
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced pillared designs for two-dimensional materials in electrochemical energy storage
Chong
Chen
a,
Nian-Wu
Li
a,
Bao
Wang
 *b,
Shuai
Yuan
cd and
Le
Yu
*a
*b,
Shuai
Yuan
cd and
Le
Yu
*a
aState Key Lab of Organic-Inorganic Composites, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: yule@mail.buct.edu.cn
bState Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: baowang@ipe.ac.cn
cShanghai University (Zhejiang Jiaxing) Emerging Industries Institute, Building 16, No. 906 Yatai Road, Nanhu District, Zhejiang 314006, P. R. China
dResearch Center of Nanoscience and Nanotechnology, Shanghai University, Shanghai 200444, P. R. China
First published on 18th August 2020
Abstract
Two-dimensional (2D) materials have attracted increased attention as advanced electrodes in electrochemical energy storage owing to their thin nature and large specific surface area. However, limited interlayer spacing confines the mass and ion transport within the layers, resulting in poor rate performance. Considerable efforts have been made to deal with this intrinsic problem of pristine 2D materials. Among them, interlayer engineering through pillared designs offers abundant electrochemical active sites and promotes ion diffusion. Synergetic effects between incorporated species and 2D hosts offer much better conductivity and surface modification. As a result, 2D materials with advanced pillared designs demonstrate great enhancement of specific capacity/capacitance and rate performance. Herein, we summarize the recent progress of pillared 2D materials in relation to the intercalated species. Moreover, we highlight their typical applications in lithium-ion storage and beyond to provide some insights on future trends towards this research area.
1. Introduction
With the rapid consumption of fossil fuel and the increasing use of portable devices, construction of electrochemical energy storage (EES) systems with long cycling life, low production cost, and high energy/power density has been considered as an urgent demand.1,2 The key challenge in realizing high-efficiency EES systems is to find suitable electrodes as the present commercialized lithium-ion batteries (LIBs) are approaching their theoretical performance limit.3,4 Therefore, considerable efforts have been made to explore appropriate materials with rationally designed structures towards diverse and emerging EES systems, such as lithium-ion capacitors (LICs),5,6 sodium-ion batteries/capacitors (SIBs or SICs),7–9 magnesium-ion batteries (MIBs)10,11 and zinc-ion batteries (ZIBs).12,13Among potential candidates, two-dimensional (2D) materials with atomic thicknesses have gained significant research interest.14,15 Commonly, 2D materials possess strong intralayer covalent bonding and weak van der Waals forces between adjacent layers. On one hand, this unique structural feature enables the diversity of 2D materials (graphene,16 some metal oxides (MOs),17–19 transition metal dichalcogenides (TMDs),20–22 layered double hydroxides (LDHs)23,24 and MXenes,5,25,26) for composition optimization. On the other hand, the ultrathin feature shortens ion diffusion path and ensures large surface area. However, critical drawbacks still exist in these pristine 2D materials such as poor conductivity limited interlayer spacing, which deteriorates mass, and ion transport, resulting in the low utilization of active materials and inferior rate performance.27 Recently, numerous structural and compositional tailoring strategies have been frequently employed to 2D materials, such as defect engineering,28,29 phase engineering,30,31 doping engineering,32 porous/hollow designs,33,34 and pillared designs.5,13,35 In particular, pillared designs refer to 2D materials with interlayers intercalated by outer molecules/ions/materials, in which these intercalated species serve as pillar-like supports.36–38 The pillaring effect leads to expanded lattice spacing, which leads to faster reaction kinetics and prevents interlayer restacking for enhanced cycling stability. Beyond that, different intercalated species could alter a series of surface physico-chemical properties such as the adsorption/desorption of electrolyte or hydrophilic/hydrophobic features. Besides, enriched chemical compositions might introduce new storage mechanisms.39
In this study, we provide a summary of the recent progress on the advanced pillared designs of 2D materials based on the incorporated species (Fig. 1). In particular, three categories based on the incorporated species are highlighted: sandwich-like structures, intercalation of ions, and incorporation of molecules or nanoparticles. Also, typical examples of these pillared 2D materials applied in EES systems such as LIBs and beyond are presented to study the synergistic effects between hosts and extraneous species. In the last part of this study, we discuss the remaining challenges and future research trends toward the pillared designs.
 | ||
| Fig. 1 An overview diagram of advanced pillared designs in electrochemical energy storage. | ||
2. Classification of pillared 2D materials
According to the intercalated species, pillared 2D materials can be generally classified into three groups:11,21,40–42 one category is sandwich-like structures composed of alternately restacked host and guest species as unilamellar nanosheets on top of one another. Usually, these layer-by-layer structures are formed through an electrostatic self-assembly strategy, wherein both the host and guest layers are inorganic species. Another important family of pillared 2D materials is produced by expanding the interlayer through the intercalation of external ions. In general, electro-driven intercalation reactions or ion–exchange reactions are employed to realize such structure. As for the third type, the interlayer spacing of 2D materials are partly expanded via the incorporation of zero-valent organic/inorganic molecules or non-layered nanoparticles.2.1. Sandwich-like structures through self-assembled methods
Sandwich-like 2D heterostructures with alternate stacking layers could demonstrate different interface features compared with the traditional heterostructures.43 For such periodic structures, each unilamellar nanosheet simultaneously acts as an active component and an interface. The active sites could largely be enhanced and synergetic effects between host and guest would be expected. To be specific, the guest layers could effectively avoid the restacking of the host layers. Moreover, the guest layers might serve as active species to participate in energy storage reactions. Owing to their ultrathin nature, charge transfer could be promoted. Structural stress during repeated cycles can be also largely buffered as each stacked nanosheet serves as a pillar to support each other. As a result, rate performance could be enhanced and severe pulverization can be avoided.44 As a typical example, Jiang et al.40 reported a hybrid sandwich-like composite via a facile hydrothermal process. Both sides of the reduced graphene oxide (rGO) are supposed to be decorated by ultrathin SnS2 nanosheets (Fig. 2a). The X-ray diffraction (XRD) pattern of the SnS2/rGO/SnS2 composite (Fig. 2b) shows the replacement of the (001) diffraction peak of pristine SnS2 by two new diffraction peaks at 2θ = 10.99° and 16.79°, indicating that the interlayer spacing of SnS2 is obviously expanded from 5.9 Å to 8.03 Å. The transmission electron microscopy (TEM) image (Fig. 2c) further confirms the successful decoration of the rGO nanosheet by the ultrathin SnS2 nanosheets. In addition to interlayer expansion, sandwich-like structures synthesized via a self-assembly method could prevent the restacking of each component. For example, He et al.41 synthesized novel three-dimensional (3D) sandwich-like graphene/1T MoS2 (3DG/TM) nanostructures through a one-pot hydrothermal method. 1T MoS2 nanosheets are formed in situ on both sides of graphene. The scanning electron microscopy (SEM) image (Fig. 2d) indicates 3DG/TM possesses an interconnected porous network. The high-resolution TEM (HRTEM) image (Fig. 2e) clearly illustrates the alternate-layered structure of 1T MoS2 and graphene. There is no obvious (002) peak of graphene in the 3DG/TM heterostructure, indicating that the agglomeration of graphene nanosheets is largely prevented (Fig. 2f).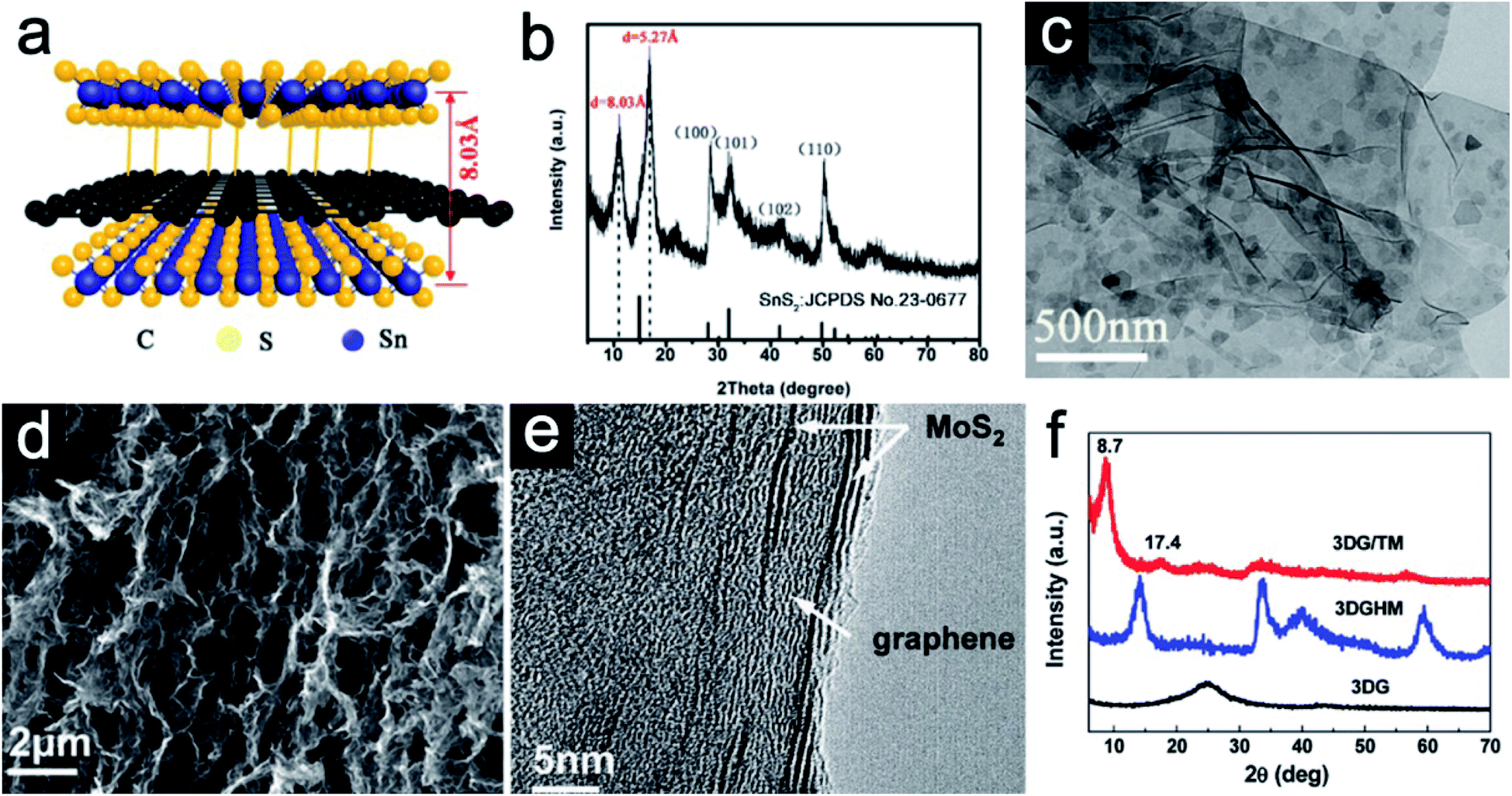 | ||
| Fig. 2 (a) Schematic of the SnS2/rGO/SnS2 sandwich-like structure. (b) XRD pattern and (c) TEM image of the SnS2/rGO/SnS2 composite. Panels (a–c) reproduced with permission.40 Copyright 2019, American Chemical Society. (d) SEM image of the porous 3DG/TM. (e) HRTEM image of 3DG/TM. (f) XRD patterns of 3DG/TM and control samples. Panels (d–f) reproduced with perpission.41 Copyright 2019, The Royal Society of Chemistry. | ||
2.2. Expanded structures via the intercalation of outer ions
Ion intercalation is a common strategy in building pillared 2D materials by electro-driven intercalation processes or ion–exchange reactions.21,26,45,46 This strategy could enhance the electron/ion transfer kinetics in host materials and stabilize the layered structure during cycling, leading to excellent rate capability and cycling stability. Moreover, the ion exchange processes might change the valence states and the surface electronegativity of the host layers. These changes could alter the chemical environments of the host species and the electrochemical behaviors. Xu et al.11 synthesized bilayer-structured vanadium oxide nanowires pillared by Mg2+ and water via chemical modification (Fig. 3a). H2O2 was chosen as the reducing agent to decrease the valence state of V species to convert α-V2O5 into VO6 octahedra. During this reconstruction, Mg2+ ions are intercalated in situ into the layer space via the coordination with lattice oxygen, while water molecules are embedded into the interlayer. As a result, the bilayer-structured Mg0.3V2O5·1.1H2O nanowires with a larger interlayer spacing are produced. Different from the usual 2D materials, the appearance of the bilayer-structured Mg0.3V2O5·1.1H2O is a one-dimensional (1D) thread-like nanowire with a smooth surface (Fig. 3b). Elemental mapping images indicate the uniform distribution of Mg, V, and O along the nanowires (Fig. 3c). To further study the location of Mg2+ ions and the state of structural water, solid-state magic angle spinning (MAS) nuclear magnetic resonance (NMR) was conducted on Mg0.3V2O5·1.1H2O in different charge and discharge stages. 25Mg MAS NMR spectra (Fig. 3d) show that the pre-intercalated Mg2+ ions act as pillars in the interlayer without distinct changes after electrochemical cycling performance. Furthermore, 1H NMR spectra (Fig. 3e) together with 13C NMR data show that pillared Mg2+ ions could serve as guards to prevent the insertion of the solvent molecules. Due to the strong bonds between Mg2+ and the oxygen in water, the water species within Mg0.3V2O5·1.1H2O evolve from the pure structural water in the pristine sample to the combination of bound water and structural water after cycling. The partial recovery of the structural water is beneficial in keeping the lattice spacing for better electrochemical kinetics. | ||
| Fig. 3 (a) Schematic of the formation of the Mg0.3V2O5·1.1H2O nanowires. (b) SEM images and (c) TEM-EDX element mapping images of Mg0.3V2O5·1.1H2O nanowires. (d) 25Mg (D) and (e) 1H (E) MAS NMR spectra collected for Mg0.3V2O5·1.1H2O in different states. Panels reproduced with permission.11 Copyright 2019, Elsevier. | ||
2.3. Pillared 2D materials through the incorporation of outer molecules or nanoparticles
Incorporation of outer zero-valent molecules can be traced back to decade ago.25,47 Water molecules in the electrode lattice could significantly accelerate the multivalent ion diffusion at the electrolyte/electrode interface.48,49 Moreover, the incorporation of long-chain molecules/polymers can alter the surface properties to reduce the diffusion barrier of an electrolyte. Some redox-active polymers could even serve as active materials to promote energy storage. As a typical example, Yu et al.50 reported the expansion of 2D VOPO4 nanosheets with tailorable interlayer distance via the intercalation of organic molecules. As shown in Fig. 4a, the organic intercalants replace the water molecules within VOPO4·2H2O bulk sheets to expand the interlayer spacing. These exchange reactions are stabilized by the hydrogen bonds formed between VOPO4 and organic molecules. Moreover, different species could change the interlayer distance. For tetrahydrofuran (THF), the spacing is enhanced from 0.74 nm to 0.88 nm. In the case of triethylene glycol (TEG), the interlayer distance further increased to 1.06 nm. XRD patterns confirm the interlayer expansion of the intercalated VOPO4 nanosheets (Fig. 4b). The HRTEM image also proves that the interlayer distance of TEG-pillared VOPO4 nanosheets is consistent with the XRD result (Fig. 4c). The intercalants could modify the chemical configuration of the VOPO4 nanosheet because the polarity or the dielectric property of the organic ligands might change the chemical value of V species. As presented in the V K-edge X-ray absorption near-edge spectroscopy (XANES), the edge shifts to a lower energy with the decrease in electronegativity for different ligands (Fig. 4d). The intensity change of the extended X-ray absorption fine structure (EXAFS) oscillations also confirms the distortion of V–O as the bonds become longer with the decrease in the intercalant electronegativity (Fig. 4e).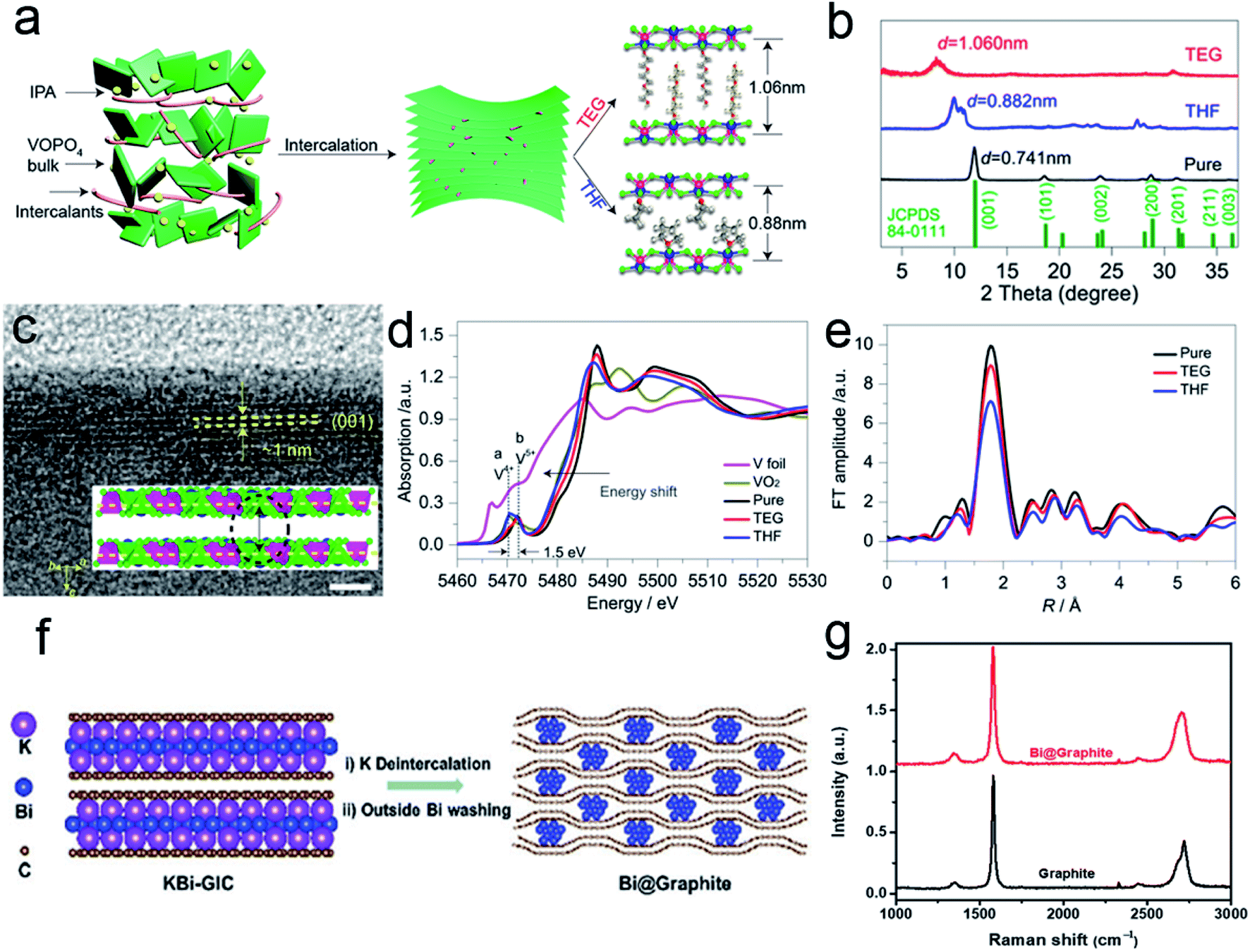 | ||
| Fig. 4 (a) Schematic of the expanded structure of VOPO4 nanosheets. (b) XRD patterns of pristine VOPO4 and pillared VOPO4 samples. (c) HRTEM image of TEG-VOPO4 nanosheets. Scale bar: 5 nm. (d) K-edge XANES of the pristine VOPO4 and pillared VOPO4 samples. (e) EXAFS at the vanadium K-edge of pristine VOPO4 and pillared VOPO4 samples. Panels (a–e) reproduced with permission.50 Copyright 2017, American Chemical Society. (f) Schematic of the synthesis of Bi@Graphite. (g) Raman spectra of the as-synthesized Bi@Graphite and graphite. Panels (f and g) reproduced with permission.42 Copyright 2018, The Royal Society of Chemistry. | ||
For the incorporation of outer nanoparticles, the heterostructures are different from the sandwich-like structures because the intercalants are not limited to 2D materials. For example, Chen et al.42 reported the synthesis of bismuth-intercalated graphite (Bi@Graphite) materials via intercalation–conversion reactions. As shown in Fig. 4f, the KBi alloy initially intercalates in two graphene layers to form binary graphite intercalation compounds (KBi-GIC). After subsequent deintercalation and acid washing, the Bi@Graphite hybrid is obtained. Raman spectra reveal that the 2D peak of Bi@Graphite is broader than that of the pure graphite, indicating that the layer number of graphene in the hybrid is less than five (Fig. 4g). After the removal of Bi nanoparticles, the graphite structure is restored. The above results indicate that the Bi species could effectively prevent the restacking of graphene sheets.
3. Applications of pillared 2D materials in EES systems
LIBs have dominated majority of the market for portable electronic devices in the last few decades. With the consumption of finite lithium resources and the increasing demand for advanced energy systems with higher power density and energy density, EES systems beyond LIBs are urgently needed to solve the above dilemma. As we mentioned before, 2D materials have proven successful in the conventional LIBs. Benefitting from their enlarged interlayer spacing with tailorable compositions, 2D materials with advanced pillared designs demonstrate apparent advantages in LIBs and beyond. Compared with the pristine structures, pillared 2D materials possess larger lattice spacing and more contact area between the electrolyte and the layers. In addition, pillar effects reduce diffusion resistance and withstand volumetric stress during cycling. In the following section, we list some typical examples in these systems to highlight the advanced pillared 2D materials.3.1. Pillared 2D materials for lithium-/sodium-ion storage
Starting from LiCoO2 cathode and graphite anode, 2D materials are the most successful electrodes in LIBs. Pillared 2D materials bring new insight for lithium-ion storage. Chen et al.51 developed nitrogen-doped graphene/MoS2/nitrogen-doped graphene (NDG/MoS2/NDG) heterostructures via thermal decomposition (Fig. 5a). The sandwich-like NDG/MoS2/NDG can protect the electrochemical intermediates such as Mo and lithium polysulfide from dissolving into the electrolyte. The synergistic effect between the MoS2 and NDG facilitates electron and Li+ transport kinetics. Compared to a pure MoS2 sample, NDG/MoS2/NDG reveals a strong oxidation peak around 1.3–1.8 V vs. Li+/Li in CV scans, indicating enhanced structural stability (Fig. 5b). In addition, the cycling performances of NDG/MoS2/NDG, NDG/MoS2 (2-times MoS2), and MoS2 clearly show the superior cycling stability of the pillared sample (Fig. 5c). NDG/MoS2/NDG retains a high and stable specific capacity of 750 mA h g−1 after 100 cycles. In contrast, NDG/MoS2 and MoS2 samples show a quick capacity decay around the early cycles.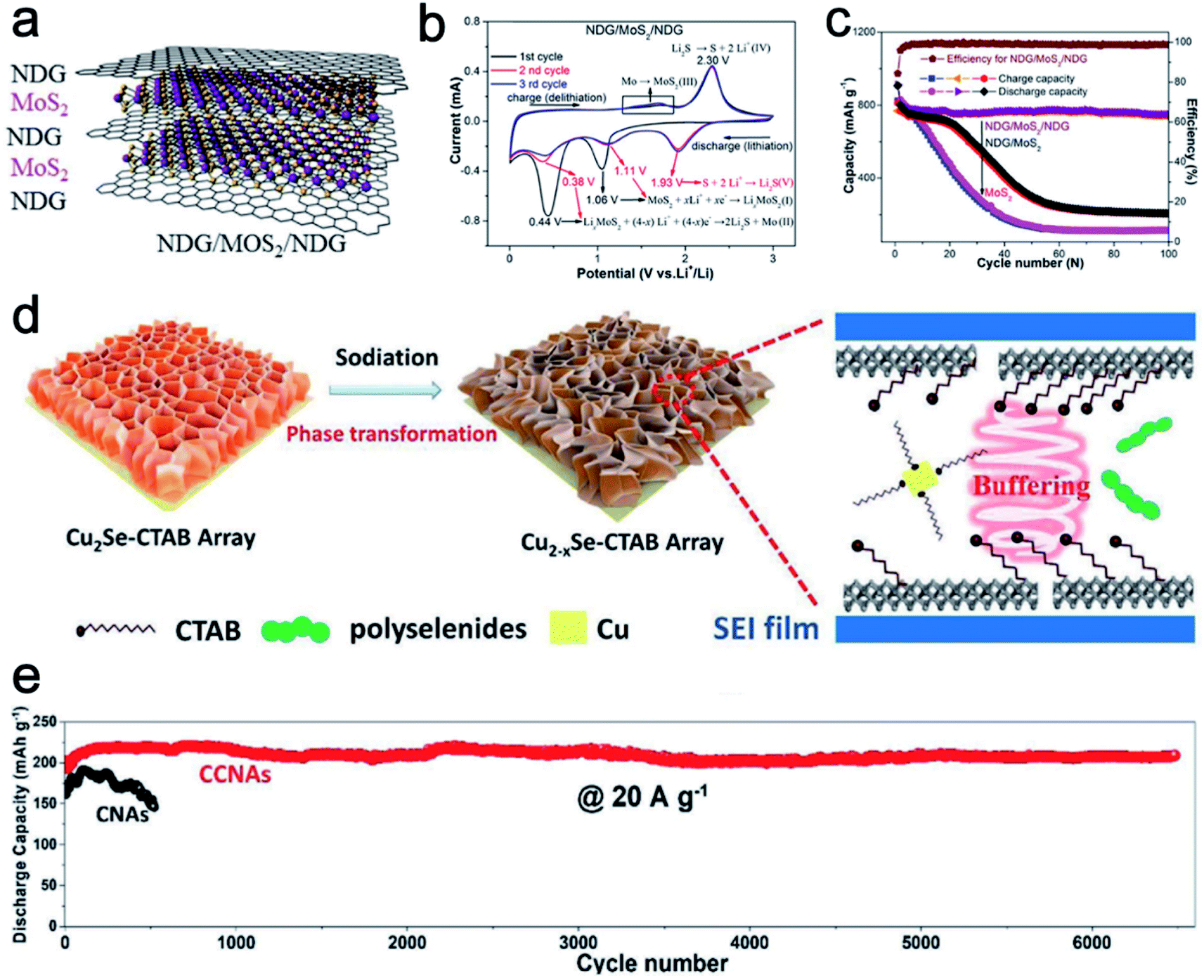 | ||
| Fig. 5 (a) Schematic of the NDG/MoS2/NDG hybrid. (b) CV curves of NDG/MoS2/NDG. (c) Cycling performance of NDG/MoS2/NDG and control samples for lithium-ion storage. Panels (a–c) reproduced with permission.51 Copyright 2017, Elsevier. (d) Schematic showing the mechanism involved in the phase transformation and enhanced performance of CCNAs for NIBs. (e) Cycling performance of CCNAs and CNAs. Panels (d and e) reproduced with permission.47 Copyright 2020, John Wiley & Sons, Inc. | ||
For the Na-ion storage, the radius of Na+ ions is larger than that of Li+ ions. Therefore, a large lattice distance is highly important to enable the ion storage and bear the structural strain during cycling. For example, Xiao et al.47 fabricated cetyltrimethyl ammonium bromide (CTAB) intercalated Cu2Se nanosheet arrays (CCNAs) through a room temperature insertion reaction. When applied as anode materials for NIBs, the intercalated CTAB molecules could not only enlarge the lattice spacing and buffer the structural strain, but also serve as the capping agent to prevent the fast growth of Cu particles (Fig. 5d). Therefore, the reaction activity of Cu particles is retained to keep high reversibility and cycling stability. As shown in Fig. 5e, CCNAs reveal better capacity retention over longer cycles than the Cu2Se nanosheet arrays without CTAB (defined as CNAs).
3.2. Pillared 2D materials for magnesium-/zinc-ion storage
Compared to the lithium-/sodium-ion storage, researches on the magnesium-/zinc-ion storage are still in their infancy as these EES systems are mostly aqueous systems. Rechargeable MIBs and ZIBs are attractive owing to rich earth abundance, high theoretical capacity, and dendrite-free plating process. However, their practical performances are hampered by the high polarity and sluggish diffusion kinetics of multivalent ions. The pillared strategy has proven its advantages to address these issues. For example, Xue et al.52 reported the synthesis of 2-ethylhexylamine intercalated VS2 nanoflowers via a solvothermal method for MIBs. The intercalated 2-ethylhexylamine molecules serve as interlayer pillars to expand the interlayer for fast ion kinetics in the coulombic interactions between Mg2+ and lattice anions of the VS2 host. Although the expanded and annealed VS2 are similar nanoflowers (Fig. 6a), the lattice distance of the (001) plane for the former is much higher (9.93 Å) than the latter (5.73 Å). The CV curves of expanded VS2 (Fig. 6b) show distinct redox peaks located at 1.37/1.18 and 0.80/0.56 V vs. Mg2+/Mg, corresponding to the Mg2+ species intercalation/deintercalation and conversion, respectively. However, there are no such obvious peaks for the annealed VS2 sample, indicating inferior electrochemical performance. Besides, the expanded VS2 sample delivers much higher rate performance at different current densities than the annealed sample (Fig. 6c). For the zinc-ion storage, Huang et al.53 reported the formation of polyaniline (PANI)-intercalated MnO2 nanolayers and investigated their electrochemical performance. As shown in Fig. 6d, the polymerization of aniline monomers and reduction of MnO42+ occur in each phase simultaneously, leading to the self-assembly of PANI and MnO2. The grainy MnO2 nanolayer has an expanded interlayer space (∼1.0 nm) due to the intercalation of PANI (Fig. 6e). Apart from the enhanced structural stability, the unusual CV curves indicate a complex ion storage mechanism for this hybrid sample (Fig. 6f). After a series of investigation, the authors assumed there is a co-insertion mechanism of H+ and Zn2+ in PANI-intercalated MnO2 nanolayers that form zinc hydroxide sulfate at the surface (Fig. 6g). Although the internal mechanism is not clear, it can be confirmed that the intercalates might bring new storage mechanism(s) for the host materials. | ||
| Fig. 6 (a) SEM images of expanded VS2 nanoflowers. (b) CV curves of the expanded VS2 electrode in MIBs. (c) Rate performance of expanded VS2 and annealed VS2 electrodes. Panels (a–c) reproduced with permission.52 Copyright 2019, John Wiley & Sons, Inc. (d) Schematic of the polyaniline (PANI)-intercalated MnO2 nanolayers. (e) HRTEM image of PANI-intercalated MnO2 nanolayers after annealing. (f) CV curves of PANI-intercalated MnO2 nanolayers. (g) Schematic of the co-insertion mechanism. Panels (d–g) reproduced with permission.53 Copyright 2018, Nature Publishing Group. | ||
4. Summary and perspectives
The increasing demand and rapid consumption of methods for powering mobile electronic devices have promoted the development of energy storage systems with high capacity and superior rate performance. 2D layered materials have been regarded as promising electrodes because of their high electrochemical activity and large specific surface. However, inherent drawbacks have strongly inhibited their practical applications. Advanced pillared designs are helpful to maximize their significant advantages and break the natural limits, leading to much higher rate capability and structural stability.In this study, various pillared design strategies (i.e., sandwich-like structure, intercalation of outer ions and incorporation of outer molecules or nanoparticles) are summarized and some typical examples applied in ion storage systems are also highlighted. 2D materials with an enlarged interlayer spacing exhibit superior electrochemical performance compared to pristine materials. Despite these progresses, further endeavors are still needed to reveal the structure-property relationship for the pillar designs. In addition, large scale production of pillared 2D nanomaterials is another challenge for practical applications.17 Future research might be carried out based on the following aspects: (1) more facile synthetic strategies should be explored for the industrial-scale manufacturing.54 (2) Apart from pillar effects, the contribution of the synthetic effect between the host and guest species deserves more research attention through in situ methods.15 (3) The strain effects of different pillared designs should be further studied as the strain is induced during the formation processes and electrochemical cycling.55
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Y. acknowledges the National Natural Science Foundation of China (Grant No. 51902016), the financial support from Fundamental Research Funds for the Central Universities (Grant No. buctrc201829). N. W. L. acknowledges the financial support from Fundamental Research Funds for the Central Universities (Grant No. buctrc201904). B. W. acknowledges National Science Foundation of China (51772296), the Foundation for State Key Laboratory of Biochemical Engineering.Notes and references
- S. Chen, L. Qiu and H.-M. Cheng, Chem. Rev., 2020, 120, 2811–2878 CrossRef CAS.
- Y. Y. Zhao, Y. S. Ye, F. Wu, Y. J. Li, L. Li and R. J. Chen, Adv. Mater., 2019, 31, 1806532 CrossRef.
- J. Wang, L. Liao, H. R. Lee, F. Shi, W. Huang, J. Zhao, A. Pei, J. Tang, X. Zheng, W. Chen and Y. Cui, Nano Energy, 2019, 61, 404–410 CrossRef CAS.
- J. L. Shi, D. D. Xiao, M. Ge, X. Yu, Y. Chu, X. Huang, X. D. Zhang, Y. X. Yin, X. Q. Yang, Y. G. Guo, L. Gu and L. J. Wan, Adv. Mater., 2018, 30, 1705575 CrossRef.
- J. Luo, W. Zhang, H. Yuan, C. Jin, L. Zhang, H. Huang, C. Liang, Y. Xia, J. Zhang, Y. Gan and X. Tao, ACS Nano, 2017, 11, 2459–2469 CrossRef CAS.
- Y. B. An, S. Chen, M. M. Zou, L. B. Geng, X. Z. Sun, X. Zhang, K. Wang and Y. W. Ma, Rare Met., 2019, 38, 1113–1123 CrossRef CAS.
- Y. Dong, Z. S. Wu, S. Zheng, X. Wang, J. Qin, S. Wang, X. Shi and X. Bao, ACS Nano, 2017, 11, 4792–4800 CrossRef CAS.
- F. X. Ding, F. Gao, X. H. Rong, K. Yang, Y. X. Lu and Y. S. Hu, Acta Phys.-Chim. Sin., 2020, 36, 1904022 Search PubMed.
- H. H. Lu, C. S. Shi, N. Q. Zhao, E. Z. Liu, C. N. He and F. He, Rare Met., 2018, 37, 107–117 CrossRef CAS.
- X. Fan, R. R. Gaddam, N. A. Kumar and X. S. Zhao, Adv. Energy Mater., 2017, 7, 1700317 CrossRef.
- Y. Xu, X. Deng, Q. Li, G. Zhang, F. Xiong, S. Tan, Q. Wei, J. Lu, J. Li, Q. An and L. Mai, Chem, 2019, 5, 1194–1209 CAS.
- X. Chen, B. Liu, C. Zhong, Z. Liu, J. Liu, L. Ma, Y. Deng, X. Han, T. Wu, W. Hu and J. Lu, Adv. Energy Mater., 2017, 7, 1700779 CrossRef.
- J. T. Huang, J. Zhou and S. Q. Liang, Acta Phys.-Chim. Sin., 2020, 2005020, DOI:10.3866/pku.Whxb202005020.
- P. Tao, S. Yao, F. Liu, B. Wang, F. Huang and M. Wang, J. Mater. Chem. A, 2019, 7, 23512–23536 RSC.
- L. Fan, M. Li, X. Li, W. Xiao, Z. Chen and J. Lu, Joule, 2019, 3, 361–386 CrossRef CAS.
- J. Chen, B. Yang, H. Hou, H. Li, L. Liu, L. Zhang and X. Yan, Adv. Energy Mater., 2019, 9, 1803894 CrossRef.
- J. Mei, T. Liao, L. Z. Kou and Z. Q. Sun, Adv. Mater., 2017, 29, 1700176 CrossRef.
- Y. Lu, L. Yu and X. W. Lou, Chem, 2018, 4, 972–996 CAS.
- Y. Guo, C. Wu, N. W. Li, S. Yuan and L. Yu, J. Mater. Chem. A, 2019, 7, 25247–25253 RSC.
- D. Sarkar, D. Das, S. Das, A. Kumar, S. Patil, K. K. Nanda, D. D. Sarma and A. Shukla, ACS Energy Lett., 2019, 4, 1602–1609 CrossRef CAS.
- H. D. Yoo, Y. Liang, H. Dong, J. Lin, H. Wang, Y. Liu, L. Ma, T. Wu, Y. Li, Q. Ru, Y. Jing, Q. An, W. Zhou, J. Guo, J. Lu, S. T. Pantelides, X. Qian and Y. Yao, Nat. Commun., 2017, 8, 339 CrossRef.
- X. Y. Yu, L. Yu and X. W. Lou, Small Methods, 2017, 1, 1600020 CrossRef.
- J. Zhang, L. Yu, Y. Chen, X. F. Lu, S. Gao and X. W. Lou, Adv. Mater., 2020, 32, 1906432 CrossRef CAS.
- Z. Li, K. Liu, K. Fan, Y. Yang, M. F. Shao, M. Wei and X. Duan, Angew. Chem., Int. Ed., 2019, 58, 3962–3966 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- P. Lian, Y. Dong, Z.-S. Wu, S. Zheng, X. Wang, S. Wang, C. Sun, J. Qin, X. Shi and X. Bao, Nano Energy, 2017, 40, 1–8 CrossRef CAS.
- M. Huang, M. Li, C. J. Niu, Q. Li and L. Q. Mai, Adv. Funct. Mater., 2019, 29, 1807847 CrossRef.
- X. H. Yao, Y. J. Ke, W. H. Ren, X. P. Wang, F. Y. Xiong, W. Yang, M. S. Qin, Q. Li and L. Q. Mai, Adv. Energy Mater., 2019, 9, 1803260 CrossRef.
- X. H. Sang, Y. Xie, M. W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS.
- Y. Zhu, L. L. Peng, Z. W. Fang, C. S. Yan, X. Zhang and G. H. Yu, Adv. Mater., 2018, 30, 1706347 CrossRef.
- Y. X. Huang, C. Y. Zhu, S. L. Zhang, X. M. Hu, K. Zhang, W. H. Zhou, S. Y. Guo, F. Xu and H. B. Zeng, Nano Lett., 2019, 19, 1118–1123 CrossRef.
- P. Luo, F. W. Zhuge, Q. F. Zhang, Y. Q. Chen, L. Lv, Y. Huang, H. Q. Li and T. Y. Zhai, Nanoscale Horiz., 2019, 4, 26–51 RSC.
- L. Yao, Q. Wu, P. X. Zhang, J. M. Zhang, D. R. Wang, Y. L. Li, X. Z. Ren, H. W. Mi, L. B. Deng and Z. J. Zheng, Adv. Mater., 2018, 30, 1706054 CrossRef.
- L. Yu, X. Y. Yu and X. W. Lou, Adv. Mater., 2018, 30, 1800939 CrossRef.
- J. Luo, C. Wang, H. Wang, X. Hu, E. Matios, X. Lu, W. Zhang, X. Tao and W. Li, Adv. Funct. Mater., 2019, 29, 1805946 CrossRef.
- V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef CAS.
- F. Gandara, J. Perles, N. Snejko, M. Iglesias, B. Gomez-Lor, E. Gutierrez-Puebla and M. A. Monge, Angew. Chem., Int. Ed., 2006, 45, 7998–8001 CrossRef CAS.
- X. Y. Zhang, D. X. Liu, D. D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS.
- C. Zhang and V. Nicolosi, Energy Storage Mater., 2019, 16, 102–125 CrossRef.
- Y. Jiang, D. Song, J. Wu, Z. Wang, S. Huang, Y. Xu, Z. Chen, B. Zhao and J. Zhang, ACS Nano, 2019, 13, 9100–9111 CrossRef CAS.
- J. R. He, G. Hartmann, M. Lee, G. S. Hwang, Y. F. Chen and A. Manthiram, Energy Environ. Sci., 2019, 12, 344–350 RSC.
- J. Chen, X. L. Fan, X. Ji, T. Gao, S. Hou, X. Q. Zhou, L. N. Wang, F. Wang, C. Y. Yang, L. Chen and C. S. Wang, Energy Environ. Sci., 2018, 11, 1218–1225 RSC.
- D. P. Zhao, H. Q. Liu and X. Wu, Nano Energy, 2019, 57, 363–370 CrossRef CAS.
- Y. F. Wang, L. X. Wang, Q. B. Yuan, J. Chen, Y. J. Niu, X. W. Xu, Y. T. Cheng, B. Yao, Q. Wang and H. Wang, Nano Energy, 2018, 44, 364–370 CrossRef CAS.
- Y. Chen, W. K. Pang, H. Bai, T. Zhou, Y. Liu, S. Li and Z. Guo, Nano Lett., 2017, 17, 429–436 CrossRef CAS.
- X. Wang, P. Hu, C. Niu, J. Meng, X. Xu, X. Wei, C. Tang, W. Luo, L. Zhou, Q. An and L. Mai, Nano Energy, 2017, 35, 71–78 CrossRef CAS.
- Y. H. Xiao, X. B. Zhao, X. Z. Wang, D. C. Su, S. Bai, W. Chen, S. M. Fang, L. M. Zhou, H. M. Cheng and F. Li, Adv. Energy Mater., 2020, 10, 2000666 CrossRef CAS.
- K. W. Nam, S. Kim, E. Yang, Y. Jung, E. Levi, D. Aurbach and J. W. Choi, Chem. Mater., 2015, 27, 3721–3725 CrossRef CAS.
- F. Wang, W. Sun, Z. Shadike, E. Y. Hu, X. Ji, T. Gao, X. Q. Yang, K. Xu and C. S. Wang, Angew. Chem., Int. Ed., 2018, 57, 11978–11981 CrossRef CAS.
- L. L. Peng, Y. Zhu, X. Peng, Z. W. Fang, W. S. Chu, Y. Wang, Y. J. Xie, Y. F. Li, J. J. Cha and G. H. Yu, Nano Lett., 2017, 17, 6273–6279 CrossRef CAS.
- B. Chen, Y. H. Meng, F. He, E. Z. Liu, C. S. Shi, C. N. He, L. Y. Ma, Q. Y. Li, J. J. Li and N. Q. Zhao, Nano Energy, 2017, 41, 154–163 CrossRef CAS.
- X. L. Xue, R. P. Chen, C. Z. Yan, P. Y. Zhao, Y. Hu, W. H. Kong, H. N. Lin, L. Wang and Z. Jin, Adv. Energy Mater., 2019, 9, 1900145 CrossRef.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef.
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Adv. Energy Mater., 2020, 10, 2000477 CrossRef CAS.
- Y. Guo, Y. Wei, H. Li and T. Zhai, Small, 2017, 13, 1701649 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |