
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed synthesis of α-ketoamides using water and dioxygen as the oxygen source†
Yuanyuan Xiaoa,
Zijuan Yib,
Xianyong Yub and
Fang Xiao *a
*a
aDepartment of Health Toxicology, Xiangya School of Public Health, Central South University, Changsha 410078, PR China. E-mail: fangxiao@csu.edu.cn
bSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
First published on 6th August 2020
Abstract
The reaction employing H2O and O2 as the co-oxygen source in the catalytic synthesis of α-ketoamides is described. This copper-catalyzed reaction is carried out in a tandem manner constituted by the hydroamination of alkyne, hydration of vinyl–Cu complex and subsequent oxidation. Isotope labeling and radical capture experiments reveal that the oxygen atom of α-ketone at α-ketoamides derives from O2 and the oxygen atom of amide group originates from H2O.
Introduction
The introduction of oxygen atoms into organic molecules to construct oxygenated compounds is one of the most fundamental subjects in organic chemistry. From a “green and sustainable chemistry” perspective, water and dioxygen are the most environmentally benign and cost-effective oxygen-containing reagents.1 Consequently, employing them as oxygen sources offers appealing access to oxygen-containing organic compounds.2,3 Herein, the example of direct utilization of H2O and O2 as the co-oxygen source to assemble α-ketoamides is reported. Isotope labeling and radical capture experiments demonstrate that the oxygen atom of α-ketone at α-ketoamide derives from dioxygen and the oxygen atom of amide group originates from water (eqn (1)).
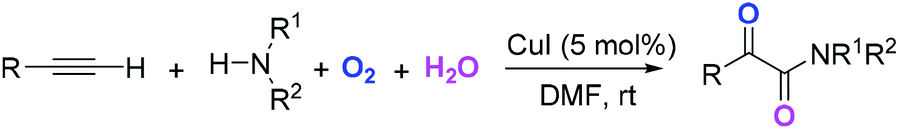 | (1) |
α-Ketoamides have attracted increasingly synthetic pursuit of chemists, as key structural motifs of many biologically active compounds and versatile building blocks.4 Various synthetic methods for the preparation of α-ketoamides have been developed over the past decades, such as amidation of α-ketoacids,5 oxidation of enamines,6 ynamines,7 arylacetamides8 and α-cyanoamides,9 Pd-catalyzed double carbonylative amination of aryl halides,10 and the oxidation of acyl cyanophosphoranes followed by amidation of the resulting α,β-diketone nitriles.11 Most of these well established approaches toward α-ketoamides often require toxic, expensive or preformed oxygen sources, such as SeO2, K2Cr2O7, CO, TBHP, and O3. Thus, the development of direct incorporation oxygen atoms from clean and cheap oxygen sources into organic frameworks to construct α-ketoamides is highly desirable.12 In 2010, Zhu reported the transformation of aldehydes with isocyanides to α-ketoamides using water as the oxygen source.13 Recently, various metal-catalyzed or metal-free methods for the oxidative synthesis of α-ketoamides from terminal alkynes,14 aryl acetaldehydes,15 ketones,16 1-arylethanols,17 phenethyl alcohol derivatives,18 and ethylarenes19 have also been developed by using dioxygen as oxygen source. In 2019, Wei and co-workers reported the transformation of α-ketoacids with isocyanides to α-ketoamides using water as the oxygen source.20
Multicomponent reaction has emerged as a powerful protocol to construct complex organic compounds.21 The present multicomponent reaction of copper-catalyzed direct oxidative transformation of alkynes and secondary amines to α-ketoamides is realized at room temperature without any ligand or additive, in which H2O and O2 were employed as the co-oxygen source (eqn (1)). Preliminary mechanistic studies suggest that this multicomponent reaction is performed in a tandem manner constituted by the hydroamination of alkyne, hydration of vinyl–Cu complex and subsequent oxidation with dioxygen. This methodology not only provides an interesting and attractive approach to α-ketoamides, but also allows an avenue to simultaneously introduce oxygen atoms from H2O and O2 into organic frameworks to access multi-oxygen containing compounds.
Results and discussion
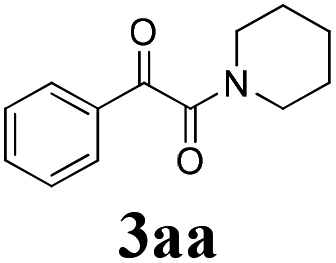
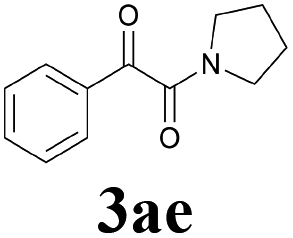
During the course of investigation on transition-metal-catalyzed oxidative transformation of alkynes and secondary amines to α-ketoamides, we found CuBr can catalyze the reaction of phenylacetylene 1a with piperidine 2a to give the product 3aa in presence of H2O (2 equiv.) under O2 without ligand or additive (Table 1, entry 1). Preliminary exploration showed that no 3aa was detected when the reaction was performed in the absence of O2 or H2O (Table 1, entries 2 and 3). These results suggested that H2O might be served as the oxygen source of 3aa and O2 as the oxidant or both of them were used as the co-oxygen source. This interesting phenomenon prompted us to optimize the reaction conditions and disclose the accurate origination of the oxygen atoms of α-ketoamides.
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | 3aab (%) |
| a Reaction conditions: 1a (2 mmol), 2a (0.5 mmol), catalyst (5 mol%), H2O (2 equiv.), O2 (balloon), solvent (0.5 mL), at room temperature, 12 h.b Isolated yields based on 2a.c Under N2.d Dry THF was used and 4 Å molecular sieve was added.e Under air. | |||
| 1 | CuBr | THF | 21 |
| 2 | CuBr | THF | 0c |
| 3 | CuBr | THF | 0d |
| 4 | CuBr2 | THF | 18 |
| 5 | CuCl2 | THF | 15 |
| 6 | CuCl | THF | 14 |
| 7 | Cu(OTf)2 | THF | 16 |
| 8 | (CH3CN)4CuPF6 | THF | 21 |
| 9 | CuI | THF | 43 |
| 10 | AgOTf | THF | 0 |
| 11 | RhCl3, | THF | 0 |
| 12 | InCl3, | THF | 0 |
| 13 | AlF3 | THF | 0 |
| 14 | AuBr3 | THF | Trace |
| 15 | — | THF | 0 |
| 16 | CuI | 1,4-Dioxane | 25 |
| 17 | CuI | MeOH | 0 |
| 18 | CuI | EtOH | 0 |
| 19 | CuI | DME | 45 |
| 20 | CuI | DCE | 32 |
| 21 | CuI | Toluene | 23 |
| 22 | CuI | DMSO | 34 |
| 23 | CuI | DMF | 71 |
| 24 | CuI | DMF | 46e |
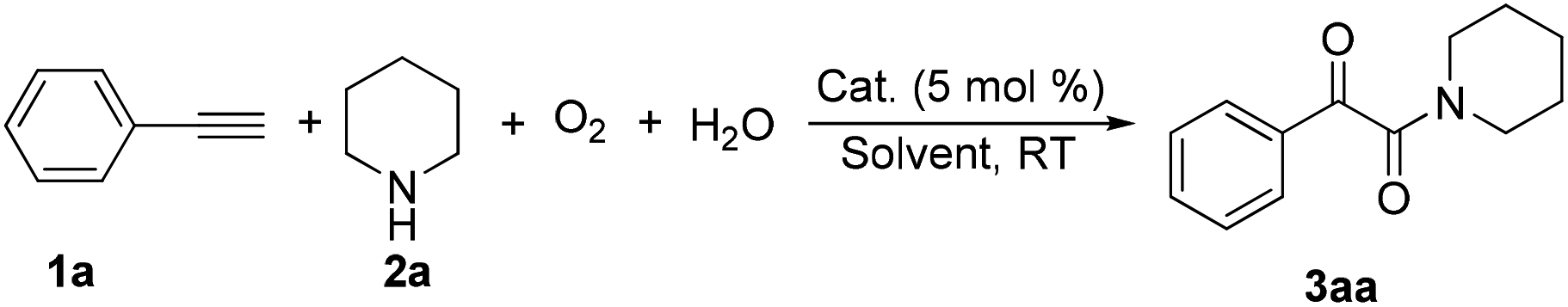
Initially, the reaction of phenylacetylene 1a with piperidine 2a was performed to examine the catalytic activity of various transition metal complexes including Au, Ag, Cu, Rh, Ni, Pd, Al, Bi, and In salts in the presence of H2O (2 equiv.) under the oxygen atmosphere. As shown in Table 1, among those metal catalysts examined (entries 4–14), CuI was found to be the best catalyst to catalyze the formation of α-ketoamide 3aa. No conversion was observed in the absence of catalyst (entry 15). The screening of solvents indicated that DMF was the optimal reaction medium (entries 16–23). This reaction could also proceed smoothly under the air atmosphere (entry 24).
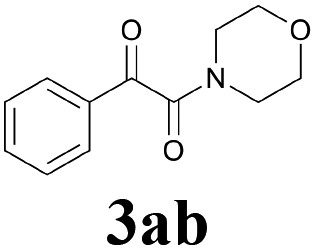
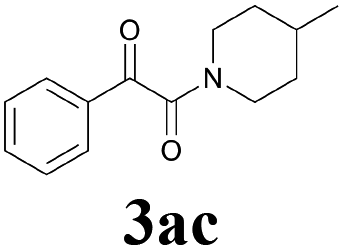
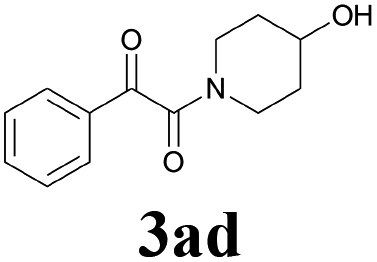
With the optimized conditions in hand, the scope of this new reaction was investigated (Table 2). Generally, the reaction tolerated electron-donating (para-, meta-, and ortho-substituted) and electron-withdrawing groups at the aromatic ring of alkynes (Table 2, entries 1–5). It was found that the reaction efficiency was affected by the steric effect. The more sterically demanding substrates such as ortho-methyl or chloro substituted arylalkynes demonstrated slightly lower activities (Table 2, entries 1–5). The present method can also apply to alkynes connected with naphthalene rings (Table 2, entries 6 and 7). The scope of amines was also examined, and both cyclic amines and linear amines were suitable substrates for this process. Cyclic amines such as piperidine, morpholine, 4-substituted piperidines, and pyrrolidine reacted with phenylacetylene or substituted phenylacetylenes to generate the corresponding products in moderate to good yields (Table 2, entries 1 and 8–17). The reaction of di-n-butylamine with 1a afforded α-ketoamide in moderate yield (Table 2, entry 18).
|
||||
|---|---|---|---|---|
| Entry | Alkyne (1) | Amine (2) | Product (3) | Yieldb (%) |
| a Reaction conditions: 1 (2 mmol), 2 (0.5 mmol), CuI (5 mol%), H2O (2 equiv.), O2 (balloon), DMF (0.5 mL), rt, 12–48 h.b Isolated yields. | ||||
| 1 |  |
 |
 |
71 |
| 2 | 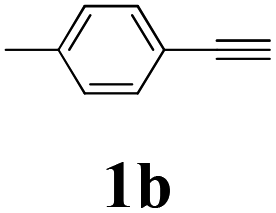 |
2a | 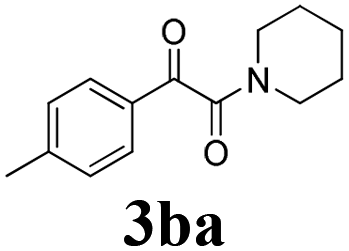 |
72 |
| 3 | 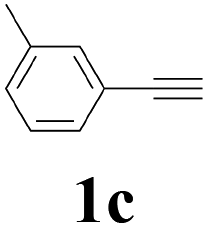 |
2a |  |
61 |
| 4 | 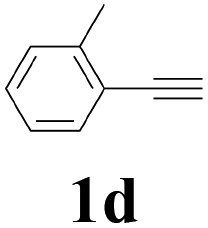 |
2a | 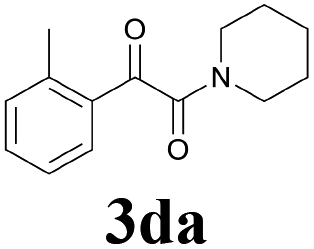 |
60 |
| 5 | 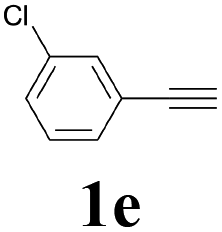 |
2a | 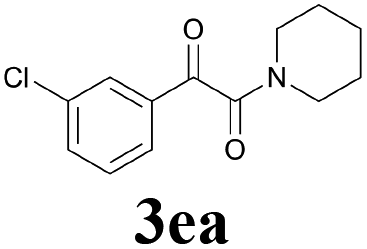 |
56 |
| 6 | 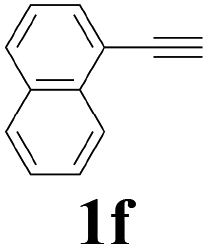 |
2a | 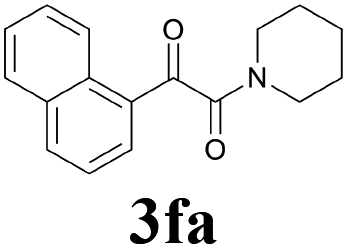 |
64 |
| 7 | 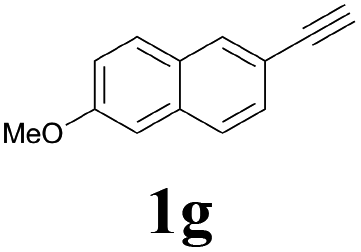 |
2a |  |
57 |
| 8 | 1a |  |
 |
65 |
| 9 | 1a | 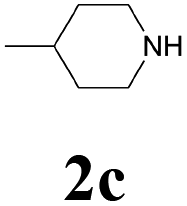 |
 |
56 |
| 10 | 1a | 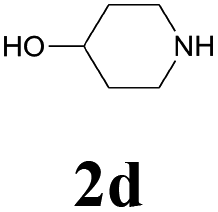 |
 |
64 |
| 11 | 1b | 2c | 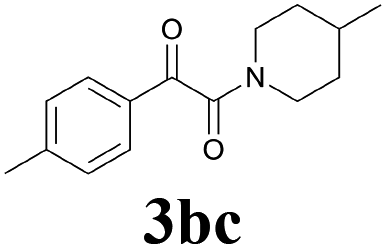 |
65 |
| 12 | 1b | 2d | 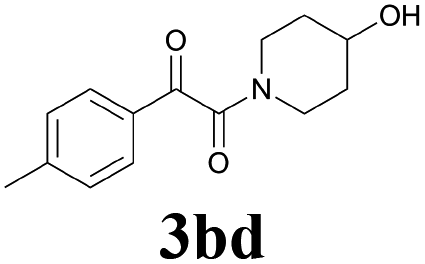 |
62 |
| 13 | 1c | 2c |  |
63 |
| 14 | 1c | 2d | 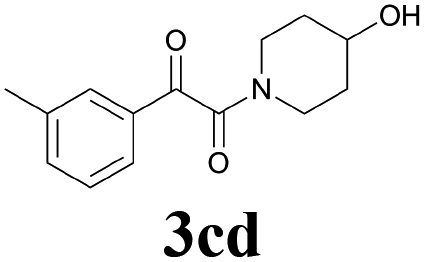 |
60 |
| 15 | 1d | 2c | 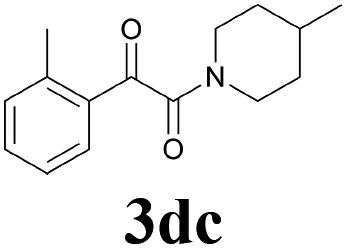 |
61 |
| 16 | 1d | 2d | 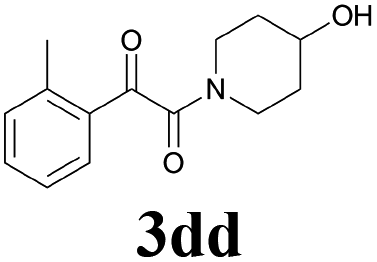 |
60 |
| 17 | 1a | 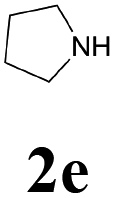 |
 |
52 |
| 18 | 1a | 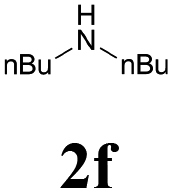 |
 |
63 |
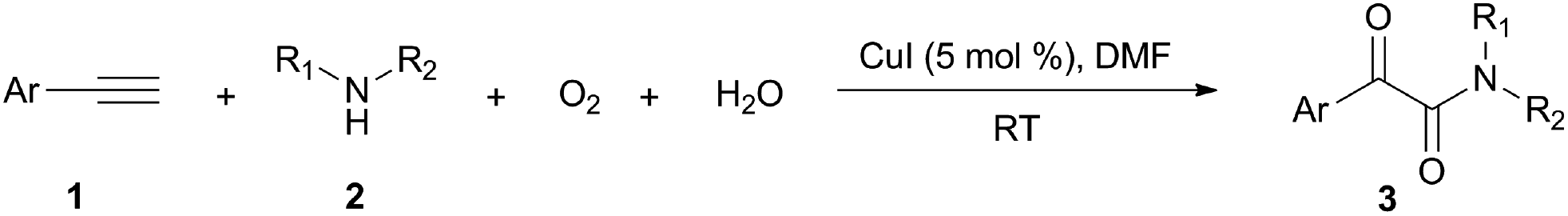
Isotope labeling and radical capture experiments were performed to elucidate the origination of the oxygen atoms of α-ketoamide. Results of these experiments demonstrate the oxygen atom of amide group originates from H2O and the oxygen atom of α-ketone at α-ketoamide derives from dioxygen.
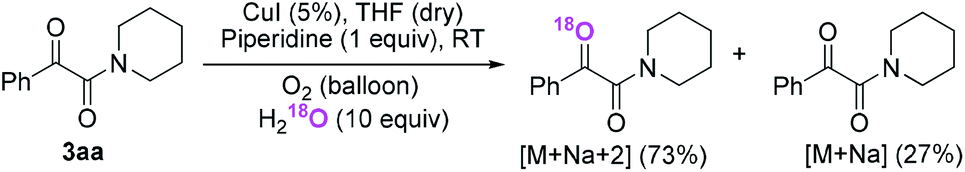
Firstly, the α-ketone group at α-ketoamide is more electrophilic than a ketone due to the electron-withdrawing effect of the amide group, thus it is possible for oxygen atom exchange with water via a hemiketal intermediate, while the amide group of α-ketoamide is stable. Indeed, as illustrated in eqn (2), when 3aa was stirred with CuI, piperidine, and H218O (10 equiv.) under oxygen atmosphere in THF,22 73% singly 18O-labeled, 27% unlabeled product were obtained and no doubly 18O-labeled α-ketoamide was detected (see HRMS in ESI†).
 | (2) |
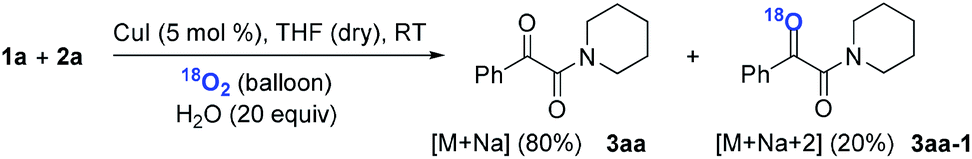
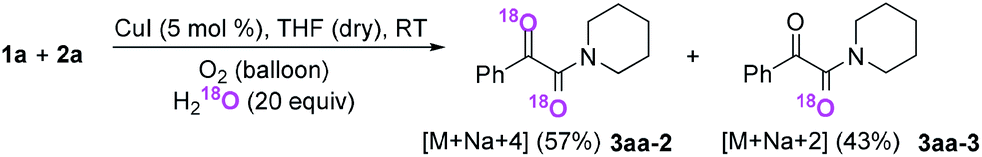
Subsequently, when the reaction of 1a and 2a was conducted in the presence of H2O (20 equiv.) under 18O2, 80% unlabeled (3aa, eqn (3)) and 20% singly 18O-labeled product (3aa-1, eqn (3)) were detected (see HRMS in ESI†). If the oxygen atom of amide group originates from 18O2, unlabeled product would not be observed via oxygen exchange with H2O. The existence of 80% unlabeled product (3aa, eqn (3)) demonstrated that the oxygen atom of amide group originated from H2O. Further control experiment showed that 57% doubly 18O-labeled (3aa-2, eqn (4)) and 43% singly 18O-labeled product (3aa-3, eqn (4)) were detected when the reaction of 1a and 2a was performed in the presence of H218O (20 equiv.) under O2 (see HRMS in ESI†). This result also revealed the oxygen atom of amide group derived from H2O (eqn (4)).
 | (3) |
 | (4) |
Radical capture experiments revealed that the oxygen atom of α-ketone at α-ketoamides derived from O2, which also indicated the possible mechanism of this transformation. As shown in eqn (5), TEMPO, a well known radical-capturing species, can remarkably suppress the formation of α-ketoamide 3aa.23 When TEMPO (30 mol%) was added to the reaction system of 1a and 2a, 69% TEMPO-trapped compound 3aa′ (isolated yield based on TEMPO) was obtained and only 4% of 3aa (isolated yield based on 2a) was detected. Furthermore, owing to the thermal instability of the covalent bond between TEMPO and the carbon free radical intermediate 3aa-4 that was easily oxidized by dioxygen to form carbonyl compounds,24,25 the transformation of 3aa′ to singly 18O-labeled α-ketoamide and TEMPO was observed in the presence of 18O2 at 70 °C (3aa-1, eqn (6)). Meanwhile, no conversion of 3aa′ to 3aa-1 was detected in the presence of H218O under N2 (eqn (7)). These results suggested that the carbonyl oxygen atom of α-ketone at α-ketoamide 3aa derived from molecular oxygen via a radical oxidation process.
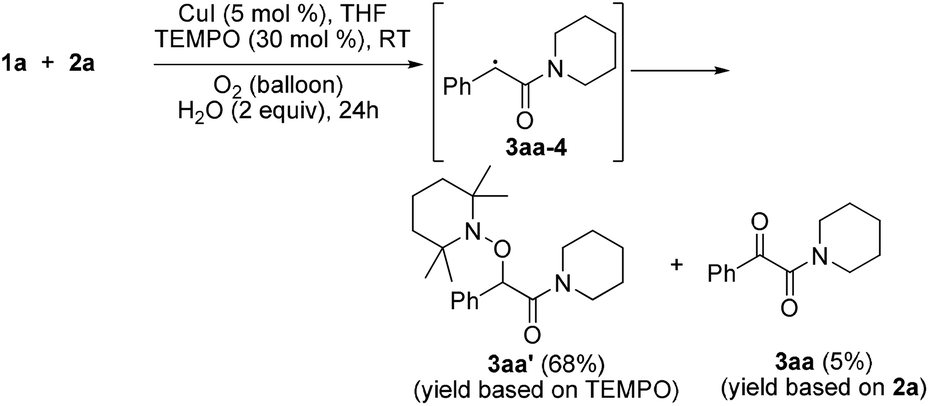 | (5) |

Based on the above experiments and previous reports,26–28 we proposed a postulated reaction pathway for this transformation as outlined in Scheme 1. Firstly, the complex 4 was formed by the reaction of the Cu species with alkyne 1 and amine 2. Then, the migration of amine to the triple bond led to the formation of the vinyl–Cu intermediate 5. Next, 5 underwent hydration to give 6. Subsequently, 7 was formed through the oxidation of 6 with dioxygen. Finally, the reductive elimination of the copper species of 727 followed by double oxidation with dioxygen would deliver the desired product 3.2,28
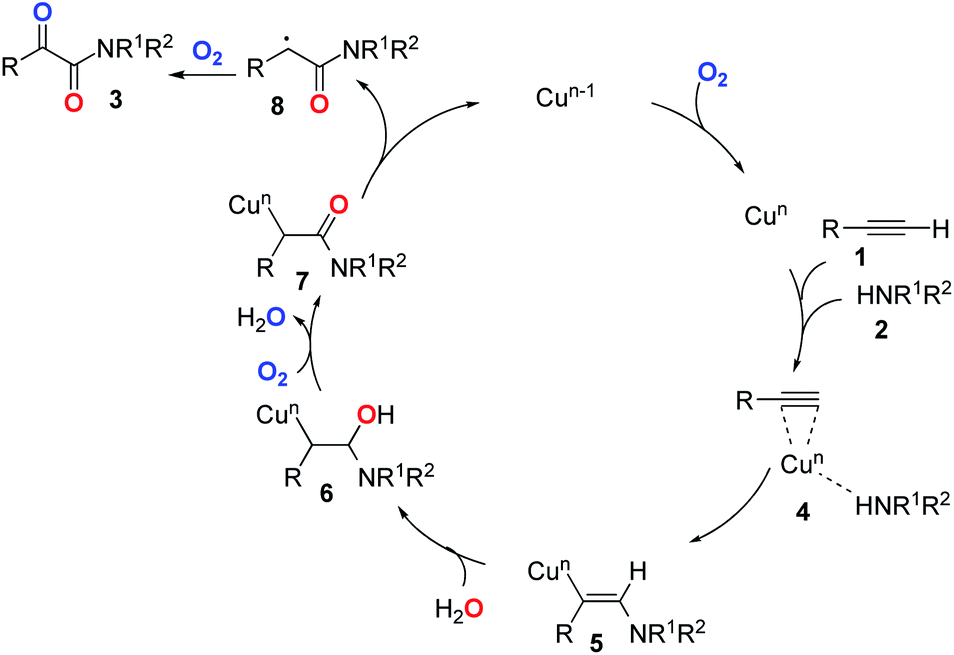 | ||
| Scheme 1 Plausible reaction pathway. | ||
Conclusions
In summary, we have successfully developed a tandem copper catalyzed approach to α-ketoamides from terminal alkynes, secondary amines, dioxygen, and water at room temperature without ligand or additive, in which O2 and H2O were used as the co-oxygen source of α-ketoamides. The present method opens a new window to construct complicated oxygen-containing compounds. Further studies of the detailed mechanism of this process and its application are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific Research Fund of Hunan Provincial Education Department (18A192).Notes and references
- (a) N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725–2750 CrossRef CAS; (b) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- (a) Q. Liu, L. Wang, H. Yue, J.-S. Li, Z. Luo and W. Wei, Green Chem., 2019, 21, 1609–1613 RSC; (b) Z. Cao, Q. Zhu, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2132–2138 CrossRef CAS; (c) W. Wei, P. Bao, H. Yue, S. Liu, L. Wang, Y. Li and D. Yang, Org. Lett., 2018, 20, 5291–5295 CrossRef CAS.
- (a) L.-Y. Xie, Y. Duan, L.-H. Lu, Y.-J. Li, S. Peng, C. Wu, K.-J. Liu, Z. Wang and W.-M. He, ACS Sustainable Chem. Eng., 2017, 5, 10407–10412 CrossRef CAS; (b) K.-J. Liu, T.-Y. Zeng, J.-L. Zeng, S.-F. Gong, J.-Y. He, Y.-W. Lin, J.-X. Tan, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 2304–2308 CrossRef CAS; (c) K.-J. Liu, J.-H. Deng, J. Yang, S.-F. Gong, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, Green Chem., 2020, 22, 433–438 RSC; (d) L. Wang, Y. Zhang, M. Zhang, P. Bao, X. Lv, H.-G. Liu, X. Zhao, J.-S. Li, Z. Luo and W. Wei, Tetrahedron Lett., 2019, 60, 1845–1848 CrossRef CAS; (e) K.-J. Liu, J.-H. Deng, T.-Y. Zeng, X.-J. Chen, Y. Huang, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2020, 31, 1868–1872 CrossRef CAS.
- (a) G. Kokotos, D. A. Six, V. Loukas, T. Smith, V. Constantinou-Kokotou, D. Hadjipavlou-Litina, S. Kotsovolou, A. Chiou, C. C. Beltzner and E. A. Dennis, J. Med. Chem., 2004, 47, 3615–3628 CrossRef CAS; (b) F. G. Njoroge, K. X. Chen, N.-Y. Shih and J. J. Piwinski, Acc. Chem. Res., 2008, 41, 50–59 CrossRef CAS; (c) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946–6948 CrossRef CAS; (d) F. R. Bou-Hamdan and J. L. Leighton, Angew. Chem., Int. Ed., 2009, 48, 2403–2406 CrossRef CAS; (e) A. Natarajan, K. Wang, V. Ramamurthy, J. R. Scheffer and B. Patrick, Org. Lett., 2002, 4, 1443–1446 CrossRef CAS; (f) Y.-X. Jia, D. Katayev and E. P. Kündig, Chem. Commun., 2010, 46, 130–132 RSC.
- (a) R. P. Singh and J. M. Shreeve, J. Org. Chem., 2003, 68, 6063–6065 CrossRef CAS; (b) A. Papanikos, J. Rademann and M. Meldal, J. Am. Chem. Soc., 2001, 123, 2176–2181 CrossRef CAS; (c) A. Chiou, T. Markidis, V. Constantinou-Kokotou, R. Verger and G. Kokotos, Org. Lett., 2000, 2, 347–350 CrossRef CAS.
- (a) Y.-H. Chen, Y.-H. Zhang, H.-J. Zhang, D.-Z. Liu, M. Gu, J.-Y. Li, F. Wu, X.-Z. Zhu, J. Li and F.-J. Nan, J. Med. Chem., 2006, 49, 1613–1623 CrossRef CAS; (b) M. Sassatelli, F. Bouchikhi, S. Messaoudi, F. Anizon, E. Debiton, C. Barthomeuf, M. Prudhomme and P. Moreau, Eur. J. Med. Chem., 2006, 41, 88–100 CrossRef CAS.
- (a) Z. F. Al-Rashid, W. L. Johnson, R. P. Hsung, Y. Wei, P.-Y. Yao, R. Liu and K. Zhao, J. Org. Chem., 2008, 73, 8780–8784 CrossRef CAS; (b) P. Müller and J. Godoy, Tetrahedron Lett., 1982, 23, 3661–3664 CrossRef.
- B. Song, S. Wang, C. Sun, H. Deng and B. Xu, Tetrahedron Lett., 2007, 48, 8982–8986 CrossRef CAS.
- J. Zhu, H. Wong, Z. Zhang, Z. Yin, J. F. Kadow, N. A. Meanwell and T. Wang, Tetrahedron Lett., 2005, 46, 3587–3589 CrossRef CAS.
- (a) F. Ozawa, H. Soyama, H. Yanagihara, I. Aoyama, H. Takino, K. Izawa, T. Yamamoto and A. Yamamoto, J. Am. Chem. Soc., 1985, 107, 3235–3245 CrossRef CAS; (b) E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS; (c) L. Huang, F. Ozawa and A. Yamamoto, Organometallics, 1990, 9, 2603–2611 CrossRef CAS.
- H. H. Wasserman and A. K. Petersen, J. Org. Chem., 1997, 62, 8972–8973 CrossRef CAS.
- D. Kumar, S. R. Vemula and G. R. Cook, ACS Catal., 2016, 6, 4920–4945 CrossRef CAS.
- M. Bouma, G. Masson and J. Zhu, J. Org. Chem., 2010, 75, 2748–2751 CrossRef CAS.
- (a) C. Zhang and N. Jiao, J. Am. Chem. Soc., 2010, 132, 28–29 CrossRef CAS; (b) A. Sagadevan, A. Ragupathi, C.-C. Lin, J. R. Hwu and K. C. Hwang, Green Chem., 2015, 17, 1113–1119 RSC; (c) M. Kumar, S. Devari, A. Kumar, S. Sultan, Q. N. Ahmed, M. Rizvi and B. A. Shah, Asian J. Org. Chem., 2015, 4, 438–441 CrossRef CAS; (d) K. Sun, G. Li, Y. Li, J. Yu, Q. Zhao, Z. Zhang and G. Zhang, Adv. Synth. Catal., 2020, 362, 1947–1954 CrossRef CAS.
- C. Zhang, Z. Xu, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11088–11092 CrossRef CAS.
- (a) F.-T. Du and J.-X. Ji, Chem. Sci., 2012, 3, 460–465 RSC; (b) J. Zhang, Y. Wei, S. Lin, F. Liang and P. Liu, Org. Biomol. Chem., 2012, 10, 9237–9242 RSC; (c) M. Zhou and Q. Song, Synthesis, 2014, 46, 1853–1858 CrossRef.
- S. Guo, Z. Fang, Z. Yang, C. Liu, Z. Dai, L. Zhao and K. Guo, RSC Adv., 2016, 6, 1503–1507 RSC.
- N. Sharma, S. S. Kotha, N. Lahiri and G. Sekar, Synthesis, 2015, 47, 726–736 CrossRef CAS.
- F. Liu, K. Zhang, Y. Liu, S. Chen, Y. Chen, D. Zhang, C. Lin and B. Wang, RSC Adv., 2017, 7, 7158–7162 RSC.
- Y. Lv, P. Bao, H. Yue, J.-S. Li and W. Wei, Green Chem., 2019, 21, 6051–6055 RSC.
- (a) H. G. O. Alvim, J. R. Correa, J. F. Assumpção, W. A. da Silva, M. O. Rodrigues, J. L. de Macedo, M. Fioramonte, F. C. Gozzo, C. C. Gatto and B. A. D. Neto, J. Org. Chem., 2018, 83, 4044–4053 CrossRef CAS; (b) D. Zhu, Y. Yao, R. Zhao, Y. Liu and L. Shi, Chem.–Eur. J., 2018, 24, 4805–4809 CrossRef CAS; (c) P. Kumari, R. Bharti and T. Parvin, Mol. Diversity, 2019, 23, 205–213 CrossRef CAS; (d) L. Zeng, B. Huang, Y. Shen and S. Cui, Org. Lett., 2018, 20, 3460–3464 CrossRef CAS; (e) G.-L. Wu and Q. P. Wu, Adv. Synth. Catal., 2018, 360, 1949–1953 CrossRef CAS; (f) K. Sun, Y. Li, R. Feng, S. Mu, X. Wang and B. Zhang, J. Org. Chem., 2020, 85, 1001–1008 CrossRef CAS.
- Water in DMF is very difficult to remove, thus we use THF instead of DMF.
- (a) A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983–4992 CrossRef CAS; (b) C. Aliaga, J. M. Juárez-Ruiz, J. C. Scaiano and A. Aspée, Org. Lett., 2008, 10, 2147–2150 CrossRef CAS; (c) M. R. Heinrich, A. Wetzel and M. Kirschstein, Org. Lett., 2007, 9, 3833–3835 CrossRef CAS; (d) A. D. Allen, B. Cheng, M. H. Fenwick, B. Givehchi, H. Henry-Riyad, V. A. Nikolaev, E. A. Shikhova, D. Tahmassebi, T. T. Tidwell and S. Wang, J. Org. Chem., 2001, 66, 2611–2617 CrossRef CAS.
- M. V. Ciriano, H.-G. Korth, W. B. van Scheppingen and P. Mulder, J. Am. Chem. Soc., 1999, 121, 6375–6381 CrossRef CAS.
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS.
- G. Huerta and L. Fomina, J. Mol. Struct.: THEOCHEM, 2006, 761, 107–112 CrossRef CAS.
- S. Murata, K. Suzuki, M. Miura and M. Nomura, J. Chem. Soc., Perkin Trans. 1, 1990, 361–365 RSC.
- Y. Usuki, X. Peng, B. Gülgeze, S. Manyem and J. Aubé, ARKIVOC, 2006,(iv), 189–199 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05921h |
| This journal is © The Royal Society of Chemistry 2020 |