
Ultrasensitive sensors based on aluminum oxide-protected reduced graphene oxide for phosphate ion detection in real water†
Guihua
Zhou‡
a,
Bing
Jin
a,
Yale
Wang
a,
Qianqian
Dong
a,
Arnab
Maity
a,
Jingbo
Chang
a,
Ren
Ren
 a,
Haihui
Pu
a,
Xiaoyu
Sui
a,
Shun
Mao
bc and
Junhong
Chen§¶
*a
a,
Haihui
Pu
a,
Xiaoyu
Sui
a,
Shun
Mao
bc and
Junhong
Chen§¶
*a
aDepartment of Mechanical Engineering, College of Engineering & Applied Science, University of Wisconsin-Milwaukee, 3200 N Cramer Street, Milwaukee, Wisconsin 53211, USA. E-mail: junhongchen@uchicago.edu
bState Key Laboratory of Pollution Control and Resource Reuse, College of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, China
cShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
First published on 16th March 2020
Abstract
Real-time detection of phosphate ions is critical for monitoring the eutrophication status of surface water. We report an ultrasensitive phosphate sensor based on a reduced graphene oxide field-effect transistor (rGO-FET). In this work, rGO nanosheets were employed as sensing channels and an aluminum oxide (Al2O3) film was applied as a passivation layer to separate the sensing channels from the sample solution. The critical thickness of the passivation layer – defining a continuous barrier film on the FET sensor – was identified, which fully covers the sensing platform and also enables sensitive responses. The detection results showed that ferritin probes have good affinity to phosphate ions in water. It was also found that providing both a high ferritin concentration and an acidic environment improves the immobilized probe density, which increases the analyte adsorption sites and improves the sensing performance. The sensing platform was used to detect phosphate ions in industrial discharge with a lower detection limit of 10 μg L−1 (105 nM). Ultimately, this inexpensive and micro-sized sensor can provide new opportunities for real-time monitoring of phosphate ions in agriculture and wastewater discharge.
Design, System, ApplicationReal-time detection of phosphate ions is critical for monitoring the eutrophication status of surface water. We propose an ultrasensitive phosphate sensor based on a reduced graphene oxide field-effect transistor (rGO-FET). In this work, rGO nanosheets were employed as sensing channels, and an aluminum oxide film (Al2O3) obtained by atomic layer deposition (ALD) was applied as a passivation layer to separate the sensing channels from the sample solution. The passivation layer critical thickness – defining a continuous barrier film on the FET sensor application – was identified, which fully covers the sensing platform, while enabling a sensitive response. It was also found that providing both a high ferritin concentration and an acidic environment improves the immobilized probe density, which increases the analyte adsorption sites and improves the sensing performance. The sensing platform realized the detection of phosphate ions in industrial discharge with a lower detection limit of 10 μg L−1 (105 nM). The combination of the nanomaterial-FET with its unique properties and specific probes sets a typical example for low-concentration monitoring of nutrients in water. Moreover, the insights gained from the study on the ALD passivation layer and ferritin–phosphate ion interactions can be used for design of many other sensing or removal systems. |
Introduction
Phosphorus is a critical mineral needed for the growth of plants and animals. It is mainly present in the form of phosphate, including orthophosphate, metaphosphate (or polyphosphate), and organically bound phosphate.1,2 In a lake ecosystem, phosphorus serves as a growth-limiting nutrient, stimulating the growth of plankton and aquatic plants while providing food for larger organisms such as mammals, fish, and zooplankton. The enrichment of phosphate accelerates the aging process of a lake or surface water ecosystem, and its overproduction will lead to an imbalance in the nutrient and material cycling process. Exposure to low levels of phosphate is not toxic to humans and animals; however, exposure to high levels of phosphate can affect their digestion systems.3 The main sources of phosphate are related to anthropogenic behaviors, including partially filtered and unfiltered sewage, runoff from agriculture sites, and the application of lawn fertilizers. Real-time monitoring of phosphate concentration in these sources is highly desired to protect water ecosystems.As one of the carbon nanomaterials in the graphene family, graphene oxide (GO) has been widely investigated for sensing applications because of its low cost and outstanding electronic properties.4–7 Although the electronic properties of GO are not comparable with those of pristine graphene due to structural defects,8 there are still many advantages for its use in real applications. In general, GO has a 2D structure similar to graphene, but the single-layer carbon sheet is abundantly decorated with oxygen-containing groups which become chemical modification sites and make the atomic-thick layers hydrophilic. Additionally, at the nanoscale, this carbon-based material exhibits a high specific surface area. The most attractive property of GO is that it can be reduced to a graphene-like nanosheet by removing oxygen-containing groups at 400 °C with the protection of a flowing Ar environment.9 After annealing, the monolayer fully-reduced graphene oxide (rGO) exhibits conductivities ranging between 0.05 and 2 S cm−1 and a field effect mobility of 2–200 cm2 V−1 s−1 at room temperature.10
rGO and graphene have been widely reported in applications such as field-effect transistors (FETs) for detecting various ions,6,11 proteins,12 DNA,13 and cells.14 These detections are based on various mechanisms, such as charge transfer, charge scattering, capacitive effect, and gating effect. The gating effect is considered to be the most reliable sensing mechanism, and it can modulate the electrical conductivity of the sensing material by generating an external electric field, induced by, for instance, the adsorbed charged species.
In our previous work15 we reported an rGO-based FET platform for orthophosphate ion detection, which achieved sensitive detection with quick responses, but the sensing mechanism was not fully understood. In addition to the gating effect induced by the adsorbed analyte ions, the sensing signal was attributed to the charge transfer between the analyte and the sensing material, since the rGO film was in direct contact with the sample solution. The conductivity changes introduced by these two effects are in opposite directions; therefore, they offset each other, leading to a reduced sensitivity. Furthermore, the probe immobilization was not well controlled, and the sensing surface had a low probe coverage density.
In this work we introduce a newly designed sensor platform based on an rGO-FET for detecting phosphate ions in real water samples. Many studies in the literature have demonstrated that graphene oxide has excellent adsorption properties due to its defects and oxygen-containing functional groups.16–18 Here, to avoid the direct adsorption of ions in water onto rGO and introducing interference signals, we first coated an Al2O3 thin film onto the rGO surface to separate the rGO from the analyte solution. There are two main stimulations that affect the FET performance: charge transfer and gating effect. The Al2O3 film eliminates the charge transfer by blocking the transfer of charge carriers from the phosphate ions to the sensing channel (rGO film), which then resolves the competing sensing mechanisms and allows for the gating effect to work as a function of phosphate concentration. The probes are immobilized to the sensing platform with reliable linkers, which enables a tunable probe coverage density. Furthermore, to deeply investigate the electronic reaction between the probes and analytes, the zeta potential was used to characterize the surface charge on the sensing platform. Finally, the stable and reliable sensor platform delivered reproducible and sensitive detection of phosphates, with a lower detection limit down to 10 μg L−1 (105 nM) in real water samples.
Experimental
Chemicals
A commercial monolayer GO suspension was ordered and used as is. Cysteamine (AET), glutaraldehyde (GA), and horse ferritin were ordered from Sigma-Aldrich, US. Industrial discharge was provided by an industry partner. Water samples of phosphate ion, sulfate ion, nitrate ion and carbonic ion were diluted from commercial standards (RICCA), which were prepared by dissolving KH2PO4, K2SO4, KNO3 and Na2CO3, respectively, in DI water.Electrode fabrication
Gold interdigitated electrodes with a gap of 5 μm were fabricated with the maskless lithography technique (MLA-150, Heidelberg Instruments). First, the electrodes were patterned on a surface-oxidized silicon wafer substrate (SiO2 thickness ∼300 nm). After developing the exposed photoresist, gold was deposited onto the patterns and the rest of the unexposed photoresist was removed. AET solution (1 mg mL−1) was pipetted onto the sensing area to functionalize the finger electrodes for 30 min. The AET-assembled electrodes were immersed in the prepared GO water suspension for 30 s, then rinsed with deionized (DI) water and gently dried with nitrogen gas. Afterwards, the GO-modified electrodes were annealed at 400 °C in an Ar environment for 10 min to thermally reduce the adhered GO sheets. To passivate the obtained rGO nanosheets, a thin Al2O3 film was deposited onto the surface using an atomic layer deposition (ALD) system (GEMStar XT, Arradiance). Au NPs were deposited onto the passivated rGO surface using a sputter coater (Q300T D, Quorum Technologies) with an average size of 4 nm in diameter and lateral spacing of ∼5 nm to provide immobilization sites for probes. AET solution was again used to functionalize the surface of the Au NPs for 30 min, and then GA solution (5% in water) was added for another 30 min. After that, the sensing platform was rinsed in DI water and dried with nitrogen gas. Lastly, ferritin solution (10 mg L−1) was pipetted on the assembled area and incubated for 3 h at room temperature. Fig. 1 shows a schematic diagram of the phosphate sensor based on the rGO-FET platform. Ferritin probes on the sensor surface were characterized and validated by energy dispersive X-ray spectroscopy (EDS) mapping and X-ray photoelectron spectroscopy (XPS) (Fig. S1 and S2†).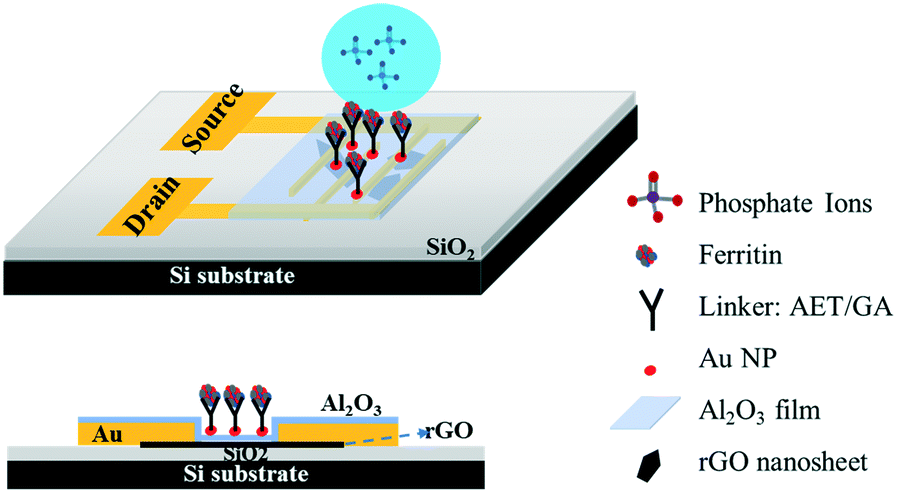 | ||
| Fig. 1 Schematic diagram and side view of the rGO-based FET device for phosphate ion detection. The gaps between the interdigitated Au electrodes are bridged by single-layer rGO nanosheets. The sensing area is covered by a thin Al2O3 film (∼4 nm) with surface-deposited Au NPs (∼4 nm diameter). The ferritin probes were immobilized on Au NPs with an AET/GA linker. | ||
Sensing test
The electrical characteristics and the sensing performance of the rGO-FET platform were measured using a Keithley 4200 semiconductor characterization system. The field-effect characteristics of rGO were tested under a back-gate voltage from −40 to 40 V at room temperature, and a bias voltage (∼0.1 V) was loaded between source/drain electrodes. As shown in Fig. 2(a), rGO presents a p-type semiconductor behavior, where current flow through the rGO channel continuously decreases as the gate voltage increases from negative to positive, because holes in p-type semiconductors are the dominant charge carriers. Applying a negative gate voltage effectively increases the concentration of hole carriers and causes a high current density to flow through the channel. The current on/off ratio of rGO in our experiments was around 1.4 or higher. The current–voltage (I–V) characterization of rGO was achieved without applying a gate voltage. Fig. 2(b) shows a linear I–V relationship, demonstrating that the contact between the rGO and gold electrodes is ohmic. The electrical conductance change in the sensing channel was recorded by monitoring the drain current (Ids) under a constant drain-source voltage (Vds) when the platform was exposed to target solutions with different analyte ion concentrations. By monitoring the resistance change of the FET platform, we achieved the electrical detection of analyte ions. Each test was repeated with 3–4 sensors to confirm the sensor's repeatability, and each had shown similar sensing responses.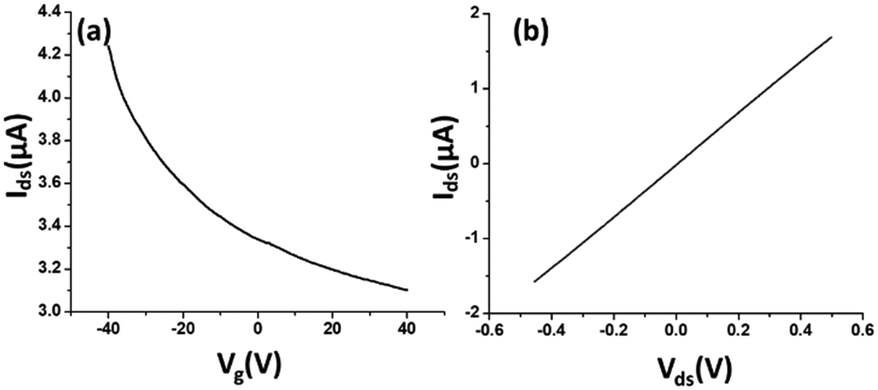 | ||
| Fig. 2 (a) Field-effect characteristics of rGO was tested with a back-gate voltage from −40 to 40 V, with a 0.1 V bias voltage loaded between the source and the drain electrodes. The identified current on/off ratio is ∼1.4. (b) I–V characterization of rGO with the source drain voltage from −0.5 to 0.5 V. | ||
Results and discussion
Platform characterization
As shown in the scanning electron microscopy (SEM) image (inset in Fig. 3(a)), an rGO sheet connected two interdigitated fingers, the source terminal and the drain terminal. The gap between these fingers is 5 μm and the size of the rGO is 5–10 μm.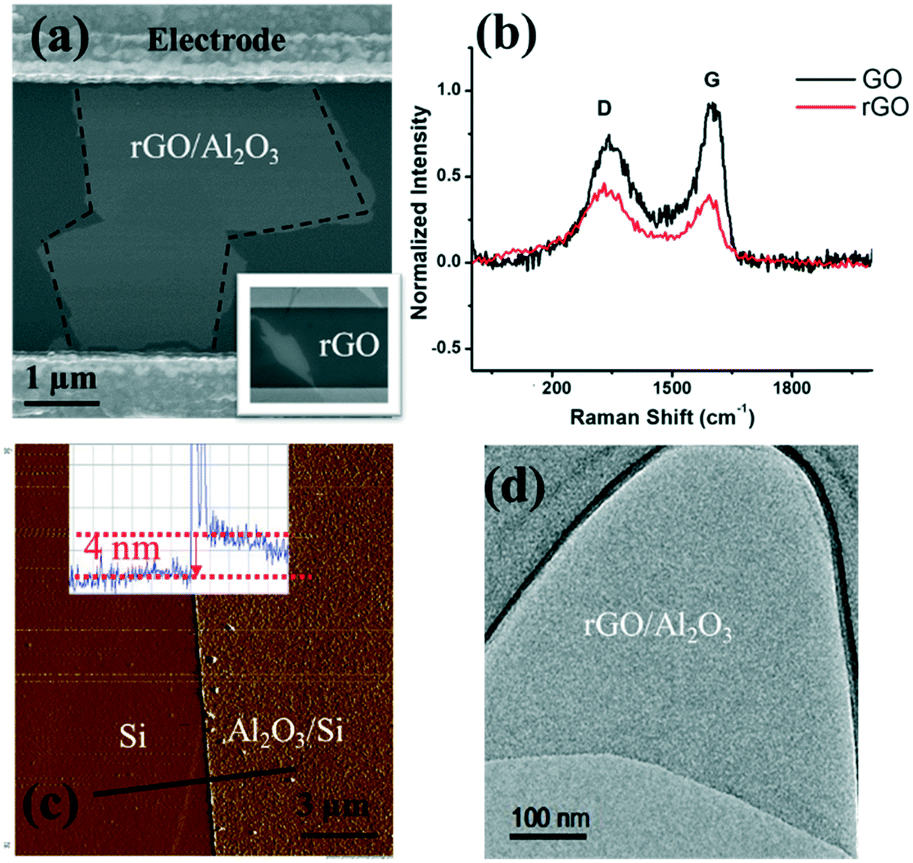 | ||
| Fig. 3 (a) SEM image of the Al2O3 film covering the rGO sheet. Inset: rGO sheet spans across the interdigitated electrode gap and is transparent under the electron beam. (b) Raman spectra of GO (black) and rGO (red). (c) AFM image of the Al2O3 film; the solid black line indicates the scanning trace; inset: height profile. (d) TEM image of the Al2O3 film covering the rGO nanosheet. | ||
To further characterize the rGO, Raman spectroscopy was used to study the sp2 domains of rGO. Fig. 3(b) shows the Raman analysis of GO and rGO, in which GO (rGO) displays a strong G peak at 1604 (1595) cm−1 due to the oxygenation of graphite. A broadened D peak at 1346 cm−1 (in contrast to 1340 cm−1 for rGO) also appears in GO due to the creation of defects, vacancies, and distortions of sp2 domains after complete oxidation.19 With thermal reduction, the ratio of the D peak intensity to G peak intensity increases in rGO, suggesting that the reduction process modifies the structure of GO with more defects.20 To reduce or eliminate the signal change introduced by the electron transfer between phosphate ions and the channel material, an Al2O3 thin film was deposited onto the rGO surface by ALD. ALD is known to be one of the best technologies to produce barriers with a low defect density under gentle process conditions. SEM, atomic force microscopy (AFM, tapping mode), and transmission electron microscopy (TEM) were used to characterize the surface morphology of the Al2O3 film on the rGO film, including its thickness and roughness. As shown in Fig. 3(a), deposition of the Al2O3 film does not alter the rGO morphology. Fig. 3(c) shows the AFM image of the Al2O3 film and its height profile. The deposited film is 4 nm thick, and the thickness investigation will be discussed in the following section. Overall, the Al2O3 film surface is flat and smooth, as shown in both Fig. 3(c) and (d). Afterwards, Au NPs were uniformly coated onto the sensing platform via a sputter-coating technology, with an average size of ∼4 nm, shown as small dots in Fig. 3.
Engineering of the passivation layer – critical thickness
It is critical to control the thickness of the passivation layer (Al2O3 film). On the one hand, it needs to be thick enough to cover the sensing platform; on the other hand, a good detection sensitivity requires a thin enough Al2O3 layer because of the sensing range limited by the gate oxide capacitance. Therefore, a very thin passivation layer is preferred to preserve a sensitive response by taking into account the Debye length of water.For the uniform deposition of Al2O3 with ALD, temperature plays a vital role. The literature has suggested the direct growth of Al2O3 on graphene layers by H2O-based ALD with temperatures ranging from 50 to 220 °C. It is reported that a uniform Al2O3 film (60 cycles) was obtained at 100 °C and the coverage of the Al2O3 film (60 cycles) was decreased at 150 °C.21 To further identify the optimum deposition temperature between 100 and 150 °C, we have conducted a comparison between films that are deposited at 100 °C, 125 °C and 150 °C. The TEM images (Fig. S3†) and AFM roughness profile (Table S1†) indicate that 125 °C provides the best uniformity with the smallest arithmetic mean deviation of the roughness profile (Ra). Hence, deposition at 125 °C was used for subsequent experiments. To identify the critical thickness – defining a continuous barrier film on the FET sensor application, control experiments were conducted on ALD Al2O3 films of various thicknesses, ranging from 2 to 20 nm. Their roughness images and profiles are shown in Fig. S4.† In Table S2,† we have listed the roughness profile. The key performance characteristic Ra shows a decreasing trend from 2 nm (Ra = 0.0524 nm) to 5 nm (Ra = 0.0295 nm), which implies that a continuous barrier film was formed at ∼5 nm, and Ra then increases along with the film thickness, which might result from the accumulation of rough points. Meanwhile, the roughness profile Rp, Rv, Rz, Rt, Rsk and Rku all show that the 4 nm Al2O3 film has the smoothest surface; therefore, 4 nm has been taken as the critical thickness for the Al2O3 passivation layer in the FET platform. The smoother surface might also suggest that the film quality is better, given that the ALD Al2O3 film is certainly not bulk-like and cavities and cracks can exist, which will allow the permeation of water upon direct contact with the rGO channel. This can lead to the surface degradation of rGO nanosheets via oxidation. As such, the sensor stability and performance will be adversely affected.
It is also observed that the deposition of the Al2O3 film tunes the semiconducting property of the FET device from p-type to bi-polar characteristic when the film thickness increases from 2 to 3, 4, and eventually to 20 nm, as shown in Fig. S5.† The same phenomenon has been reported by Thakur et al.22 and Schmid et al.23 We speculate that the deposition of the Al2O3 film introduced electrons to the underlying rGO layer, since the remaining oxygen groups in rGO can participate in the Al2O3 growth through migration during the ALD process, leaving the dangling carbon bond intact and leading to a decrease in the hole concentration. Therefore, the FET sensor demonstrates a bi-polar property with the deposition of the Al2O3 film, and the n-type property gradually manifests along with the increase of the film thickness. It is noteworthy that the zeta potential on the FET platform surface (which is equivalent to the top gate Vgs and can be converted to the bottom gate voltage) is negatively charged, which will be discussed in Table 1. Therefore, the sensor functions in the p-type zone.
| Sample | Zeta potential (mV) |
|---|---|
| Ferritin sensing surface in DI water | −26.0 |
| Ferritin sensing surface in phosphate solution (1 mg L−1) | −13.2 |
Effects of ferritin concentration and pH on probe immobilization
The sensitive and selective detection of the FET platform is achieved by the specific adsorption of analytes onto probes. Therefore, to optimize the sensing performance, a high probe density is desired, which also should increase the adsorption capacity. In the probe immobilization step, we performed a set of control experiments with prepared ferritin concentrations ranging from 1–100 mg mL−1. Ferritin is a hollow, globular, iron storage protein, with internal and external diameters typically between 8 and 12 nm,24 shown as the bright dots in Fig. 4. | ||
| Fig. 4 SEM images of immobilized ferritin with a ferritin concentration of 5 mg mL−1 at pH values of (a) 7.83, (b) 6.98, (c) 5.10, and (d) 4.26. | ||
The coverage density of immobilized ferritin was identified by the SEM image of the sensing surface by using the software ImageJ to quantify the ferritin probes. The relationship between the prepared ferritin concentration and ferritin coverage density is presented in Fig. 5(a), which indicates that increasing the ferritin concentration improves the ferritin coverage density, and that the coverage density gradually reaches a slope of saturation. Further investigation revealed that the coverage density is affected by the concentration of the prepared ferritin as well as its pH condition.
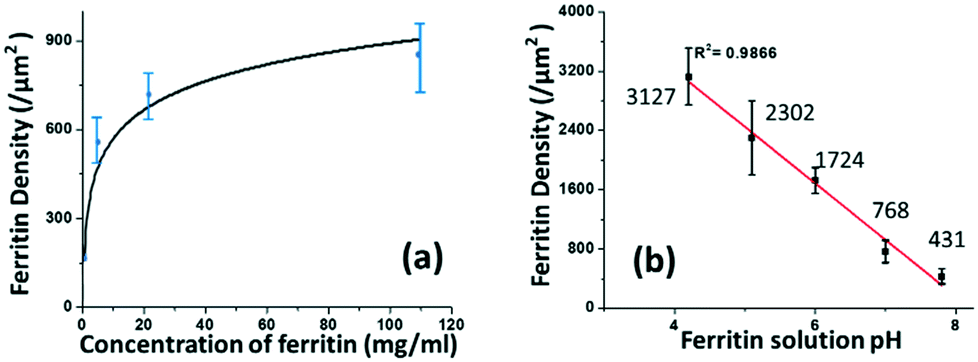 | ||
| Fig. 5 The correlation between the prepared ferritin solution and immobilized ferritin density on the sensing surface in terms of its (a) concentration and (b) pH. | ||
To study the effect of pH on ferritin immobilization, the concentration of ferritin solution was fixed at 5 mg mL−1 while the pH value was modulated between 4 and 8 with 0.1 nM NaOH and 0.1 nM HCl. The ferritin density was quantified by normalizing the counts of ferritin cores on the SEM image. The prepared 5 mg mL−1 ferritin solution had a pH value of 6.95, with an immobilized ferritin density of ∼768 μm−2. Fig. 5(b) shows a reverse linear relationship between the pH value of the prepared ferritin solutions and the immobilized ferritin density. The maximum density (3127 μm−2) was found at pH 4.26; the minimum density (431 μm−2) was found at pH 7.83. This indicates that surface immobilization is greatly affected by the pH condition and an acidic environment enhances the immobilization of ferritin probes. A similar result was achieved even when a lower ferritin concentration (1 mg mL−1) was used for probe fabrication. The underlying mechanism would be the surface charge on the Au NP surface under different pH conditions.
It was reported that the zeta potential of stabilized Au NPs is a function of pH, and the stabilized Au NPs exhibited an isoelectric point (when the zeta potential is zero) at pH 4.6.25 Between pH 4.6 and 12.0, the Au NP surface is negatively charged, and the higher pH value of the Au NP solution leads to the stronger negative charge on the particle surface. In this pH range, the ferritin is also negatively charged since the isoelectric point of horse ferritin is 4.4.26 The negatively charged AuNPs repel the negatively charged ferritin molecules from adsorbing onto the sensing surface, and the repulsive force increases along with the increase of pH, according to Coulomb's law. The response between the Au NPs and ferritin molecules under different pH conditions results in different ferritin coverage densities. A relatively acidic environment results in the highest ferritin coverage density. Thereafter, in our study, all the sensors used for detection were prepared by immobilizing pH 5 ferritin solutions.
Detecting phosphate ions
The rGO-based FET device was used for real-time detection of phosphate ions in a liquid phase. A bias voltage was loaded between the source and the drain electrodes (∼0.1 V) to supply power on the sensing platform. Fig. 6(a) shows the dynamic response of the rGO–ferritin sensor to phosphate ions in concentrations ranging from 25–100 μg L−1. The test was conducted in a drop-cast manner and DI water was used as the detection background. A stable current signal was obtained prior to the phosphate ion test. The prepared phosphate solution in the lab was pH 7.4, which mainly contains HPO42− and H2PO4−.27 The zeta potential of the ferritin sensing surface in the DI water was −26.0 mV, as shown in Table 1. In this case, the immobilized ferritin molecules generated a negative electric field on the platform which attracted the charge carriers (holes) from the p-type rGO channel to the channel surface; this process is known as the gating effect. When phosphate ions are introduced, the current on the platform decreases in real-time depending on the concentration levels, as shown in Fig. 6(a). Many studies have reported that inside the ferritin shell iron ions form mineral crystallites together with phosphate and hydroxide ions, and the crystallites are stored between the ferritin core and the protein shell.28 It is likely that when the introduced phosphate ions attach onto the ferritin probes, the structure and configuration of ferritin are deformed, causing chemical polarity alteration on the ferritin surface. Therefore, the gating effect generated by the ferritin probes becomes weaker and thus decreases the current signal on the sensing channel. This analysis was confirmed by the surface zeta potential measurement.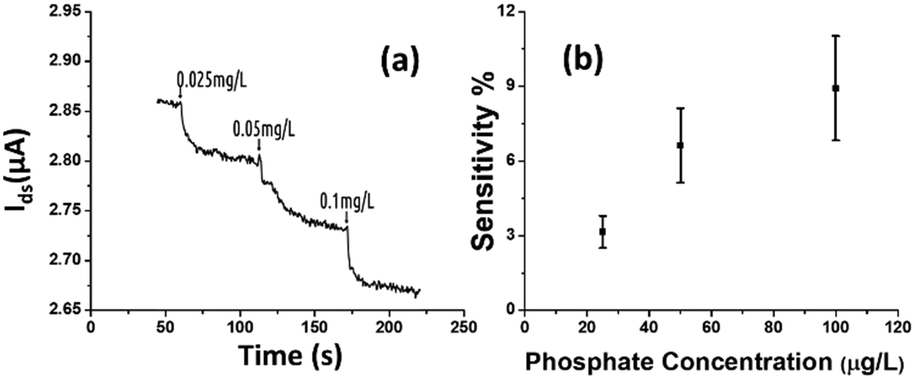 | ||
| Fig. 6 (a) Dynamic responses of the rGO-FET sensor to phosphate ions with concentrations of 25 μg L−1, 50 μg L−1, and 100 μg L−1. (b) The relationship between the phosphate concentration and the corresponding sensitivity shown by the sensing platform. The lower detection limit could reach down to 10 μg L−1. | ||
As shown in Table 1, the introduction of phosphate ions (1 mg L−1) changed the ferritin surface zeta potential from −26 to −13.2 mV. Fig. 6(b) shows the outstanding sensing responses introduced by the phosphate ions, which ranged between 25 μg L−1 (263 nM) and 100 μg L−1 (1.05 μM). Although there is no published maximum contaminant level (MCL) for phosphate in water, the recommended MCL of total phosphate in rivers and streams is 0.1 mg L−1;29 our sensing test focused on this threshold value. Compared with the sensing performance reported in the literature,15 we obtained much higher sensitivities with the newly designed rGO-FET platform, as shown in Fig. 6(b), which is attributed to the application of the Al2O3 passivation layer and the optimization of probe immobilization. Further experiments demonstrated that the lower detection limit of the sensor could reach 10 μg L−1 (105 nM).
Real water test
The tested phosphate solutions above were prepared in the lab with DI water. Here, the rGO-based FET platform was used to detect phosphate ions in real water, specifically industrial discharge. DI water was again used as the background for the testing. Originally, the phosphate concentration of the initial industrial discharge was 0.97 mg L−1 (tested with ion chromatograph (IC)), and its pH was 7.69. Then the original industrial discharge was diluted 100, 50, 10, and 5 times with DI water, and these diluted solutions were introduced to the sensing platform. The response is shown in Fig. 7(a). A current decrease signal was demonstrated, as discussed above, due to the reducing gating effect introduced by the phosphate ions. This test demonstrates that the platform sensor could be used to detect phosphate ions in real water with a lower detection limit of 10 μg L−1 (105 nM).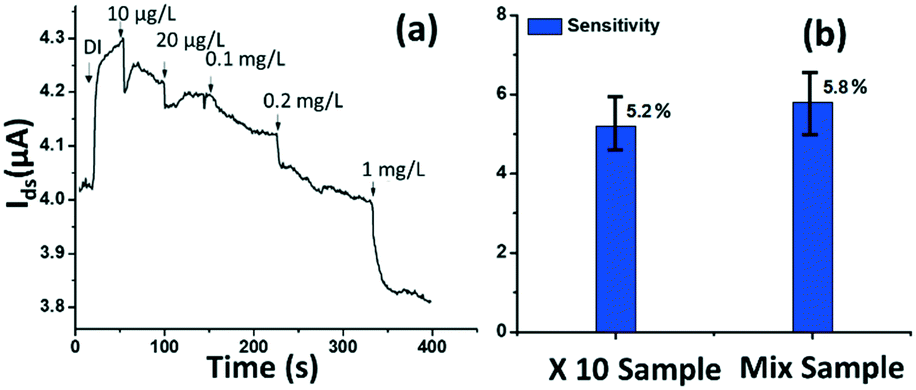 | ||
| Fig. 7 (a) Dynamic response of the rGO-FET platform to real water containing 10 μg L−1–1 mg L−1 phosphate ions. (b) Selectivity investigation of phosphate detection in real water. The 10 times diluted real phosphate water sample contains ∼0.1 mg L−1 phosphate ions. The mix sample contains 0.1 mg L−1 Na2CO3, 0.1 mg L−1 KNO3, 0.1 mg L−1 K2SO4, and 0.1 mg L−1 real water phosphate ions. | ||
To investigate the selectivity performance of the rGO-FET platform, three interference ions were introduced, SO42−, CO32−, and NO3−. A mixture solution was prepared by adding interference ions to 10 times diluted real water that contains ∼0.1 mg L−1 phosphate. The response of the mix sample was compared with that of the 10 times diluted phosphate real water, in the same test. The results are shown in Fig. 7(b). The two samples showed comparable responses, which indicates that the introduction of SO42−, CO32−, and NO3− ions didn't generate distinct interfering signals, except limiting coulombic adsorption to the sensing surface. This confirms that the rGO-FET can differentiate phosphate ions from other interference anions. The application of the Al2O3 passivation layer also likely contributed to the realization of selective phosphate detection in real water samples. Note that the sensor selectivity is due to the chemical binding between ferritin and phosphate ions (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) FeO–OH + HPO42− → FeO–O–PO32− + H2O). Phosphate ions react with hydroxyl groups in the ferritin and chemically bond to iron oxides by releasing H2O molecules. As a result, the sensor platform is capable of detecting various organophosphate species under the condition that at least one hydroxyl group is present in the probe.
FeO–OH + HPO42− → FeO–O–PO32− + H2O). Phosphate ions react with hydroxyl groups in the ferritin and chemically bond to iron oxides by releasing H2O molecules. As a result, the sensor platform is capable of detecting various organophosphate species under the condition that at least one hydroxyl group is present in the probe.
For future real applications, the sensor should have a long shelf life. To realize this, the sensors can be stored either in inert gas or dry air. On the one hand, the thin passivation layer (i.e., 4 nm Al2O3) is permeable to the moisture in air under ambient conditions, which can oxidize the senor channel and lead to the degradation of sensor performance. On the other hand, the hydroxyl groups on the ferritin surface can also be consumed by potential hydrocarbon pollutants in air through releasing water molecules, resulting in the reduced coverage of reactive sites for sensing. While the protection by improving the storage condition can elongate the life of the single use sensor, preserving the sensor stability for continuous monitoring in water can be challenging. This is because the Al2O3 film may experience corrosion over time, especially at high temperatures. Further research is warranted to address such a challenge.
Conclusions
In this work we demonstrated an rGO-FET sensor for ultrasensitive and real-time detection of phosphate ions, both in lab-prepared ideal water and in real industrial water samples provided by an industry partner. The sensor platform is promising for low-concentration detection and real-time monitoring of phosphate ions. The combination of the nanomaterial-FET with its unique properties and specific probes sets a typical example for low-concentration monitoring of nutrients in water. Yet, further development is needed for real-world applications. The sensor can be further improved by tuning the platform configuration and optimizing the surface modification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the U.S. National Science Foundation through the NSF GOALI grant (CBET-1606057). The authors thank Dr. Heather Owen, Mr. Donald Robertson, Dr. Steven Hardcastle, and Mr. Patrick Anderson for technical support with SEM, TEM, AFM, and IC analyses, respectively. We also want to thank Dr. Guanglong Tian and Dr. Heng Zhang from the Metropolitan Water Reclamation District of Greater Chicago, Dr. Matthew Diebel and Mr. Brian Weigel from the Wisconsin Department of Natural Resources and Mr. Patrick Cardiff from Grande Cheese Company for helpful discussions.Notes and references
- S. Chang and M. L. Jackson, Soil Sci., 1957, 84, 133–144 CrossRef CAS.
- X. Jin, X. Jiang, Y. Yao, L. Li and F. Wu, Water Environ. Res., 2006, 78, 2405–2411 CrossRef CAS.
- G. A. Block, T. E. Hulbert-Shearon, N. W. Levin and F. K. Port, Am. J. Kidney Dis., 1998, 31, 607–617 CrossRef CAS PubMed.
- S. Cui, H. Pu, S. A. Wells, Z. Wen, S. Mao, J. Chang, M. C. Hersam and J. Chen, Nat. Commun., 2015, 6, 8632 CrossRef CAS PubMed.
- Y. Chen, R. Ren, H. Pu, X. Guo, J. Chang, G. Zhou, S. Mao, M. Kron and J. Chen, Sci. Rep., 2017, 7, 10974 CrossRef PubMed.
- J. Chang, G. Zhou, E. R. Christensen, R. Heideman and J. Chen, Anal. Bioanal. Chem., 2014, 406, 3957–3975 CrossRef CAS PubMed.
- S. Mao, J. Chang, H. Pu, G. Lu, Q. He, H. Zhang and J. Chen, Chem. Soc. Rev., 2017, 46, 6872–6904 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- G. Zhou, J. Chang, S. Cui, H. Pu, Z. Wen and J. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19235–19241 CrossRef CAS PubMed.
- C. Gómez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef PubMed.
- X. Chen, G. Zhou, S. Mao and J. Chen, Environ. Sci.: Nano, 2018, 5, 837–862 RSC.
- Y. Ohno, K. Maehashi and K. Matsumoto, J. Am. Chem. Soc., 2010, 132, 18012–18013 CrossRef CAS PubMed.
- C. T. Lin, P. T. K. Loan, T. Y. Chen, K. K. Liu, C. H. Chen, K. H. Wei and L. J. Li, Adv. Funct. Mater., 2013, 23, 2301–2307 CrossRef CAS.
- L. H. Hess, M. Jansen, V. Maybeck, M. V. Hauf, M. Seifert, M. Stutzmann, I. D. Sharp, A. Offenhäusser and J. A. Garrido, Adv. Mater., 2011, 23, 5045–5049 CrossRef CAS PubMed.
- S. Mao, H. Pu, J. Chang, X. Sui, G. Zhou, R. Ren, Y. Chen and J. Chen, Environ. Sci.: Nano, 2017, 4, 856–863 RSC.
- L. Fan, C. Luo, X. Li, F. Lu, H. Qiu and M. Sun, J. Hazard. Mater., 2012, 215, 272–279 CrossRef PubMed.
- G. Srinivas, J. Burress and T. Yildirim, Energy Environ. Sci., 2012, 5, 6453–6459 RSC.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5689 RSC.
- K. Krishnamoorthy, M. Veerapandian, R. Mohan and S.-J. Kim, Appl. Phys. A: Mater. Sci. Process., 2012, 106, 501–506 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef PubMed.
- Y. Zhang, Z. Qiu, X. Cheng, H. Xie, H. Wang, X. Xie, Y. Yu and R. Liu, J. Phys. D: Appl. Phys., 2014, 47, 055106 CrossRef.
- B. Thakur, G. Zhou, J. Chang, H. Pu, B. Jin, X. Sui, X. Yuan, C.-H. Yang, M. Magruder and J. Chen, Biosens. Bioelectron., 2018, 110, 16–22 CrossRef CAS PubMed.
- M. Schmid, M. Shishkin, G. Kresse, E. Napetschnig, P. Varga, M. Kulawik, N. Nilius, H. P. Rust and H. J. Freund, Phys. Rev. Lett., 2006, 97, 046101 CrossRef CAS PubMed.
- W. C. Roberts, W. R. Hartley, Drinking Water Health Advisory: Munitions (illustrated ed.), CRC Press, 1992 Search PubMed.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS PubMed.
- A. Mazur, I. Litt and E. Shorr, J. Biol. Chem., 1950, 187, 473–484 CAS.
- M. M. Benjamin, Water chemistry, Waveland Press, 2014 Search PubMed.
- J. F. Jacobs, M. N. Hasan, K. H. Paik, W. R. Hagen and M. C. vanLoosdrecht, Biotechnol. Bioeng., 2010, 105, 918–923 CAS.
- B. Oram, Water Quality Terms GLOSSARY, https://www.water-research.net/index.php/glossary.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI:10.1039/c9me00156e |
| ‡ These two authors contributed equally to this work. |
| § Current address: Pritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60637,USA. E-mail: E-mail: junhongchen@uchicago.edu. |
| ¶ Current address: Argonne National Laboratory, Argonne, IL 60439, USA. E-mail: E-mail: junhongchen@anl.gov. |
| This journal is © The Royal Society of Chemistry 2020 |