
Recent progress in the synthesis of limonoids and limonoid-like natural products
Shaomin
Fu
and
Bo
Liu
 *
*
Key Laboratory of Green Chemistry &Technology of the Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: chembliu@scu.edu.cn; Fax: +8628 85413712; Tel: +86 28 85413712
First published on 19th May 2020
Abstract
Limonoid and limonoid-like natural products have demonstrated diverse bioactive properties and attracted considerable synthetic efforts. They provide a good platform to examine the viability of methodologies in the efficient construction of complex skeletons. In this review, we have discussed the recent progress in the syntheses of limonoids and limonoid-like natural products, covering from January 2011 to March 2020. The key transformations are highlighted. Finally, we summarize current “state-of-art” advance on novel synthetic strategy in syntheses of linonoids and limonoid-like molecules. Besides, future outlook are also presented.
 Shaomin Fu | Shaomin Fu grew up in Fujian, China and received his B.S. from South China Agriculture University in 2009. He completed his graduate study at South China University of Technology, where he received his Ph.D. under the supervision of Prof. Wei Zeng. In 2015, he began work as a postdoctoral researcher in Tsinghua University in the laboratory of Prof. Qiang Liu. In May 2018, he became an Associate Professor in the College of Chemistry of Sichuan University. His research interests focus on synthetic methodology and its application in total synthesis. He has published over 20 research papers, including two highly cited papers. |
 Bo Liu | Bo Liu was born in Chongqing, China and received his BSc from Southwest Normal University (now merged into Southwest University) in 1998. He then obtained his MSc at Chengdu Institute of Organic Chemistry of the Chinese Academy of Sciences in 2001 under the supervision of Prof. Xiaoming Feng. Then, he earned his Ph.D. in 2004 at the Shanghai Institute of Organic Chemistry (SIOC) under the supervision of Prof. Wei-Shan Zhou. In 2004, he moved to UT Southwestern Medical Center, Dallas as a postdoctoral fellow in the laboratory of Prof. Jef K. De Brabander. In 2007, Prof. Liu joined the faculty at Sichuan University. His research interests focus on the total synthesis of bioactive natural products and NPS-oriented methodology. He has published over 60 research papers. |
1 Introduction
Limonoids are widely distributed in nature and have diverse biological activities.1 To date, various limonoids have been identified in nature. Structurally, the typical limonoid skeleton is characterized as 4,4,8-trimethyl-17-furanylsteroid. Besides, intact limonoids are defined as the unaltered bond of the parent nucleus, a pentacyclic carbon skeleton. Degraded limonoids commonly result from the oxidative cleavage of the parent nucleus, featured by fused bicyclic γ- or δ-lactones.2 Seco-limonoids are the oxidative fragmentation of one or more androstane cores. Finally, highly decorated limonoids are generally acknowledged as a high degree of ring fission and rearrangement of the parent nucleus. Considerable endeavours have been devoted toward the syntheses of limonoids.2,3 Consequently, a 22-year synthetic campaign from Ley's group led to the historical synthesis of Azadirachtin,4 with the discovery of new chemistry, thus encouraging more challenging syntheses from researchers. To maintain the coherence and focus of this review, degraded limonoids and synthetic studies toward limonoids are not highlighted herein.3 Besides, the successful syntheses of limonoid-like natural products, especially cardenolides and nortriterpenoids, will be included in this review.5 This review describes the syntheses of limonoids and limonoid-like natural products covering from January 2011 to March 2020 to avoid duplicating the beautiful review released by Heasley in 2011 (Fig. 1).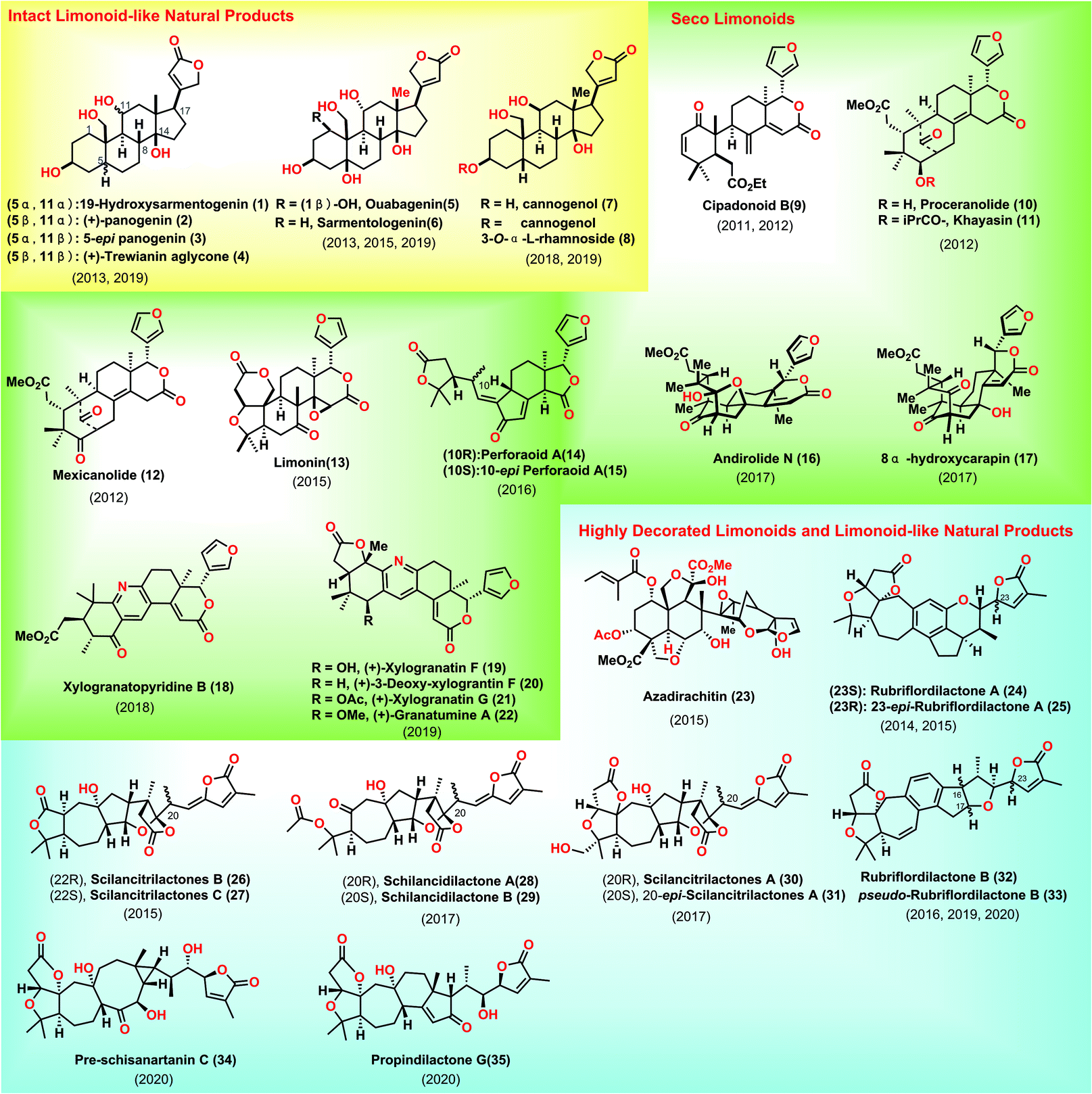 | ||
| Fig. 1 Selected limonoids and limonoid-like natural products that have been synthesized since 2011. | ||
2. Intact limonoid-like natural products
2.1 Inoue's synthesis of 19-hydroxysarmentogenin (2013)
Cardiotonic steroids can be considered as a group of intact limonoid-like natural products,3 which contain cardenolides and bufadienolides. Their structural variations are attributed to the oxygenation patterns located on C1, C3, C5, C11, C14 and C19 or epoxide. Cardiotonic steroids have demonstrated diverse biological activities.79-Hydroxysarmentogenin-3β-O-β-6-deoxyguloside (1), a cardiotonic steroid, was isolated from Crossopetalum gaumeri (Loes.) in the Yucatec Mayan community.7 It possesses eight continuous stereo-centres, cis A/B and C/D ring fusion, a tertiary 14 β-hydroxyl group, and a 17β-unsaturated lactone, posing a considerable challenge for its total synthesis. In 2013, Inoue and co-workers described the total synthesis of 19-hydroxysarmentogenin 1 (Scheme 1).8
 | ||
| Scheme 1 Inoue's synthesis of 19-hydroxysarmentogenin (2013). | ||
The pursuit of 1 commenced with the intermolecular Diels–Alder reaction of (S)-perillaldehyde 36 with the Rawal diene 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 followed by acidic treatment, resulting in the desired enone 38 with cis selectivity at ring fusion (Scheme 1a). The two carbonyl groups in 38 were subsequently reduced, followed by chemo-selective oxidation of the resultant allylic alcohol at C7 to give 39, which was further acetylated and oxidatively cleaved to generate ketone 40. Compound 40 was subjected to a Bayer–Villiger reaction, leading to the C3-acetylated 41. With 41 in hand, the removal of the two acetylated alcohols followed by treatment with TBSOTf furnished 42. PPTS mediated the de-protection of the TBS ether at C7, furnishing ketone alcohol 43. At this stage, their task was to prepare fragment 49. They attempted to utilize a six-step sequence from the known di-ketone 4410 to obtain 49, which involved the stereo-selective reduction of bis-ketone, alcohol protection, alkene oxidative cleavage, Horner–Wadsworth–Emmons reaction, and conversion of the TBS protecting group into an acetyl group.
9 followed by acidic treatment, resulting in the desired enone 38 with cis selectivity at ring fusion (Scheme 1a). The two carbonyl groups in 38 were subsequently reduced, followed by chemo-selective oxidation of the resultant allylic alcohol at C7 to give 39, which was further acetylated and oxidatively cleaved to generate ketone 40. Compound 40 was subjected to a Bayer–Villiger reaction, leading to the C3-acetylated 41. With 41 in hand, the removal of the two acetylated alcohols followed by treatment with TBSOTf furnished 42. PPTS mediated the de-protection of the TBS ether at C7, furnishing ketone alcohol 43. At this stage, their task was to prepare fragment 49. They attempted to utilize a six-step sequence from the known di-ketone 4410 to obtain 49, which involved the stereo-selective reduction of bis-ketone, alcohol protection, alkene oxidative cleavage, Horner–Wadsworth–Emmons reaction, and conversion of the TBS protecting group into an acetyl group.
Subsequently, they planned to merge fragments 43 and 49 (Scheme 1b). Pleasingly, exposure of enol ether 49 to bromine afforded intermediate dibromide 50, which was in situ treated with alcohol 43via SN2 bromine displacement11 to furnish acetal 51. Treating bromide 51 with BEt3/(TMS)3SiH12 initiated a carbon radical 52, which underwent a radical addition to the C8–C9 double bond from the less hindered top face of the molecule, assembling tricycle 53. With 53 in hand, the acid-mediated elimination of MeOH followed by the TBS protection of the C3 alcohol provided 54. The removal of acetyl in 54 followed by the oxidation of the free hydroxyl groups gave ketone 55. The authors closed ring C of the target via one-pot chemo-selective C8 enol formation followed by treatment with C14 ketone, delivering 56 as the major product with good regio- and stereo-selectivity. Coupling precursor 64 could be prepared from 56via a six-step operation, including removal of ketone, oxidative cleavage of alkene/hemi-acetylation, alcohol protection, stereo-selective Birch reduction and iodination.13 Equipped with iodide 64, they envisioned a Pd-catalysed Stille coupling reaction to append butanolide onto the steroid core. In practice, they observed that the treatment of 64 with stannane 65 under [Pd(PPh3)4/CuCl/LiCl] produced adduct 66. To complete the synthesis of 1, the desired β-oriented stereochemistry at C17 should be established. Initial attempts for the straightforward hydrogenation of C16![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C17 in 66 were found to be unfeasible, which only led to the undesired 17-epi-19-hydroxysarmentogenin after removal of TBS-protection. Ultimately, they discovered that introducing a bulky TMS group at C14 (67) was vital in shielding the convex face of the CD ring, thus providing the inverted α face selectivity of hydrogenation (dr = 6:1). Finally, the acidic-mediated de-protection of the primary alcohol achieved 19-hydroxysarmentogenin (1).
C17 in 66 were found to be unfeasible, which only led to the undesired 17-epi-19-hydroxysarmentogenin after removal of TBS-protection. Ultimately, they discovered that introducing a bulky TMS group at C14 (67) was vital in shielding the convex face of the CD ring, thus providing the inverted α face selectivity of hydrogenation (dr = 6:1). Finally, the acidic-mediated de-protection of the primary alcohol achieved 19-hydroxysarmentogenin (1).
In Inoue's synthesis, they successfully completed the target from the commercially available (S)-perillaldehyde 36. They tactfully adopted radical cyclization to install the desired C9 stereo-chemistry via clever substrate control. The ring C was fashioned stereo-selectively via judicious application of an aldol reaction. Smart manipulation of the steric hinder at the tertiary alcohol in 67 allowed for the installation of the desired the stereo-chemistry at C17, which would definitely inspire the syntheses of related natural products.
2.2 Baran's synthesis of ouabagenin (2013)
Ouabain was identified from the bark and roots of the ouabaio tree. It is widely utilized for the treatment of congestive heart failure.14 It is also used as an adrenal hormone that naturally occurs in mammals. Besides, biological evaluation revealed that it demonstrated high inhibitory activity toward Na+/K+-ATPase.15 Ouabain is constituted of ten stereo-genic centres, six hydroxyl groups and a β-oriented butanolide. The total synthesis of ouabain was first achieved by Deslongchamps's group (2007)16 through a polyanionic cyclization methodology, which was highlighted in Heasley's review.2 In 2013, Baran and co-workers described the scalable semi-synthesis of ouabagenin (5) from cortisone acetate 68.17 Key to Baran's synthesis is the strategic use of a redox relay and oxidative stereochemical relay (Scheme 2).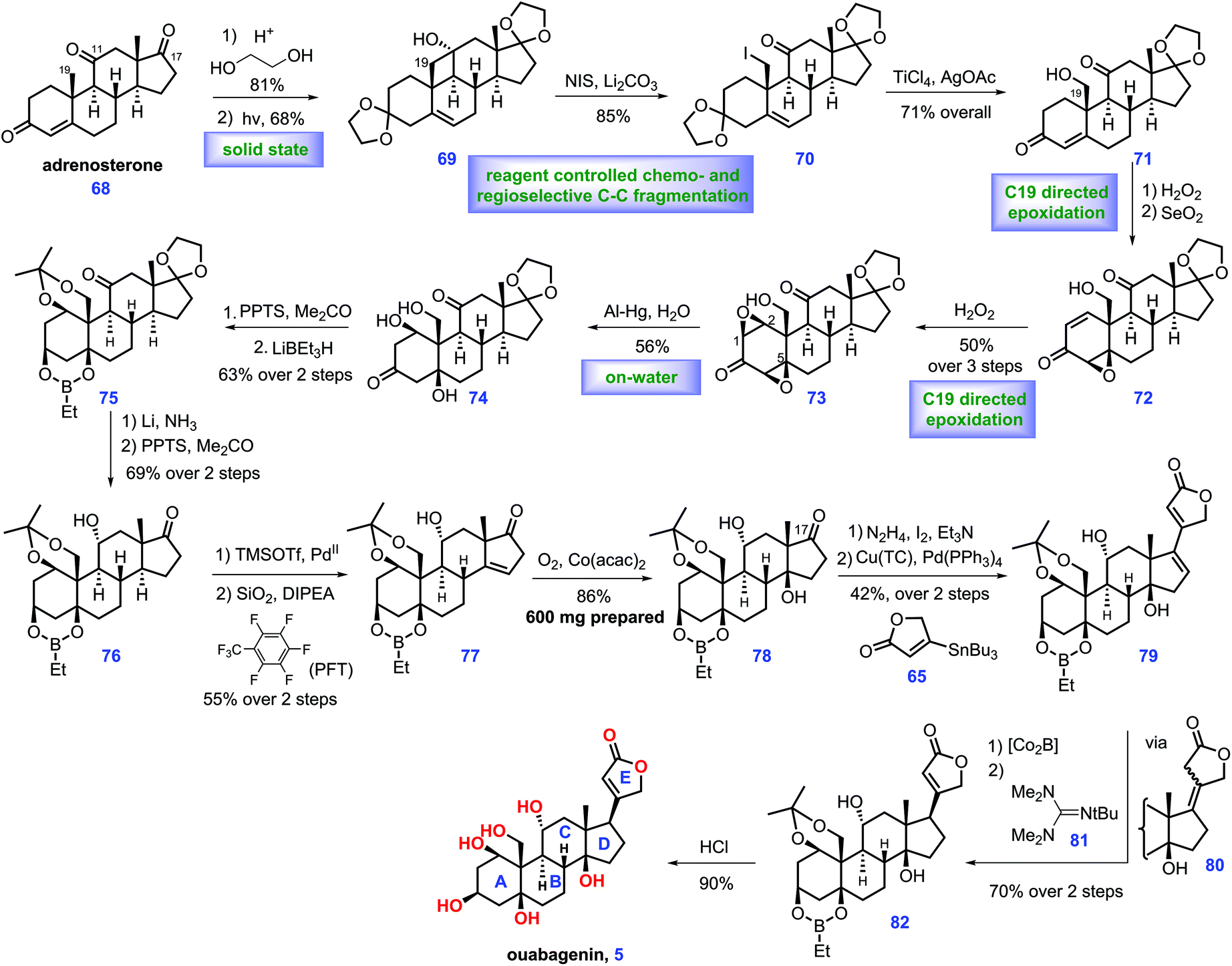 | ||
| Scheme 2 Baran's synthesis of ouabagenin (2013). | ||
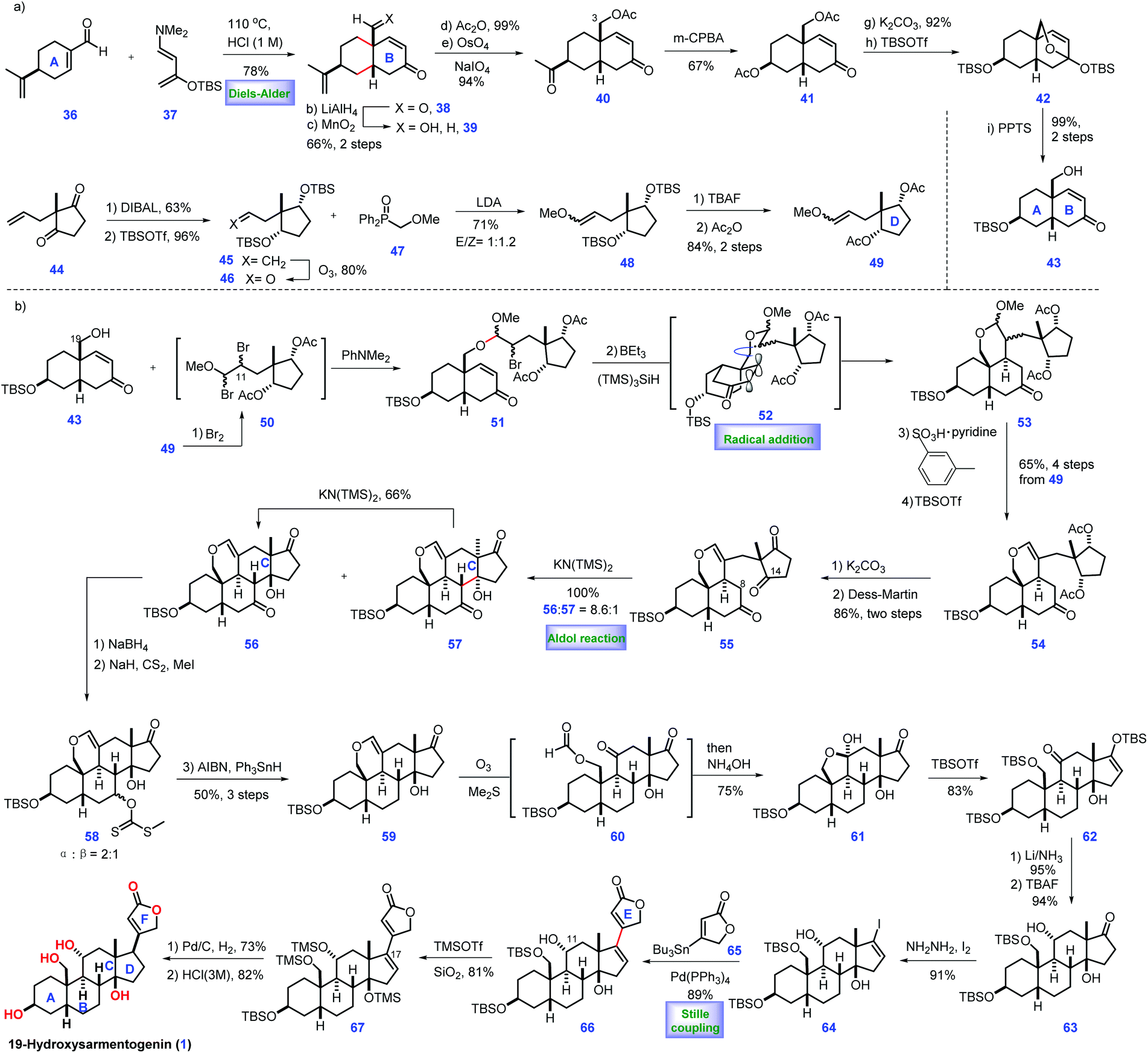
Their endeavours commenced with the commercially available cortisone acetate, which was converted into adrenosterone 68via a known procedure.17 After ketalization and recrystallization, the first redox-relay reaction was devised, which proceeded via the Norrish type II photochemical functionalization18 of the angular C19 methyl group aided by C11 ketone. Notably, the above transformation only led to a moderate yield (43%) in the conventional solution (EtOH), which was attributed to the competitive formation of side products via the Norrish type I cleavage of the C9–C11 bond. Inspired by the work of Garcia-Garibay,19 the authors successfully developed a solid-state (sodium dodecyl sulfate, SDS) irradiation of ketalized compound in aqueous suspension, which resulted in improved yield (68%). Subsequently, initial experiments to conduct oxidative fragmentation of the C11–C19 bond of 69 were unfruitful. Eventually, the authors identified that 69 could be transformed into 70 under Barluenga's reagent,20 which, however, is costly. Accordingly, an inexpensive reagent, NIS,21 was selected as an alternative. Then, TiCl4-mediated selective deketalization of C3 ketone followed by AgOAc-promoted hydrolysis of C19 iodide provided enone 71. At this juncture, the authors envisioned a next series of relay transformations, where the C19 hydroxyl moiety was switched into the β face of ring A via a diastereoselective stereochemical relay. In practice, H2O2 induced C19 hydroxyl-directed β-epoxidation of 71 followed by dehydrogenation of the C1–C2 bond, giving epoxide 72, which served as the substrate for a second hydroxyl-directed epoxidation. Using this method, diepoxide 73 was accessible. Moving forward, the authors found that the reductive opening of diepoxide 73 on scale proved challenging. Although numerous conditions were screened, only a mixture of enones were obtained. Fortunately, the authors finally discovered that the aluminum amalgam-mediated reductive opening of 73 smoothly achieved the desired triol 74 under “on-water” conditions22 (aqueous suspension). The selective protection of 74 to an acetonide followed by LiBEt3H-induced reduction of the C3 ketone furnished 75, where the hydroxyl at C1 and C5 was protected as an ethyl boronic ester. Li/NH3 triggered the thermodynamic reduction of the C11 ketone followed by PPTS-mediated deketalization of the C17 ketone, affording 76, thus setting the stage for the final redox-relay process. In real experiment, the authors attempted dehydrogenation of ketone 76 followed by olefin isomerization23 to form enone 77, which, however, resulted in low conversion and epimerization of the C14 stereo-centre. Pleasingly, the authors identified that fluorinated solvents could secure the reaction, while minimizing the undesired epimerization. Exposure of 77 to Mukaiyama hydration24 conditions produced 78 with the desired tertiary alcohol installed (dr = 8:1, favored 78). 78 is useful as it could be employed as a precursor to achieve plentiful analogues varied at C17. To complete the synthesis, it was necessary to attach a butenolide subunit to advance 78. In practice, 78 was first promoted to vinyl iodide via Barton's method.13 Then Pd-catalysed Stille-coupling reaction of the iodide compound with stannane 65 provided 79 with a full ring system established. In the final stage of the synthesis, direct reduction of the C16–C17 olefin of dienoate 79 in a productive manner failed since this reduction proceeded from the undesirable convex face of the molecule. Finally, the authors found that exposure of 79 to Co2B25 generated an intermediate, tetrasubstituted olefin 80, which was further treated with Barton's base2681 under heat. The resulting enolate 82 was obtained with the desired stereochemistry at C17 (dr = 3:1) favored. Finally, deprotection of all the hydroxyl groups in 82 completed ouabagenin (5).
In Baran's work, the novel and outstanding redox relay and oxidative stereochemical relay showcase their potential in the elaboration of complex steroids. Notably, the key precursor of ouabagenin, 78, could be prepared on scale, thus providing a versatile route to steroids analogues varied at C17. Most importantly, this work resulted in the discovery of new chemistry, such as “on-water” epoxide fragmentation and chemo-selective dienoate reduction, which will accelerate the synthesis of related natural products.
2.3 Inoue's synthesis of ouabagenin (2015)
In 2015, Inoue and co-workers also described the total synthesis of ouabagenin 5, involving intramolecular radical cyclization and aldol reaction as key steps (Scheme 3).27 Their pursuit of ouabagenin 5 began with the installation of a ring AB system of the target. Treatment of (R)-perillaldehyde 36 with Rawal's diene 37 under heat triggered the credible Diels–Alder reaction, leading to the desired cis-decalin with good regio- and diastereoselectivity, which was followed by deprotection via aqueous acid to give 38. LiAlH4-mediated reduction of two ketone groups, followed by chemo-selective oxidation of the allylic alcohol provided ketone 39. TBS protection of the C3-enol and C19-hydroxy groups in 39 followed by Saegusa reaction28 introduced dienone 83. Equipped with 83, the authors turned their attention toward the installation of the stereochemistry of C1- and 5-oxygen functional groups. Pleasingly, C3-β-alcohol-directed stereo-selective reduction of ketone 83 by chiral reagent 8429 arrived at desired 85 as a 3:1 diastereomeric mixture.
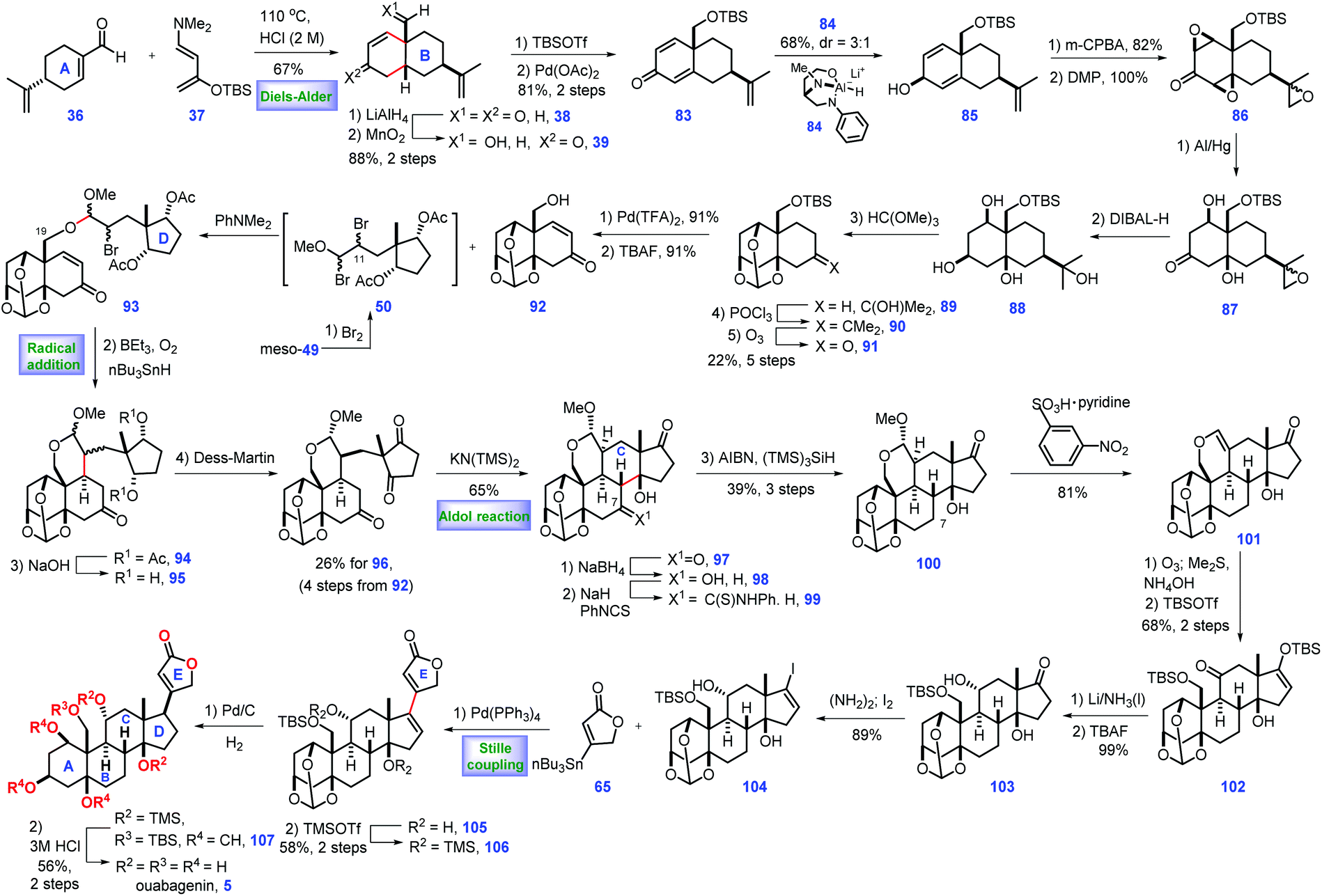 | ||
| Scheme 3 Inoue's synthesis of ouabagenin (2015). | ||
Moving forward, the epoxidation of 85via m-CPBA followed by oxidation furnished tri-epoxide 86. Sequential reductive opening of the two epoxides via Al/Hg,30 stereo-selective reduction of C3 ketone, ortho-esterification of three cis-hydroxyl groups, dehydration of the tertiary alcohol and ozonolysis of the tetra-substituted olefin furnished orthoester 91. Pd-catalysed dehydrogenation31 of 91 followed by TBAF-mediated deprotection of TBS-ether provided the desired alcohol 92 with the requisite ring system AB installed. Learned from their previous work,8 Inoue conducted dibromination of meso-4932 that furnished intermediate 50, which was in situ merged with 92via SN2 bromine displacement, thus giving 93 as a diastereomeric mixture. The remaining C11-bromo group of 93 allowed the key radical cyclization reaction (Et3B/n-Bu3SnH/O2) from the β-face of the C9-olefin,33 leading to product 94 with the required C9 stereo-centre constructed. Removal of two Ac groups followed by DMP oxidation produced diketone 96. With 96 in hand, the authors sought to develop conditions for construction of ring C of the target. They found 97 was subsequently fashioned via a KN(TMS)2 (30 mol%) catalysed intramolecular aldol reaction where the ring C was formed. In the forward sense, their goal was to develop conditions for the deoxygenation of C7 at 97, which was achieved via a three-step operation involving chemo-selective reduction at C7 ketone, derivation into thio-carbamate with thio-isocyanate and AIBN/(TMS)3SiH-induced reductive cleavage of the C7-thiocarbamate. Using this method, 100 was accessible from 97 in 39% yield (3 steps).
Their efforts were then focused on the construction of the C11-functional group. Acid-mediated elimination of MeOH in 100 gave 101. Ozonolysis of 101 followed by reductive workup and de-formylation gave the resultant hemiacetal product, C17- and C19-oxygen of which was further capped as TBS ether 102 together with the liberation of the C11-ketone. Then they planned to install an equatorial C11-OH. In practice, they identified that the Birch reducing conditions (Li/NH3) followed by deprotection of the C19 enol silyl ether could achieve the desired 103. 103 was further developed to vinyl iodide 104, setting the stage for a cross-coupling reaction. Inspired by their previous work,8,17 they conducted a Pd-catalysed Stille coupling reaction34 with stannane 65 to afford 105, where the full ring system was installed.
With access to 105, what remained was the establishment of the C17 stereo-centre. Learning from their previous work,8 the protection of the C14-hydroxy group with TMS-ether enabled the Pd-catalysed 1,4-reduction of enone 106 from the less steric concave α-face, thus yielding the desired 107. To complete the synthesis, global removal of the protecting groups in 107 furnished the ouabagenin (5).
In Inoue's synthesis, the core strategy of ring formation was similar with their aforementioned work in the total synthesis of 19-hydroxysarmentogenin 1,8 both of which involved the Diels–Alder reaction to construct the ring AB system, a tactful SN2 bromine displacement to connect ring AB and ring D, the intramolecular Aldol reaction to install the ring C and a Pd-catalysed Stille coupling reaction to introduce lactone ring E. Besides, a clever C3-β-alcohol-induced di-epoxidation, followed by a reductive ring-opening reaction nicely installed both C1 and C5 hydroxyl groups. The triol was protected to orthoester 89, which could minimize side reactions in the subsequent transformations.
2.4 Nagorny's synthesis of ouabagenin, sarmentologenin, 19-hydroxysarmentogenin, and 5-epi-panogenin and their derivatives (2019)
In 2016, Nagorny's group reported synthetic endeavours toward the total synthesis of cardiotonic steroids 19-hydroxysarmentogenin 1 and trewianin aglycone 4via Cu(II)-catalysed asymmetric Michael addition/intramolecular aldol cyclization reactions as the core protocol.35 In 2018, they documented the synthesis of cannogenol-3-O-α-L-rhamnoside 8 utilizing an identical strategy.36 In 2019, they further described a full report on the total syntheses of ouabagenin 5, sarmentologenin 6, 19-hydroxysarmentogenin 1, 5-epi-panogenin 3 and other cardiotonic steroid involved the above-mentioned Cu(II)-catalyzed Michael addition/intramolecular aldol cyclization reactions as key protocols (Scheme 4) and other cardiotonic steroids,37 which has included their aforementioned work in 201635 and 2018.36 Herein, we discuss their full paper (2019).37
 | ||
| Scheme 4 Nagorny's synthesis of ouabagenin, sarmentologenin, 19-hydroxysarmentogenin, and 5-epi-panogenin and their derivatives (2019)-1. | ||
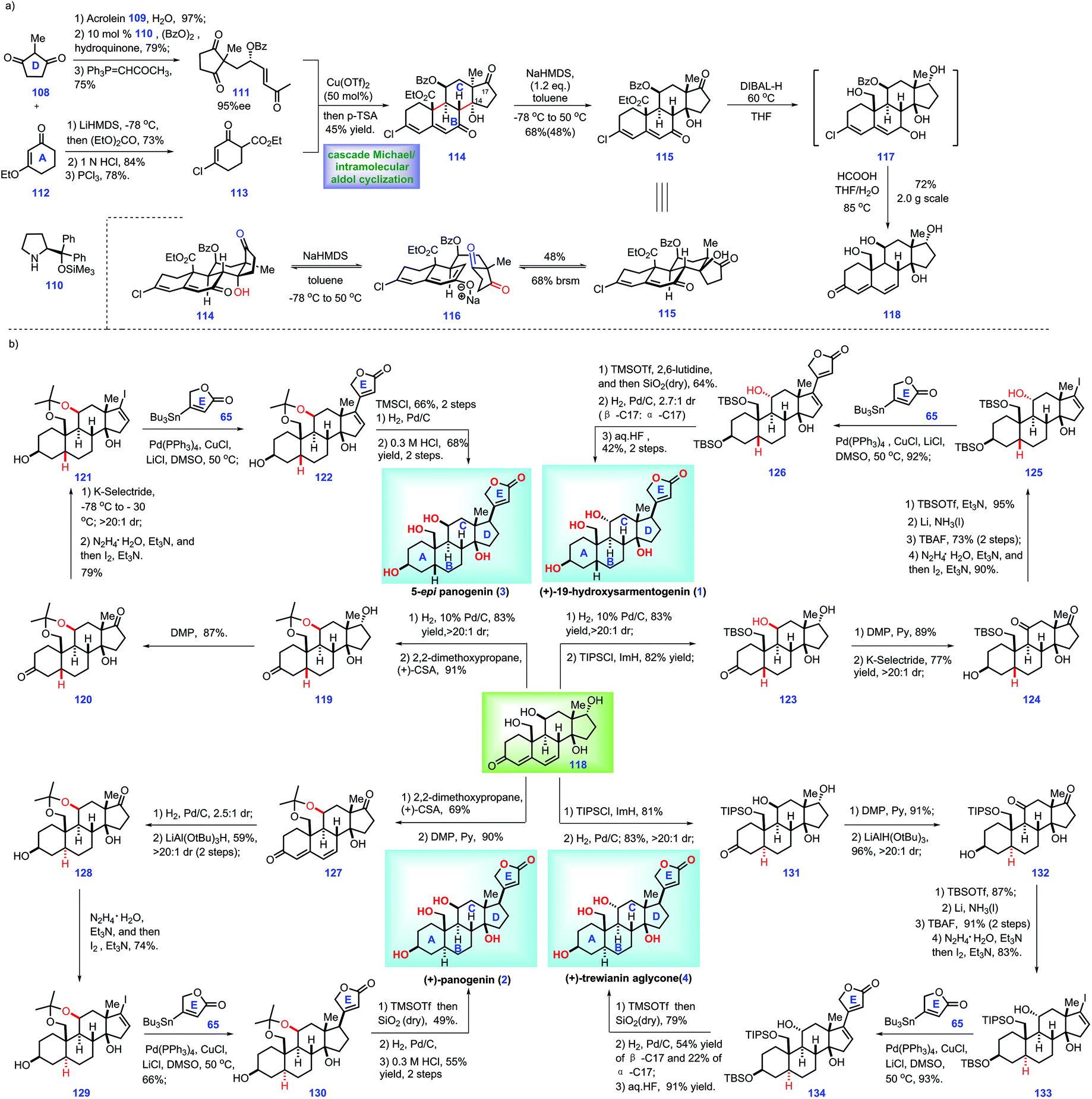
In this work, the authors envisioned a convergent strategy to complete a series of cardiotonic steroids from a common core, steroid 118. Accordingly, their project commenced with the assembly of 118 (Scheme 4a). Initially, 1,3-di-ketone 108 was advanced to enone 111 through a three-step manipulation involving a Michael addition38 with acrolein 109, an organo-catalyst 110 catalysed enantioselective oxidation reaction39 with benzoyl peroxide and a Wittig reaction with 1-(triphenylphosphoranylidene)-2-propanone. Instead, ethyl-vinyl ether 112 was progressed to β-ketoester 113via acylation with diethylcarbonate, acid-hydrolysis of ethyl-vinyl ether and chlorination. Here, the authors sought to install the ring BCDE system of core 118. In practice, key fragments enone 111 and β-ketoester 113 were successfully merged via a Cu(II)-catalysed asymmetric Michael addition reaction.40 The resultant Michael adduct allowed for a subsequent p-TsOH-mediated double aldol cyclization cascade, leading to 114 with a tetra-cycle ring system installed.
Moving forward, they turned their attention toward the elaboration of 113 into common core 118, which involved intermediate 115. DFT calculations showed that diastereomer 115 was more stable than 114. Hence, the authors planned to isomerize 114 into 115via thermodynamic equilibrium. After screening extensive conditions, the author identified that the treatment of NaHMDS (1.2 eq.) with 114 at −78 °C to 50 °C initiated a retro-aldolization reaction, leading to 116, which followed by intramolecular aldol condensation to fashion 115. Notably, Na+-containing bases demonstrated better reactivity than Li+ and K+ counter-cations. With access to 115, the author devised a global reducing strategy followed by acidic workup to fulfil 118. However, early attempts to employ LiAlH4 led to complex results, while the reduction of C19 ester proceeded slowly with the use of LiBH4 and DIBALH. Fortunately, treatment of 115 with DIBAL-H under heat (60 °C) afforded intermediate 117, which was further treated with aqueous formic acid at elevated temperature (80 °C), thus allowing the core 118.
With core 118 constructed, the authors then developed conditions for the convergent synthesis of cardiotonic steroids 1, 2, 3 and 4 (Scheme 4b). The authors envisioned that these four syntheses held some common characteristics as follows: (1) the α-C5 configuration (1 and 3) and β-C5 configuration (4 and 2) could be both established via diastereoselective reduction of 118. (2) The C11 configuration position could be inverted (4 and 6) or retained (2 and 3). (3) A C17 butenolide moiety should be attached via Stille coupling in the final stage.17
With this in mind, the authors first targeted 5-epi-panogenin (3). Global hydrogenation of the diene motif in 118 followed by protection of the resultant diol (C11, C19) with 2,2-dimethoxypropane afforded acetonide 119 with a β-C5 stereo-centre built. In a parallel experiment, the site-selective protection of C19 hydroxyl in 118 with TBSCl furnished 123.
Moving forward, DMP oxidation of 119 yielded 120, which was then processed through the site-selective reduction of C3 ketone and a Shapiro reaction to give rise to vinyl iodide 121. 121 was served as a substrate for the Pd-catalysed Stille cross-coupling reaction with stannane 65, thus affording 122 with a full ring system fashioned. With access to 122, what remained was to build the C17 stereo-centre. Similar to the work of Inoue and Baran,8,16,17 the authors also failed to direct the hydrogenation of the Δ-olefin.16,17 Pleasingly, when the C14 hydroxyl in 122 was protected with TMS ether, it allowed a Pd-catalysed hydrogenation reaction with a much higher stereo-selective mixture (25:1.0:2.8). Exposure of the resulting mixture to acidic conditions removed the acetonide protecting group, thus completing the 5-epi-panogenin (3).
Moving forward, the authors then aimed for 19-hydroxysarmentogenin 1. Compared with 5-epi-panogenin 3, accomplishment of 19-hydroxysarmentogenin (1) was performed via similar operations but called for requisite manipulations to construct the α-C11 stereo-centre. In practice, global oxidation of alcohol in 123 followed by site-selective reduction of C3-ketone gave rise to 124. Treating 124 with TBSOTf induced selective protection of C17 ketone, thus generating a TBS ether product. Li/NH3 initiated the stereo-selective reduction of the C11 ketone followed by removal of the TBS protecting group, providing 125 with the requisite α-C11 hydroxyl group fashioned. Fragment 125 and stannane 65 were then connected via a similar Pd-catalysed Stille coupling to achieve 126. With the full ring system in place, all that remained was to elaborate 126 into target 1. Finally, 19-hydroxysarmentogenin (1) was procured via three additional manipulations, including TBS-protection of the C13 alcohol, hydrogenation of the Δ-alkene16,17 and global deprotection of the alcohols.
Their goal then was to accomplish the panogenin (2) and trewianin aglycone (4), which featured a trans-A/B ring fusion and α-C5 stereo-centre. Initially, they targeted panogenin (2). The project commenced with the protection of 118 to form the acetonide product, which followed by C17 alcohol oxidation, thus affording ketone 127. Stereoselective hydrogenation of 127 delivered the product with a fashioned trans-A/B ring (2.5:1 dr at C5). LiAl(OtBu)3H-mediated stereoselective reduction of the C3 ketone in the above product gave rise to 128 (59%, two steps). Then 128 was advanced to vinyl iodide 129, which was further transformed into panogenin (2) via the aforementioned protocol for 5-epi-panogenin (3).
Next, the authors focused their attention toward the synthesis of trewianin aglycone (4). Exposure of 118 to TBSCl/ImH masked the C19 alcohol of 118 as the TBS ether, thus allowing the following stereoselective hydrogenation. Pleasingly, the resultant trans-A/B ring fused 131 was generated with high diastereoselectivity (>20:1). Then, 131 underwent a two-step redox manipulation to form 132. Finally, 132 was advanced to trewianin aglycone (4) via the abovementioned operations to panogenin (2).
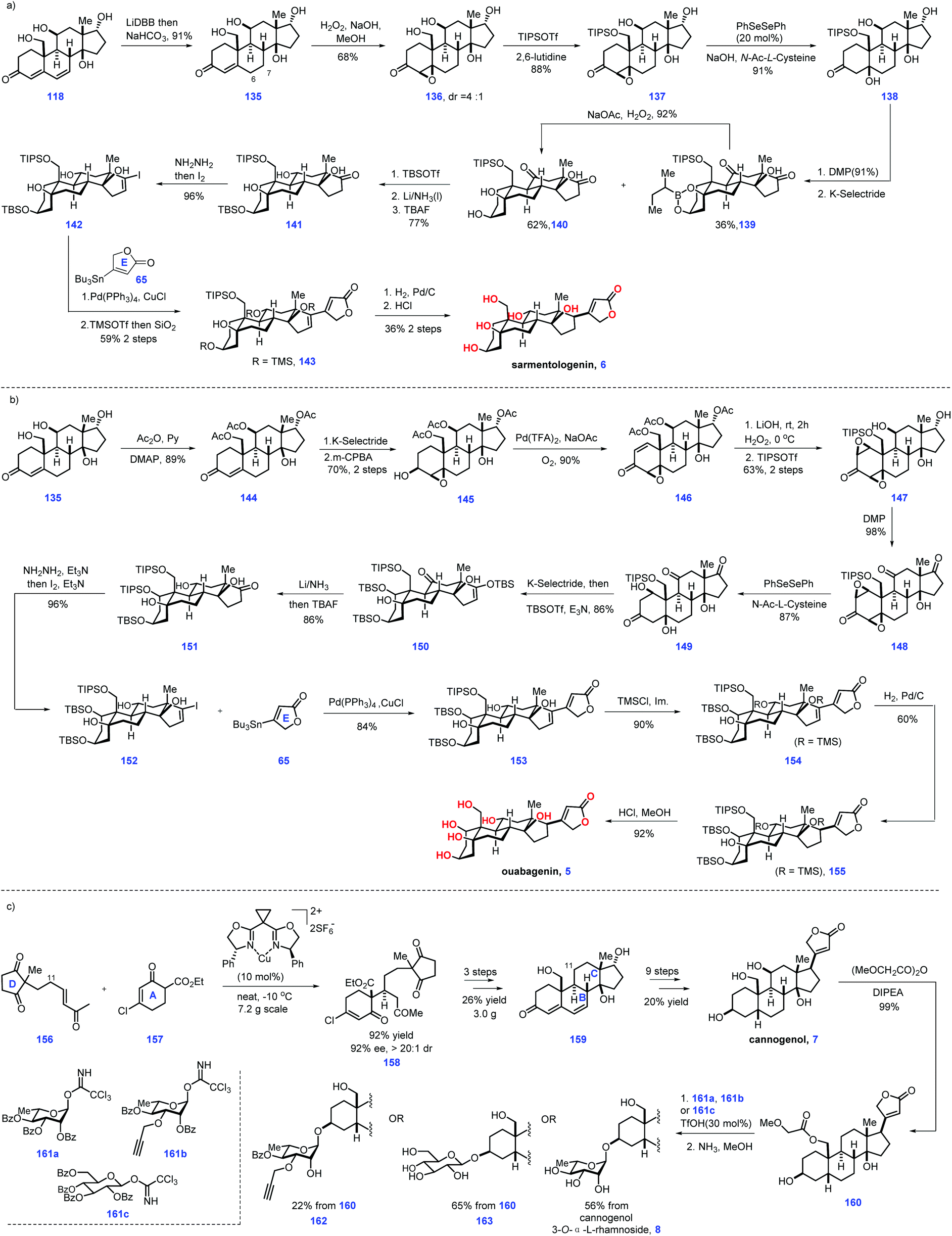
Moving forward, the authors then envisioned an “oxidase phase” strategy to conquer sarmentologenin (6) and ouabagenin (5) (Scheme 5a). Initial experiments to expose 118 to the conventional reducing conditions such as selective hydrogenation, conjugate reduction and diimide reduction failed to give the target product. Fortunately, they eventually identified LiDBB was engaged in the reduction of the Δ alkene6,7 in 118, leading to the desired 135 after aq. NaHCO3 workup. Epoxidation of enone 135 fashioned β-epoxide 136 as the major product (dr = 4:1). However, further advancement of 136 into 137 proved challenging since product 137 was feasible to undergo dehydration, thus returning to enone 135. Unfortunately, common reducing conditions only led to decomposition of the reaction. Finally, the authors discovered that oxidase-mediated reducing conditions (PhSeSePh, NaOH, N-Ac-L-cysteine) could form the alcohol product. However, they failed to isolate the alcohol product due to its high hydrophilicity. To address this issue, the C19 alcohol of 136 was protected to form TIPS ether 137, which was then smoothly advanced to sarmentologenin core 138. Global oxidation of 138 afforded the ketone product, which was used as a precursor for a C3 ketone reduction, thus arriving at boronic ester 139 (36%) together with the desired 140 (62%). Note that boronic ester 139 could be converted into 140via H2O2-mediated oxidation. 140 was progress to 141via three additional sequences involving TBS protection of the C3 alcohol, β-selective reduction of the C13 ketone and de-protection of the C5, C14 TBS ether. Subjecting 141 to Shapiro reaction gave vinyl iodide 142, which was followed by Pd-catalysed Stille coupling with stannane 65 and site-selective alcohol protection to construct 143. To complete the synthesis of sarmentologenin 6, the requisite C17 stereochemistry should be fashioned. In practice, Pd-catalysed stereoselective hydrogenation of 143 followed by an acid-mediated global deprotection reaction secured sarmentologenin 6.
 | ||
| Scheme 5 Nagorny's synthesis of ouabagenin, sarmentologenin, 19-hydroxysarmentogenin, and 5-epi-panogenin and their derivatives (2019)-2. | ||
After that, their task turned to ouabagenin (5), which called for the installation of the C1 hydroxyl motif from sarmentologenin (6) (Scheme 5b). The authors conceived a bis-epoxidation of 118 or 135 followed by epoxide-reopening strategy to simultaneously introduce the desired double hydroxyl group. In real experiment, attempts to utilize this strategy to fulfil the target failed, which was attributed to the feasible aromatization of the steroidal ring A system.
After considerable experiments, the authors eventually identified a stepwise sequence to enable the introduction of C1 and C5 oxygenation. Site-selective acetylation of the C11, C17 and C19 hydroxyls of 135 gave 144, which was subjected to a two-step transformation involved α-face reduction of the ketone, followed by C3-directed epoxidation of alkene, resulting in 145. Exposure of 145 to Pd-catalysed oxidative conditions furnished enone 146. LiOH mediated the global removal of the acetate ester in 146 followed by C1–C2 epoxidation (H2O2) and selective protection of the C19 alcohol as TIPS ether furnished bis-epoxide 147. The authors guessed that the TIPS protecting group could enhance the hydrophobicity of 147, thus allowing the following steps to proceed smoothly. Oxidation of 147 led to triketone 148, thus setting the stage for an epoxide opening reaction. The initial experiment showed the product 149 was sensitive to dehydrating conditions (basic or acidic media), leading to aromatization of ring A. Pleasingly, epoxide opening reactions could be achieved under the condition of (PhSeSePh, N-Ac-L-Cys, NaOH), which gave target 149 in good yield (87%). Note that cautious adjustment of the ratio of N-Ac-L-Cys to NaOH was crucial in minimizing the side reactions. Stereoselective reduction of the C3-ketone moiety of 150via K-selectride generated the C3-hydroxyl product, which was advanced to the C3- and C17-silylated enol ether 151. Following a similar protocol, 151 was processed through a five-step sequence, including stereoselective reduction of ketone, desilylation of the enol ether, Shapiro reaction, a Pd-catalysed coupling reaction with stannane 65, TMS protection of the C5 and C11 alcohol and Pd-catalysed stereo-selective hydrogenation of the Δ alkene16,17 to deliver 155, which was subjected to global deprotection conditions (HCl/MeOH) to complete the ouabagenin (5).
Finally, the authors focused on cannogenol glycosides 8, 162 and 163, which lacked C11 oxygenation (Scheme 5c). Treatment of enone 156 with β-keto-ester 157via a Cu-catalysed enantioselective Michael addition gave 158. Based on the aforementioned protocol for its C11-hydroxyl analogue 118, 158 could be smoothly advanced to enone 159via three additional operations. Then, 159 was subjected to a 9-step transformation to produce the cannogenol (7). Treatment of the more reactive C19 hydroxyl in 7 with (MeOCHCO)2O led to 160, thus setting the stage for the subsequent glycosylation of the C3 alcohol via trichloroacetimidate 161a. The corresponding α-L-rhamnoside secured. Moving forward, removal of both the sugar benzoates and C19 methoxyacetate provided the cannogenol α-L-rhamnoside (8). Note that subjecting 160 to the condition of 50% ammonia in methanol initiated selective hydrolysis of the ester motif, leaving butanolide intact. Correspondingly, glycosides 162 and 163 could be made from 160. Notably, α-L-rhamnosides of the steroids digitoxigenin,41 bufalin,42 and strophanthidol43 could also be fashioned via a similar protocol. Then, the synthetic steroids were subjected to biological investigation. Pleasingly, the cannogenol-3-O-α-L-rhamnoside (8) and strophanthidol-3-O-α-L-rhamnoside exhibited excellent anticancer activity in the concentration range of 10–100 nM.
In Nagorny's synthetic campaign, the ring ABCD system was efficiently and elegantly established via a novel copper-catalysed intermolecular asymmetric cascade Michael/Aldol reaction with four desired stereocentres installed. More importantly, the key cascade reaction could be scaled up, thus allowing the preparation of enough materials for the later biological investigation. According to their ingenious design, the authors tactfully envisioned the key common intermediate 118, which set the stage for the rapid and convergent syntheses of a series of related natural products. Additionally, enzyme chemistry was successfully utilized to promote the ring opening of the epoxide in the complex molecule synthesis, which provided enlightening value to conquer other limonoid-like compounds.
3. Sec limonoids
3.1 Williams's synthesis of (−)-cipadonoid B, khayasin, proceranolide, and mexicanolide (2012)
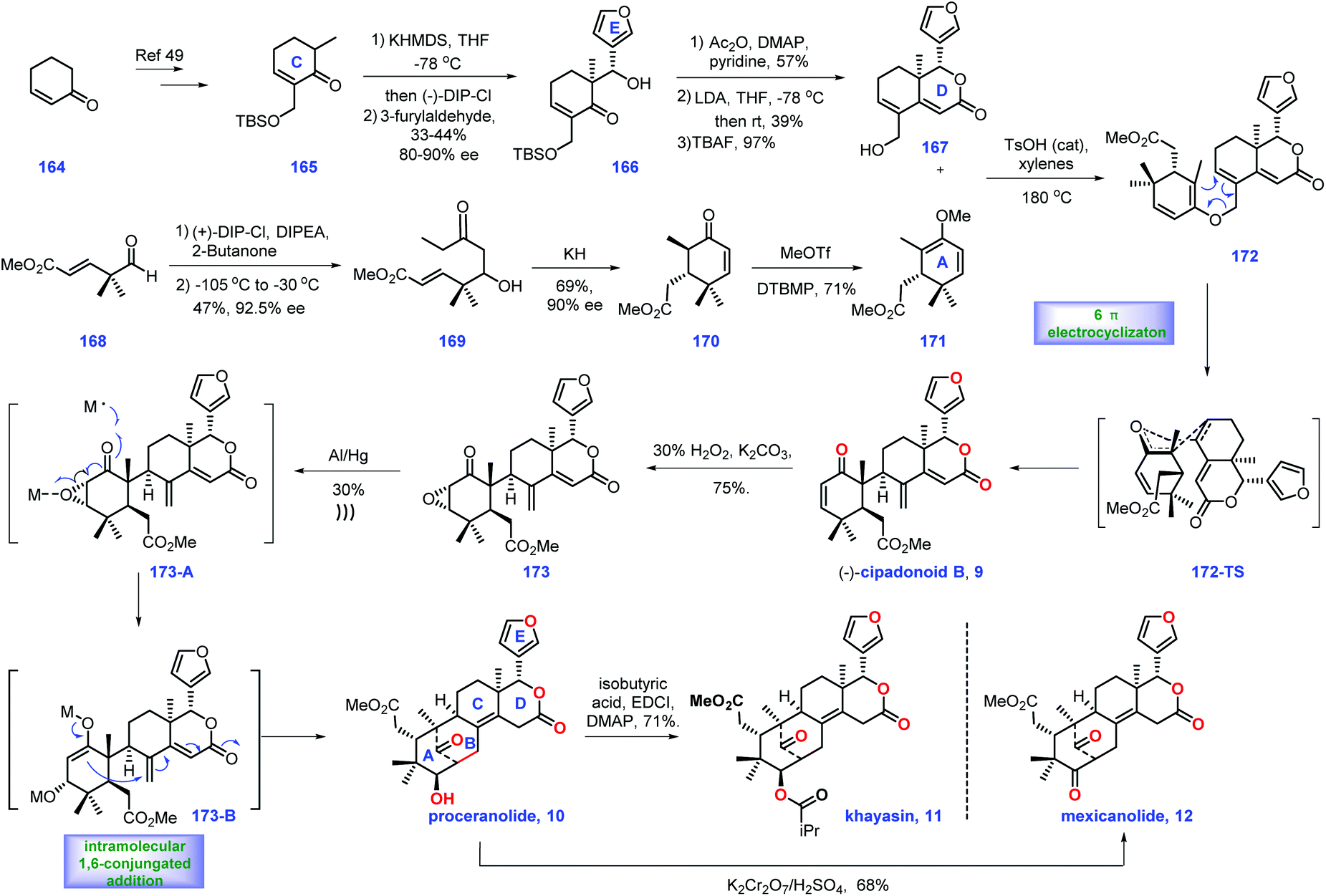
In 1966, Taylor's group isolated tetranortriterpenoid khayasin 11, which belongs to the mexicanolide class of limonoids.44 Khayasin 11 was utilized as an insecticide to kill coconut leaf beetle Brontispa longissima.45,46 In 2011, Williams's group disclosed the racemic synthesis of cipadonoid B 9.47 In 2012, they further reported convergent asymmetric total synthesis of mexicanolide limonoids including (−)-cipadonoid B 9.48In 2012, Williams and co-workers reported the asymmetric total synthesis of khayasin 11 together with (−)-cipadonoid B 9, proceranolide 10, and mexicanolide 12via a convergent strategy (Scheme 6).48 Initially, they developed conditions for the asymmetric synthesis of intermediate (+)-azedaralide 167. Enone 164 was firstly converted into 165via known procedures.49 Then [(−)-DIP-Cl] induced the asymmetric aldol reaction of 165 with 3-furylaldehyde, giving alcohol 166 in 33–44% yield with 80–90% ee. Then 166 was utilized as a substrate for a three-step sequence, including acetylation of alcohol, an aldol reaction and removal of TBS ether to form (+)-azedaralide 167.
 | ||
| Scheme 6 Williams's synthesis of (−)-cipadonoid B, khayasin, proceranolide, and mexicanolide (2012). | ||
With access to (+)-167, their goal was to install the diene fragment 171. Treatment of (+)-DIP-Cl and DIPEA with 2-butanone followed by addition to aldehyde 168 furnished 169 with 47% yield and 92.5% ee. With 169 in hand, KH initiated an intramolecular Michael reaction to construct cyclohexanone 173/170 as a 4:1 mixture of diastereomers (epimeric at C6). However, the above mixture was inconsequential, as both diastereomers could be transformed into (−)-vinyl ether 171via treatment with methyl triflate.
Equipped with (+)-azedaralide 167 and (−)-vinyl ether 171, they next devised to the asymmetric synthesis of (−)-cipadonoid B 10via a cascade ketal-Claisen rearrangement reaction. In practice, subjecting 171 and 167 to acidic conditions under heat (TsOH, 180 °C, 4 h) initiated the cascade condensation to enol ether 172-TS, which was in situ followed by rearrangement to arrive at the desired enantiopure (−)-cipadonoid B 9.47 DFT calculation demonstrated that the ketal-Claisen cascade favored the proposed transition state to minimize steric hinderance. Consequently, the authors turned their attention to other mexicanolide natural products. The regio- and stereoselective epoxidation of (−)-cipadonoid 9 yielded 173 with β oxygenation at C-3 installed. Al/Hg induced ring-opening of the epoxide50 in 173 followed by intramolecular 1,6-conjugate addition of 173B produced (−)-proceranolide 10. Oxidation of (−)-proceranolide 10 formed (−)-mexicanolide 12. Acylation of (−)-proceranolide 10 gave rise to (−)-khayasin 11.
In this synthesis, the clever one-pot cascade ketal-Claisen rearrangement rapidly afforded (−)-cipadonoid B; however, its efficiency was unsatisfactory (25% yield). An impressive solving metal-induced radical skeleton rearrangement reaction smartly constructed the desired [3.3.1] framework, which enabled the collective synthesis of a series of mexicanolide natural products.
3.2 Yamashita's synthesis of limonin (2015)
Limonin 13 was discovered from the bitter components of the citrus fruit in 1841,51 but its structure was confirmed until 1960, which contains an intact skeleton of 4,4,8-trimethyl-17-furyl-13a-androstane. Currently, hundreds of limonoids have been identified.52 In 2015, Yamashita and co-workers first completed their synthetic efforts toward limonin 13. Key to their success involved a tandem radical cyclization to install a ring CDF system as the core strategy (Scheme 7).53 Their endeavours began with the installation of the ring CDF system from the commercially available geraniol 174. Sequential alcohol chlorination, alkene region-selective epoxidation, propargylation and Ti(III)-mediated epoxide opening reaction generated allylic alcohol 175. Exposure of 175 to SOCl2 followed by treatment with the dianion of ethyl 2-chloroacetoacetate 176 and TBAF, gave alkynyl β-ketoester 178. | ||
| Scheme 7 Yamashita's synthesis of limonin (2015). | ||
With access to 178, the authors focused on an efficient tandem radical cyclization strategy to fashion the ring CDF system. Eventually, they found a manganese-mediated condition [Mn(OAc)3·2H2O] was capable of engaging the cascade cyclization, leading to the desired 179 with moderate diastereoselectivity at C7 (dr = 2.2:1).54 Consequently, the authors then sought to assemble ring A of the target. Elimination of chloride in 179via Zn/AcOH followed by Robinson cyclization with methyl vinyl ketone (MVK) smoothly forged tetracyclic 180 with ring A constructed. Double methylation of the C4 position of 180 afforded 181 in good yield (80%).
Here, the authors devised a cross coupling strategy to append a furan motif onto the core structure. Thus, the protocols for coupling precursor should be established. Initially, experiments to convert the exo methylene group in 181 into a ketone group via conventional oxidative cleavage conditions were found to be unfeasible. Instead, they switched to a five-step manipulation including reduction of the ester and C3-ketone, separate protection of the diol, epoxidation of the exo methylene and NaCN-mediated ring opening of the epoxide followed by elimination of CH3CN to afford ketone 184.55 The copper-catalysed oxidation of 184 gave enone 185,56 which served as a substrate for three additional operations, including Birch reduction, DMP oxidation and acetyl removal, to furnish hemiacetal 186 with an installed C5 stereocentre. The C7 ketone of 186 was chemoselectively reduced via LiAlH(OtBu)3 and the resultant bis-alcohol was further protected as TBS ether 187. Ketone 187 was further advanced to enone 188 by Ito–Saegusa oxidation. Exposing this material to triflation conditions (DTBMP, Tf2O) followed by a Pd-catalysed Stille coupling reaction with butenolide 65gave the coupled product 189 with ring G established.
Moving forward, their next task was to set up the lactone moiety. In practice, 189 was subjected to singlet oxygen (1O2), triggering a [4 + 2] cycloaddition reaction, which was followed by two additional operations involving butanolide reduction and acetylation/elimination. The target endoperoxide 190 secured.57 Exposure of 190 to [(Ph3P)3RuCl2] initiated isomerization of the endo-peroxide moiety,58 affording bis-(epoxide) 191, which is in situ quenched by silica gel, inducing a 1,2-hydride shift to arrive at the undesired 192. Notably, 192 could be isomerized under base (DBU) to form 193/192 as a 5.4:1 mixture in 61% yield (2 steps).
With the desired 193 in hand, a Baeyer–Villiger rearrangement was conducted followed by removal of TES ether, constructing epoxylactone 194 with lactone F formed. To complete the synthesis, installation of ring AB system of the target was necessary. 194 was exposed to Suarez reaction conditions,59 [PhI(OAc)2/I2/O2(10 atm)/hv], triggering the homolysis of the C3–C4 bond followed by C4 oxidation to give rise to alkoxy radical intermediate 195. Intermediate 195 underwent a cascade 1,5-hydrogen abstraction, iodination and THF-ring formation to give 218 with the full ring skeleton established. Finally, only redox manipulations were required to access limonin 13. TBAF-mediated de-protection of TES ether in 196 followed by Ley's oxidation60 completed the synthesis of (−)-13.
In Yamashita's synthesis, a well-designed Mn(OAc)3-promoted radical cyclization rapidly assembled the CDF ring system with the desired stereochemistry installed, thus definitely accelerating the synthesis of intact limonoid natural products. A clever O2-induced [4 + 2] cycloaddition followed by Ru-catalysed isomerization of endo-peroxide ingeniously assembled the required epoxide and subsequent lactone. The impressive Suarez reaction nicely fashioned the requisite ring AB system. Furthermore, some clever transformations (189 → 190 and 194 → 196) were also vital to the overwhelming success of their work.
3.3 Yang, Shen and Hao's synthesis of perforanoid A (2016)
Hao's group isolated a series of limonoids from Meliaceae and Simaroubaceae species, which showed diverse biological acitivities.61,62 In 2016, they further identified a new limonoid, perforanoid A 14, from this plant, which contains two lactone moieties, one all-carbon quaternary stereo-centre and a novel BCD tricyclic skeleton. Together with the isolation of perforanoid A 14, Yang, Shen and Hao disclosed their efforts toward the first total synthesis of this molecule involving Rh-catalysed Pauson–Khand reaction as the key transformation (Scheme 8).63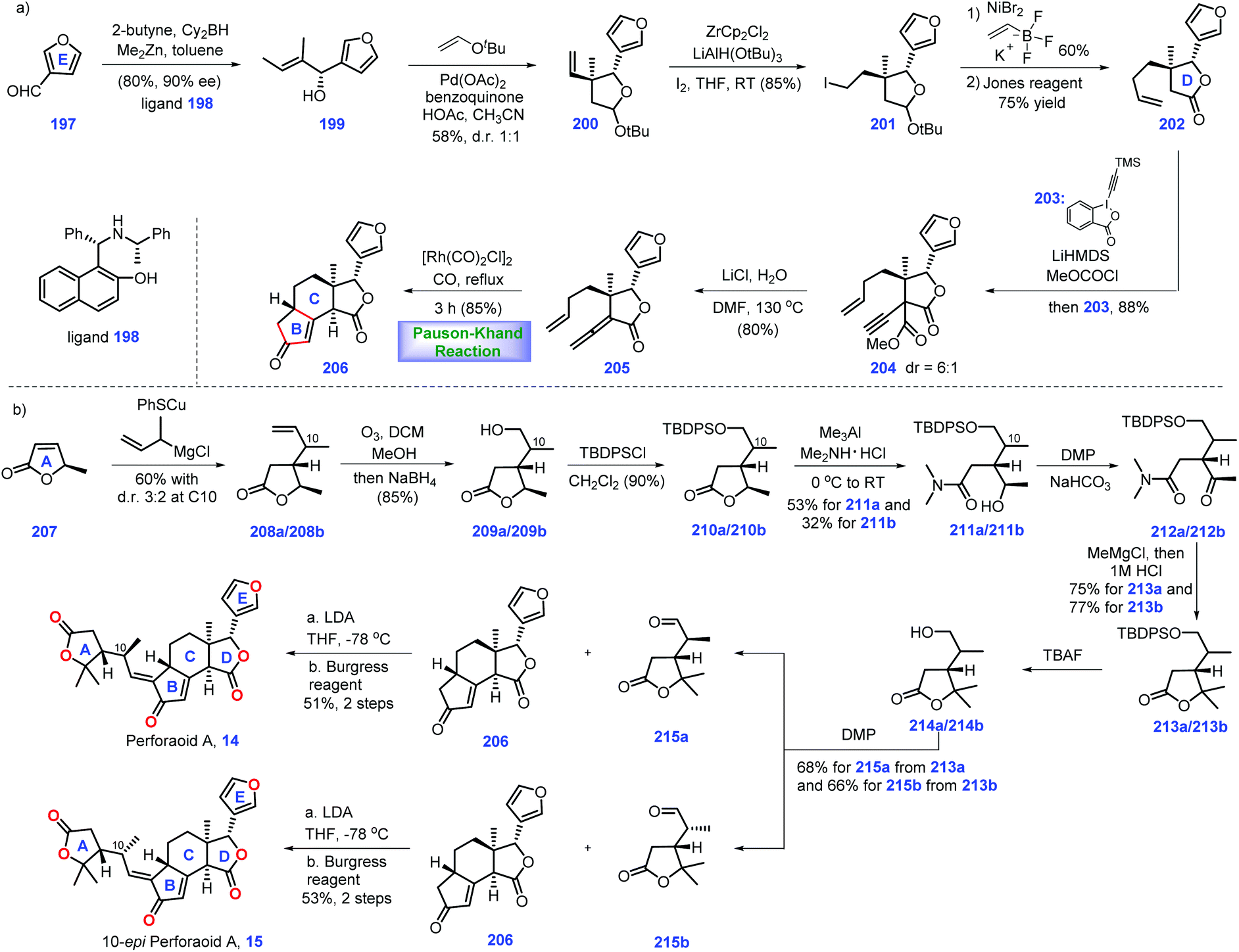 | ||
| Scheme 8 Yang, Shen and Hao's synthesis of perforanoid A (2016). | ||
Initially, their efforts focused on the establishment of the ring BCD system of the target (Scheme 8a). Treatment of furan-3-carbaldehyde 197 with 2-butenyl methyl zinc in the presence of chiral ligand 19864 initiated an enantioselective alkenylation reaction. The chiral secondary alcohol 199 was generated with good yield (80%) and high ee (90%). Taking inspiration from the work of Morken,65 treatment of 199 with 2-methyl-2-(vinyloxy)-propane under Pd-catalysed oxidative cyclization,66 [Pd(OAc)2/BQ/AcOH], formed 200 with ring D installed. Then 200 was advanced to iodide 201via Zr-mediated iodination. At this stage, the authors envisioned a cross-coupling reaction to introduce the terminal olefin. However, early efforts utilizing standard Suzuki, Negishi, and Kumada coupling conditions to transform iodide 201 into terminal olefin proved troublesome. Ultimately, it was found that Ni-catalysed coupling conditions67 could furnish the target olefin, which subsequently underwent Jones oxidation to deliver γ-butyrolactone 202. Moving forward, their goal was to set up an allene moiety. Exposure of 202 to excess LiHMDS generates a transient enolate, thus allowing the sequential coupling reaction with methyl carbonochloridate and Waser's reagent 20368 to furnish 204 as a 6:1 diastereomeric mixture. Note that the mixture of diastereomers in 204 proved inconsequential since both isomers could be converted into allene 205via decarboxylation.
In the key step of the synthesis, the authors speculated a metal-catalysed Pauson–Khand reaction strategy to create the ring BC system of the target. Pleasingly, subjecting allene 205 to Rh-catalysed Pauson–Khand reaction conditions {[Rh(CO)2Cl]2/CO/toluene/120 °C} furnished tetra-cycle 206 (85%). Note that a low reaction concentration (0.008 M) was crucial to this transformation.
With tetra-cycle 206 in hand, their next task was the construction of ring A system, lactone 215a/215b (Scheme 8b). The project commenced with the copper-mediated 1,4-addition of Grignard reagent to furanone 207, leading to adduct 208a/208b in 60% yield with a 3:2 dr value at C10. The mixture of 208a/208b was subjected to olefin ozonolysis followed by aldehyde reduction, furnishing corresponding alcohol 209a/209b, which was protected to TBS ether 210a/209b. With access to 210a/209b, AlMe3-mediated amidation with Me2NH·HCl allowed 211a (53%) and 211b (32%). To identify the stereochemistry of C10 in perforanoid A 14, the authors transformed 211a and 211b into 215a and 215b, respectively. Dess–Martin oxidation of 212a afforded ketone 212a, which set the stage for the subsequent one-pot addition of MeMgCl followed by acid-mediated lactonization, forming lactone 213a with ring A established. Notably, lactone 213b could also be prepared from 213bvia the abovementioned routes. Equipped with 213a/213b, redox manipulations were required to access 215a/215b. Removal of TBDPS ether in 213a followed by DMP oxidation gave rise to aldehyde 215a. Identical manipulations were conducted for the synthesis of 215a from 213b. It is worth noting that the aldehyde was obtained without epimerization under DMP oxidation. Moving forward, the authors employed Mosher's method to determine the absolute stereochemistry at C10 in 215a and 215b, which was S and R, respectively.
To complete the synthesis of perforanoid A 14, they sought to append lactone ring A onto tetra-cycle 206. Pleasingly, the authors found LDA-mediated aldol reaction of key fragment enone 206 with aldehyde 215b followed by dehydration accomplished perforanoid A 14. In a parallel experiment, 215b provided 10-epi-perforanoid A 15. Then the synthesized perforanoid A 14 was subjected to biological investigation, which demonstrated potent anti-tumor activities with IC50 values ranging from 3.91 to 6.17 μM.
In their synthesis, the successful and tactful utilization of the key PKR reaction ensured efficient completion of this synthesis in 10 steps. A clever Pd-catalysed oxidative cyclization strategy was also tactfully executed to establish ring D. The efficient PKR reaction together with elegant Pd-catalysed oxidative cyclization will undoubtedly enrich the synthetic strategy in limonoid synthesis.
3.4 Newhouse's synthesis of (±)-andirolide N and 8α-hydroxycarapin (2017)
Screening the extracts of flowers of a mahogany tree from the Amazonian rainforest (Carapa guianensis) by Tanaka and co-workers resulted in the isolation of andirolide N 16,69,70 a limonoid tetranortriterpenoid natural product, which featured a bicyclo[3.3.1]nonane core with bridged tetrahydrofuran ring system. In 2017, Newhouse and co-workers reported their efforts toward the total synthesis of (±)-andirolide N 16 and 8α-hydroxycarapin 17via intermolecular Diels–Alder reaction and stereoselective intramolecular Michael addition as the key protocols (Scheme 9).71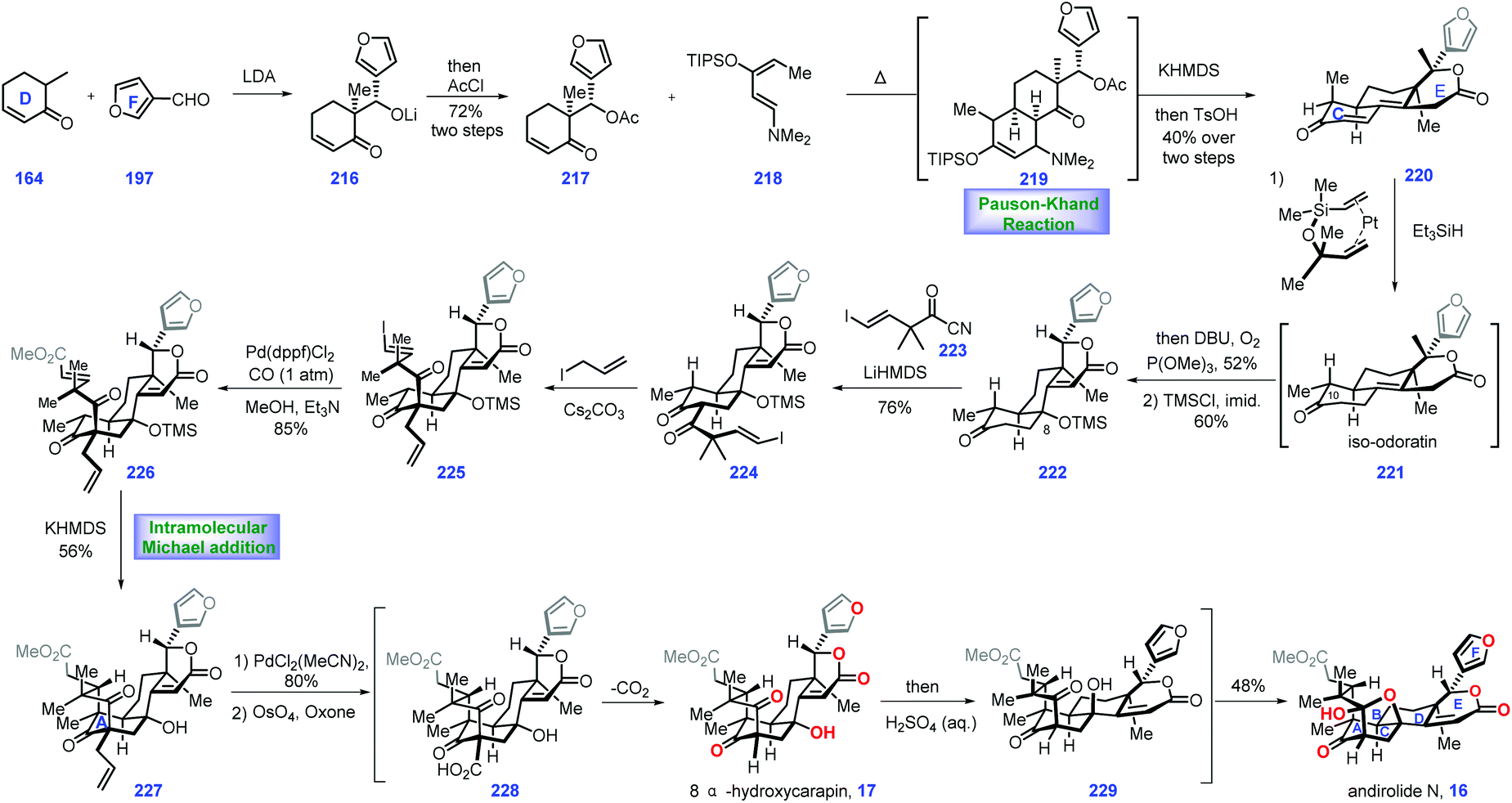 | ||
| Scheme 9 Newhouse's synthesis of (±)-andirolide N and 8α-hydroxycarapin (2017). | ||
The project started at the synthesis of iso-odoratin 221. Treatment of the known compound 16472 with LDA led to a transient lithium enolate, thus allowing a diastereoselective aldol reaction with 3-furaldehyde 197. The resultant alkoxide 216 was protected with AcCl to obtain furan 218, which served as a precursor for a thermal Diels–Alder reaction with Rawal-type diene 218. The cyclo-adduct 219 was generated as a mixture of diastereomers. Treating 219 with KHMDS triggered an intramolecular aldol reaction followed by β-elimination of the dimethylamino group. The resulting product was desilylated and dehydrated under TsOH to deliver lactone 220 as a mixture of diastereomers at C10 (1:1 dr), which proved inconsequential since both of these mixtures could be transformed into iso-odoratin 221 as a single diastereomer via a hydrosilylation reaction (Karstedt's catalyst, Et3SiH).73
With 221 in hand, their attention turned toward completion of (±)-andirolide N 16. Installation of a hydroxyl group at C8 in the limonoid framework posed a challenge in the semi-synthesis.74 Fortunately, the authors identified that subjecting the crude iso-odoratin 221 to alkene hydration conditions [P(OMe)3/DBU/O2] followed by alcohol protection furnished 222 with the desired C8 hydroxyl secured. With access to 222, they sought to construct the bicyclo-[3.3.1]nonane framework of the target. Exposing 222 to LiHMDS initiated the transient lithium enolate, which was in situ treated with acyl cyanide 223 to achieve di-ketone 224. The authors speculated that the devised acylation strategy75 minimized the retro-aldol and aldol condensation side reactions. Furthermore, it was found that the blocked α-position of the 1,3-diketone76 is crucial in the subsequent Michael addition to form the bicyclo-[3.3.1]nonane skeleton. After extensive attempts, the authors discovered that a carbon-based blocking group could be engaged in the formation of the bridged bicycle. In practice, 224 was processed through α-allylation to deliver 225. In this key transformation, the allylic group approached di-ketone 224 from the less hindered convex face, adopting the favored geometry for the synthesis of the bridged ring system.
Stepping forward, their next objective was to install the Michael addition precursor. Here, the authors devised a palladium-catalysed carbomethoxylation reaction, which successfully assembled 226 from 225. In the key step of this synthesis, exposure of 226 to KHMDS triggered an intramolecular Michael addition. The sterically congested 227 was generated with the bicyclo[3.3.1]nonane motif installed. With 227 in hand, a Pd-catalysed alkene isomerization77 followed by oxidative cleavage78 gave rise to 228, which served as a substrate for spontaneous decarboxylation, resulting in 8α-hydroxycarapin 17. To complete the synthesis of (±)-andirolide N 16, inversion of the tertiary alcohol was requisite. Pleasingly, exposing 17 to acidic aqueous conditions furnished the diastereomeric alcohol of 8α-hydroxycarapin, 229. The authors reasoned that a stabilized carbocation conjugated to the neighboring enolate accounted for this stereo-chemical inversion. Finally, ketalization of alcohol 229 completed the synthesis of the thermodynamically favored (±)-andirolide N 16.
In Newhouse's synthesis, both the stereoselective Diels–Alder reaction and well-designed intramolecular Michael addition enabled completion of the target in only 12 steps from commercially available materials. Some ingenious stereo-control reactions (224 → 225 and 17 → 229) were vital to the overwhelming success of this work. The advanced intermediate iso-odoratin 221 could be obtained in a batch of 15 g, which supplies sufficient materials to fulfil the target molecules.
3.5 Newhouse's synthesis of xylogranatopyridine B (2018)
In 2014, xylogranatopyridine B 18, a structurally unique limonoid identified from the leaves of a Chinese mangrove tree (Xylocarpus granatum),79 which featured a pyridine ring in its core. In 2018, Newhouse and co-workers reported the total synthesis of (−)-xylogranatopyridine B 18, involving aza-6π-electro-cyclization/oxidative aromatization as a key transformation (Scheme 10).80 Initially, they targeted an enone functionalization reaction, where reliable conditions were established to achieve the construction of oxime 234. Treatment of 3-methyl-2-cyclohexenone 230 with MeCu initiated the 1,4-addition. The stannane precursor facilitated the synthesis of xylogranatopyridine B. In practice, the project commenced with the resulting copper enolate in situ trapped with TMSCl to afford enoxysilane 231, which served as a precursor for Yamamoto's oxime formation reaction (AgNO2/TIPSCl),81 leading to α-keto oxime 232. Then, 232 was advanced to oxime 234via a two-step conversion, including ketone methylenation and O-benzoylation.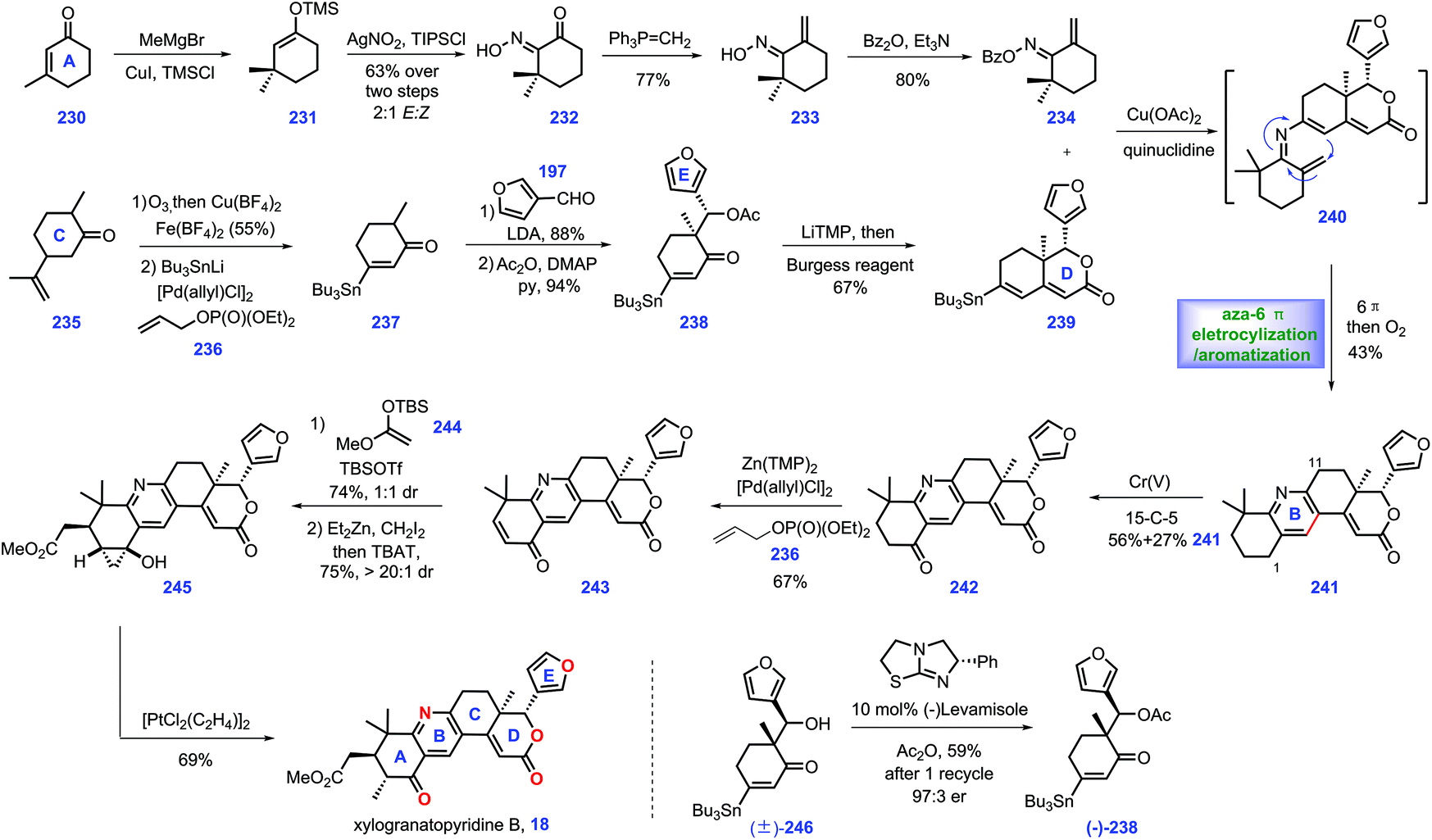 | ||
| Scheme 10 Newhouse's synthesis of xylogranatopyridine B (2018). | ||
Then, their synthetic efforts turned to assembly of fragment stannane 239. The project began with the commercially available dihydrocarvone 235. Ozonolytic fragmentation72 of 235 followed by Pd-catalysed oxidative stannylation82 generated stannane 237.
Exposure of 237 to LDA yielded lithium enolate, which was subsequently treated with 3-furaldehyde 219 to undergo a diastereoselective aldol reaction, followed by its capture by acylation to furnish 238. With 238 in hand, enone fragment 239 was assembled via LiTMP-promoted intramolecular aldol reaction followed by trapping with Burgess reagent.
With fragments 234 and 239 in hand, the authors envisioned an aza-6π-electrocyclization and aromatization strategy to install pyridine ring C. However, initial screenings to merge these two fragments via Chan–Lam coupling conditions83,84 proved challenging since oxime 234 and stannane 239 decomposed during the reaction. Fortunately, it was discovered that when the aforementioned amine additives85 were replaced with quinuclidine, the key aza-6π electrocyclization/aromatization ensued, thus forming 241 with the full ring system established. Notably, when each of the oxime isomers were separately evaluated in the Chan–Lam coupling, both reactions proceeded smoothly, which was attributed to the thermal isomerization of either 234 or 240 in the reaction. Accordingly, the authors applied the isomeric mixture of 234 in this transformation.
With installed 241, they next focused on developing conditions to achieve the selective oxidation of benzylic at C1 over C11. They speculated that the meta-position (C1) of pyridine was more electron-rich than its ortho-position (C11).86 Hence, various oxidants were evaluated to achieve this transformation. Ultimately, they identified that a Cr(V) complex87 was capable of engaging this chemo-selective oxidation, yielding ketone 242 in 56% yield together with 27% recovered 241. According to the protocol from their previous work,82 a Pd-catalysed α,β-dehydrogenation of ketone 242 afforded enone 243, which served as substrate for a Mukaiyama–Michael reaction with enoxysilane 244, resulting in a 1:1 diastereomeric mixture of enolate at C5. However, the subsequent capture of the transient enolate via methylation failed since the target 18 was less stable in basic conditions. Alternatively, they devised a Simmons–Smith cyclopropanation strategy, a neutral condition, to circumvent this issue. Exposure of the transient enolate to ZnEt2/CH2I2 initiated cyclopropanation with excellent diastereoselectivity (>20:1 dr). Then, the TBAT-promoted deprotection of the siloxycyclopropane gave a single diastereomer 245 together with a mixture of C5 epimers (combined 75% yield).
Finally, they conducted a cyclopropanol opening reaction via Zeise's dimer, [PtCl2(C2H4)]2,88 to complete xylogranatopyridine B 18. It is worth noting that they further prepared chiral acetate (−)-238 (43%, 94% ee) on a decagram scale via Birman's acylative kinetic resolution of alcohol (±)-246.89 The chiral (−)-238 secured (−)-xylogranatopyridine B 18. In Newhouse's synthesis, xylogranatopyridine B 18 was achieved via the longest linear 11 steps from commercially available materials. A credible aldol reaction was devised to join ring D and ring E. A novel enone functionalization strategy was devised to prepare the stannane compound (ring A), which was successfully utilized to merge with ring CDE via a Pd-catalysed Chan–Lam coupling reaction. The impressive aza-6π electrocyclization/oxidative aromatization nicely installed the core pyridine ring C, thus providing a reliable platform for the synthesis of other pyridine-based limonoids.
3.6 Newhouse's synthesis of (+)-granatumine A and related bislactone (2019)
In 2014, Guo and co-workers identified a bislactone limonoid alkaloid named granatumine A 22 from Chinese mangrove (Xylocarpus granatum). Biological investigation demonstrated that it exhibited moderate inhibitory activity against PTP1B.79 In 2019, Newhouse's group reported the first total synthesis of (+)-granatumine A 22 and related bislactone90 employing Pd-catalysed ketone α,β-dehydrogenation and convergent Knoevenagel condensation/carbonyl-selective electro-cyclization as key protocols (Scheme 11). At the beginning, the authors targeted azedaralide derivative 250 (Scheme 11a). Dehydrogenation of ketone 247 to 248via known literature procedures,101 such as Birch reduction or bromination/elimination methods,91 proved unsuccessful.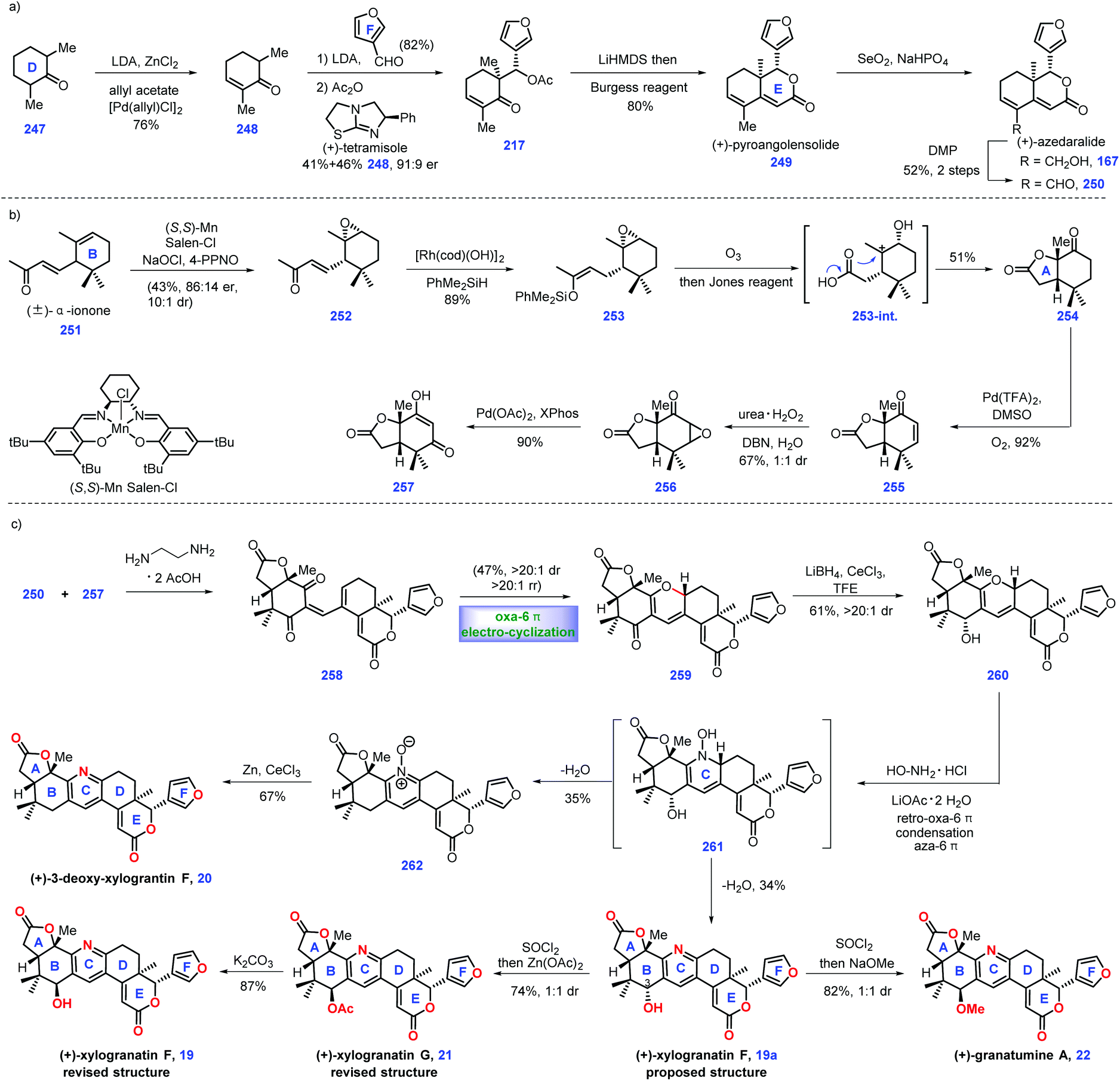 | ||
| Scheme 11 Newhouse's synthesis of (+)-granatumine A and related bislactone (2019). | ||
Inspired by their original work,82 palladium-catalysed α,β-dehydrogenation of ketone 247 successfully delivered enone 248. In this transformation, they successfully developed a less expensive reaction condition, which utilized allyl acetate as an oxidant, LDA as a base and 1 mol% loading of palladium salt as catalyst. Treatment of enone 248 with 3-furaldehyde under (+)-tetramisole89 induced an acylative kinetic resolution, leading to acetoxy 217 in 41% yield with 82% ee. Note that the undesired enantiomer could be recovered as 248via a retro-aldol reaction. LiHMDS-initiated intramolecular aldol reaction of 217 followed by dehydration (Burgess Reagent) furnished (+)-pyroangolensolide 249, which served as a precursor for the subsequent methyl site-selective allylic oxidation, thus generating (+)-azedaralide 167. Dess–Martin oxidation of the resulting alcohol in 167 formed aldehyde 250.
With aldehyde 250 in hand, the authors set about for the construction of 1,3-diketone 257 (Scheme 11b). Upon subjecting (±)-α-ionone 25192 to Jacobsen's epoxidation conditions,93 direct kinetic resolution ensued, thus providing the desired epoxide (−)-252 (43%, 86:14 er, 10:1 dr). [RhCl(OH)]2 catalysed the hydrosilylation94 of enone 252 to give enoxysilane 253. Treatment of 253 with ozone followed by Jones oxidation furnished ketone 254. The authors envisioned that intermediate 253-int was responsible for this transformation, where acid-mediated opening of the epoxide followed by its capture by carboxylic acid and oxidation of the resultant secondary alcohol occurred. Inspired by the work of Stahl,31 the Pd-catalysed oxidative dehydrogenation of 254 furnished enone 255, which was subjected to nucleophilic epoxidation conditions (urea H2O2, DBN), forming epoxide 256 as an inconsequential 1:1 diastereomeric mixture.
Here, they sought to install the core tetra-substituted pyridine ring of the target (Scheme 11c). In practice, they anticipated to utilize a Pd-mediated α,β-epoxy ketone opening reaction95 of epoxide 256 to construct which, however, 257 was unsuccessful. After extensive experiments, they found that phosphine ligands XPhos were capable of engaging this transformation, thus affording 257 in good yield (90%).
Pressing forward, key fragments di-ketone 257 and aldehyde 250 were then merged via Knoevenagel condensation96 to give enedione intermediate 258, followed by the vital oxa-6π electrocyclization97 under thermal conditions to install 2H-pyran 259 as the kinetically and thermodynamically favored product, which was further confirmed via DFT calculation. The C3 ketone in 259 was converted into allylic alcohol 260via site-selective Luche reduction. Notably, the utilization of less nucleophilic solvents, such as CF3CH2OH, was crucial in minimizing the decomposition of the reactants.
With alcohol 260 in hand, the authors then constructed the final pyridyl ring through treatment with hydroxylamine under reflux, giving the originally proposed structure of (+)-xylogranatin F 19a. Then, they turned their synthetic efforts toward (+)-granatumine A 22. Initially, their attempts to treat derivative 19a with methoxide via SN2 displacement proved challenging since an SN1 solvolysis pathway dominated in this transformation. Ultimately, the one-pot chlorination of benzylic alcohol 19a followed by methanolysis completed the synthesis of (+)-granatumine A 22. Although the pyridine N-oxide in 262 could be smoothly reduced98 to 3-deoxy-xylogranatin F 20, the following benzylic oxidation of 20 into (+)-xylogranatin F 19a proved troublesome due to the increased steric demand and enhanced deactivation energy by inductive effects. Note that the authors discovered the NMR structure prediction calculations were not in accordance with the originally proposed structure of xylogranatin F 19a. Accordingly, they sought to its chemical synthesis to address this unclarified issue. Learning from their proposed biosynthesis, they hypothesized that the correct structure was the C3-epimer of 19. In practice, the SOCl2-mediated chlorination of alcohol 19 followed by Zn(OAc)2-promoted substituted reaction completed the synthesis of (+)-xylogranatin G 21. Ester hydrolysis of 21 furnished 19, whose spectral data was in accordance with that reported for (+)-xylogranatin F 19.
In Newhouse's synthesis, the elegant utilization of Knoevenagel condensation/carbonyl-selective electrocyclization in conjunction with a modified Pd-catalysed ketone α,β-dehydrogenation provided an efficient and credible platform for the synthetic campaign of related limonoid molecules. A clever ring-opening of epoxide followed by trapping with carboxylic acid rapidly enabled the desired cis-lactone. Impressively, the key precursor 250 could be rapidly constructed in 6 steps, thus allowing the complete synthesis of (+)-xylogranatin F 19 in 10 steps.
4. Highly decorated limonoids and limonoid-like natural products
4.1 Watanabe's formal synthesis of azadirachtin (2015)
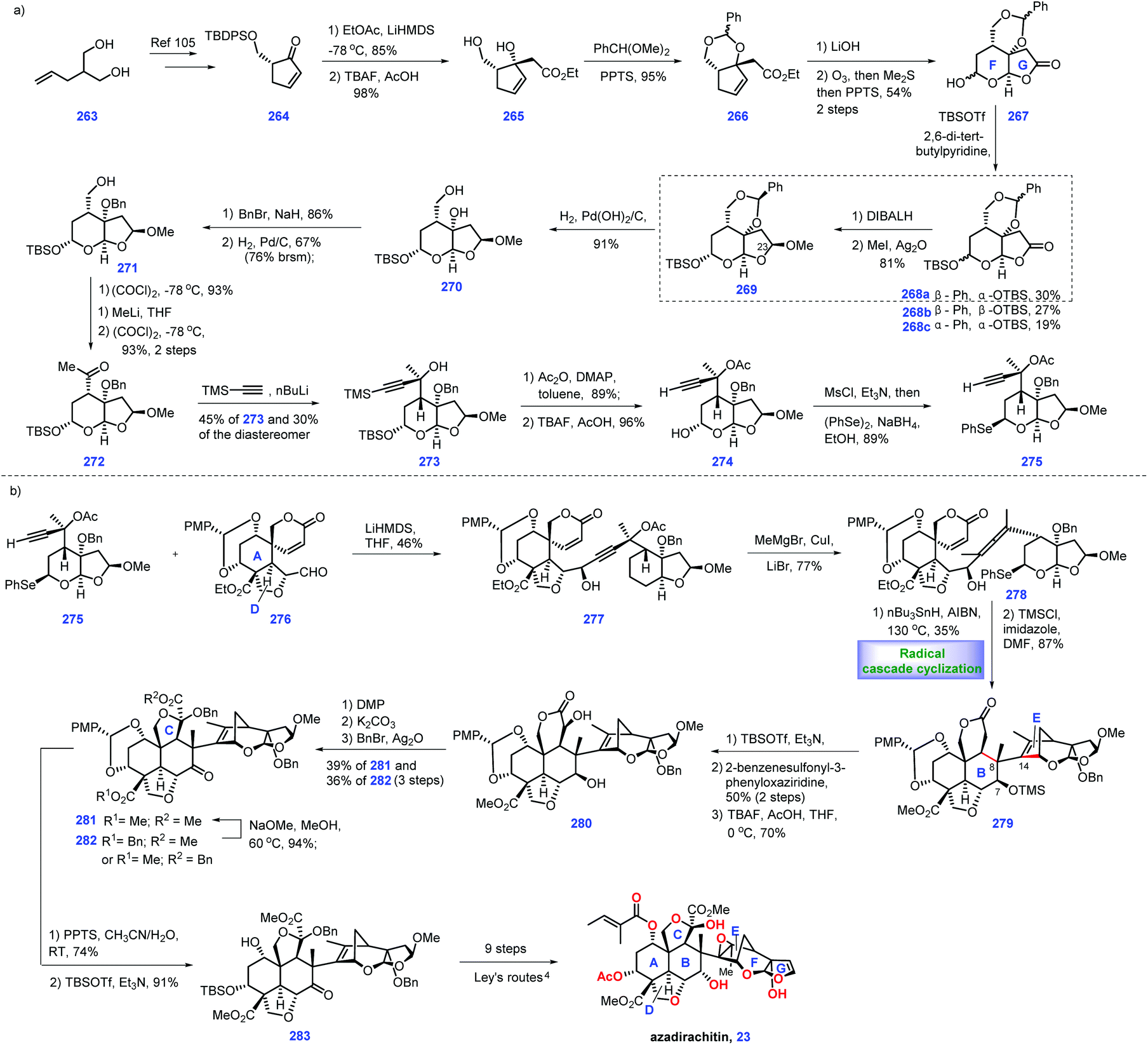
In 1968, azadirachtin 23 was isolated from the neem tree Azadirachta indica A. Juss (Meliaceae). It featured sixteen contiguous stereogenic centers with multiple oxygen-containing functional groups.99 However, its accurate structure was not determined until 1987.100 Biological evaluations have demonstrated that it possesses antifeedant and growth disruption properties against insect species.101 The key challenge in the synthesis of 23 involves the installation of the sterically hindered C8–C14 bond. Numerous synthetic endeavours for azadirachtin 23 have been demonstrated.102 However, only Ley's group achieved its landmark total synthesis in 2007, which has been discussed in Heasley's review (2011).102 In 2015, Watanabe and workers disclosed another protocol to fulfil the asymmetric formal synthesis of azadirachtin 23, involving a novel radical initiated cascade cyclization reaction as a key step to install the challenging C8–C14 bond (Scheme 12).103 Knowing that Watanabe's synthesis of fragment 276104 has been discussed in Heasley's review,2 it will not be duplicated here.
 | ||
| Scheme 12 Watanabe's formal synthesis of azadirachtin (2015). | ||
Watanabe's efforts to construct fragment 275 commenced with diol 263, which was first converted into the known (R)-264105 (Scheme 12a). 264 served as substrate for an aldol reaction with ethyl acetate to arrive at 265 (Scheme 16a). Initial experiments to protect the tertiary alcohol with benzyl ether proved troublesome. In comparison, the authors devised an alternative route where the TBS ether was deprotected followed by masking the resultant diol as cyclic acetal 266. Hydrolysis of ester 266 resulted in an acid product, thus allowing ozonolysis of the double bond, followed by PPT-promoted hemi-acetylation. Compound 267 was obtained with four inseparable diastereomers. Treatment of 267 with TBSOTf afforded the desired 268a and undesired 268b/268c. The authors noted that a bulkier base, 2,6-di-tert-butylpyridine, played a key role in generating the product with good yield.
Stepping forward, the authors planned to establish conditions for the conversion of the lactone moiety into methyl acetal. According to the protocol of Ley's work,4 utilization of β-acetal at C23 268a was engaged in a one-pot reduction/methylation to give 269. However, treating 268b and 268c with the same conditions as 268a only gave the products as 1.1:1 and 1.3:1 diastereomeric mixtures, respectively. Initially, exposure of 269 to DIBAL failed to give 271. Instead, 269 could be processed through three additional manipulations, involving Pd-catalysed de-protection of cyclic acetyl, benzyl protection of diol and selective de-protection of primary alcohol to form 271. Sequential Swern oxidation of the primary alcohol, addition of MeLi to the aldehyde and Swern oxidation of the second alcohol afforded ketone 272. Reacting 272 with lithium trimethylsilylacetylide delivered tertiary alcohol 273 (45%), together with its diastereomer (30%). 273 was treated with Ac2O and the resultant product was subjected to the global deprotection of silyl ether to yield 274. Mesylation of the alcohol in 274 followed by displacement with a phenylseleno group provided fragment 275.
Pressing forward, their goal was to incorporate fragment 276 and selenium 275 (Scheme 13b). Treatment of 275 with LiHMDS generated lithium acetylide, thus allowing for a nucleophilic addition with aldehyde 276.104 Pleasingly, the coupling product 277 was obtained in 46% yield. Exposing 277 to a methyl-copper reagent triggered an SN2′ reaction, furnishing allene 278,106,107 which functioned as a radical cyclization precursor. After extensive investigation, the authors discovered that DMF was the optimal solvent for this reaction.
 | ||
| Scheme 13 Li's synthesis of rubriflordilactone A (2014). | ||
Here, their next task was to develop conditions for the crucial radical cyclization. After extensive screening, the authors eventually identified the conditions (nBu3SnH, AIBN, DMF/130 °C) was capable of engaging the key radical cyclization, leading to the cyclized product with the key C8–C14 bond established. TMS protection of the alcohol in above cyclized product generated 279, which could be utilized as a substrate for treatment with TBSOTf, thus providing the resultant silyl ketene acetal compound, which was subsequently oxidized by the Davis reagent to form the alcohol product. The authors noted that the free hydroxyl group at C7 resulted in the decomposition of the reactants. Subsequently, global removal of the silyl ether protecting group provided 280. DMP oxidation of 280 followed by methanolysis furnished the cyclic hemi-acetyl product, which was further treated with BnBr/Ag2O to furnish 281 together with benzyl ester 282. It is notable that 282 could be recycled to 281via methanolysis. In the final stages of the synthesis, PPTS promoted deprotection of p-methoxy-benzylidene acetal followed by the site-selective protection of C3 alcohol with TBSOTf to complete 283. According to the protocol of Ley's work,4283 could be advanced to azadirachtin 23via a 9-step sequence. One contribution of Watanabe's synthesis was the development of a rapid route to obtain the optically active right-hand segment. Their effort qualifies as a landmark in the synthesis of azadirachtin due to its successful connection of the highly steric ring B and ring E system via a genius key intramolecular tandem radical cyclization.
4.2 Li's synthesis of rubriflordilactone A (2014)
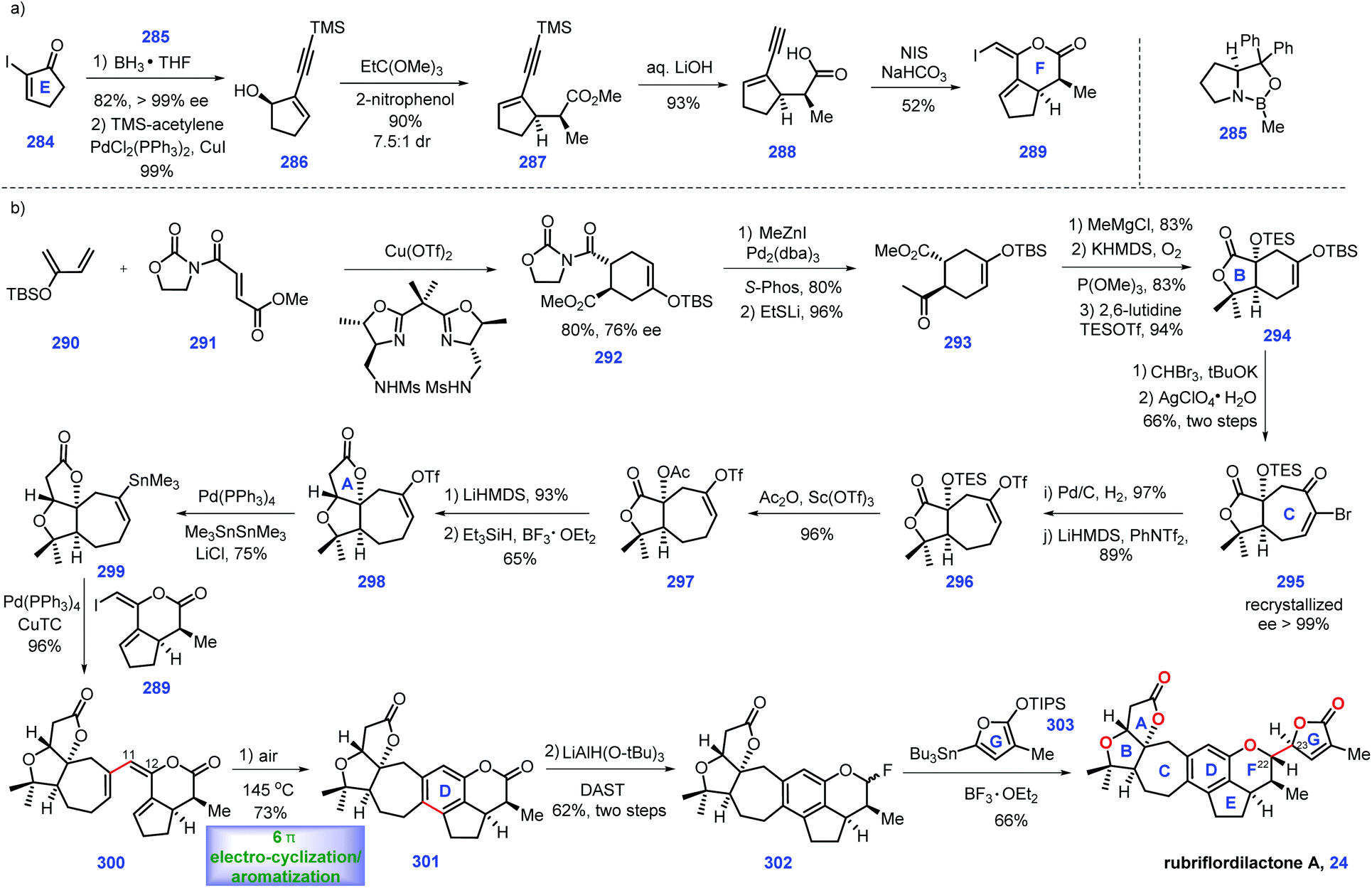
Some of nortriterpenoids can be considered as limonoid-like natural products, which are widely distributed in Schisandra plants.108 They have attracted increasing synthetic attention from the researchers109 due to their diverse and excellent biological activities. Yang's group has made significant progress in the total syntheses of several schinortriterpenoids,110–113 including schindilactone A (2011),110 propindilactone G (2015),111 lancifodilactone G acetate (2017)112 and 19-dehydroxyarisandilactone A (2017),113 all of which were highlighted in their recent report (2019).114 Hence, we will not duplicate the abovementioned works in this review.In 2006, Sun and co-workers reported isolation of rubriflordilactones A and B from Schisandra rubriflora,115 which have widespread use in traditional Chinese medicine to treat illness. Structurally, they feature a 5/7/5/5/5-fused pentacyclic ring system. Biological investigation showed that rubriflordilactone B 32 was found to show potent anti-HIV activity.
In 2014, Li and co-workers described the total synthesis of rubriflordilactone A 24 (Scheme 13).116 According to their successful experience117 in utilizing the 6π-electrocyclization/aromatization strategy to install the complex natural products, they managed to employ this protocol in the synthesis of rubriflordilactone A 24. Their efforts began by forming fragment 289 (Scheme 13a). CBS (285)-mediated 1,2-reduction of enone 284 followed by Sonogashira coupling with TMS-acetylene gave 286. Subjecting alcohol 286 to John–Claisen rearrangement conditions118 [2-nitrophenol (2 mol%), EtC(OMe)3, 100 °C] gave methyl ester 287, which was employed as a substrate for a saponification reaction, leading to acid 288. Stereospecific iodolactonization119 of 288 transformed it into E-alkene 289 with the desired ring F installed. The authors noted that 289 should be prepared in situ since it is unstable when preserved.
Equipped with 289, their goal was to assemble fragment 299 (Scheme 13b). Treating diene 290 with dienophile 291via a Cu-catalysed asymmetric Diels–Alder reaction120 furnished chiral 292 with 76% ee. LiSEt-mediated removal of the oxazolidinone moiety in 292 followed by a Pd-catalysed Negishi cross coupling reaction121 with Me2Zn resulted in ketone 293. Inspired by Yang's synthesis of schindilactone A,110293 was then processed through three additional operations, involving nucleophilic addition of methyl Grignard reagent to ketone followed by in situ lactonization, α-hydroxylation of lactone and TBS protection of the resultant alcohol to form lactone 294. 294 was then converted into bromoenone 295via a two-step transformation. Notably, 295 could be recrystallized to receive the near optically pure 295 (>99% ee). Subjecting enone 295 to 1,4-reduction conditions (Pd/C, H2) allowed in situ debromination to transform it into cycloheptanone which served as a precursor for a triflation reaction, thus generating 296 with excellent regioselectivity. The authors speculated that the steric difference between the α and α′ positions of the carbonyl accounted for this selectivity. Replacement of the TES ether in 296 with an acetate group yielded acetate 297, thus setting the stage for intramolecular Dieckmann condensation. BF3·OEt2/Et3SiH-mediated122 deoxygenation of the resultant hemi-ketal species gave 298. With access to 298, Pd-catalysed Stille cross-coupling reaction123 with Me3SnSnMe3 afforded stannane 299.
Equipped with 299 and 289, the authors devised a coupling reaction to merge fragments 299 and 289. After extensive screenings, they identified that Pd(0)/CuTC124 was capable of engaging the Stille-Migita reaction, providing target triene 300 in high yield (96%). Here, their goal was to develop conditions to fulfil the key π-electrocyclization/aromatization protocol. The authors envisioned that rigid E-geometry of the C11–C12 bond was crucial to the cyclization. Pleasingly, exposure of triene 300 under heat at air atmosphere triggered cascade 6π-electrocyclization/aromatization, thus forming arene 301. LiAlH(Ot-Bu)3125 mediated the regioselective reduction of C26 ketone, which generated a mixture of 301 together with its ring-opened aldehyde. With access to 301, the final step was to attach the lactone motif appendage onto C22. However, exposure of the abovementioned mixture 301 with siloxy furan did not deliver the coupling product but an open-chain alcohol. The author reasoned that the ring strain of the 6,5,6-tricyclic system in conjunction with the easily dissociated tendency of the phenoxide moiety accounted for this outcome. Here, they added DAST126 to this mixture of lactol/aldehyde, which successfully gave fluoride 302. Meanwhile siloxy furan was elaborated to the electron-richer donor stannane 303.127 Ultimately, the treatment of fluoride 302 with stannane 303via BF3·OEt2-mediated128 SN2 displacement gave rubriflordilactone A. To confirm the stereochemistry of the final step, a model reaction was conducted, and the resultant product was confirmed by X-ray crystallographic analysis.
In Li's synthesis, the authors showcased the powerful convergent synthetic strategy in the quick assembly of complex molecules. Relying on the authors' insightful consideration, the tricky lactone F was successfully incorporated onto the core substructure. It is of great significance that the elegant application of the 6-π-electrocyclization/aromatization in this synthesis demonstrates the viability of this strategy in total synthesis.
4.3 Anderson's synthesis of (+)-rubriflordilactone A (2015)
After Li's synthesis (2014), Anderson and co-workers also reported the total synthesis of (+)-rubriflordilactone A10924 (2015). Critical to the success of their endeavour was the strategic use of palladium- or cobalt-catalysed cyclotrimerization as key steps (Scheme 14).115,129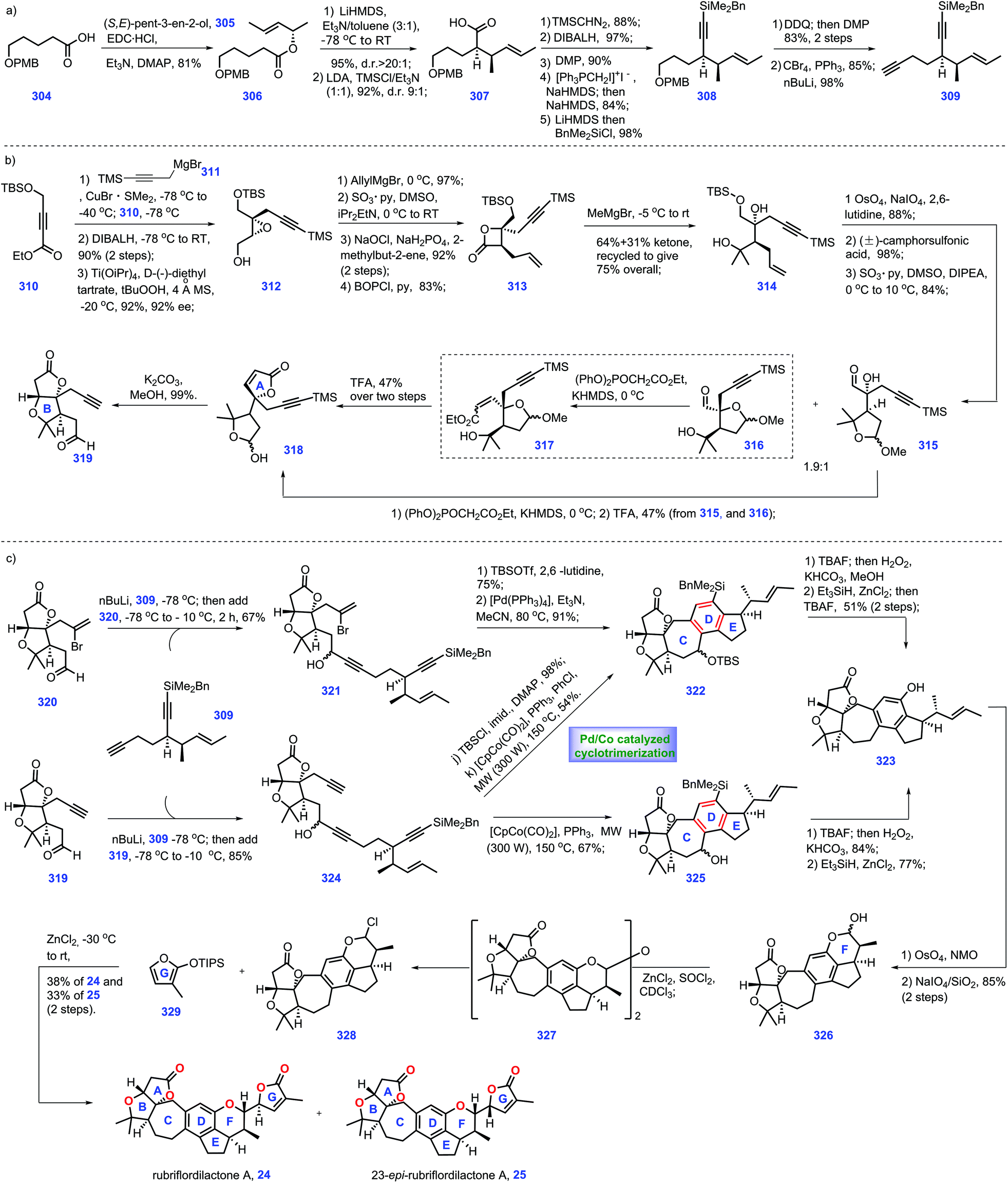 | ||
| Scheme 14 Anderson's synthesis of (+)-rubriflordilactone A (2015). | ||
The project started with the construction of fragment terminal alkyne 309 (Scheme 14a). Esterification of (S,E)-pent-3-en-2-ol 305 with carboxylic acid 304 gave ester 306, which was subjected to Ireland–Claisen rearrangement conditions followed by workup. The target acid 307 was obtained in high yield (96%) and excellent diastereoselectivity (dr > 20:1). Then 307 was transformed into benzyldimethylsilyl alkyne 308,116 the para-methoxybenzyl ether moiety of which was subsequently converted into terminal alkyne 309.
With fragment 309 in hand, they turned their attention to the synthesis of bicyclic alkyne-bearing aldehyde 319 (Scheme 14b). Their efforts began with ester 310, which could be transformed into epoxide 312via three additional operations involving alkyne carbocupration,33,130 ester reduction and Sharpless asymmetric epoxidation.131 Then epoxide 312 was advanced to lactone 313 through epoxide ring-opening, alcohol oxidation and β-lactonization. Treatment of 313 with methylmagnesium bromide initiated double nucleophilic addition, affording 314 with the gem-dimethyl group of the ring B installed. Alkene 314 was then processed through alkene oxidative cleavage, acetalization and alcohol oxidation to deliver aldehyde regioisomers 315 and 316. Although this transformation was generated as a mixture of acetyl isomers, it proved inconsequential since both isomers could be converted into aldehyde 319. In a real experiment, upon treatment of aldehyde 315 with (PhO)2POCH2CO2Et, Ando olefination ensued, thus affording the alkenyl product, which served as a precursor for the acid-promoted hydrolysis of the acetal. The desired 318 was secured. Meanwhile conversion of 316 into lactol 317 was accomplished by Ando olefination.132 TFA-mediated hydrolysis of acetal in 317 followed by lactonization led to aldehyde 318. Exposure of 318 to base conditions (K2CO3/MeOH) initiated intramolecular oxy-Michael addition spontaneous lactonization to form 319 with ring AB established. Note that bromide 320 could also be prepared via the abovementioned protocol.
Stepping forward, their next task was to merge fragment 320 with 309 (Scheme 14c). Exposure of diyne 309 to nBuLi followed by treatment with aldehyde 319 furnished alcohol 321. Here, they arrived at the key step of the synthesis. Inspired by their previous work,133 protected propargylic alcohol was crucial to enable this key cyclization. Accordingly, TBS protection of the free alcohol in 321, followed by a Pd-catalysed cyclization reaction installed the core ring ABCDE pentacycle 322, which was then advanced to the fully functionalized 323via phenol oxidation and benzyl deoxygenation.134 Alternatively, the authors also envisioned a cobalt-catalysed cyclotrimerization strategy.135 In practice, treating diyne 319 with aldehyde 320 gave rise to triyne 324, thus allowing a subsequent cobalt-catalysed cyclotrimerization reaction under microwave heating, which led to pentacycle 325. 325 could also be processed through a two-step process via Tamao oxidation followed by benzylic deoxygenation to arrive at penta-cycle 323. Meanwhile, 324 could also be advanced to 322via silylation of the alcohol followed by cobalt-catalysed cyclotrimerization.
With access to penta-cycle 323, what remained was to install the ring FG system of the target. A two-step oxidative elaboration of 323 furnished lactol 326, which was subsequently treated with a mixture of SOCl2/ZnCl2, thus generating chloropyran 328, presumably via the quick formation of dimeric intermediate 327. To complete the synthesis, a butenolide motif should be attached onto the core structure, chloropyran 328. Pleasingly, treatment of 328 with siloxyfuran 329 under zinc(II) chloride136 secured the desired cross-coupling reaction, thus providing rubriflordilactone A 24 (38%) together with its C23-epimer 25 (33%). In Anderson's synthesis, it is of significance that several stereocentres were wisely installed via credible transformations, such as substrate-controlled Claisen rearrangement and hydroxyl-induced titanium-catalysed asymmetric epoxidation, both of which enabled the preparation of the chiral building block in a large quantity. An ingenious metal-catalysed cyclotrimerization efficiently assembled the ring CDE system which is believed to pave the way for achieving other related natural products.
4.4 Tang's synthesis of (+)-rubriflordilactones B and C (2015)
In 2012, Sun's group identified schilancitrilactones B (26) and C (27) from the stems of Schisandra Lancifolia.137 Biological investigation revealed that schilancitrilactone C demonstrated good inhibiting activity toward HIV-1, while schilancitrilactone B was inactive. In 2016, Tang and co-workers described the first total synthesis of schilancitrilactones B and C. Critical to the success of this synthesis is the beautiful use of intramolecular radical cyclization to assemble a seven-member ring, late-stage iodination, and intermolecular radical C–C bond formation (Scheme 15).138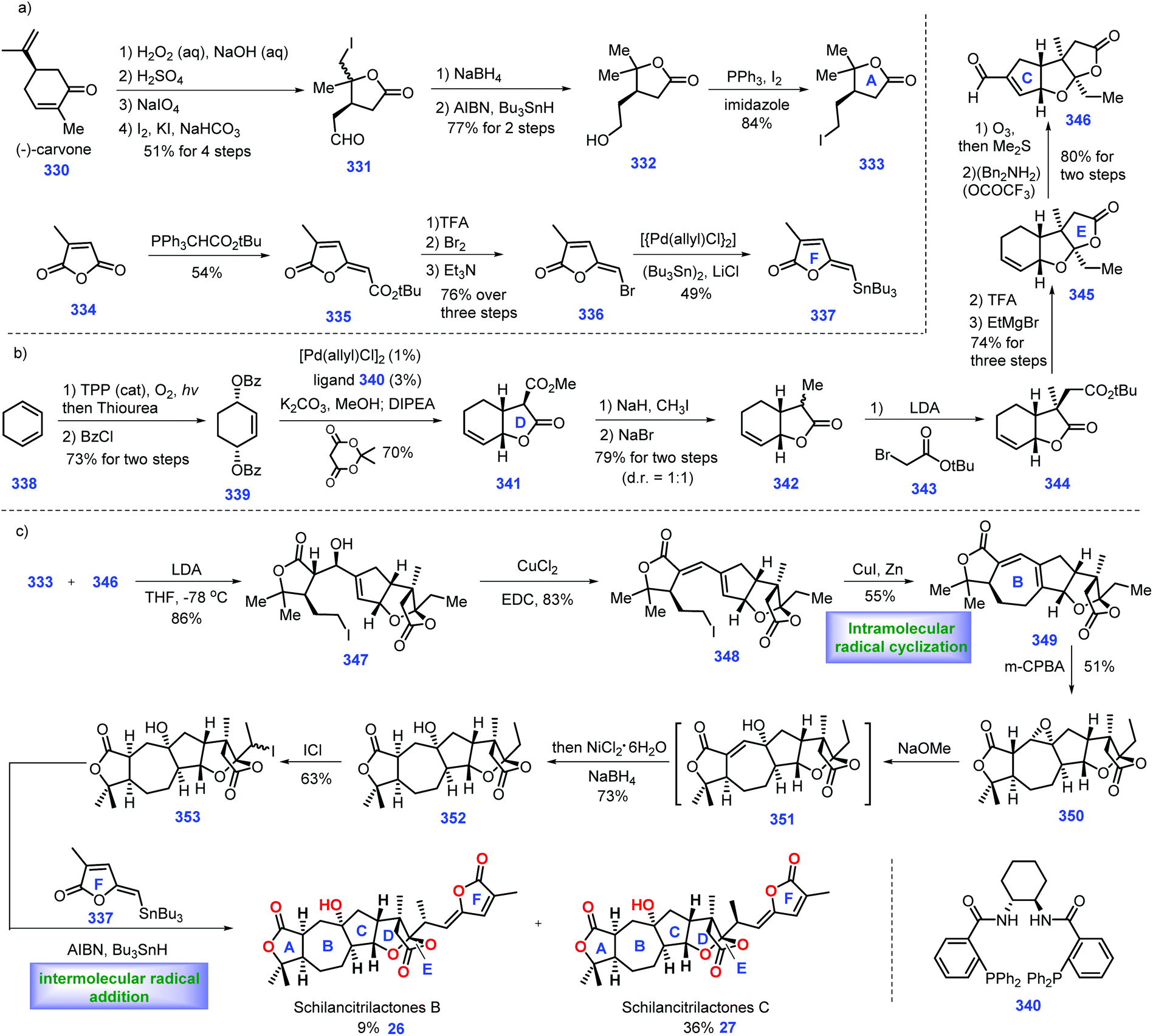 | ||
| Scheme 15 Tang's synthesis of schilancitrilactones B and C (2015). | ||
Their synthetic efforts began with the construction of alkyl iodide 333 (Scheme 15a). Inspired by the work from Fukuyama's group,139L-carvone (330) was first converted into aldehyde 331via a four-step manipulation,140 including enone epoxidation, epoxide hydrolysis, diol oxidative cleavage and alkene iodo-lactonization.
Reduction of aldehyde 331 gave an alcohol compound, which served as a substrate for deiodination reaction under AIBN/Bu3SnH. The resulting 332 was obtained in 90% yield. 332 was then subjected to Appel conditions to produce iodide 333.
With 333 in hand, the authors turned their attention toward the construction of lactone 337 (Scheme 15a). Initially, citraconic anhydride was accessed through a four-step reliable transformation to generate bromide 336. Pd-catalysed stannylation141 of 336 with (Bu3Sn)2 led to stannane 337.
Moving forward, they next planned to establish fragment 346 (Scheme 15b). Taking inspiration from the work of Trost,142 1,3-cyclohexadiene 338 was funnelled into lactone 341via a three-step sequence. 341 was then exposed to NaH/CH3I, inducing C23 methylation, then followed by an NaBr-triggered decarboxylation reaction. The resulting 342 was accessible in 79% yield (dr = 1:1).
Exposure of 342 to LDA generated transient lithium enolates, which in situ reacted with tert-butyl bromoacetate 343via alkylation to furnish 344. Trifluoroacetic acid-mediated hydrolysis of the esters followed by treatment with ethyl magnesium bromide yielded 345 with the desired ethyl group installed. Ozonolysis of 345 followed by intramolecular aldol condensation produced aldehyde 346 with ring C installed.
Equipped with fragments 333, 337, and 346, the authors then developed conditions for the total synthesis of schilancitrilactones B 26 and C 27 (Scheme 15c). The authors first envisioned to merge 333 and 346. In practice, exposing iodo compound 333 under LDA followed by treatment with aldehyde 346 generated alcohol 347 with an excellent dr value (17:1). Then CuCl2-mediated dehydration143 of 347 formed enone 348 as a 2:1 inseparable mixture, thus setting the stage for the key radical cyclization. Early screening to employ AIBN/nBu3SnH only resulted in the decomposition of 348. Besides, photoredox catalysis144 was also found to be unfeasible. Ultimately, they discovered that Luche's condition145 (CuI, Zn under ultrasound) was capable of being converted 348 into the desired cyclized product 349 with the formation of ring B. m-CPBA-mediated epoxidation of 349 resulted in 350, which served as precursor for a cascade ring opening reaction/reduction (NaOMe, NiCl2·6H2O/NaBH4), thus constructing alcohol 352. In the final stage of the synthesis, the authors focused on appending a lactone motif onto the advanced core 352. ICl-mediated iodination146 at C20 in 352 provided iodo 353, which was further treated with stannane 337 under AIBN/Bu3SnH to complete the synthesis of schilancitrilactones B (26, 9%) and C (27, 36%).
In Tang's synthesis, the seven-membered ring was assembled brilliantly by judicious application of an intramolecular radical cyclization. A tactful late-stage iodination set the stage for the following clever intermolecular radical addition reaction, thus nicely appending the lactone motif. The above impressive strategy provides an attractive and viable alternative to the challenging late-stage connection of the fully functionalized core and lactone motif in the synthesis of complex limonoid-like natural products.
4.5 Li's synthesis of (+)-rubriflordilactone B (2016)
Following their brilliant work on the synthesis of (+)-rubriflordilactone A 24,116 Li and co-workers further documented their efforts toward the first and asymmetric total synthesis of (+)-rubriflordilactone B 32.147 Critical to the success of their endeavour was the strategic use of the 6π-electrocyclization–aromatization strategy as the key step (Scheme 16).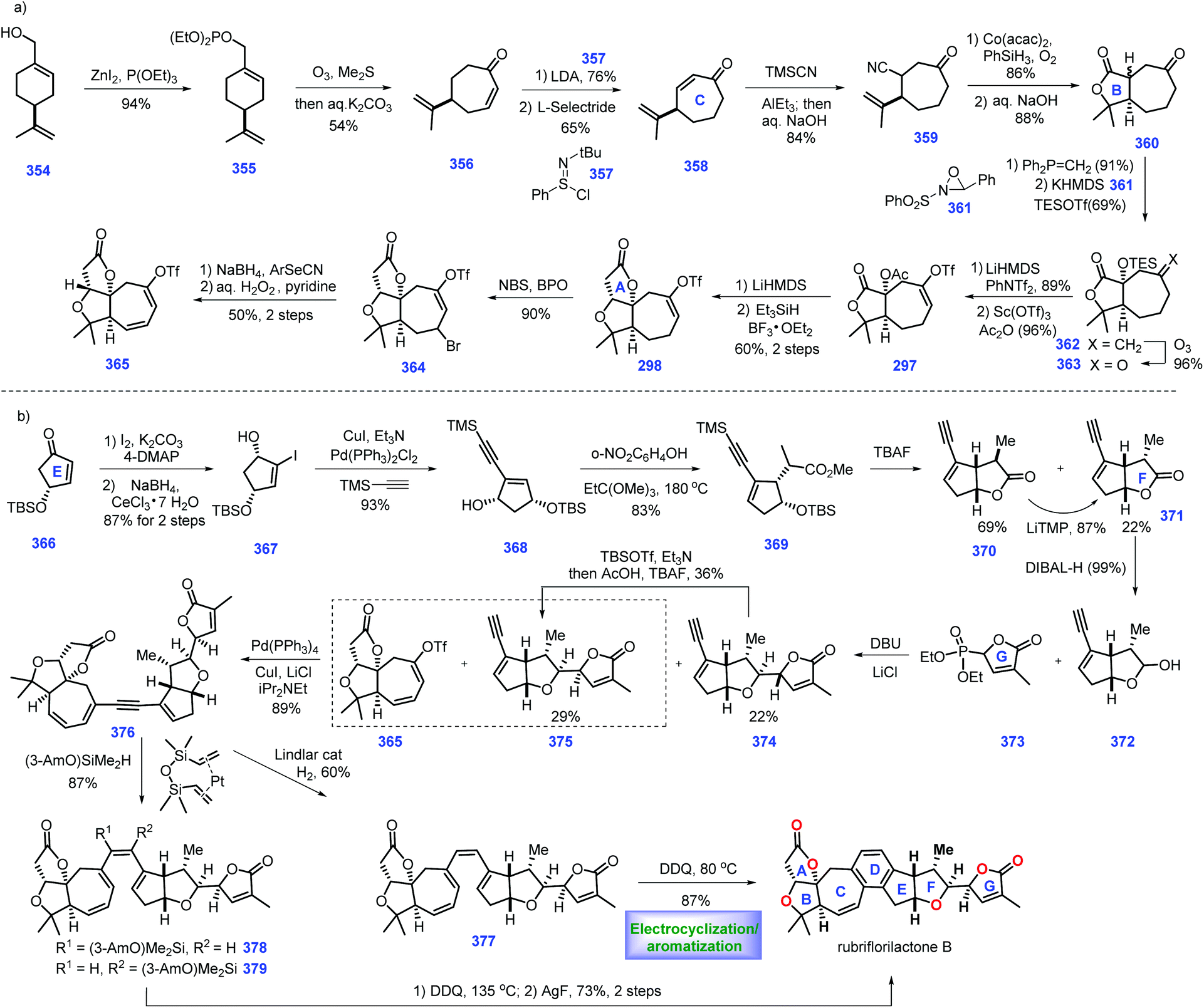 | ||
| Scheme 16 Li's synthesis of (+)-rubriflordilactone B (2016). | ||
Their pursuit of 32 began with the preparation of fragment 365 (Scheme 16a). Subjecting 354 to Arbuzov-type reaction conditions gave rise to phosphonate 355, which served as substrate for an ozonolysis and intramolecular HWE reaction. The resulting product, cycloheptenone 356, was obtained in 54% yield. Mukaiyama dehydrogenation148 of 356 generated dienone, which was followed by L-selectride-induced regioselective 1,4-reduction, thus giving the desired enone 358. Exposure of 358 to in situ-prepared Et2AlCN followed by base treatment furnished nitrile 359. Mukaiyama hydration of 359 provided a tertiary alcohol,24 which served as a precursor for a lactonization reaction under base conditions to transform it into lactone 360 with ring B installed. Notably, a low concentration of 359 was vital for inhibiting the undesirable bimolecular radical addition process. With 360 in hand, sequential ketone methylenation, α-hydroxylation of the lactone enolate and silylation of the alcohol gave 362 over three steps. Exposing this material to ozonolytic conditions afforded ketone 363. Triflation (KHMDS, PhNTf2) of 363 followed by displacement of silyl ether with an acetate group gave 297. LiHMDS-mediated intramolecular Dieckmann condensation of 297 followed by BF3·OEt2-induced 1,4-reduction furnished lactone 298 with the ring A formed. Subjecting 298 to the conditions of NBS/BPO/85 °C triggered radical bromination at the less hindered C7, thus generating bromide 364, which served as a precursor for selenidation, followed by oxidation and elimination reaction to afford diene 365.
With diene 365 in hand, their attention then turned to assembly of fragment 375 (Scheme 16b). In practice, iodination of known enone 366 followed by Luche reduction delivered alcohol 367. Pd-catalysed Sonogashira coupling of 367 with TMS–acetylene led to 368, which was utilized as a precursor for a Johnson–Claisen rearrangement149 to furnish ester 369. TBAF-mediated cascade desilylation/lactonization of 369 resulted in a pair of chromatographically separable epimers, 370 and 371. Fortunately, the authors found that the undesirable 371 could be converted into 370. DIBAL-H induced reduction of 370 generated lactol 372, which was treated with phosphonate150 under Masamune–Roush conditions to construct a pair of epimers, 374 and 375 with ring G system attached. Pleasingly, the undesired 374 could be partially advanced to 375, which functioned as a coupling precursor.
Subsequently, the authors sought to develop conditions for the completion of rubriflordilactone B 32. In practice, the authors first attempted to connect key fragments 365 with 375via a Pd-catalysed Sonogashira coupling reaction, which readily provided triene-yne 376 in 89% yield. However, initial efforts to semi-hydrogenate 376 suffered from low reaction reproducibility and less product stability. Here, hydrosilylation of 376 using Karsted catalyst151 was successfully introduced, thus allowing 378 and 379 to be obtained as a 1:1 mixture. This proved inconsequential since both mixtures could be converted into rubriflordilactone B. In a real experiment, upon subjecting 378 and 379 to heat (135 °C), 6π-electrocyclization/aromatization ensued, thus providing mixed penta-substituted arenes, which were then desilylated via AgF to complete the target with the key ring B generated. However, the authors discovered the spectra of the synthetic product were not identical to the authentic material. Although they further synthesized 23-epi-rubriflordilactone B, its spectra also did not match that of the isolated material. From a strategic perspective, the clever electrocyclization–aromatization sequence exemplified in this work is efficient and tactful, which provides a creative strategy to assemble a benzene ring in complex molecules. The successful utilization of the hydrosilylation approach is critical to the overwhelming success of this process. Although the synthetic products did not match the authentic material, the established strategies in this work showcase the potent access to various diastereomeric structures of 32, which may help elucidate the true structure of isolated pseudo-rubriflordilactone B in the future.
4.6 Tang's syntheses of schilancidilactones A and B, schilancitrilactone A, and 20-epischilancitrilactone A (2017)
In 2009, Sun's group identified schilancidilactones A and B and schilancitrilactone A (1–3), from the stems of Schisandra lancifolia.137 Schilancidilactone A was reported to demonstrate inhibiting activity toward HIV-1, while schilancitrilactone A exhibited antifeedant activity. Following their previous successful work on the total syntheses of schilancitrilactones B 26 and C 27,138 Tang's group further disclosed their synthetic efforts toward the total synthesis of schilancidilactones A 28 and B 29, schilancitrilactone A 30, and 20-epi-schilancitrilactone A 31, involving a late-stage nickel-catalysed cross coupling strategy as a novel step (Scheme 17).152 | ||
| Scheme 17 Tang's synthesis of schilancidilactones A and B, schilancitrilactone A, and 20-epischilancitrilactone A (2017). | ||
The task commenced with the known compound 349,138 which could be prepared via diene 328 (Scheme 17a). 349 was then treated with LDA followed by exposure to O2/P(OMe)3. Consequently, alcohol 380 was accessible in 77% yield. VO(acac)2/TBHP-mediated epoxidation of 380 gave rise to epoxide 381, which was reacted with methyl magnesium bromide, inducing a nucleophilic addition reaction. The resulting alcohol 382 was in situ oxidized by PDC to transform it into 383. SmI2-induced reductive ring opening153 of 383 afforded 384, which served as a substrate for the PyHBr3-mediated154,155 late stage C(sp3)–H bromination at C20, generating bromide 384.
With access to bromide 384, the authors then sought to complete the total synthesis of schilancidilactones A 28 and B 29. Initially, they anticipated to utilize the radical addition strategy to generate the C20–C22 bond.138 Unfortunately, their attempts to employ typical radical conditions and photoredox catalysis144 did not succeed in delivering schilancidilactones A 28 and B 29. Taking inspiration from recent progress in the nickel-catalysed cross coupling of alkyl halides,156 the authors speculated whether key fragments 384 and stannane 385 could be merged via a nickel catalysed cross-coupling reaction. In practice, the authors pleasingly identified that Ni(cod)2/dppm was capable of engaging the cross-coupling reaction, thus providing schilancidilactones A 28 and B29 in 36% and 7% yield, respectively.
Here, their goal then was to complete the synthesis of schilancitrilactone A 30 and 20-epi-schilancitrilactone A 31. Their efforts commenced with the formation of fragment iodide 390. The known compound 386 was prepared from (−)-carvone 330 (Scheme 17b).138 Then 386 was converted into iodide 387via Appel reaction. NaOMe-mediated methanolysis of 387 gave rise to epoxide 388,157 which was rearranged to lactone 389via acid-promoted selective epoxide opening and cyclization. Alcohol 389 was further protected to TBS ether 390. Moving forward, the authors turned their attention to complete the synthesis of schilancitrilactone A 30 and 20-epi-schilancitrilactone A 31. Learning from their previous experiences in the synthesis of schilancitrilactones B 26 and C 27,138 exposure of 390 to LDA induced transient lithium enolate, which was in situ treated with known aldehyde 346, thus providing alcohol 391. Then 391 was advanced to allylic alcohol 392.138 Alcohol 392 was protected to acetate 393, which was utilized as a precursor for LDA-promoted Dieckmann-type condensation.111 The desired lactone 394 was received in 90% yield. Martin's sulfurane-mediated dehydration143 of 394 furnished enone 395. L-Selectride-induced selective reduction of 395 produced 396. Exposing 396 to Mukaiyama conditions [Co(acac)2, PhSiH3, and O2]24 installed a tertiary alcohol in C9. PyHBr3-mediated C(sp3)–H bromination of 397 afforded 398. To complete the synthesis, the installation of a butenolide motif onto the core structure was required. Pleasingly, the authors found that the nickel-catalysed cross-coupling of 398 with stannane 385 successfully delivered schilancitrilacetone A 30 together with its C20-epimer 31.
In Tang's synthesis, the authors devised a clever nickel-catalysed cross-coupling to merge the advanced core and lactone motif, which showcased an attractive alternative to synthesize related natural products. Moreover, skillful utilization of late-stage C(sp3)–H halogenation provides an efficient and credible platform for the synthetic campaign of related Schisandraceae triterpenoids, thus facilitating their further biological evaluation.
4.7 Anderson's synthesis of (−)-rubriflordilactone B and (−)-pseudo-rubriflordilactone B (2019)
Li et al. noted in their original work that their synthetic rubriflordilactone B 32, which was confirmed by X-ray crystallographic analysis,147 did not match that isolated by Sun's group.115 Then DFT calculation by Kaufman and Sarotti showed that the difference between these two forms of rubriflordilactone B 32 lies at C16 and C17 in ring EF.158 To address this issue, Anderson and co-workers reported synthetic endeavours toward the total synthesis of (−)-rubriflordilactone B 32 and (−)-pseudo-rubriflordilactone B 33, utilizing rhodium-catalysed alkyne cyclotrimerization as the key strategy (Scheme 18).159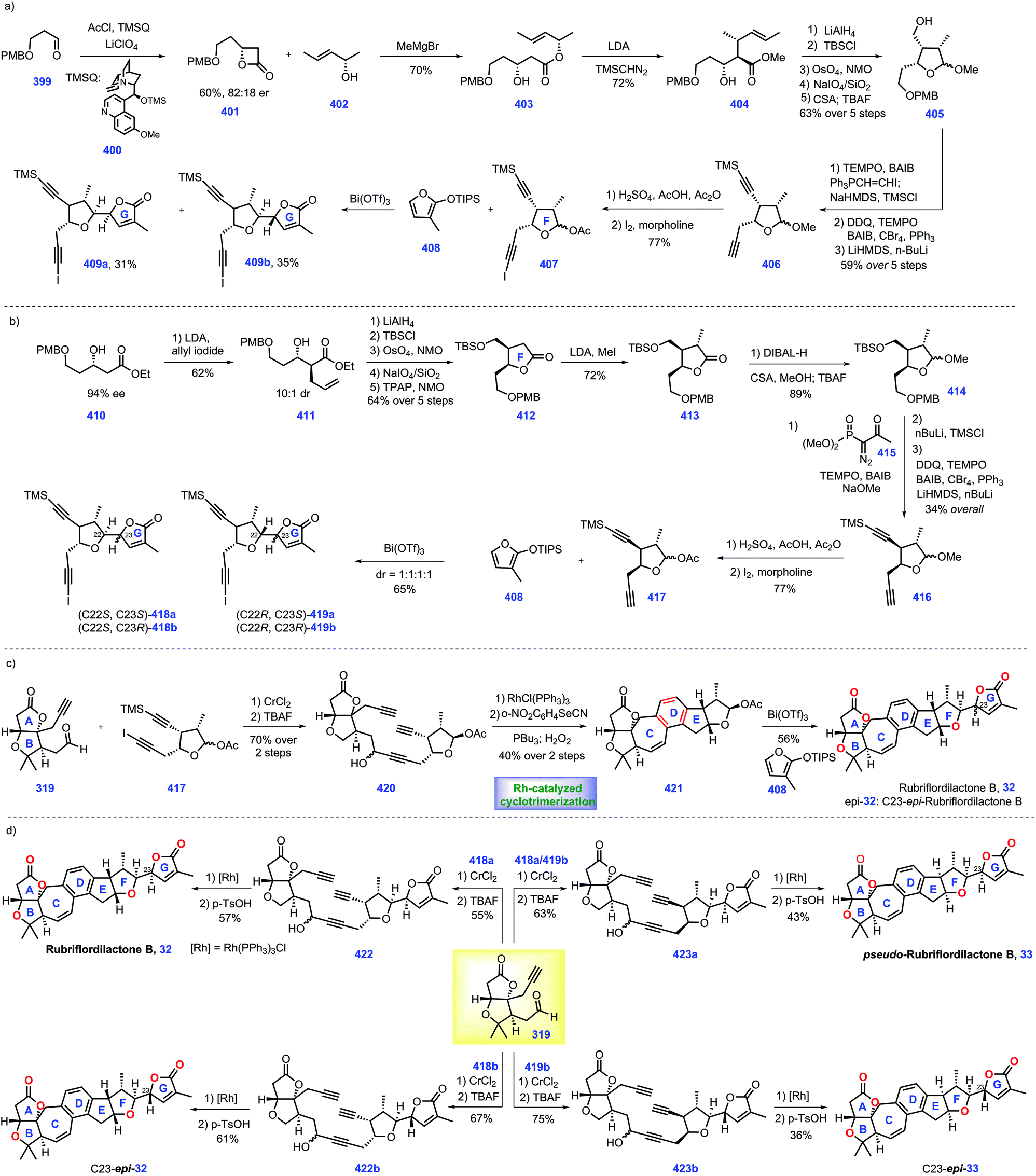 | ||
| Scheme 18 Anderson's synthesis of (−)-rubriflordilactone B and (−)-pseudo-rubriflordilactone B (2019). | ||
The project commenced with the installation of the ring FG system of the target (Scheme 18a). Aldehyde 399 was treated with ketene via TMSQ 400-mediated enantioselective [2 + 2] cycloaddition160 to produce chiral β-lactone 401 (60%, 64% ee). Exposure of chiral allylic alcohol 402 to MeMgCl led to magnesium alkoxide, thus allowing an in situ ring-opening reaction with 401. The desired enantio-enriched alcohol 403 was obtained. Dianionic Ireland–Claisen rearrangement of 403 followed by quenching with TMS-diazomethane gave ester 404 with two desired stereocentres in ring-F. Then sequential ester reduction, TBS-protection of alcohol, oxidative cleavage of alkene, hemi-acetylation and removal of TBS ether provided furan 405. Equipped with 405, the two oxygen-containing edge chains were then converted into diyne 406via several requisite functional group transformations. Acetoxylation of diyne 406 led to the acetal product, which was then treated with I2/morpholine, generating iodoalkyne 407.
With access to 407, the authors then sought to assemble butenolide ring G. In the experiment, they identified that the treatment of 407 with siloxyfuran 408161 in the presence of Bi(OTf)3 led to 409a (31%) and 409b (35%). Although the desired stereocentre at ring F (C22) could be well controlled, it proved challenging in inducing the stereocenter on ring G (C23), which only generated a mixture of stereoisomers (1:1). Considering that 409a only secured rubriflordilactone B 32, the synthesis of pseudo-rubriflordilactone B 33 called for an alternative synthetic protocol (Scheme 18b).
Then the project began with enantioenriched β-hydroxyl ester 410, which was obtained from a known β-ketoester162via Noyori's asymmetric hydrogenation.163 Treatment of 410 with LDA followed by allylation furnished 411 with good diastereoselectivity (10:1), which was transformed into lactone 412via the aforementioned sequences. Diastereoselective methylation of lactone 412 with CH3I in the presence of LDA afforded 413 with a pleasing dr value (10:1). Reduction of lactone in 413 followed by TBAF-mediated deprotection of TBS ether delivered acetal 414. With access to 414, alkyne 417 was achieved via similar manipulations, which was employed for synthesis of 409a/b. Note that exposure of 417 and 408 to Bi(OTf)3 failed to induce stereo-control, thus leading to four diastereomeric diynes 418a/b and 419a/b with an approximately equimolar ratio (1:1:1:1) where 418a and 419a were confirmed via X-ray analysis, while 418a and 419b were inseparable.
Here, the authors arrived at the key stage of this work. To evaluate the viability of core alkyne cyclotrimerization and furan addition, a model reaction was conducted, which proceeded smoothly with the desired ring CDEFG installed. In a real experiment, treatment of 407 with aldehyde 319109via Nozaki–Hiyama–Kishi conditions (CrCl2) followed by TBAF-mediated deprotection of TMS ether gave cyclized precursor 420, which was subjected to the key rhodium-catalysed cyclotrimerization reaction (Scheme 18c),164 successfully installing the desired hexacycle. However, upon subsequent treatment of the hexacycle with Martin sulfurane, it failed to undergo dehydration, which may be attributed to steric hindrance. Fortunately, they found that exposing the hexacycle to Grieco's protocol165 induced a dehydration reaction to provide 421. In the final step, treating 421 with furan 408 in the presence of Bi(OTf)3 completed the total synthesis of rubriflordilactone B 32 together with its C23-epimer (epi-32) as a 1:1 mixture. Unfortunately, the authors found it difficult to isolate 32 and C23-isomer 32.
To address this issue, they envisioned attachment of a butenolide motif in an earlier stage, thus allowing for a more convergent synthesis (Scheme 16d). In practice, subjecting 319 with 408a or 408b to Nozaki–Hiyama–Kishi reaction followed by the removal of silyl enol ether gave 422a and 422b, respectively. Moving forward, Rh-catalysed cyclotrimerization of 422a and 422b followed by dehydration resulted in the formation of rubriflordilactone B 32 and epi-32, respectively. Exposure of aldehyde 319 with 408a/409b and 408b to the abovementioned protocol completed the synthesis of pseudo-rubriflordilactone B 33 and epi-33. Pleasingly, they found that the NMR spectroscopic data of synthetic pseudo-rubriflordilactone B 33 was identical to that of the isolated material, thus demonstrating the DFT-predicted stereochemistry.
In Anderson's synthesis, the key contribution led to the identification of the synthetic pseudo-rubriflordilactone B 33, which match well with the isolated material. Their successful implementation of the key rhodium-catalysed cyclotrimerization strategy in the late-stage synthesis offered an appealing and viable protocol to obtain the related complex molecules. In their work, the ingenious reorganized synthetic strategy allowed for a more convergent synthesis.
4.8 Li's synthesis of pseudo-rubriflordilactone B (2020)
In 2016, Li and co-workers disclosed the first total synthesis of pseudo-rubriflordilactone B 33, involving 6π-electrocyclization/aromatization as the key step.147 Unfortunately, the synthetic material did not match the natural material. The authors reasoned that the isolated sample of “rubriflordilactone B” may contain two components, that is 33 and its congener “pseudo-rubriflordilactone B”. In 2019, Anderson's synthetic efforts resulted in the elucidation of the true structure of pseudo-rubriflordilactone B 33.145 Soon after, Li's group also successfully documented their detailed investigation aimed at elucidating the complete structure of pseudo-rubriflordilactone B 33 (Scheme 19).166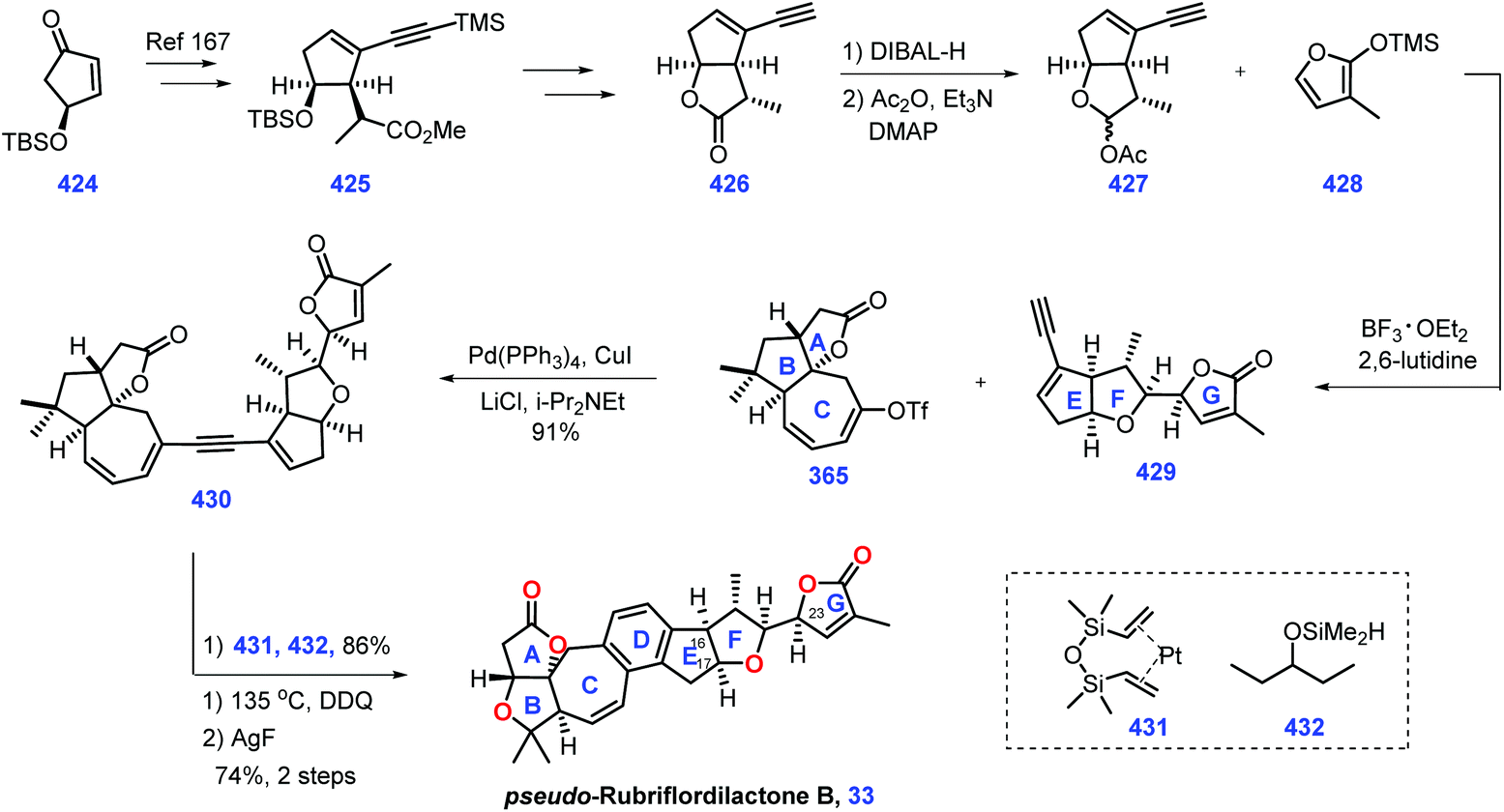 | ||
| Scheme 19 Li's synthesis of pseudo-rubriflordilactone B (2020). | ||
To pursue the real structure of 33, Li and co-workers carefully prepared 33 and its fifteen stereoisomers. Considering that these stereoisomers share similar synthetic pathways, we will comment on the route shown in Scheme 20 since the obtained product was identical to that of the authentic pseudo-rubriflordilactone B.
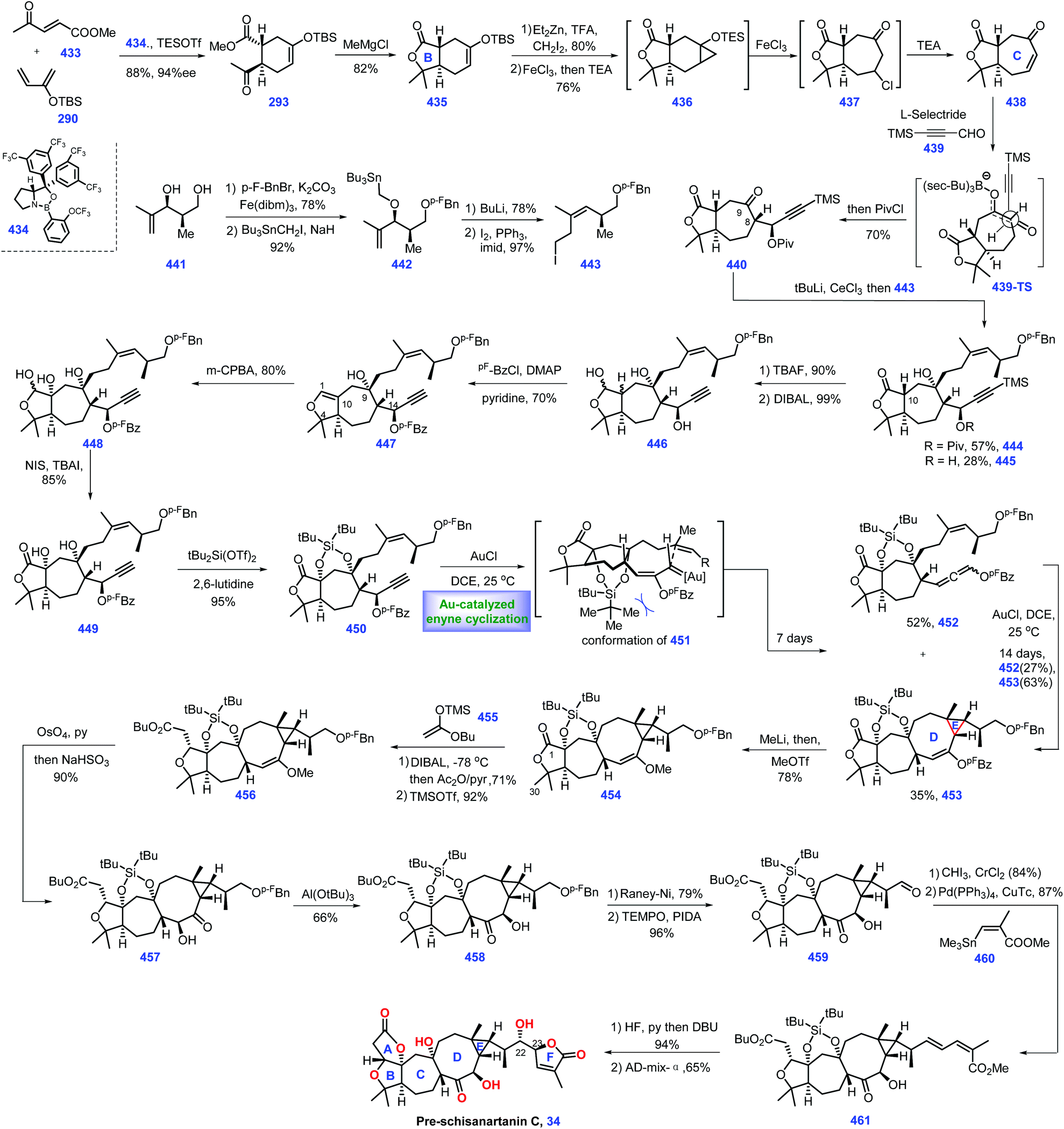 | ||
| Scheme 20 Yang's synthesis of pre-schisanartanin C (2020). | ||
Their synthetic endeavours commenced with chiral enone 424,167 which was processed through a four-step sequence involving α-iodination, Sonogashira coupling, Luche reduction, and Johnson–Claisen rearrangement to enyne 425. Then, the TBAF-mediated deprotection of silyl ether followed by lactonization provided lactone 426. 426 was converted into 427via a two-step manipulation, including reduction of lactone and protection of the resulting semi-acetal. Treatment of 427 with 1-siloxyfuran 428168via BF3·OEt2-mediated vinylogous Mukaiyama aldol reaction furnished 429. Notably, 2,6-lutidine was crucial for securing the good efficiency of this transformation. Then, key fragments 429 and known 365 were then merged via a Pd-catalysed Sonogashira coupling reaction. Using this method, 430 was accessible in 91% yield.
With 430 in hand, the final step was the installation of the benzene ring of the target. According to their previous work,147430 was then processed through a four-step sequence, involving platinum-catalysed cis-hydrosilylation,169,170 6π-electro-cyclization, DDQ-promoted oxidative aromatization, and AgF-mediated desilylation to provide synthetic rubriflordilactone B. Pleasingly, the NMR spectra of the above synthetic material matched well with that of the authentic pseudo-rubriflordilactone B. Notably, they found that the synthetic material demonstrated good inhibiting activity toward HIV infection (EC50 = 0.288 μM).
In Li's work, the comprehensive synthesis of a series of pseudo-rubriflordilactone B 33 and its 15 stereoisomers was vital to its overwhelming success. Obviously, this effort qualifies as a landmark in identifying the complete structure of pseudo-rubriflordilactone B 33. Also, the confirmed structure of pseudo-rubriflordilactone B 33 is crucial for further investigation of its structure–activity relationship.
4.9 Yang's synthesis of pre-schisanartanin C (2020)
In 2010, Sun's group reported the isolation of a new Schisandra nortriterpenoid, called pre-schisanartanin C 34,108 from Schisandra propinqua var. propinqua. Structurally, it contained a bicyclo[6.1.0]nonane framework, a quaternary stereogenic center at C13 and a labile α-hydroxyl ketone, which posed a considerable challenge for its total synthesis. Notably, its absolute configuration was still not determined. The biological investigation showed that it exhibited promising anti-cancer, anti-HIV and anti-hepatitis activities.171 Recently, Yang and Chen reported the completion of their synthetic efforts toward pre-schisanartanin C.172 In 2017, the authors accomplished the synthesis of one of the Schisandra nortriterpenoids, arisandilactone A,112 where an ideal aldehyde intermediate was achieved via somewhat lengthy routes (31 steps) from (R)-carvone. Considering that the authors had gained sufficient experiences from Au-catalysis in total synthesis,173,174 they envisioned Au-catalysed enyne cyclization as the core step to provide a more concise platform to the bicyclo[6.1.0]nonane skeleton,175 thus allowing efficient completion of pre-schisanartanin C 34 (Scheme 20). Their project commenced with the construction of fragment ketone 440 and iodide 441. Initially, the authors planned to treat diene 290 with dienophile 433via an enantioselective Diels–Alder reaction. Using this method, ketoester 293 was obtained. However, utilizing 433 as a precursor was challenging since both the ketone and ester groups in 433 may coordinate to the chiral catalyst, thus leading to regioselectivtiy issue.176 Hence, the authors anticipated the meticulous evaluation of various chiral Lewis acids could enable the Diels–Alder reaction. Fortunately, chiral Lewis acid 434 was identified to catalyse the enantioselective Diels–Alder reaction, generating chiral ketoester 293 in 88% yield with 94% ee. Treatment of 293 with MeMgCl initiated a cascade Grignard addition/lactonization, giving lactone 435. Simmon–Smith cyclopropanation of the silyl enol ether in 435 afforded 436, which upon in situ treatment with FeCl3, cyclopropanol oxidative fragmentation/elimination177 ensued, thus giving enone 438. L-Selectride-mediated178 1,4-reduction of 438 followed by an aldol reaction179 with aldehyde 439 and Piv protection of the resultant alcohol generated 440. In this work, the author reasoned that the formation of the less steric-demand complex 439-TS180 accounted for the observed diastereoselectivity of aldol condensation.
Subsequently, their next task was to construct iodide 443. Initially, the authors anticipated to protect diol 441via treatment with p-F-BnBr in the presence of Fe(dibm)3181 to yield the benzyl ether product, which was followed by exposure to Bu3SnCH2I/NaI. The desired stannane 442 was secured. Exposing 442 to nBuLi induced a Wittig–Still182 rearrangement, and the resultant alcohol was then processed through Appel iodination183 to provide iodide 443. Moving forward, key fragments 443 and 440 were then connected via a stereo-selective alkylation reaction, which delivered 444 (57%) together with 445 (28%) without the Piv group. The authors guessed that the observed diastereoselectivity was attributed to the approach of the alkylating agent to the carbonyl C9 of 443 from the less hindered face.
Then, the authors sought to install the C10 hydroxyl with the requisite stereochemistry. The mixture of 444 and 445 was advanced to 446via lactone reduction, alkyne desilylation and secondary alcohol deacylation. Chemoselective protection of C1 alcohol followed by elimination resulted in vinyl ether 447. Epoxidation of 447 followed by hydration afforded diol 448 with C10 hydroxyl installed. Notably, the methyl group at C4 masked the top face of the double bond (C1–C10), thus allowing epoxidation of the vinyl ether with the desired stereoselectivity. The C1 hydroxyl group in diol 448 was then oxidized to lactone 449. Here, the authors envisioned that a strongly biased conformation184 would be crucial to the key Au-catalysed annulation reaction. Hence, the cis-diol was capped as cyclic silylidene 450,185 which was confirmed by X-ray crystallography. Pleasingly, the X-ray structure demonstrated that the enyne components approached each other, thus feasibly allowing the next Au-catalysed cyclization reaction.
With access to 450, the crucial cyclization was then explored. In this experiment, the authors speculated that the benzoyl group in 450 would first undergo a 1,2-migration186 to give gold carbene 451, which served as an intermediate for the key Au-catalysed intramolecular cyclopropanation to construct 453 with the challenging ring DE system installed. In practice, exposure of 450 to AuCl under strict anhydrous conditions for one week installed pentacycle 453 (35%) together with allenyl ester 452. It is worth noting that exposing 452 to AuCl for a prolonged reaction time (14 days) gave rise to 453 in 27% yield, albeit with the recovered allenyl ester 452 (63%). The author discovered that when the hydroxyl group was protected with benzyl ether, the reaction was complicated, which was attributed to the migration of the benzylic hydrogen to form vinyl gold carbene complexes.187 Instead, installation of pF-Bz as a protecting group prevented the undesirable benzylic hydrogen-shift process. Removal of the pF-Bz protecting group followed by treatment with MeOTf formed methyl ether 399, which was advanced to 456 through three additional manipulations, involving DIBAL-H mediated lactone reduction, hemiacetal protection and TMSOTf-promoted stereoselective Mukaiyama aldol reaction with enol silyl ether 455. The authors proposed that the axial methyl group at C30 in 454 guided the enol silyl ether 455 to access an in situ oxonium ion from the opposite orientation, which elucidated the newly installed C1 stereogenic center. Subjecting 456 to dihydroxylation condition188 produced 457, thus allowing the key Al(OtBu)3-mediated189 hydroxyl ketone isomerization to obtain the thermodynamically more stable 458. Then 458 was converted into aldehyde 459via RANEY® Ni catalysed deprotection of the pFBn group and TEMPO/PIDA-mediated alcohol oxidation.190 Takai–Utimoto olefination191 of aldehyde 459 afforded a vinyl iodide, which served as an intermediate for the Pd-catalysed Stille cross-coupling reaction with stannane 460, yielding diene 461. To complete the synthesis, the conditions for the requisite ring A and F needed to be established. Treatment of 461 with HF/py followed by DBU-promoted lactonization furnished the lactone product with ring A formed, which was subsequently subjected to Sharpless asymmetric dihydroxylation conditions (AD mix-α) to achieve the synthesis of pre-schisanartanin C 34.
In this synthetic campaign, the novel and impressive Au-catalysed enyne cyclization strategy proceeded in an efficient and stereo-selective manner to establish the critical bicyclo[6.1.0]nonane core. An Al(OtBu)3-mediated isomerization strategy tactfully formed the desired core hydroxy ketone structure in ring D. An elegant Sharpless asymmetric dihydroxylation nicely constructed the desired ring F albeit with the desired stereocentres at C22 and C23. Definitely, the successful implementation of creative strategies in this synthesis will encourage further synthetic endeavours toward other total syntheses involving bicyclo[6.1.0]nonane cores.
4.10 Gui's synthesis of propindilactone G (2020)
In 2008, propindilactone G, a C29 schinortriterpenoid, was isolated by Sun's group from the stems of Schisandra propinqua var. propinqua.192 Structurally, propindilactone G bears a 5/5/7/6/5-pentacyclic skeleton, 10 stereocentres and multiple oxygen-containing functional groups. In 2015, Yang and co-workers disclosed an excellent synthesis of propindilactone G 35. Key to their success was an asymmetric Diels–Alder reaction, Pauson–Khand reaction and an oxidative hetero-coupling reaction to append the side motif.111 Notably, Yang's synthesis corrected the originally proposed synthetic compound to be C17-epi- propindilactone G. Recently, Gui and co-workers reported the beautiful bioinspired synthesis of nortriterpenoid propindilactone G 35.193Gui's synthesis was inspired by the biosynthetic mechanism proposed by Sun and co-workers. Sun supposed that the core framework of schinortriterpenoid III may be derived from IIvia a smart transesterification/oxa-Michael addition (Scheme 21a).108II could be traced back to Ivia 1,10-oxidative cleavage. I was obtained from cycloartane 462via dehydrogenation and Baeyer–Villiger oxidation. Notably, the authors found that propindilactone G 35 and steroidal lactone 463 possessed the same stereo-centres at C5, C8, C13, and C20 and similar C/D-ring systems. Moreover, steroidal lactone 463 could be feasibly acquired from the degradation of tigogenin.
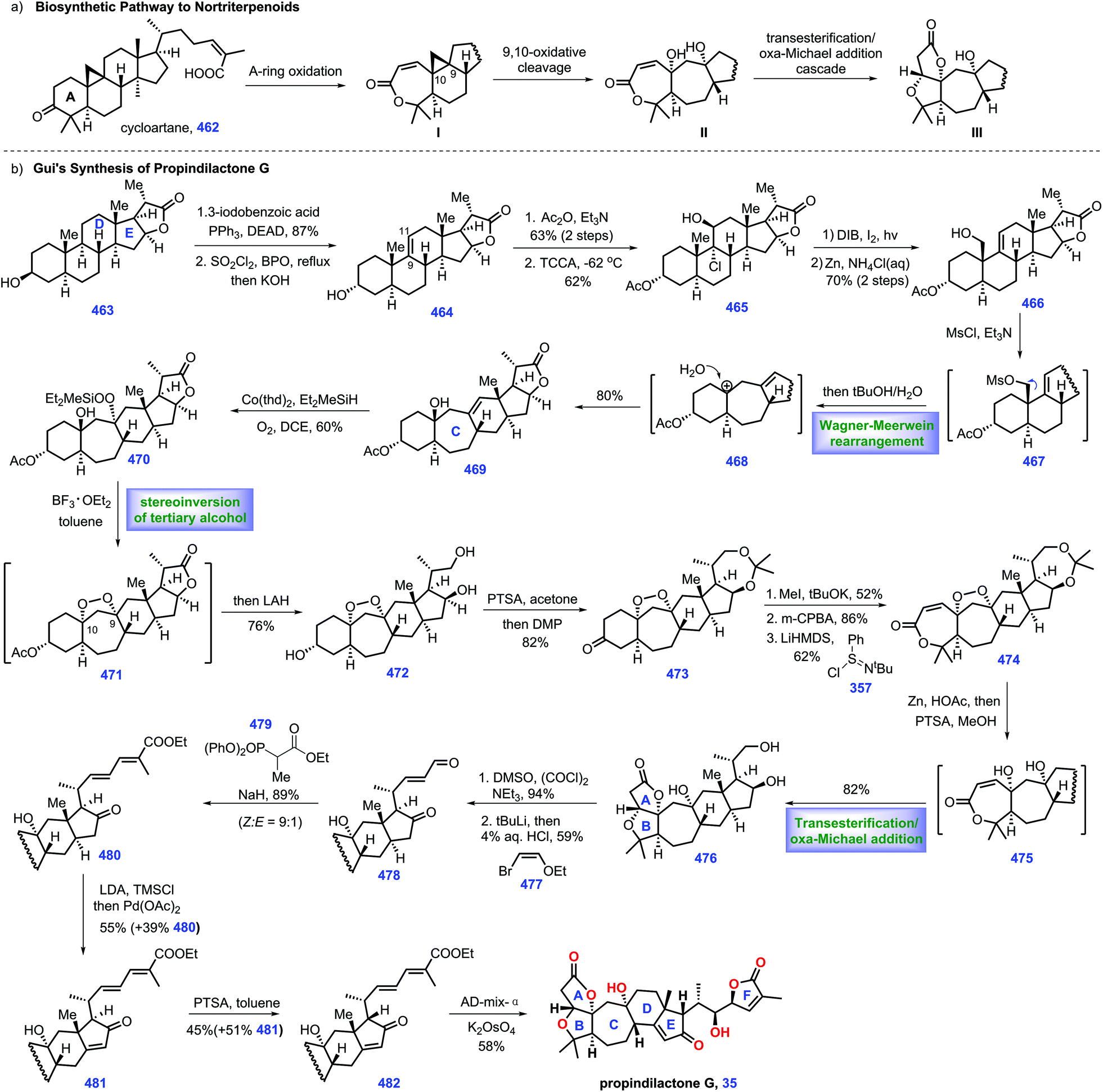 | ||
| Scheme 21 Gui's synthesis of propindilactone G (2020). | ||
To experimentally confirm this speculated biosynthetic pathway, their synthetic efforts began with installation of the C9–C11 alkene from steroidal lactone 463 (Scheme 21b). Taking inspiration from Breslow's remote functionalization strategy,194 treatment of 463 with 3-iodobenzoic acid under Mitsunobu reaction conditions provided a C3 α-iodobenzoate, which was employed as a precursor for selective chlorination at C9 followed by E2 elimination,195 thus providing the desired olefin 464.
Moving forward, the authors attempted the functionalization of the C19-Me group. They considered Suárez's remote radical functionalization strategy directed by the β-OH group at C11, which was capable of the site-selective oxidation of the C19-Me group. In practice, subjecting 464 to Ac2O/NEt3 followed by treatment with trichloroisocyanuric acid196 gave chlorohydrin 465 with the desired β-OH at C11 installed.197 Exposing 465 to Suárez's conditions (DIB/I2, hv) followed by reduction with Zn and aq. NH4Cl furnished C19-alcohol 466.
Equipped with 466, the authors turned their attention toward the construction of ring C via a ring expansion reaction. According to the modified work by Barton et al.,198 the authors expected to introduce a good leaving group to facilitate Wagner–Meerwein rearrangement. After extensive screenings, they eventually identified mesylation of 466 followed by treatment with THF/H2O at 80 °C triggered Wagner–Meerwein rearrangement via the proposed tertiary cation 468. The desired C10-alcohol 469 was obtained in 60% yield. Pleasingly, the authors discovered that this ring expansion reaction could be scaled up (4 g) with a higher yield (80%) using a mixed solvent of tBuOH/H2O. Then 469 was subjected to Mukaiyama hydration conditions. The resulting diol was generated. However, the stereochemistry of the formed hydroxyl group at C10 was opposite to that of propindilactone G. Initially, the authors attempted to invert the stereocenter of C10, which was found to be unfeasible.199 Dussault et al. reported200 that the opening of oxetanes by alkyl hydroperoxides could be achieved by Lewis acids. Inspired by Dussault's work, the authors then conducted Co(thd)2-catalysed Mukaiyama hydroperoxysilylation201 of 469 to provide peroxysilyl alcohol 470, which was induced by BF3·Et2O via intramolecular cyclization.202 Pleasingly, endoperoxide 471 was generated with the desired α-configuration of C10 established. Interestingly, the endoperoxide moiety here could be utilized as a temporary protecting group of C9,C10-diol in the following transformations. The LiAlH4-mediated reduction of both the ester and lactone in 471 gave rise to triol 472.
Moving forward, the authors then sought to develop conditions for the installation of the key 5/5-fused bicyclic lactone via a key biomimetic transesterification/oxa-Michael addition cascade. Initially, the hydroxyl groups at C16 and C22 in 472 were protected to form an acetonide, which served as a substrate for the following Dess–Martin oxidation of the C3-hydroxyl group. The resulting ketone 473 was produced in 82% yield. Then 473 was converted to 474via a three-step manipulation, involving selective α-dimethylation, Baeyer–Villiger oxidation, and Mukaiyama dehydrogenation.148 The authors speculated that 474 would be reduced to diol 475, thus setting the stage for the proposed biomimetic cascade. In the real experiment, the Zn/HOAc-mediated reductive opening of endoperoxide 474 afforded diol 476. Interestingly, the authors discovered that 475 partially underwent the cascade transesterification/oxa-Michael addition to form the desired 5/5-fused bicyclic lactone skeleton (AB ring system). Fortunately, they identified that the addition of p-toluenesulfonic acid and MeOH to the above conditions secured the reaction, thus leading to diol 476 in 82% yield from 474.
To complete the synthesis of propindilactone G, the final step was to attach the challenging side moiety onto core 476. The authors anticipated to employ the C16 and C22 hydroxyl groups to introduce the butenolide side chain and the C16-enone moiety. Accordingly, exposure of alcohol 476 to Swern oxidation conditions followed by a homologation203 reaction with 477 transformed it into 478. The Z-selective Horner–Wadsworth–Emmons reaction204 of 478 with 479 generated diene 480, which was subjected to Saegusa oxidation,28 thus affording enone 481. However, initial screenings to achieve dihydroxylation of the C22–C23 olefin of 481 suffered from low efficiency. According to the work from Yang's group,111 the authors found that the configuration at C17 of 481 was essential to this reaction. Accordingly, the configuration at C17 was epimerized as a 1:1 ratio via catalytic TsOH. Pleasingly, subjecting the desired 482 to Sharpless asymmetric dihydroxylation conditions successfully completed the synthesis of propindilactone G.
In general, this beautiful work showcased the experimental evidence of the proposed biosynthesis pathway of propindilactone G 35. The clever use of stereoinversion of tertiary alcohol-mediated BF3·OEt2 nicely constructed the desired stereocentre at C10. The impressive biomimetic transesterification/oxa-Michael addition cascade efficiently installed the 5/5-fused bicyclic lactone moiety. It is worthy of praise that the entire propindilactone G synthetic campaign only required 20 steps and some sequences could be scaled up. It is believed that the Wagner–Meerwein rearrangement, Suárez remote radical functionalization, in conjunction with the transesterification/oxa-Michael addition cascade, may have broader heuristic value for the synthesis of related natural product.
5. Conclusions
We summarized the recent advancements in the synthesis of limonoids and limonoid-like natural products, where novel and elegant synthetic methodologies, including copper-catalysed cascade Michael addition/cyclization,35–37 metal catalysed cycloisomerization,109,159 Au-catalysed enyne cyclization,172 metal-mediated radical cascade cyclization,53 6π-electro-cyclization,90,116 Pauson–Khand reaction,63 nickel-catalysed sp2–sp3 cross-coupling, and biomimetic transesterification/oxa-Michael cascades, have been successfully employed to conquer the challenging target molecules. Considering the diverse structure of limonoids, particularly the highly decorated limonoid-related molecules, sometimes novel and unique synthetic strategies are desirable, such as Watanabe's installation of the C8–C14 bond of azadirachtin via an ingenious radical cascade cyclization strategy.53 Several limonoids share a common core structure, thus the rational design of synthetic strategies to establish a common intermediate will allow the convergent syntheses of limonoids and limonoid-like natural products with higher efficiency. The carbocation-mediated polyene cyclization is common in biosynthesis; however, it is seldom utilized in the total syntheses of limonoid-related molecules. In addition, scalability poses a challenge for most synthetic limonoid-related compounds, which is a key limitation for molecules with clinical potential. Protecting-group-free synthesis is also a challenge in the syntheses of limonoids. Definitely, excess protecting groups and redox operations involved will reduce the step economy. Hence, efficient syntheses are possible when powerful synthetic methodologies are leveraged on judiciously chosen reactants in conjunction with optimal reaction conditions. We believe that researchers can draw enough experiences from emerging limonoids and limonoid-like total syntheses, thus guiding their future endeavours towards the total syntheses of these compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the NSFC (21672153, 21901170, U19A2014, 21925106), and the Fundamental Research Funds for the Central Universities.Notes and references
- (a) Y.-L. Lan, J.-C. Lou, X.-W. Jiang, X. Wang, J.-S. Xing, S. Li and B. Zhang, A research update on the anticancer effects of bufalin and its derivatives, Oncol. Lett., 2019, 17, 3635 CAS; (b) F. K. Khalaf, I. Tassavvor, A. Mohamed, Y. Chen, D. Malhotra, Z. Xie, J. Tian, S. T. Haller, K. Westfall, W. H. W. Tang and D. J. Kennedy, Epithelial and endothelial adhesion of immune cells is enhanced by cardiotonic steroid signaling through Na+/K+-ATPase-alpha-1, J. Am. Heart Assoc., 2020, 9, e013933 CrossRef PubMed; (c) A. V. Lopachev, D. A. Abaimov, T. N. Fedorova, O. M. Lopacheva, N. V. Akkuratova and E. E. Akkuratov, Cardiotonic steroids as potential endogenous regulators in the nervous system, Neurochem. J., 2018, 12, 1 CrossRef CAS.
- B. Heasley, Synthesis of limonoid natural products, Eur. J. Org. Chem., 2011, 2011, 19 CrossRef.
- M. Michalak, K. Michalak and J. Wicha, The synthesis of cardenolide and bufadienolide aglycones, and related steroids bearing a heterocyclic subunit, Nat. Prod. Rep., 2017, 34, 361 RSC.
- (a) G. E. Veitch, E. Beckmann, B. J. Burke, A. Boyer, S. L. Maslen and S. V. Ley, Synthesis of Azadirachtin: a long but successful journey, Angew. Chem., Int. Ed., 2007, 46, 7629 CrossRef CAS PubMed; (b) G. E. Veitch, E. Beckmann, B. J. Burke, A. Boyer, C. Ayats and S. V. Ley, A relay route for the synthesis of Azadirachtin, Angew. Chem., Int. Ed., 2007, 46, 7633 CrossRef CAS PubMed; (c) S. V. Ley, A. Abad-Somovilla, J. C. Anderson, C. Ayats, R. Bänteli, E. Beckmann, A. Boyer, M. G. Brasca, A. Brice, H. B. Broughton, B. J. Burke, E. Cleator, D. Craig, A. A. Denholm, R. M. Denton, T. Durand-Reville, L. B. Gobbi, M. Göbel, B. L. Gray, R. B. Grossmann, C. E. Gutteridge, N. Hahn, S. L. Harding, D. C. Jennens, L. Jennens, P. J. Lovell, H. J. Lovell, M. L. de la Puente, H. C. Kolb, W.-J. Koot, S. L. Maslen, C. F. McCusker, A. Mattes, A. R. Pape, A. Pinto, D. Santafianos, J. S. Scott, S. C. Smith, A. Q. Somers, C. D. Spilling, F. Stelzer, P. L. Toogood, R. M. Turner, G. E. Veitch, A. Wood and C. Zumbrunn, The synthesis of Azadirachtin: a potent insect antifeedant, Chem. – Eur. J., 2008, 14, 10683 CrossRef CAS PubMed.
- B. Heasley, Chemical synthesis of the cardiotonic steroid glycosides and related natural products, Chem. – Eur. J., 2012, 18, 3092 CrossRef CAS PubMed.
- (a) N. F. Z. Schneider, C. Cerella, C. M. O. Simões and M. Diederich, Anticancer and immunogenic properties of cardiac glycosides, Molecules, 2017, 22, 1932 CrossRef CAS PubMed; (b) L. Q. De Sousa, K. d. C. Machado, S. F. d. C. Oliveira, L. d. S. Aradjo, E. d. S. Moncao-Filho, A. A. C. Melo-Cavalcante, G. M. Vieira Jr. and P. M. P. Ferreira, Bufadienolides from amphibians: A promising source of anticancer prototypes for radical innovation, apoptosis triggering and Na+/K+-ATPase inhibition, Toxicon, 2017, 127, 63 CrossRef PubMed; (c) L. H. A. Cavalcante-Silva, E. d. A. Lima, D. C. M. Carvalho, J. M. de Sales-Neto, A. K. d. A. Alves, J. G. F. M. Galvao, J. S. d. F. d. Silva and S. Rodrigues-Mascarenhas, Much more than a cardiotonic steroid: modulation of inflammation by Ouabain, Front. Physiol., 2017, 8, 895 CrossRef PubMed.
- (a) B. Kopp and W. Kubelka, New cardenolides from convallaria-majalis, Planta Med., 1982, 45, 195 CrossRef CAS PubMed; (b) A. Ankli, J. Heilmann, M. Heinrich and O. Sticher, Cytotoxic cardenolides and antibacterial terpenoids from Crossopetalum gaumeri, Phytochemistry, 2000, 54, 531 CrossRef CAS PubMed.
- K. Mukai, D. Urabe, S. Kasuya, N. Aoki and M. Inoue, A Convergent total synthesis of 19-Hydroxysarmentogenin, Angew. Chem., Int. Ed., 2013, 52, 5300 CrossRef CAS PubMed.
- S. A. Kozmin, J. M. Janey and V. H. Rawal, 1-amino-3-siloxy-1, 3-butadienes: Highly reactive dienes for the Diels-Alder reaction, J. Org. Chem., 1999, 64, 3039 CrossRef CAS PubMed.
- (a) H. J. Bestmann, G. Schade, H. Lüike and T. Mçnius, Cumulated ylides.17. Synthesis of cyclic-compounds from triphenyl[(phenylmino)-ethenylidene]phosphorene and oxocarboxylic acid- A novel method for anellation, Chem. Ber., 1985, 118, 2640 CrossRef CAS; (b) T. Mandai, Y. Kaihara and J. Tsuji, A New candidate for a properly substituted CD ring component of Vitamin D3 via intramolecular asymmetric olefination of a 1,3-cyclopentanedione derivative, J. Org. Chem., 1994, 59, 5847 CrossRef CAS.
- (a) Y. Ueno, K. Chino, M. Watanabe, O. Moriya and M. Okawara, Homolytic carbocyclization by use of a heterogeneous supported organotin catalyst. A new synthetic route to 2-alkoxytetrahydrofurans and γ-butyrolactones, J. Am. Chem. Soc., 1982, 104, 5564 CrossRef CAS; (b) G. Stork, R. Mook Jr., S. A. Biller and S. D. Rychnovsky, Free-radical cyclization of bromo acetals. Use in the construction of bicyclic acetals and lactones, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS; (c) X. J. Salom-Roig, F. Denes and P. Renaud, Radical cyclization of haloacetals: The Ueno-Stork reaction, Synthesis, 2004, 1903 CAS.
- (a) K. Nozaki, K. Oshima and K. Utimoto, Et3B-induced radical addition of R3SnH to acetylenes and its application to cyclization reaction, J. Am. Chem. Soc., 1987, 109, 2547 CrossRef CAS; (b) C. Ollivier and P. Renaud, Organoboranes as a source of radicals, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed.
- D. H. R. Barton, R. E. OBrien and S. Sternhell, A new reaction of hydrazones, J. Chem. Soc., 1962, 470 RSC.
- S. H. Rahimtoola and T. Tak, The use of digitalis in heart failure, Curr. Probl. Cardiol., 1996, 21, 781 CrossRef CAS PubMed.
- (a) I. Prassas and E. P. Diamandis, Novel therapeutic applications of cardiac glycosides, Nat. Rev. Drug Discovery, 2008, 7, 926 CrossRef CAS PubMed; (b) R. A. Newman, P. Yang, A. D. Pawlus and K. I. Block, Cardiac glycosides as novel cancer therapeutic agents, Mol. Interventions, 2008, 8, 36 CrossRef CAS PubMed; (c) J. A. R. Salvador, J. F. S. Carvalho, M. A. C. Neves, S. M. Silvestre, A. J. Leitao, M. M. C. Silva and M. L. Sáe Melo, Anticancer steroids: linking natural and semi-synthetic compounds, Nat. Prod. Rep., 2013, 30, 324 RSC.
- (a) H. Zhang, M. S. Reddy, S. Phoenix and P. Deslongchamps, Total synthesis of Ouabagenin and Ouabain, Angew. Chem., Int. Ed., 2008, 47, 1272 CrossRef CAS PubMed; (b) M. S. Reddy, H. Zhang, S. Phoenix and P. Deslongchamps, Total synthesis of Ouabagenin and Ouabain, Chem. – Asian J., 2009, 4, 725 CrossRef CAS PubMed.
- (a) R. Hans, Q. Zhou and P. S. Baran, Strategic redox relay enables a scalable synthesis of ouabagenin, a bioactive cardenolide, Science, 2013, 339, 59 CrossRef PubMed; (b) H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, Development of a concise synthesis of Ouabagenin and hydroxylated corticosteroid analogues, J. Am. Chem. Soc., 2015, 137, 1330 CrossRef CAS PubMed.
- H. Wehrli, M. S. Heller, K. Schaffner and O. Jäger, About steroids and sexual hormones. 223 UV Control of 11-oxo- steroids in the manufacture of 11β, 19-C, 9β, 19-cyclo- and 19-hydroxy-5α-pregnan compounds, Helv. Chim. Acta, 1961, 44, 2162 CrossRef CAS.
- D. Ng, Z. Yang and M. A. Garcia-Garibay, Total synthesis of (±)-herbertenolide by stereospecific formation of vicinal quaternary centers in a crystalline ketone, Org. Lett., 2004, 6, 645 CrossRef CAS PubMed.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Bis(pyridine)iodonium tetrafluoroborate (IPy2BF4): A versatile oxidizing reagent, Chem. – Eur. J., 2004, 10, 4206 CrossRef CAS PubMed.
- C. E. McDonald, H. Holcomb, T. Leathers, F. Ampadu-Nyarko and J. Frommer Jr., N-iodosuccinimide mediated oxidative cleavage of vicinal, monoprotected diols, Tetrahedron Lett., 1990, 31, 6283 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, “On Water”: Unique reactivity of organic compounds in aqueous suspension, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- U. Egner, K.-H. Fritzemeier, W. Halfbrodt, N. Heinrich, J. Kuhnke, A. Müller-Fahrnow, G. Neef, K. Schöllkopf and W. Schwede, 7α,15α-Ethano bridged steroids. Synthesis and progesterone receptor interaction, Tetrahedron, 1999, 55, 11267 CrossRef CAS.
- S. Isayama and T. Mukaiyama, A new method for preparation of alcohols from olefins with molecular oxygen and phenylsilane by the use of Bis(acetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 1071 CrossRef.
- S.-K. Chung, Selective reduction of mono- and disubstituted olefins by sodium borohydride and cobalt(II), J. Org. Chem., 1979, 44, 1014 CrossRef CAS.
- D. H. R. Barton, J. D. Elliott and S. D. Géro, Synthesis and properties of a series of sterically hindered guanidine bases, J. Chem. Soc., Perkin Trans. 1, 1982, 2085 RSC.
- (a) K. Mukai, S. Kasuya, Y. Nakagawa, D. Urabe and M. Inoue, A convergent total synthesis of ouabagenin, Chem. Sci., 2015, 6, 3383 RSC; (b) D. Urabe, Y. Nakagawa, K. Mukai, K. Fukushima, N. Aoki, H. Itoh, M. Nagatomo and M. Inoue, Total synthesis and biological evaluation of 19-Hydroxysarmentogenin-3-O-α-l-rhamnoside, Trewianin, and their Aglycons, J. Org. Chem., 2018, 83, 13888 CrossRef CAS PubMed.
- Y. Ito, T. Hirao and T. Saegusa, Synthesis of α, β-unsaturated carbonyl compounds by Palladium(II)- catalyzed dehydrosilylation of silyl enol ethers, J. Org. Chem., 1978, 43, 1011 CrossRef CAS.
- T. Sato, Y. Goto and T. Fujisawa, Asymmetric reduction of alkyl phenyl ketones with a chiral hydride reagent prepared from lithium aluminum hydride and (S)-4-anilino-3-methylamino-1-butanol, Tetrahedron Lett., 1982, 23, 4111 CrossRef CAS.
- (a) M. E. Jung and G. Piizzi, First synthesis of the A/B ring of Ouabain, Org. Lett., 2003, 5, 137 CrossRef CAS PubMed; (b) E. J. Corey and H. E. Ensley, Highly stereoselective conversion of prostaglandin A2 to the 10, 11α-oxido derivative using a remotely placed exogenous directing group, J. Org. Chem., 1973, 38, 3187 CrossRef CAS.
- (a) R. J. Theissen, Preparation of α, β-unsaturated carbonyl compunds, J. Org. Chem., 1971, 36, 752 CrossRef CAS; (b) T. Diao and S. S. Stahl, Synthesis of cyclic enones via direct Palladium-catalyzed aerobic dehydrogenation of ketones, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- (a) H. J. Bestmann, G. Schade, H. Like and T. Mçnius, Chem. Ber., 1985, 118, 2640 CrossRef CAS; (b) T. Mandai, Y. Kaihara and J. Tsuji, A new candidate for a properly substituted CD ring component of Vitamin D3 via intramolecular asymmetric llefination of a 1,3-cyclopentanedione derivative, J. Org. Chem., 1994, 59, 5847 CrossRef CAS.
- (a) J. Wegner, S. V. Ley, A. Kirschning, A.-L. Hansen, J. M. Garcia and I. R. Baxendale, A total synthesis of millingtonine A, Org. Lett., 2012, 14, 696 CrossRef CAS PubMed; (b) M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, Total syntheses of anominine and Tubingensin A, J. Am. Chem. Soc., 2012, 134, 8078 CrossRef CAS PubMed.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1 CAS.
- W. Kaplan, H. R. Khatri and P. Nagorny, Concise enantioselective total synthesis of cardiotonic steroids 19-Hydroxysarmentogenin and Trewianin aglycone, J. Am. Chem. Soc., 2016, 138, 7194 CrossRef CAS PubMed.
- B. Bhattarai and P. Nagorny, Enantioselective total synthesis of cannogenol-3-O-α-l-rhamnoside via sequential Cu(II)-catalysed michael addition/intramolecular aldol cyclization reactions, Org. Lett., 2018, 20, 154 CrossRef CAS PubMed.
- H. R. Khatri, B. Bhattarai, W. Kaplan, Z. Li, M. J. Curtis Long, Y. Aye and P. Nagorny, Modular total synthesis and cell-based anticancer activity evaluation of Ouabagenin and other cardiotonic steroids with varying degrees of oxygenation, J. Am. Chem. Soc., 2019, 141, 4849 CrossRef CAS PubMed.
- (a) T. Kano, H. Mii and K. Maruoka, Direct asymmetric benzoyloxylation of aldehydes catalyzed by 2-tritylpyrrolidine, J. Am. Chem. Soc., 2009, 131, 3450 CrossRef CAS PubMed; (b) M. J. P. Vaismaa, S. C. Yau and N. C. O. Tomkinson, Organocatalytic α-oxybenzoylation of aldehydes, Tetrahedron Lett., 2009, 50, 3625 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jorgensen, Enantioselective organocatalyzed α sulfenylation of aldehydes, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed.
- (a) N. Cichowicz, W. Kaplan, I. Khomutnik, B. Bhattarai, Z. Sun and P. Nagorny, Concise enantioselective synthesis of oxygenated steroids via sequential Copper(II)- catalyzed Michael addition/intramolecular aldol cyclization reactions, J. Am. Chem. Soc., 2015, 137, 14341 CrossRef CAS PubMed; (b) J. Lee, S. Wang, M. Callahan and P. Nagorny, Copper(II)-catalysed tandemdecarboxylative Michael/Aldol reactions leading to the formation of functionalized cyclohexenones, Org. Lett., 2018, 20, 2067 CrossRef CAS PubMed; (c) H. R. Khatri, N. Carney, R. Rutkoski, B. Bhattarai and P. Nagorny, Recent progress in steroid synthesis triggered by the emergence of new catalytic methods, Eur. J. Org. Chem., 2020, 755 CrossRef CAS.
- (a) A. K. V. Iyer, M. Zhou, N. Azad, H. Elbaz, L. Wang, D. K. Rogalsky, Y. Rojanasakul, G. A. O'Doherty and J. M. Langenhan, A direct comparison of the anticancer activities of Digitoxin MeON-Neoglycosides and O-Glycosides, ACS Med. Chem. Lett., 2010, 1, 326 CrossRef CAS PubMed; (b) X. Li, Y. Ren, Y. Bao, J. Liu, X. Zhang, Y. Zhang, X. Sun, X. Yao and J. Tang, Synthesis of C3-Neoglycosides of digoxigenin and their anticancer activities, Eur. J. Med. Chem., 2018, 145, 252 CrossRef CAS PubMed.
- Y. Moreno, L. Banuls, E. Urban, M. Gelbcke, F. Dufrasne, B. Kopp, R. Kiss and M. Zehl, Structure–activity relationship analysis of bufadienolide-induced in vitro growth inhibitory effects on mouse and human cancer cells, J. Nat. Prod., 2013, 76, 1078 CrossRef PubMed.
- (a) L.-S. Shi, S.-C. Kuo, H.-D. Sun, S. L. Morris-Natschke, K.-H. Lee and T.-S. Wu, Cytotoxic cardiac glycosides and coumarins from Antiaris toxicaria, Bioorg. Med. Chem., 2014, 22, 1889 CrossRef CAS PubMed; (b) D.-M. Tian, H.-Y. Cheng, W.-Z. Shen, J.-S. Tang and X.-S. Yao, Cardiac glycosides from the seeds of Thevetia peruviana, J. Nat. Prod., 2016, 79, 38 CrossRef CAS PubMed; (c) J.-H. Tay, V. Dorokhov, S. Wang and P. Nagorny, Regioselective single pot C3-glycosylation of strophanthidol using methylboronic acid as a transient protecting group, J. Antibiot., 2019, 72, 437 CrossRef CAS PubMed.
- E. K. Adesogan, C. W. L. Bevan, J. W. Powell and D. A. H. Taylor, Extractives from West African timbers. Part XV. The structure of the low-melting compound from Khaya senegalensis, Chem. Commun., 1966, 27 RSC.
- J. Wu, M. Y. Li, J. Zhang, G. Feng and T. Satyanandamurty, Khayasin and 2′S-methylbutanoylproceranolide: Promising candidate insecticides for the control of the coconut leaf beetle, Brontispa longissimi, J. Pestic. Sci., 2011, 36, 22 CrossRef CAS.
- C. W. L. Bevan, J. W. Powell and D. A. H. Taylor, West African timbers. Part X. The structure of Cedrela odorata substance B, Chem. Commun., 1965, 281 RSC.
- J. M. Faber and C. M. Williams, A concise total synthesis of (±)-cipadonoid B from synthetic azedaralide, Chem. Commun., 2011, 47, 2258 RSC.
- J. M. Faber, W. A. Eger and C. M. Williams, Enantioselective total synthesis of the Mexicanolides: Khayasin, Proceranolide, and Mexicanolide, J. Org. Chem., 2012, 77, 8913 CrossRef CAS PubMed.
- L. A. Baker, C. M. Williams, P. V. Bernhardt and G. W. Yanik, Azedaralide: total synthesis, relative and absolute stereochemical assignment, Tetrahedron, 2006, 62, 7355 CrossRef CAS.
- (a) E. J. Corey and H. E. Ensley, Highly stereoselective conversion of prostaglandin A2 to the 10, 11α-oxido derivative using a remotely placed exogenous directing group, J. Org. Chem., 1973, 38, 3187 CrossRef CAS; (b) T. Pouplin, B. Tolon, P. Nuhant, B. Delpech and C. Marazano, Synthetic studies towards bridgehead diprenyl-substituted bicyclo[3.3.1]nonane-2,9-diones as models forpolyprenylated acylphloroglucinol construction, Eur. J. Org. Chem., 2007, 5117 CrossRef CAS.
- Bernays, Limonin, Justus Liebigs Ann. Chem., 1841, 40, 317 Search PubMed.
- (a) A. Roy and S. Saraf, Limonoids: overview of significant bioactive triterpenes distributed in plants kingdom, Biol. Pharm. Bull., 2006, 29, 191 CrossRef CAS PubMed; (b) D. E. Champagne, O. Koul, M. B. Isman, G. G. E. Scudder and G. H. N. Towers, Biological activity of limonoids from the rutales, Phytochemistry, 1992, 31, 377 CrossRef CAS.
- S. Yamashita, A. Naruko, Y. Nakazawa, L. Zhao, Y. Hayashi and M. Hirama, Total synthesis of Limonin, Angew. Chem., Int. Ed., 2015, 54, 8538 CrossRef CAS PubMed.
- (a) B. B. Snider, J. J. Patricia and S. A. Kates, Lipase-catalyzed irreversible transesterification for preparative synthesis of chiral glycerol derivatives, J. Org. Chem., 1988, 53, 2137 CrossRef CAS; (b) B. B. Snider, R. M. Mohan and S. A. Kates, Manganese(III)-based oxidative free-radical cyclization. Synthesis of (+)-podocarpic acid, J. Org. Chem., 1985, 50, 3659 CrossRef CAS; (c) B. B. Snider, Manganese(III)-Based oxidative free-radical cyclizations, Chem. Rev., 1996, 96, 339 CrossRef CAS PubMed.
- (a) P. A. Wade and J. F. Bereznak, Sulfonylisoxazolines: reliable intermediates for the preparation of β-hydroxy nitriles, J. Org. Chem., 1987, 52, 2973 CrossRef CAS; (b) S. Scholz, H. Hofmeister, G. Neef, E. Ottow, C. Scheidges and R. Wiechert, Synthese von 14,17-überbrückten 11β-Arylsteroiden, Liebigs Ann. Chem., 1989, 151 CrossRef CAS.
- (a) J. A. R. Salvador, M. L. S. Melo and A. S. C. Neves, Copper-catalysed allylic oxidation of Δ5-steroids by t-butyl hydroperoxide, Tetrahedron Lett., 1997, 38, 119 CrossRef CAS; (b) M. S. Kharasch and G. Sosnovsky, The reactions of t-Butyl perbenzoate and olefins-a stereospecific reaction, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS.
- W. Li and P. L. Fuchs, Synthesis of functionalized sulfonamides via 1, 3-dipolar cycloaddition of pentafluorophenyl vinylsulfonate, Org. Lett., 2003, 5, 2849 CrossRef CAS PubMed.
- M. Suzuki, H. Ohtake, Y. Kameya, N. Hamanaka and R. Noyori, Ruthenium(II)-catalyzed reactions of 1,4-epiperoxides, J. Org. Chem., 1989, 54, 5292 CrossRef CAS.
- A. Boto, R. Freire, R. Hernández and E. Suárez, Tandem β-fragmentation−hydrogen abstraction reaction of alkoxy radicals in steroidal systems, J. Org. Chem., 1997, 62, 2975 CrossRef CAS PubMed.
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Tetrapropylammonium Perruthenate, Pr4N+RuO4−, TPAP: A Catalytic Oxidant for Organic Synthesis, Synthesis, 1994, 639 CrossRef.
- (a) K.-H. Qui, C. Angele, R. Claude, N.-N. Hanh, N.-V. Kinh and K.-H. Francoise, New rearranged limonoids from Harrisonia perforata. III, J. Nat. Prod., 2001, 64, 634 CrossRef PubMed; (b) X. Fang, Y.-T. Di, H.-P. He, H.-Y. Liu, Z. Zhang, Y.-L. Ren, Z.-L. Gao, S. Gao and X.-J. Hao, Cipadonoid A, a novel limonoid with an unprecedented skeletonfrom Cipadessa cinerasecns, Org. Lett., 2008, 10, 1905 CrossRef CAS PubMed; (c) X. Fang, Q. Zhang, C.-J. Tan, S.-Z. Mu, Y. Lv, Y.-B. Lu, Q.-T. Zheng, Y.-T. Di and X.-J. Hao, Cipadonoids B–G, six new limonoids from Cipadessa cinerascens, Tetrahedron, 2009, 65, 7408 CrossRef CAS; (d) X.-H. Yan, Y.-T. Di, X. Fang, S.-Y. Yan, H.-P. He, S.-L. Li, Y. Lv and X.-J. Hao, Chemical constituents from fruits of Harrisonia perforate, Phytochemistry, 2011, 72, 508 CrossRef CAS PubMed; (e) C.-M. Yuan, Y. Zhang, G.-H. Tang, Y.-T. Di, M.-M. Cao, X.-Y. Wang, G.-Y. Zuo, S.-L. Li, H.-M. Hua, H.-P. He and X.-J. Hao, Khayseneganins A–H, limonoids from Khaya senegalensis, J. Nat. Prod., 2012, 76, 327 CrossRef PubMed.
- (a) G. E. Brandt, M. D. Schmidt, T. E. Prisinzano and B. S. Blagg, Gedunin, a novel Hsp90 inhibitor: semisynthesis of derivatives and preliminary structure−activity relationships, J. Med. Chem., 2008, 51, 6495 CrossRef CAS PubMed; (b) J. Cui, Z. Deng, M. Xu, P. Proksch, Q. Li and W. Lin, Protolimonoids and limonoids from the chinese mangrove plant Xylocarpus granatum, Helv. Chim. Acta, 2009, 92, 139 CrossRef CAS; (c) R.-Y. Kuo, K. Qian, S. L. Morris-Natschke and K. H. Lee, Plant-derived triterpenoids and analogues as antitumor and anti-HIV agents, Nat. Prod. Rep., 2009, 26, 1321 RSC; (d) S.-E. Lee, M.-R. Kim, J.-H. Kim, G.-R. Takeoka, T.-W. Kim and B.-S. Park, Antimalarial activity of anthothecol derived from Khaya anthotheca (Meliaceae), Phytomedicine, 2008, 15, 533 CrossRef CAS PubMed.
- (a) C. Lv, X. Yan, Q. Tu, Y. Di, C. Yuan, X. Fang, Y. Ben-David, L. Xia, J. Gong, Y. Shen, Z. Yang and X. Hao, Isolation and asymmetric total synthesis of Perforanoid A, Angew. Chem., Int. Ed., 2016, 55, 7539 CrossRef CAS PubMed; (b) C. Lv, Q. Tu, J. Gong, X. Hao and Z. Yang, Asymmetric total synthesis of (−)-perforanoid A, Tetrahedron, 2017, 73, 3612 CrossRef CAS; (c) Y. Han, Y. Zhao and S. Ma, Rhodium-catalysed Pauson–Khand-type cyclization of 1,5-allene-alkynes: a chirality transfer strategy for optically active bicyclic ketones, Chem. – Eur. J., 2019, 25, 9529 CrossRef CAS PubMed.
- J.-X. Ji, L.-Q. Qiu, C. W. Yip and A. S. C. Chan, A convenient, one-step synthesis of optically active tertiary aminonaphthol and its applications in the highly enantioselective alkenylations of aldehydes, J. Org. Chem., 2003, 68, 1589 CrossRef CAS PubMed.
- S. Trudeau and J. P. Morken, Short and efficient total synthesis of fraxinellone limonoids using the stereoselective Oshima−Utimoto reaction, Org. Lett., 2005, 7, 5465 CrossRef CAS PubMed.
- (a) M. A. Evans and J. P. Morken, Stereoselective synthesis of furans by the Pd-catalyzed Oshima−Utimoto reaction, Org. Lett., 2005, 7, 3367 CrossRef CAS PubMed; (b) M. A. Evans and J. P. Morken, Asymmetric synthesis of (−)-Dihydroxanthatin by the stereoselective Oshima−Utimoto reaction, Org. Lett., 2005, 7, 3371 CrossRef CAS PubMed.
- G. A. Molander and O. A. Argintaru, Stereospecific Ni-catalyzed cross-coupling of potassium alkenyltrifluoroborates with alkyl halides, Org. Lett., 2014, 16, 1904 CrossRef CAS PubMed.
- D. Fernández González, J. P. Brand and J. Waser, Ethynyl-1,2-benziodoxol-3(1H)-one (EBX): an exceptional reagent for the ethynylation of keto, cyano, and nitro esters, Chem. – Eur. J., 2010, 16, 9457 CrossRef PubMed.
- Y. Tanaka, A. Sakamoto, T. Inoue, T. Yamada, T. Kikuchi, T. Kajimoto, O. Muraoka, A. Sato, Y. Wataya, H.-S. Kim and R. Tanaka, Andirolides H–P from the flower of andiroba (Carapa guianensis, Meliaceae), Tetrahedron, 2012, 68, 3669 CrossRef CAS.
- B.-D. Lin, T. Yuan, C.-R. Zhang, L. Dong, B. Zhang, Y. Wu and J.-M. Yue, Structurally diverse limonoids from the fruits of Swietenia mahagoni, J. Nat. Prod., 2009, 72, 2084 CrossRef CAS PubMed.
- A. W. Schuppe and T. R. Newhouse, Assembly of the limonoid architecture by a divergent approach: total synthesis of (±)-Andirolide N via (±)-8α-Hydroxycarapin, J. Am. Chem. Soc., 2017, 139, 631 CrossRef CAS PubMed.
- D. Huang, A. W. Schuppe, M. Z. Liang and T. R. Newhouse, Scalable procedure for the fragmentation of hydroperoxides mediated by copper and iron tetrafluoroborate salts, Org. Biomol. Chem., 2016, 14, 6197 RSC.
- (a) L. N. Mander and S. P. Sethi, Regioselective synthesis of β-ketoesters from lithium enolates and methyl cyanoformate, Tetrahedron Lett., 1983, 24, 5425 CrossRef CAS; (b) C. R. Johnson and R. K. Raheja, Hydrosilylation of enones: platinum divinyltetramethyldisiloxane complex in the preparation of triisopropylsilyl and triphenylsilyl enol ethers, J. Org. Chem., 1994, 59, 2287 CrossRef CAS; (c) Q. Tang and S. E. Sen, carbomethoxypropionyl cyanide: A regioselective C-acylation reagent for the preparation of β-dicarbonyl compounds, Tetrahedron Lett., 1998, 39, 2249 CrossRef CAS; (d) J. Stein, L. N. Lewis, Y. Gao and R. A. Scott, In situ determination of the active catalyst in hydrosilylation reactions using highly reactive Pt(0) catalyst precursors, J. Am. Chem. Soc., 1999, 121, 3693 CrossRef CAS.
- W. R. Chan, D. R. Taylor and R. T. Aplin, Extracts of cedrela odorata L.-IV: The structure of odoratin, an undecanortriterpene, Tetrahedron, 1972, 28, 431 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Redox economy in organic synthesis, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- (a) E. J. Corey, W. Li and T. Nagamitsu, An efficient and concise enantioselective total synthesis of Lactacystin, Angew. Chem., Int. Ed., 1998, 37, 1676 CrossRef CAS; (b) A. Sen and T.-W. Lai, Electron-transfer catalysis of substitution, Inorg. Chem., 1981, 20, 4036 CrossRef CAS; (c) J. Yu, M. J. Gaunt and J. B. Spencer, Convenient preparation of trans-arylalkenes via Palladium(II)-catalyzed isomerization of cis-arylalkenes, J. Org. Chem., 2002, 67, 4627 CrossRef CAS PubMed; (d) K. V. Petrova, J. T. Mohr and B. M. Stoltz, Enantioselective total synthesis of (+)-Cassiol, Org. Lett., 2009, 11, 293 CrossRef CAS PubMed.
- (a) A. Schmidt, A. R. Nödling and G. Hilt, An alternative mechanism for the Cobalt-catalyzed isomerization of terminal alkenes to (Z)-2-alkenes, Angew. Chem., Int. Ed., 2015, 54, 801 CrossRef CAS PubMed; (b) H. J. Lim, C. R. Smith and T. V. RajanBabu, Facile Pd(II)- and Ni(II)-catalyzed isomerization of terminal alkenes into 2-alkenes, J. Org. Chem., 2009, 74, 4565 CrossRef CAS PubMed.
- B. R. Travis, R. S. Narayan and B. Borhan, Osmium tetroxide-promoted catalytic oxidative cleavage of olefins: an organometallic ozonolysis, J. Am. Chem. Soc., 2002, 124, 3824 CrossRef CAS PubMed.
- Z.-F. Zhou, H. L. Liu, W. Zhang, T. Kurtan, A. Mandi, A. Benyei, J. Li, O. Taglialatela-Scafati and Y.-W. Guo, Bioactive rearranged limonoids from the Chinese mangrove Xylocarpus granatum Koenig, Tetrahedron, 2014, 70, 6444 CrossRef CAS.
- A. W. Schuppe, D. Huang, Y. Chen and T. R. Newhouse, Total synthesis of (−)-Xylogranatopyridine B via a Palladium-catalyzed oxidative stannylation of enones, J. Am. Chem. Soc., 2018, 140, 2062 CrossRef CAS PubMed.
- M. Baidya and H. Yamamoto, Metal nitrite: A powerful oxidizing reagent, J. Am. Chem. Soc., 2011, 133, 13880 CrossRef CAS PubMed.
- (a) Y. Chen, J. P. Romaire and T. R. Newhouse, Palladium-catalyzed α, β-dehydrogenation of esters and nitriles, J. Am. Chem. Soc., 2015, 137, 5875 CrossRef CAS PubMed; (b) Y. Chen, A. Turlik and T. R. Newhouse, Amide α, β-dehydrogenation using allyl-Palladium catalysis and a hindered monodentate anilide, J. Am. Chem. Soc., 2016, 138, 1166 CrossRef CAS PubMed; (c) A. Turlik, Y. Chen and T. R. Newhouse, Dehydrogenation adjacent to carbonyls using Palladium–allyl intermediates, Synlett, 2016, 27, 331 CAS; (d) Y. Chen, D. Huang, Y. Zhao and T. R. Newhouse, Allyl-Palladium-catalyzed ketone dehydrogenation enables telescoping with enone α,β-vicinal difunctionalization, Angew. Chem., Int. Ed., 2017, 56, 8258 CrossRef CAS PubMed; (e) Y. Zhao, Y. Chen and T. R. Newhouse, Allyl-Palladium-catalyzed α, β-dehydrogenation of carboxylic acids via enediolates, Angew. Chem., Int. Ed., 2017, 56, 13122 CrossRef CAS PubMed.
- S. Liu and L. S. Liebeskind, A simple, modular synthesis of substituted pyridines, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed.
- (a) S. Liu, Y. Yu and L. S. Liebeskind, N-substituted imines by the Copper-catalyzed N-imination of boronic acids andorganostannanes with O-acyl ketoximes, Org. Lett., 2007, 9, 1947 CrossRef CAS PubMed; (b) Z. Zhang, Y. Yu and L. S. Liebeskind, N-amidation by Copper-mediated cross-coupling of organostannanes or boronic acids with O-acetyl hydroxamic acids, Org. Lett., 2008, 10, 3005 CrossRef CAS PubMed.
- (a) J. X. Qiao and P. Y. S. Lam, Copper-promoted carbon-heteroatom bond cross-coupling with boronic acids and derivatives, Synthesis, 2011, 829 CrossRef CAS; (b) J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, Spectroscopic studies of the Chan–Lam amination: A mechanism-inspired solution to boronic ester reactivity, J. Am. Chem. Soc., 2017, 139, 4769 CrossRef CAS PubMed.
- T. R. Newhouse and P. S. Baran, If C-H bonds could talk: selective C-H bond oxidation, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed.
- (a) M. Krumpolc and J. Rocěk, Synthesis of stable chromium(V) complexes of tertiary hydroxy acids, J. Am. Chem. Soc., 1979, 101, 3206 CrossRef CAS; (b) N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, Two-phase synthesis of (−)-Taxuyunnanine D, J. Am. Chem. Soc., 2014, 136, 4909 CrossRef CAS PubMed.
- (a) K. Ikura, I. Ryu, A. Ogawa, N. Sonoda, S. Harada and N. Kasai, Synthesis, characterization, and properties of ethylene-coordinated complexes of β-platinum(II) ketones, Organometallics, 1991, 10, 528 CrossRef CAS; (b) J. O. Hoberg and P. W. Jennings, Platinum(II)-catalyzed Isomerization of alkoxycyclopropanes to alkylated ketones, Organometallics, 1996, 15, 3902 CrossRef CAS.
- (a) V. B. Birman and X. Li, Benzotetramisole: A remarkably enantioselective acyl transfer catalyst, Org. Lett., 2006, 8, 1351 CrossRef CAS PubMed; (b) A. Ortiz, T. Benkovics, G. L. Beutner, Z. Shi, M. Bultman, J. Nye, C. Sfouggatakis and D. R. Kronenthal, Scalable synthesis of the potent HIV inhibitor BMS-986001 by non - enzymatic dynamic kinetic asymmetric transformation (DYKAT), Angew. Chem., Int. Ed., 2015, 54, 7185 CrossRef CAS PubMed.
- A. W. Schuppe, Y. Zhao, Y. Liu and T. R. Newhouse, Total synthesis of (+)-Granatumine A and related bislactone limonoid alkaloids via a pyran to pyridine interconversion, J. Am. Chem. Soc., 2019, 141, 9191 CrossRef PubMed.
- A. S. Kende, P. Fludzinski and J. H. Hill, Chloroacetylenes as Michael acceptors. Mechanism and synthetic utility of enolate reactions with halogenated olefins and chloroacetylenes, J. Am. Chem. Soc., 1984, 106, 3551 CrossRef CAS.
- (a) C. Fehr and O. Guntern, Efficient synthesis of enantiomerically pure α-Ionone from (R)- and (S)-α-Damascone, Helv. Chim. Acta, 1992, 75, 1023 CrossRef CAS; (b) D. Soorukram and P. Knochel, Enantioselective synthesis of α-Ionone derivatives using an anti SN2′ substitution of functionalized zinc organometallics, Org. Lett., 2004, 6, 2409 CrossRef CAS PubMed; (c) M. Bovolenta, F. Castronovo, A. Vadalà, G. Zanoni and G. Vidari, A simple and efficient highly enantioselective synthesis of α-Ionone and α-Damascone, J. Org. Chem., 2004, 69, 8959 CrossRef CAS PubMed.
- (a) E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane, J. Am. Chem. Soc., 1991, 113, 7063 CrossRef CAS; (b) S. L. Vander Velde and E. N. Jacobsen, Kinetic resolution of racemic Chromenes via asymmetric epoxidation: synthesis of (+)-Teretifolione B, J. Org. Chem., 1995, 60, 5380 CrossRef CAS; (c) T. Hamada, R. Irie and T. Katsuki, How does chiral oxo(salen)manganese(V) complex discriminate the enantioface of simple olefins? Scope and limitation of salen-catalyzed epoxidation, Synlett, 1994, 479 CrossRef CAS; (d) Y. Noguchi, R. Irie, T. Fukuda and T. Katsuki, Mn-salen catalysed asymmetric epoxidation of (±)-3-alkylindene: Reagent-dependent stereoselectivity, Tetrahedron Lett., 1996, 37, 4533 CrossRef CAS.
- A. Mori and T. Kato, [Rh(OH)(cod)]2 (cod = 1,5-Cyclooctadiene): A highly efficient catalyst for 1,4-hydrosilylation of α,β-unsaturated carbonyl compounds, Synlett, 2002, 1167 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Dialkylbiaryl phosphines in Pd-catalyzed amination: a user's guide, Chem. Sci., 2011, 2, 27 RSC.
- (a) M. J. Riveira, G. N. Quiroga, E. G. Mata, V. Gandon and M. P. Mischne, Cycloisomerization of conjugated trienones and isomeric 2H-Pyrans: unified strategy toward cyclopenta[b]furans, J. Org. Chem., 2015, 80, 6515 CrossRef CAS PubMed; (b) J. P. Malerich, T. J. Maimone, G. I. Elliott and D. Trauner, Biomimetic synthesis of antimalarial naphthoquinones, J. Am. Chem. Soc., 2005, 127, 6276 CrossRef CAS PubMed.
- (a) H. Cerfontain, J. A. Geenevasen and P. C. M. van Noort, Photochemistry of dienones. Part 7. On the photosensitized isomerization of (E)-β-ionone and its isomeric α-pyran. Evidence for exciplex formation between the α-pyran and fluoren-9-one, J. Chem. Soc., Perkin Trans. 2, 1980, 1057 RSC; (b) C. S. López, R. Álvarez, M. Domínguez, O. N. Faza and Á. R. de Lera, Complex thermal behavior of 11-cis-retinal, the ligand of the visual pigments, J. Org. Chem., 2009, 74, 1007 CrossRef PubMed; (c) M. N. V. Sastry, S. Claessens, P. Habonimana and N. De Kimpe, Synthesis of the natural products 3-Hydroxymollugin and 3-Methoxymollugin, J. Org. Chem., 2010, 75, 2274 CrossRef CAS PubMed; (d) A. J. Hall, S. P. Roche and L. M. West, Synthesis of Briarane diterpenoids: biomimetic transannular oxa-6π electrocyclization induced by a UVA/UVC photoswitch, Org. Lett., 2017, 19, 576 CrossRef CAS PubMed; (e) L. A. M. Murray, T. Fallon, C. J. Sumby and J. H. George, Total synthesis of Naphterpin and Marinone Natural Products, Org. Lett., 2019, 21, 8312 CrossRef CAS PubMed.
- B. W. Yoo, H. Jung II, S. H. Kim, Y. S. Ahn and J. Y. Choi, Selective and efficient deoxygenation of amine-N-Oxides with CeCl3·7H2O/Zinc System, Bull. Korean Chem. Soc., 2013, 34, 359 CrossRef CAS.
- J. H. Butterworth and E. D. Morgan, Isolation of a substance that suppresses feeding in locusts, Chem. Commun., 1968, 23 RSC.
- (a) C. J. Turner, M. S. Tempesta, R. B. Taylor, M. G. Zagorski, J. S. Termini, D. R. Schroeder and K. Nakanishi, An NMR spectroscopic study of azadirachtin and its trimethyl ether, Tetrahedron, 1987, 43, 2789 CrossRef CAS; (b) J. N. Bilton, H. B. Broughton, P. S. Jones, S. V. Ley, Z. Lidert, E. D. Morgan, H. S. Rzepa, R. N. Sheppard, A. M. Z. Slawin and D. J. Williams, An x-ray crystallographic, mass spectroscopic, and NMR study of the limonoid insect antifeedant azadirachtin and related derivatives, Tetrahedron, 1987, 43, 2805 CrossRef; (c) W. Kraus, M. Bokel, A. Bruhn, R. Cramer, I. Klaiber, A. Klenk, G. Nagl, H. Pçhnl, H. Sadlo and B. Volger, Structure determination by nmr of azadirachtin and related compounds from azadirachta indica a. Juss (Meliaceae), Tetrahedron, 1987, 43, 2817 CrossRef CAS.
- (a) S. V. Ley, A. A. Denholm and A. Wood, The chemistry of azadirachtin, Nat. Prod. Rep., 1993, 10, 109 RSC; (b) S. R. Fernandes, L. Barreiros, R. F. Oliveira, A. Cruz, C. Prudencio, A. I. Oliveira, C. Pinho, N. Santos and J. Morgado, Chemistry, bioactivities, extraction and analysis of azadirachtin: State-of-the-art, Fitoterapia, 2019, 134, 141 CrossRef CAS PubMed.
- (a) T. Durand-Reville, L. B. Gobbi, B. L. Gray, S. V. Ley and J. S. Scott, Highly selective entry to the Azadirachtin skeleton via a Claisen rearrangement/radical cyclization sequence, Org. Lett., 2002, 4, 3847 CrossRef CAS PubMed; (b) T. Fukuzaki, S. Kobayashi, T. Hibi, T. Ikuma, J. Ishihara, N. Kanoh and A. Murai, Studies aimed at the total synthesis of Azadirachtin. A modeled connection of C-8 and C-14 in Azadirachtin, Org. Lett., 2002, 4, 2877 CrossRef CAS PubMed; (c) K. C. Nicolaou, M. Follmann, A. J. Roecker and K. W. Hunt, Model studies towards Azadirachtin: Part 1. construction of the crowded C8-C14 bond by radical chemistry, Angew. Chem., Int. Ed., 2002, 41, 2103 CrossRef CAS; (d) K. C. Nicolaou, A. J. Roecker, M. Follmann and R. Baati, Model studies towards Azadirachtin: Part 2. construction of the crowded C8-C14 bond by transition metal chemistry, Angew. Chem., Int. Ed., 2002, 41, 2107 CrossRef CAS; (e) K. C. Nicolaou, A. J. Roecker, H. Monenschein, P. Guntupalli and M. Follmann, Studies towards the synthesis of Azadirachtin: enantioselective entry into the Azadirachtin framework through cascade reactions, Angew. Chem., Int. Ed., 2003, 42, 3637 CrossRef CAS PubMed; (f) K. C. Nicolaou, P. K. Sasmal, T. V. Koftis, A. Converso, E. Loizidou, F. Kaiser, A. J. Roecker, C. C. Dellios, X.-W. Sun and G. Petrovic, Model studies towards Azadirachtin. Part 2. construction of the crowded C8—C14 bond by transition metal chemistry, Angew. Chem., Int. Ed., 2005, 44, 3447 CrossRef CAS PubMed; (g) S. V. Ley, Development of methods suitable for natural product synthesis: The azadirachtin story, Pure Appl. Chem., 2005, 77, 1115 CAS; (h) D. Nakagawa, M. Miyashita and K. Tanino, Synthetic studies on azadirachtin: stereoselective construction of the ABCE ring system, Tetrahedron Lett., 2010, 51, 2771 CrossRef CAS; (i) K. Sakurai and K. Tanino, Synthetic studies on azadirachtin: construction of the ABC ring system via the Diels–Alder reaction of a vinyl allenylsilane derivative, Tetrahedron Lett., 2015, 56, 496 CrossRef CAS; (j) C. Tan, W. Chen, X. Mu, Q. Chen, J. Gong, T. Luo and Z. Yang, Efficient large scale syntheses of 3-Deoxy-d-manno-2-octulosonic acid (Kdo) and its derivatives, Org. Lett., 2015, 17, 2338 CrossRef CAS PubMed; (k) H. Shi, C. Tan, W. Zhang, Z. Zhang, R. Long, T. Luo and Z. Yang, Synthetic progress toward Azadirachtins. 1. enantio- and diastereoselective synthesis of the left-wing fragment of 11-epi-Azadirachtin I, Org. Lett., 2015, 17, 2342 CrossRef CAS PubMed.
- N. Mori, T. Kitahara, K. Mori and H. Watanabe, Asymmetric formal synthesis of Azadirachtin, Angew. Chem., Int. Ed., 2015, 54, 14920 CrossRef CAS PubMed.
- H. Watanabe, N. Mori, D. Itoh, T. Kitahara and K. Mori, Synthetic study towards Azadirachtin: an efficient and stereoselective construction of the AB rings with full functionality, Angew. Chem., Int. Ed., 2007, 46, 1512 CrossRef CAS PubMed.
- R. Rej, N. Jana, S. Kar and S. Nanda, Stereoselective synthesis of a novel natural carbasugar and analogues from hydroxymethylated cycloalkenone scaffolds, Tetrahedron: Asymmetry, 2012, 23, 364 CrossRef CAS.
- T. L. Macdonald and D. R. Reagan, Reaction of propargylic substrates with organocopper species. Synthetic aspects, J. Org. Chem., 1980, 45, 4740 CrossRef CAS.
- M. D. Clay and A. G. Fallis, Acetylenic allenophanes: an asymmetric synthesis of a bis(alleno)-bis(butadiynyl)-meta-cyclophane, Angew. Chem., Int. Ed., 2005, 44, 4039 CrossRef CAS PubMed.
- (a) Y.-M. Shi, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Triterpenoids from the Schisandraceae family: an update, Nat. Prod. Rep., 2015, 32, 367 RSC; (b) Y.-G. Xia, B.-Y. Yang and H.-X. Kuang, Schisandraceae triterpenoids: a review, Phytochem. Rev., 2015, 14, 155 CrossRef CAS.
- S. S. Goh, G. Chaubet, B. Gockel, M.-C. A. Cordonnier, H. Baars, A. W. Phillips and E. A. Anderson, Total synthesis of (+)-Rubriflordilactone A, Angew. Chem., Int. Ed., 2015, 54, 12618 CrossRef CAS PubMed.
- Q. Xiao, W.-W. Ren, Z.-X. Chen, T.-W. Sun, Y. Li, Q.-D. Ye, J.-X. Gong, F.-K. Meng, L. You, Y.-F. Liu, M.-Z. Zhao, L.-M. Xu, Z.-H. Shan, Y. Shi, Y.-F. Tang, J.-H. Chen and Z. Yang, Diastereoselective total synthesis of (±)-Schindilactone A, Angew. Chem., Int. Ed., 2011, 50, 7373 CrossRef CAS PubMed.
- (a) L. You, X.-T. Liang, L.-M. Xu, Y.-F. Wang, J.-J. Zhang, Q. Su, Y.-H. Li, B. Zhang, S.-L. Yang, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Propindilactone G, J. Am. Chem. Soc., 2015, 137, 10120 CrossRef CAS PubMed; (b) L.-M. Xu, L. You, Z.-H. Shan, R.-C. Yu, B. Zhang, Y.-H. Li, Y. Shi, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Propindilactone G, Part 1: initial attempts towards the synthesis of Schiartanes, Chem. – Asian. J., 2016, 11, 1406 CrossRef CAS PubMed; (c) J.-J. Zhang, L. You, Y.-F. Wang, Y.-H. Li, X.-T. Liang, B. Zhang, S.-L. Yang, Q. Su, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Propindilactone G, Part 2: enantioselective construction of the fully functionalized BCDE ring system, Chem. – Asian. J., 2016, 11, 1414 CrossRef CAS PubMed; (d) X.-T. Liang, L. You, Y.-H. Li, H.-X. Yu, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Propindilactone G, Part 3: The final phase and completion of the synthesis, Chem. – Asian. J., 2016, 11, 1425 CrossRef CAS PubMed.
- Y.-X. Han, Y.-L. Jiang, Y. Li, H.-X. Yu, B.-Q. Tong, Z. Niu, S.-J. Zhou, S. Liu, Y. Lan, J.-H. Chen and Z. Yang, Biomimetically inspired asymmetric total synthesis of (+)-19-dehydroxyl arisandilactone A, Nat. Commun., 2017, 8, 14233 CrossRef PubMed.
- (a) D.-D. Liu, T.-W. Sun, K.-Y. Wang, Y. Lu, S.-L. Zhang, Y.-H. Li, Y.-L. Jiang, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Lancifodilactone G Acetate, J. Am. Chem. Soc., 2017, 139, 5732 CrossRef CAS PubMed; (b) T.-W. Sun, D.-D. Liu, K.-Y. Wang, B.-Q. Tong, J.-X. Xie, Y.-L. Jiang, Y. Li, B. Zhang, Y.-F. Liu, Y.-X. Wang, J.-J. Zhang, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Lancifodilactone G Acetate. 1. diastereoselective synthesis of CDEFGH ring system, J. Org. Chem., 2018, 83, 6893 CrossRef CAS PubMed; (c) K.-Y. Wang, D.-D. Liu, T.-W. Sun, Y. Lu, S.-L. Zhang, Y.-H. Li, Y.-X. Han, H.-Y. Liu, C. Peng, Q.-Y. Wang, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Lancifodilactone G Acetate. 2. Final phase and completion of the total synthesis, J. Org. Chem., 2018, 83, 6907 CrossRef CAS PubMed.
- Z. Yang, The journey of Schinortriterpenoid total syntheses, Acc. Chem. Res., 2019, 52, 480 CrossRef CAS PubMed.
- W. L. Xiao, L. M. Yang, N. B. Gong, L. Wu, R. R. Wang, J. X. Pu, X. L. Li, S. X. Huang, Y. T. Zheng, R. T. Li, Y. Lu, Q. T. Zheng and H. D. Sun, Rubriflordilactones A and B, Two novel Bisnortriterpenoids from Schisandra rubriflora and their biological activities, Org. Lett., 2006, 8, 991 CrossRef CAS PubMed.
- J. Li, P. Yang, M. Yao, J. Deng and A. Li, Total synthesis of Rubriflordilactone A, J. Am. Chem. Soc., 2014, 136, 16477 CrossRef CAS PubMed.
- R. J. Anderson, V. L. Corbin, G. Cotterrell, G. R. Cox, C. A. Henrick, F. Schaub and J. B. Siddall, Stereoselective addition of organocopper reagents to acetylenic esters and amides. Synthesis of juvenile hormone analogs, J. Am. Chem. Soc., 1975, 97, 1197 CrossRef CAS PubMed.
- (a) Y. Gao, J. M. Klunder, R. M. Hanson, H. Masamune, S. Y. Ko and K. B. Sharpless, Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS; (b) I. Larrosa, M. I. Da Silva, P. M. Gýmez, P. Hannen, E. Ko, S. R. Lenger, S. R. Linke, A. J. P. White, D. Wilton and A. G. M. Barrett, Highly convergent three component benzyne coupling: the total synthesis of ent-Clavilactone B, J. Am. Chem. Soc., 2006, 128, 14042 CrossRef CAS PubMed.
- K. Ando, Highly selective synthesis of Z-unsaturated esters by using new Horner−Emmons reagents, ethyl (diarylphosphono)acetates, J. Org. Chem., 1997, 62, 1934 CrossRef CAS PubMed.
- (a) S. S. Goh, H. Baars, B. Gockel and E. A. Anderson, Metal-catalysed syntheses of abridged CDE rings of Rubriflordilactones A and B, Org. Lett., 2012, 14, 6278 CrossRef CAS PubMed.
- (a) S. Bracegirdle and E. A. Anderson, Arylsilane oxidation—new routes to hydroxylated aromatics, Chem. Commun., 2010, 46, 3454 RSC; (b) E. J. Rayment, N. Summerhill and E. A. Anderson, Synthesis of phenols via fluoride-free oxidation of arylsilanes and arylmethoxysilanes, J. Org. Chem., 2012, 77, 7052 CrossRef CAS PubMed; (c) M. Suginome and H. Ihara, Impact of perfluorination on the charge-transport parameters of oligoacene crystals, J. Am. Chem. Soc., 2009, 131, 7502 CrossRef PubMed.
- N. Nicolaus, S. Strauss, J. M. Neudorfl, A. Prokop and H. G. Schmalz, A [2+2+2]-cycloaddition approach toward 6-oxa-allocolchicinoids with apoptosis-inducing activity, Org. Lett., 2009, 11, 341 CrossRef CAS PubMed.
- (a) V. D. Grob, T. G. Squires and J. R. Vercellotti, Glycosyl chlorides through reaction with zinc chloride-thionyl chloride, Carbohydr. Res., 1969, 10, 595 CrossRef CAS; (b) L. P. Egan, T. G. Squires and J. R. Vercellotti, Acetylated aldosyl chlorides by reaction of aldose peracetates with zinc chloride-thionyl chloride, Carbohydr. Res., 1970, 14, 263 CrossRef CAS.
- (a) G. D. Allred and L. S. Liebeskind, Copper-mediated cross-coupling of organostannanes with organic iodides at or below room temperature, J. Am. Chem. Soc., 1996, 118, 2748 CrossRef CAS; (b) D. R. Williams and K. G. Meyer, Total synthesis of (+)-Amphidinolide K, J. Am. Chem. Soc., 2001, 123, 765 CrossRef CAS PubMed; (c) A. Fürstner, J. A. Funel, M. Tremblay, L. C. Bouchez, C. Nevado, M. Waser, J. Ackerstaff and C. C. Stimson, A versatile protocol for Stille–Migita cross coupling reactions, Chem. Commun., 2008, 44, 2873 RSC; (d) H. N. Lim and K. A. Parker, Total Synthesis of the potent androgen receptor antagonist (−)-Arabilin: A strategic, biomimetic [1,7]-hydrogen shift, J. Am. Chem. Soc., 2011, 133, 20149 CrossRef CAS PubMed.
- (a) W. E. Parham and L. D. Huestis, The reaction of dichlorocarbene with 2, 3-Chromene and 3, 4-Chromene, J. Am. Chem. Soc., 1962, 84, 813 CrossRef CAS; (b) X. Xiong, Y. Li, Z. Lu, M. Wan, J. Deng, S. Wu, H. Shao and A. Li, Synthesis of the 6, 6, 5, 7-tetracyclic core of daphnilongeranin B, Chem. Commun., 2014, 50, 5294 RSC.
- (a) G. H. Posner and S. R. Haines, A convenient, one-step, high-yield replacement of an anomeric hydroxyl group by a fluorine atom using dast. Preparation of glycosyl fluorides, Tetrahedron Lett., 1985, 26, 5 CrossRef CAS; (b) K. Toshima, Glycosyl fluorides in glycosidations, Carbohydr. Res., 2000, 327, 15 CrossRef CAS PubMed.
- (a) S. Liras, C. L. Lynch, A. M. Fryer, B. T. Vu and S. F. Martin, Applications of vinylogous mannich reactions. Total syntheses of the ergot alkaloids Rugulovasines A and B and Setoclavine, J. Am. Chem. Soc., 2001, 123, 5918 CrossRef CAS PubMed; (b) S. W. Elmore, M. J. Coghlan, D. D. Anderson, J. K. Pratt, B. E. Green, A. X. Wang, M. A. Stashko, C. W. Lin, C. M. Tyree, J. N. Miner, P. B. Jacobson, D. M. Wilcox and B. C. Lane, Nonsteroidal selective glucocorticoid modulators: the effect of C-5 alkyl substitution on the transcriptional activation/repression profile of 2,5-dihydro-10-methoxy-2,2,4- trimethyl-1H-[1] benzopyrano [3,4-f] quinolones, J. Med. Chem., 2001, 44, 4481 CrossRef CAS PubMed.
- (a) N. A. Petasis and I. Akritopoulou, The boronic acid mannich reaction: A new method for the synthesis of geometrically pure allylamines, Tetrahedron Lett., 1993, 34, 583 CrossRef CAS; (b) S. Lee and D. W. C. MacMillan, Organocatalytic vinyl and Friedel−Crafts alkylations with trifluoroborate Salts, J. Am. Chem. Soc., 2007, 129, 15438 CrossRef CAS PubMed; (c) J. Zeng, S. Vedachalam, S. Xiang and X.-W. Liu, Direct C-Glycosylation of organotrifluoroborates with glycosyl fluorides and its application to the total synthesis of (+)-Varitriol, Org. Lett., 2011, 13, 42 CrossRef CAS PubMed; (d) J. Kim and M. Movassaghi, Concise total synthesis and stereochemical revision of (+)-Naseseazines A and B: regioselective arylative dimerization of diketopiperazine alkaloids, J. Am. Chem. Soc., 2011, 133, 14940 CrossRef CAS PubMed; (e) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Iridium-catalyzed enantioselective allylic vinylation, J. Am. Chem. Soc., 2013, 135, 994 CrossRef CAS PubMed.
- W.-L. Xiao, R.-T. Li, S.-X. Huang, J.-X. Pu and H.-D. Sun, Triterpenoids from the Schisandraceae family, Nat. Prod. Rep., 2008, 25, 871 RSC.
- (a) Z. Lu, Y. Li, J. Deng and A. Li, Total synthesis of the Daphniphyllum alkaloid daphenylline, Nat. Chem., 2013, 5, 679 CrossRef CAS PubMed; (b) Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, D. J. Edmonds, J. Zhong and A. Li, Biomimetically inspired asymmetric total synthesis of (+)-19-dehydroxyl arisandilactone A, Nat. Commun., 2015, 6, 6096 CrossRef CAS PubMed.
- R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, The orthoester Johnson–Claisen rearrangement in the synthesis of bioactive molecules, natural products, andsynthetic intermediates – recent advances, Eur. J. Org. Chem., 2014, 2833 CrossRef CAS.
- M. J. Sofia and J. A. Katzenellenbogen, 3-(Acylamido)-4-phenyl-6(E)-(iodomethylene)tetrahydro-2- pyranones. Synthesis of novel amino acid analogs, J. Org. Chem., 1985, 50, 2331 CrossRef CAS.
- (a) A. Sakakura, R. Kondo, Y. Matsumura, M. Akakura and K. Ishihara, Rational design of highly effective asymmetric Diels−Alder catalysts bearing 4,4′-sulfonamidomethyl groups, J. Am. Chem. Soc., 2009, 131, 17762 CrossRef CAS PubMed; (b) I. B. Seiple, S. Su, I. S. Young, A. Nakamura, J. Yamaguchi, L. Jørgensen, R. A. Rodriguez, D. P. O′Malley, T. Gaich, M. Kock and P. S. Baran, Enantioselective total syntheses of (−)-Palau'amine, (−)-Axinellamines, and (−)-Massadines, J. Am. Chem. Soc., 2011, 133, 14710 CrossRef CAS PubMed.
- (a) H. Tokuyama, S. Yokoshima and T. Fukuyama, A novel ketone synthesis by a palladium-catalyzed reaction of thiol esters and organozinc reagents, Tetrahedron Lett., 1998, 39, 3189 CrossRef CAS; (b) S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Catalysts for Suzuki—Miyaura coupling processes: scope and studies of the effect of ligand structure, Angew. Chem., Int. Ed., 2004, 43, 1871 CrossRef CAS PubMed.
- (a) G. A. Kraus, K. A. Frazier, B. D. Roth, M. J. Taschner and K. Neuenschwander, Conversion of lactones into ethers, J. Org. Chem., 1981, 46, 2417 CrossRef CAS; (b) M. D. Lewis, K. C. Cha and Y. Kishi, Highly stereoselective approaches to α- and β-C-glyco-pyranosides, J. Am. Chem. Soc., 1982, 104, 4976 CrossRef CAS.
- R. C. Winstead, T. H. Simpson, G. A. Lock, M. D. Schiavelli and D. W. Thompson, A regioselective entry to vinyl lithiums from unsymmetrical ketones via enol triflates, J. Org. Chem., 1986, 51, 277 CrossRef.
- (a) X. Luo, Y. Chang, X.-J. Zhang, J.-X. Pu, X.-M. Gao, Y.-L. Wu, R.-R. Wang, W.-L. Xiao, Y.-T. Zheng, Y. Lu, G.-Q. Chen, Q.-T. Zheng and H.-D. Sun, Schilancidilactones A and B: two novel tetranortriterpenoids with an unprecedented skeleton from Schisandra lancifolia, Tetrahedron Lett., 2009, 50, 5962 CrossRef CAS; (b) X. Luo, Y.-M. Shi, R.-H. Luo, S.-H. Luo, X.-N. Li, R.-R. Wang, S.-H. Li, Y.-T. Zheng, X. Du, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Schilancitrilactones A–C: three unique nortriterpenoids from Schisandra lancifolia, Org. Lett., 2012, 14, 1286 CrossRef CAS PubMed.
- L. Wang, H. Wang, Y. Li and P. Tang, Total synthesis of Schilancitrilactones B and C, Angew. Chem., Int. Ed., 2015, 54, 5732 CrossRef CAS PubMed.
- (a) S. Takita, S. Yokoshima and T. Fukuyama, A practical synthesis of (−)-Kainic Acid, Org. Lett., 2011, 13, 2068 CrossRef CAS PubMed; (b) S. Takita, S. Yokoshima and T. Fukuyama, A practical synthesis of (-)-Kainic Acid, Synthesis, 2011, 3848 CAS.
- T. Benneche, Z. Hussain, A. A. Scheie and J. Lönn-Stensrud, Synthesis of 5-(bromomethylene) furan-2(5H)-ones and 3-(bromomethylene)isobenzofuran-1(3H)-ones as Inhibitors of microbial quorum sensing, New J. Chem., 2008, 32, 1567 RSC.
- W. D. Wulff, G. A. Peterson, W. E. Bauta, K.-S. Chan, K.-L. Faron, S. R. Gilbertson, R. W. Kaesler, D. C. Yang and C. K. Murray, A regioselective entry to vinyl lithiums from unsymmetrical ketones via enol triflates, J. Org. Chem., 1986, 51, 277 CrossRef CAS.
- B. M. Trost, S. Tanimori and P. T. Dunn, A simple divergence from asymmetric cyclopropane to lactone annulation, J. Am. Chem. Soc., 1997, 119, 2735 CrossRef CAS.
- (a) H. Sai and H. Ohmizu, Facile stereoselective syntheses of (E)- and (Z) -α-substituted cinnamates by stereoselective dehydration reaction with 1-ethyl-3-(3-dimethyl- aminopropyl)carbodiimide (EDC), Tetrahedron Lett., 1999, 40, 5019 CrossRef CAS; (b) H. Sai, T. Ogiku and H. Ohmizu, Facile stereoselective synthesis of (E)- and (Z)-α-substituted cinnamates: stereospecific dehydration reaction with 1-ethyl-3-(3-dimethylaminopropyl)- carbodiimide (EDC) and copper(II) chloride, Tetrahedron, 2007, 63, 10345 CrossRef CAS.
- For selected reviews, see: (a) K. Zeitler, Photoredox catalysis with visible light, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Visible light photocatalysis as a greener approach to photochemical synthesis, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W. Xiao, Visible-light photoredox catalysis, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- J. L. Luche and C. Allavena, Ultrasound in organic synthesis 16. Optimisation of the conjugate additions to α, β-unsaturated carbonyl compounds in aqueous media, Tetrahedron Lett., 1988, 29, 5369 CrossRef CAS.
- T. Satoh, K. Nanba and S. Suzuki, Reduction of organic compounds with NaBH4-transition metal salt systems. IV. Selective hydrogenation of olefines in unsaturated esters, Chem. Pharm. Bull., 1971, 19, 817 CrossRef CAS.
- P. Yang, M. Yao, J. Li, Y. Li and A. Li, Total synthesis of rubriflordilactone B, Angew. Chem., Int. Ed., 2016, 55, 6964 CrossRef CAS PubMed.
- T. Mukaiyama, J. Matsuo and H. Kitagawa, A new and one-pot synthesis of α, β-unsaturated ketones by dehydrogenation of various ketones with N-tert-Butyl phenylsulfinimidoyl chloride, Chem. Lett., 2000, 1250 CrossRef CAS.
- M. Toyota, T. Asoh, M. Matsuura and K. Fukumoto, Stereoselective transformation of enantiopure cyclohexenol into cis-Hydrindan. An enantioselective formal total synthetic route to (+)-Pumiliotoxin C, J. Org. Chem., 1996, 61, 8687 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, A general entry to antifeedant Sesterterpenoids: Total synthesis of (+)-Norleucosceptroid A, (−)-Norleucosceptroid B, and (−)-Leucosceptroid K, Angew. Chem., Int. Ed., 2014, 53, 11351 CrossRef CAS PubMed.
- M. Yang, X. Yang, H. Sun and A. Li, Total synthesis of Ileabethoxazole, Pseudopteroxazole, and seco-Pseudopteroxazole, Angew. Chem., Int. Ed., 2016, 55, 2851 CrossRef CAS PubMed.
- (a) H. Wang, X. Zhang and P. Tang, Total syntheses of schilancidilactones A and B, schilancitrilactone A, and 20-epi-schilancitrilactone A via late-stage nickel-catalyzed cross coupling, Chem. Sci., 2017, 8, 7246 RSC; (b) H. Wang, L. Wang, Y. Li, X. Zhang and P. Tang, Collective synthesis of Schilancidilactones A, B and Schilancitrilactones A, B, C, 20-epi-Schilancitrilactone A, Chin. J. Chem., 2019, 37, 255 CAS.
- G. A. Molander and G. Hahn, Lanthanides in organic synthesis. 4. Reduction of α, β-epoxy ketones with Samarium Diiodide. A route to chiral, nonracemic aldols, J. Org. Chem., 1986, 51, 2596 CrossRef CAS.
- K. Hagiya, A. Yamasaki, T. Okuyama and T. Sugimura, Asymmetric meta-arene–alkene photocycloaddition controlled by a 2, 4-pentanediol Tether, Tetrahedron: Asymmetry, 2004, 15, 1409 CrossRef CAS.
- E. Mernyak, E. Kozma, A. Hetenyi, L. Mark, G. Schneider and J. Woelfling, Stereoselective synthesis of spiro and condensed pyrazolines of steroidal α,β-unsaturated ketones and nitrilimines by 1,3-dipolar cycloaddition, Steroids, 2009, 74, 520 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. Jana, T. P. Pathak and M. S. Sigman, Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent advances in homogeneous nickel catalysis, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- P. A. Bartlett and J. Myerson, Stereoselective epoxidation of acyclic olefinic carboxylic acids via Iodolactonization, J. Am. Chem. Soc., 1978, 100, 3950 CrossRef CAS.
- N. Grimblat, T. S. Kaufman and A. M. Sarotti, Computational chemistry driven solution to Rubriflordilactone B, Org. Lett., 2016, 18, 6420 CrossRef CAS PubMed.
- (a) M. Mohammad, V. Chintalapudi, J. Carney, S. Mansfield, P. Sanderson, K. Christensen and E. A. Anderson, Convergent total syntheses of (-)-Rubriflordilactone B and (-)-pseudo-Rubriflordilactone B, Angew. Chem., Int. Ed., 2019, 58, 18177 CrossRef CAS PubMed; (b) O. Torres, M. Fernandez, A. Diaz-Jimenez, A. Pla-Quintana, A. Roglans and M. Sola, Examining the factors that govern the regioselectivity in Rhodium-catalyzed alkyne cyclotrimerization, Organometallics, 2019, 38, 2853 CrossRef CAS; (c) S. Nishigaki, Y. Shibata, A. Nakajima, H. Okajima, Y. Masumoto, T. Osawa, A. Muranaka, H. Sugiyama, A. Horikawa, H. Uekusa, H. Koshino, M. Uchiyama, A. Sakamoto and K. Tanaka, synthesis of belt- and Möbius-Shaped cycloparaphenylenes by rhodium-Catalyzed alkyne cyclotrimerizationm, J. Am. Chem. Soc., 2019, 141, 14955 CrossRef CAS PubMed.
- (a) C. Zhu, X. Shen and S. G. Nelson, Cinchona alkaloid-Lewis acid catalyst systems for enantioselective ketene−aldehyde cycloadditions, J. Am. Chem. Soc., 2004, 126, 5352 CrossRef CAS PubMed; (b) X. Shen, A. S. Wasmuth, J. Zhao, C. Zhu and S. G. Nelson, Catalytic asymmetric assembly of stereodefined propionate units: An enantioselective total synthesis of (−)-Pironetin, J. Am. Chem. Soc., 2006, 128, 7438 CrossRef CAS PubMed.
- (a) Y. Peng, S.-M. Duan and Y.-W. Wang, Concise synthesis of the DEFG ring system in rubriflordilactone B, Tetrahedron Lett., 2015, 56, 4509 CrossRef CAS; (b) T. Ollevier, J.-E. Bouchard and V. Desyroy, Diastereoselective Mukaiyama aldol reaction of 2-(trimethylsilyloxy)furan catalyzed by bismuth triflate, J. Org. Chem., 2008, 73, 331 CrossRef CAS PubMed.
- C. Herb and M. E. Maier, A formal total synthesis of the Salicylihalamides, J. Org. Chem., 2003, 68, 8129 CrossRef CAS PubMed.
- R. Noyori, T. Ohkuma, M. Kitamura, H. Takaya, N. Sayo, H. Kumobayashi and S. Akutagawa, Asymmetric hydrogenation of β-keto carboxylic esters. A practical, purely chemical access to β-hydroxy esters in high enantiomeric purity, J. Am. Chem. Soc., 1987, 109, 5856 CrossRef CAS.
- (a) C. Alayrac, D. Schollmeyer and B. Witulski, First total synthesis of antiostatin A1, a potent carbazole-based naturally occurring antioxidant, Chem. Commun., 2009, 1464 RSC; (b) M. R. Tracey, J. Oppenheimer and R. P. Hsung, Rhodium(I)-catalyzed [2+2+2] cycloadditions of ynamides in the synthesis of amide-substituted chiral Biaryls, J. Org. Chem., 2006, 71, 8629 CrossRef CAS PubMed; (c) E. A. Anderson, E. J. Alexanian and E. J. Sorensen, Synthesis of the furanosteroidal antibiotic Viridin, Angew. Chem., Int. Ed., 2004, 43, 1998 CrossRef CAS PubMed.
- P. A. Grieco, S. Gilman and M. Nishizawa, Organoselenium chemistry. A facile one-step synthesis of alkyl aryl selenides from alcohols, J. Org. Chem., 1976, 41, 1485 CrossRef CAS.
- P. Yang, J. Li, L. Sun, M. Yao, X. Zhang, W.-L. Xiao, J.-H. Wang, P. Tian, H.-D. Sun, P.-T. Puno and A. Li, Elucidation of the structure of Pseudorubriflordilactone B by chemical synthesis, J. Am. Chem. Soc., 2019 DOI:10.1021/jacs.9b09699.
- L. F. Tietze, C. Stadler, N. Böhnke, G. Brasche and A. Grube, Synthesis of enantiomerically pure cyclopentene building blocks, Synlett, 2007, 485 CrossRef CAS.
- E. Yoshii, T. Koizumi, E. Kitatsuji, T. Kawazoe and T. Kaneko, Preparation and some reactions of 2-trimethylsilyloxyfurans, Heterocycles, 1976, 4, 1663 CrossRef CAS.
- M. Wan, M. Yao, J.-Y. Gong, P. Yang, H. Liu and A. Li, Synthesis of the tetracyclic core of chlorospermines, Chin. Chem. Lett., 2015, 26, 272 CrossRef CAS.
- D. A. Rooke and E. M. Ferreira, Platinum-catalyzed hydrosilylations of internal alkynes: harnessing substituent effects to achieve high regioselectivity, Angew. Chem., Int. Ed., 2012, 51, 3225 CrossRef CAS PubMed.
- C. Lei, W.-L. Xiao, S.-X. Huang, J.-J. Chen, J.-X. Pu and H.-D. Sun, Pre-schisanartanins C–D and propintrilactones A–B, two classes of new nortriterpenoids from Schisandra propinqua var. propinqua, Tetrahedron, 2010, 66, 2306 CrossRef CAS.
- Y.-L. Jiang, H.-X. Yu, Y. Li, P. Qu, Y.-X. Han, J.-H. Chen and Z. Yang, Asymmetric total synthesis of Pre-schisanartanin C, J. Am. Chem. Soc., 2020, 142, 573 CrossRef CAS PubMed.
- (a) C. Aubert, O. Buisine and M. Malacria, The behavior of 1, n-enynes in the presence of transition metals, Chem. Rev., 2002, 102, 813 CrossRef CAS PubMed; (b) A. Marinetti, H. Jullien and A. Voituriez, Enantioselective, transition metal catalyzed cycloisomerizations, Chem. Soc. Rev., 2012, 41, 4884 RSC; (c) Y. Zhang, T. Luo and Z. Yang, Strategic innovation in the total synthesis of complex natural products using gold catalysis, Nat. Prod. Rep., 2014, 31, 489 RSC; (d) C. I. Stathakis, P. L. Gkizis and A. L. Zografos, Metal-catalyzed cycloisomerization as a powerful tool in the synthesis of complex sesquiterpenoids, Nat. Prod. Rep., 2016, 33, 1093 RSC; (e) Y. Hu, M. Bai, Y. Yang and Q. Zhou, Metal-catalyzed enyne cycloisomerization in natural product total synthesis, Org. Chem. Front., 2017, 4, 2256 RSC.
- Y.-Q. Gu, C.-H. Tan, J.-X. Gong and Z. Yang, Diversity-oriented synthesis of natural products via gold-catalyzed cascade reactions, Synlett, 2018, 29, 1552 CrossRef CAS.
- (a) F.-D. Boyer, X. Le Goff and I. Hanna, Gold(I)-Catalyzed cycloisomerization of 1,7- and 1,8-enynes: application to the synthesis of a new allocolchicinoid, J. Org. Chem., 2008, 73, 5163 CrossRef CAS PubMed; (b) X. Moreau, A. Hours, L. Fensterbank, J.-P. Goddard, M. Malacria and S. Thorimbert, Use of ionic liquids in the platinum- and gold-catalyzed cycloisomerization of enyne systems, J. Organomet. Chem., 2009, 694, 561 CrossRef CAS; (c) I. D. G. Watson, S. Ritter and F. D. Toste, Asymmetric synthesis of medium-sized rings by intramolecular Au(I)-catalyzed cyclopropanation, J. Am. Chem. Soc., 2009, 131, 2056 CrossRef CAS PubMed; (d) D. J. Gorin, I. D. G. Watson and F. D. Toste, Fluorenes and styrenes by Au(I)-catalyzed annulation of enynes and alkynes, J. Am. Chem. Soc., 2008, 130, 3736 CrossRef CAS PubMed; (e) P.-C. Zhang, Y. Wang, Z.-M. Zhang and J. Zhang, Gold(I)/Xiang-Phos-catalyzed asymmetric intramolecularcyclopropanation of indenes and trisubstituted alkenes, Org. Lett., 2018, 20, 7049 CrossRef CAS PubMed; (f) C. Obradors and A. M. Echavarren, Gold-catalyzed rearrangements and beyond, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed.
- (a) L. Kong, X. Han and P. Jiao, Catalytic asymmetric Diels–Alder reactions involving aryl vinyl ketones, Chem. Commun., 2014, 50, 14113 RSC; (b) S. Thamapipol, B. Ludwig, C. Besnard, C. Saudan and E. P. Kündig, Ruthenium Lewis Acid-catalyzed asymmetric Diels–Alder reactions: reverse-face selectivity for α, β-unsaturated aldehydes and ketones, Helv. Chim. Acta, 2016, 99, 774 CrossRef CAS.
- Y. Ito, S. Fujii and T. Saegusa, Reaction of 1-silyloxybicyclo [n.1.0]alkanes with iron(III) chlorides. A facile synthesis of 2-cycloalkenones via ring enlargement of cyclic ketones, J. Org. Chem., 1976, 41, 2073 CrossRef CAS.
- (a) E. J. Corey, Catalytic enantioselective Diels–Alder reactions: methods, mechanistic fundamentals, pathways, and applications, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS; (b) E. J. Corey, Enantioselective catalysis based on cationic oxazaborolidines, Angew. Chem., Int. Ed., 2009, 48, 2100 CrossRef CAS PubMed.
- A. K. Ghosh, J. Kass, D. D. Anderson, X.-M. Xu and C. Marian, L-Selectride-mediated, highly diastereoselective asymmetric reductive Aldol reaction: access to an important subunit for bioactive molecules, Org. Lett., 2008, 10, 4811 CrossRef CAS PubMed.
- R. Noyori, I. Nishida and J. Sakata, Erythro-selective aldol reaction via tris(dialkylamino)sulfonium enolates, J. Am. Chem. Soc., 1981, 103, 2106 CrossRef CAS.
- B. Ren, O. Ramström, Q. Zhang, J. Ge and H. Dong, An Iron(III) catalyst with unusually broad substrate scope in regioselective alkylation of diols and polyols, Chem. – Eur. J., 2016, 22, 2481 CrossRef CAS PubMed.
- W. C. Still and A. Mitra, A highly stereoselective synthesis of Z-trisubstituted olefins via [2, 3]-sigmatropic rearrangement. Preference for a pseudoaxially substituted transition state, J. Am. Chem. Soc., 1978, 100, 1927 CrossRef CAS.
- R. Appel, Tertiary Phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and P-N linkage, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef.
- (a) D. Ikuta, Y. Hirata, S. Wakamori, H. Shimada, Y. Tomabechi, Y. Kawasaki, K. Ikeuchi, T. Hagimori, S. Matsumoto and H. Yamada, Conformationally supple glucose monomers enable synthesis of the smallest cyclodextrins, Science, 2019, 364, 674 CrossRef CAS PubMed; (b) C. E. Madu and C. J. Lovely, A Pauson−Khand approach to the Hamigerans, Org. Lett., 2007, 9, 4697 CrossRef CAS PubMed.
- E. J. Corey and P. B. Hopkins, Diisopropylsilyl ditriflate and di-tert-butysilyl ditriflate: new reagents for the protection of diols, Tetrahedron Lett., 1982, 23, 4871 CrossRef CAS.
- V. Mamane, T. Gress, H. Krause and A. Fürstner, Platinum- and gold-catalyzed cycloisomerization reactions of hydroxylated enynes, J. Am. Chem. Soc., 2004, 126, 8654 CrossRef CAS PubMed.
- (a) I. D. Jurberg, Y. Odabachian and F. Gagosz, Hydroalkylation of alkynyl ethers via a gold(I)-Catalyzed 1,5-hydride shift/cyclization sequence, J. Am. Chem. Soc., 2010, 132, 3543 CrossRef CAS PubMed; (b) B. Bolte and F. Gagosz, Gold and brønsted Acid catalyzed hydride shift onto allenes: divergence in product selectivity, J. Am. Chem. Soc., 2011, 133, 7696 CrossRef CAS PubMed; (c) P. Morán-Poladura, E. Rubio and J. M. González, Intramolecular C-H activation through gold(I)- catalyzed reaction of iodoalkynes, Angew. Chem., Int. Ed., 2015, 54, 3052 CrossRef PubMed; (d) X. L. Lu, M. Y. Lyu, X. S. Peng and H. N. C. Wong, Gold(I)-catalyzed tandem cycloisomerization of 1,5-enyne ethers by hydride transfer, Angew. Chem., Int. Ed., 2018, 57, 11365 CrossRef CAS PubMed; (e) N. Kim and R. A. Widenhoefer, Ionization of gold (γ-methoxy)vinyl complexes generates reactive gold vinyl carbene complexes, Chem. Sci., 2019, 10, 6149 RSC; (f) C. Shu, C.-Y. Shi, Q. Sun, B. Zhou, T.-Y. Li, Q. He, X. Lu, R.-S. Liu and L.-W. Ye, Generation of endocyclic vinyl carbene complexes via gold-catalyzed oxidative cyclization of terminal diynes: toward naphthoquinones and carbazolequinones, ACS Catal., 2019, 9, 1019 CrossRef CAS.
- (a) W. A. Herrmann, S. J. Eder and W. Scherer, Catalytic oxidation of partially and fully fluorinated olefins with osmium tetroxide, Angew. Chem., Int. Ed. Engl., 1992, 31, 1345 CrossRef; (b) M. R. Sivik, J. C. Gallucci and L. A. Paquette, Crystal structure analysis of an osmium(VI) bisglycolate produced by reaction of a sterically hindered chiral nonracemic alkene with osmium tetroxide, J. Org. Chem., 1990, 55, 391 CrossRef CAS.
- L. A. Paquette, F. J. Montgomery and T.-Z. Wang, An abbreviated, highly stereocontrolled route to precursors of Taxol. Elaboration of a fully functionalized C ring by means of intramolecular aldol cyclization, J. Org. Chem., 1995, 60, 7857 CrossRef CAS.
- R. Siedlecka, J. Skarzewski and J. Mlochowski, Selective oxidation of primary hydroxy groups in prinary-secondary diols, Tetrahedron Lett., 1990, 31, 2177 CrossRef CAS.
- K. Takai, K. Nitta and K. Utimoto, Simple and selective method for aldehydes (RCHO)→ (E)-haloalkenes (RCH:CHX) conversion by means of a haloform-chromous chloride system, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS.
- C. Lei, S.-X. Huang, J.-J. Chen, L.-B. Yang, W.-L. Xiao, Y. Chang, Y. Lu, H. Huang, J.-X. Pu and H.-D. Sun, Propindilactones E − J, Schiartane nortriterpenoids from Schisandra propinqua var. propinqua, J. Nat. Prod., 2008, 71, 1228 CrossRef CAS PubMed.
- Y. Wang, B. Chen, X. He and J. Gui, Bioinspired synthesis of nortriterpenoid Propindilactone G, J. Am. Chem. Soc., 2020, 142, 5007 CrossRef CAS PubMed.
- R. Breslow, R. J. Corcoran, B. B. Snider, R. J. Doll, P. L. Khanna and R. Kaleya, Selective halogenation of steroids using attached aryl iodide templates, J. Am. Chem. Soc., 1977, 99, 905 CrossRef CAS PubMed.
- K. Mori, K. Fukamatsu and M. Kido, Synthesis of chlorinated steroids related to the structures proposed for blattellastanosides A and B, the aggregation pheromone of the German cockroach, Blattella germanica L, Liebigs Ann. Chem., 1993, 657 CrossRef CAS.
- M. Wengert, A. M. Sanseverino and M. C. S. d. Mattos, Trichloroisocyanuric acid: an alternate green toute for the transformation of alkenes into epoxides, J. Braz. Chem. Soc., 2002, 13, 700 CrossRef CAS.
- P. d. Armas, J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Intramolecular hydrogen abstraction. Hypervalent organoiodine compounds, convenient reagents for alkoxyl radical generation, J. Chem. Soc., Perkin Trans. 1, 1989, 405 RSC.
- (a) D. H. R. Barton, D. Kumari, P. Welzel, L. J. Danks and J. F. McGhie, Photochemical transformations. Part XXV. The synthesis of cycloartenol, J. Chem. Soc. C, 1969, 332 RSC; (b) D. H. R. Barton, R. P. Budhiraja and J. F. McGhie, Photochemical transformations. Part XXVI. Some 19-substituted lanostane derivatives, J. Chem. Soc. C, 1969, 336 RSC.
- (a) P. T. Marcyk, L. R. Jefferies, D. I. AbuSalim, M. Pink, M.-H. Baik and S. P. Cook, Stereoinversion of unactivated alcohols by tethered sulfonamides, Angew. Chem., Int. Ed., 2019, 58, 1727 CrossRef CAS PubMed; (b) R. A. Watile, A. Bunrit, J. Margalef, S. Akkarasamiyo, R. Ayub, E. Lagerspets, S. Biswas, T. Repo and J. S. M. Samec, Intramolecular substitutions of secondary and tertiary alcohols with chirality transfer by an iron(III) catalyst, Nat. Commun., 2019, 10, 3826 CrossRef PubMed.
- (a) P. H. Dussault, T. K. Trullinger and F. Noor-e-Ain, Opening of substituted oxetanes with H2O2 and alkyl hydroperoxides: stereoselective approach to 3-peroxyalcohols and 1, 2, 4-trioxepanes, Org. Lett., 2002, 4, 4591 CrossRef CAS PubMed; (b) P. Dai and P. H. Dussault, Intramolecular reactions of hydroperoxides and oxetanes: stereoselective synthesis of 1, 2-dioxolanes and 1,2-dioxanes, Org. Lett., 2005, 7, 4333 CrossRef CAS PubMed.
- I. Shigeru and M. Teruaki, Novel method for the preparation of triethylsilyl peroxides from olefins by the reaction with molecular oxygen and triethylsilane catalyzed by bis(1,3-diketonato)cobalt(II), Chem. Lett., 1989, 18, 573 CrossRef.
- M. Brindisi, S. Gemma, S. Kunjir, L. Di Cerbo, S. Brogi, S. Parapini, S. D′ Alessandro, D. Taramelli, A. Habluetzel, S. Tapanelli, S. Lamponi, E. Novellino, G. Campiani and S. Butini, Synthetic spirocyclic endoperoxides: new antimalarial scaffolds, MedChemComm, 2015, 6, 357 RSC.
- (a) R. H. Wollenberg, K. F. Albizati and R. Peries, A nucleophilic acetaldehyde equivalent. Preparation and synthetic applications of cis-2-ethoxyvinyllithium, J. Am. Chem. Soc., 1977, 99, 7365 CrossRef CAS; (b) K. S. Y. Lau and M. Schlosser, (Z)-2-Ethoxyvinyllithium: a remarkably stable and synthetically useful 1,2-counterpolarized species, J. Org. Chem., 1978, 43, 1595 CrossRef CAS.
- K. Ando, Z-Selective Horner − Wadsworth − Emmons reaction of α -substituted ethyl (diarylphosphono)acetates with aldehydes, J. Org. Chem., 1998, 63, 8411 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |