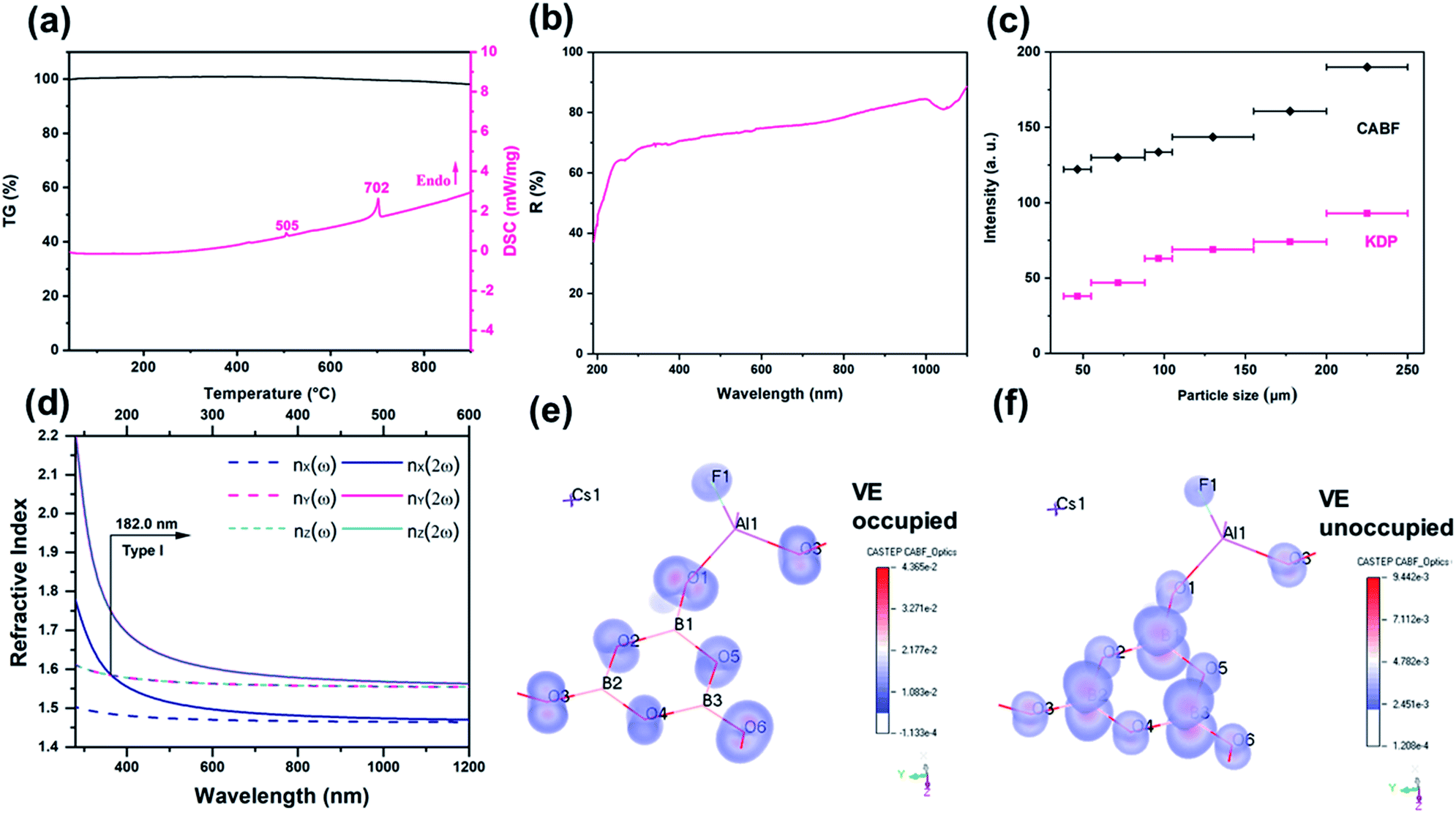 | ||
| Fig. 2 (a) Thermal behaviour of CABF. (b) The diffuse reflectance spectrum of CABF. (c) PSHG measurements at 1064 nm. (d) Calculated type I phase-matching condition of CABF. Dashed lines: refractive-indices of fundamental light. Solid lines: refractive-indices of second-harmonic light. The λSH of CABF is estimated by satisfying nZ(ω) = nX(2ω). The SHG-weighted electron density maps of the occupied (e) and unoccupied (f) states in the VE process. | ||
CABF exhibits a wide transparency window from 300 to 1100 nm, with a short UV cut-off edge below 190 nm (Fig. 2b). Even at 190 nm, the reflectance is nearly 40%. DFT calculations of the band gap using the HSE06 hybrid functional with high accuracy result in 7.49 eV (corresponding to 166 nm), further indicating that CABF could be an excellent candidate for optical materials operating in the DUV region. As described earlier,26,28 the large band gap of CABF could be attributed to the elimination of dangling bonds in [B3O6] units and the introduction of fluorine.
Powder SHG (PSHG) measurement using the Kurtz–Perry method32 reveals that CABF has SHG intensities of 2.0× KDP (particle size range: 200–250 μm) under 1064 nm fundamental wave laser radiation. It also suggests that CABF is phase matchable as the SHG intensities continue to rise until it attains the maximum (Fig. 2c). Under 532 nm fundamental wave laser radiation, CABF also shows phase matching behavior and has an SHG response about 1/6 times that of BBO in the particle size range of 200–250 μm (Fig. S7†). The SHG response of CABF is larger than that of KBBF (∼1.2× KDP) and suitable for DUV NLO applications. Applying Kleinmann symmetry in point group mm2, there are three nonzero and independent SHG coefficients, and our calculated SHG coefficients for CABF are d15 = −0.027 pm V−1, d24 = 0.941 pm V−1, and d33 = −0.955 pm V−1, which are consistent with the PSHG results. The SHG ability of CABF is comparable to those of other DUV NLO materials, including CBF (1.9× KDP),26 ABF (3× KDP),25 MB5O7F3 (M = Ca and Sr, 2.3–2.5× KDP),29–31 (NH4)2PO3F (1.0× KDP),12 and NaNH4PO3F·H2O (1.1× KDP).16 Compared with the CBF archetype, the increased SHG response of CABF can be understood by considering the geometry factor of NLO-active [B3O6] units. Structurally, CABF is pretty much like CBF; however, the substitution of [BO3F] by [AlO3F] results in structural modulation of the [B3O6] groups. As shown in Fig. S8,† the rotation angle (φ) and deviation angle (θ) of the B3O6 groups decrease from 33 and 6.8° in CBF, to 25 and 4.4° in CABF, respectively. The smaller φ and θ angles in CABF result in more “coplanar and aligned” [B3O6] units, that is, a favorable arrangement for generating the SHG response according to the anionic group approach.6
The phase-matching ability is a key factor that should never be ignored for any practical DUV NLO application, which relies on both birefringence (Δn) and its dispersion of the crystal.33 The calculated Δn of CABF is 0.091 at 1064 nm, large enough to satisfy DUV phase-matching (Fig. S10†). Based on the dispersion of the refractive indices, the shortest SHG phase-matching wavelength (λSH) of CABF can reach 182 nm (Fig. 2d), which is comparable to that of KBBF and other recent reported NLO materials (Table S4†). To the best of our knowledge, the phase-matching wavelength of CABF is the shortest among the SHG-active aluminum borates and aluminum borate fluorides (Table S4†).
To correlate the observed optical properties and electronic structure of CABF, band structure calculations were performed. The partial density of states (PDOS) projected on the constitutional atoms (see Fig. S9†) demonstrates that B-2p, Al-3p, O-2p, and F-2p states occupy the frontier orbitals (the top of the valence band and the bottom of the conduction band). The high overlap of these orbitals indicates that both the [B3O6] and [AlO3F] groups determine the SHG response and optical birefringence in CABF, while the Cs+ cations show a very small contribution. This observation was also supported by the SHG-weighted electron density analysis.34 The directly perceived images (Fig. 2e, f, and S11†) indicate that the 2p orbitals of O/F atoms in the occupied states and the anti π-orbitals of the [BO3] groups in the unoccupied states dominate the SHG contribution. In particular, the O or F atoms in [AlO3F] groups reveal considerable SHG density values, which further suggests that the [AlO3F] group could be a promising NLO-active unit.
Conclusions
In summary, the first B3O6–AlO3F based NLO crystal, CABF, has been obtained through a structural evolution approach. CABF exhibits a short UV cut-off edge (obv. < 190 nm; calc. 166 nm) and good SHG performance (2.0× KDP at 1064 nm). Our measurement and calculations of the refractive index show that the shortest SHG phase matching wavelength of CABF is down to ∼182 nm. Specifically, CABF is one of the very few materials (see Table S4†) that can break the so called “200 nm wall” of DUV and simultaneously grow crystals with fewer disadvantages (e.g. toxic raw materials, layer growth habit, requirement of a closed system). We thus believe that the design and successful synthesis of Be-free CABF expand the frontiers of DUV NLO materials.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21975062, 61835014, and 51425206) and the Advanced Talents Incubation Program of the Hebei University (Grant No. 521000981271).Notes and references
- N. Savage, Nat. Photonics, 2007, 1, 83 CrossRef CAS.
- D. F. Eaton, Science, 1991, 253, 281–287 CrossRef CAS PubMed.
- D. Cyranoski, Nature, 2009, 457, 953–955 CrossRef CAS.
- T. T. Tran, H. W. Yu, J. M. Rondinelli, K. R. Poeppelmeier and P. S. Halasyamani, Chem. Mater., 2016, 28, 5238–5258 CrossRef CAS.
- P. S. Halasyamani and J. M. Rondinelli, Nat. Commun., 2018, 9, 2972 CrossRef PubMed.
- C. T. Chen, G. L. Wang, X. Y. Wang and Z. Y. Xu, Appl. Phys. B: Lasers Opt., 2009, 97, 9–25 CrossRef CAS.
- G. H. Zou, C. S. Lin, H. Jo, G. Nam, T.-S. You and K. M. Ok, Angew. Chem., Int. Ed., 2016, 55, 12078–12082 CrossRef CAS PubMed.
- C. Wu, G. Yang, M. G. Humphrey and C. Zhang, Coord. Chem. Rev., 2018, 375, 459–488 CrossRef CAS.
- D. H. Lin, M. Luo, C. S. Lin, F. Xu and N. Ye, J. Am. Chem. Soc., 2019, 141, 3390–3394 CrossRef CAS.
- G. H. Zou, Z. E. Lin, H. M. Zeng, H. Jo, S.-J. Lim, T.-S. You and K. M. Ok, Chem. Sci., 2018, 9, 8957–8961 RSC.
- B. B. Zhang, G. Q. Shi, Z. H. Yang, F. F. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS.
- B. B. Zhang, G. P. Han, Y. Wang, X. L. Chen, Z. H. Yang and S. L. Pan, Chem. Mater., 2018, 30, 5397–5403 CrossRef CAS.
- L. Xiong, J. Chen, J. Lu, C. Y. Pan and L. M. Wu, Chem. Mater., 2018, 30, 7823–7830 CrossRef CAS.
- M. Mutailipu, M. Zhang, Z. H. Yang and S. L. Pan, Acc. Chem. Res., 2019, 52, 791–801 CrossRef CAS.
- G. P. Han, Y. Wang, B. B. Zhang and S. L. Pan, Chem.–Eur. J., 2018, 24, 17638–17650 CrossRef CAS.
- J. Lu, J. N. Yue, L. Xiong, W. K. Zhang, L. Chen and L. M. Wu, J. Am. Chem. Soc., 2019, 141, 8093–8097 CAS.
- M. Luo, F. Liang, Y. X. Song, D. Zhao, N. Ye and Z. S. Lin, J. Am. Chem. Soc., 2018, 140, 6814–6817 CrossRef CAS.
- H. P. Wu, H. W. Yu, S. L. Pan, Z. J. Huang, Z. H. Yang, X. Su and K. R. Poeppelmeier, Angew. Chem., Int. Ed., 2013, 52, 3406–3410 CrossRef CAS.
- H. W. Yu, W. G. Zhang, J. Young, J. M. Rondinelli and P. S. Halasyamani, Adv. Mater., 2015, 27, 7380–7385 CrossRef CAS.
- S. G. Zhao, L. Kang, Y. G. Shen, X. D. Wang, M. A. Asghar, Z. S. Lin, Y. Y. Xu, S. Y. Zeng, M. C. Hong and J. H. Luo, J. Am. Chem. Soc., 2016, 138, 2961–2964 CrossRef CAS.
- K. M. Ok, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef CAS.
- T. T. Tran, N. Z. Koocher, J. M. Rondinelli and P. S. Halasyamani, Angew. Chem., Int. Ed., 2017, 56, 2969–2973 CrossRef CAS.
- Z. G. Hu, Y. C. Yue, X. A. Chen, J. Y. Yao, J. N. Wang and Z. S. Lin, Solid State Sci., 2011, 13, 875–878 CrossRef CAS.
- S. G. Zhao, P. F. Gong, S. Y. Luo, S. J. Liu, L. Li, M. A. Asghar, T. Khan, M. C. Hong, Z. S. Lin and J. H. Luo, J. Am. Chem. Soc., 2015, 137, 2207–2210 CrossRef CAS PubMed.
- G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS.
- X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS.
- Z. Fang, X. X. Jiang, M. H. Duan, Z. Y. Hou, C. C. Tang, M. J. Xia, L. J. Liu, Z. S. Lin, F. D. Fan, L. Bai and C. T. Chen, Chem.–Eur. J., 2018, 24, 7856–7860 CrossRef CAS.
- M. Mutailipu, M. Zhang, B. B. Zhang, L. Y. Wang, Z. H. Yang, X. Zhou and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 6095–6099 CrossRef CAS.
- M. Luo, F. Liang, Y. X. Song, D. Zhao, F. Xu, N. Ye and Z. S. Lin, J. Am. Chem. Soc., 2018, 140, 3884–3887 CrossRef CAS.
- Z. Z. Zhang, Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Inorg. Chem., 2018, 57, 4820–4823 CrossRef CAS PubMed.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- W. G. Zhang, H. W. Yu, H. P. Wu and P. S. Halasyamani, Chem. Mater., 2017, 29, 2655–2668 CrossRef CAS.
- M. H. Lee, C. H. Yang and J. H. Jan, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235110 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1913159. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04862f |
| This journal is © The Royal Society of Chemistry 2020 |