
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the ORR selectivity for H2O2 electrogeneration by modification of Printex L6 carbon with 1,4-naphthoquinone: a theoretical, experimental and environmental application study†
Matheus S.
Kronka
 *ab,
Fernando L.
Silva
a,
Alysson S.
Martins
ab,
Michell O.
Almeida
a,
Káthia M.
Honório
c and
Marcos R. V.
Lanza
*ab
*ab,
Fernando L.
Silva
a,
Alysson S.
Martins
ab,
Michell O.
Almeida
a,
Káthia M.
Honório
c and
Marcos R. V.
Lanza
*ab
aSão Carlos Institute of Chemistry, University of São Paulo, Avenida Trabalhador São-Carlense 400, São Carlos, SP 13566-590, Brazil. E-mail: matheus.kronka@gmail.com; marcoslanza@usp.br
bNational Institute of Alternative Technologies for Detection, Toxicological Evaluation and Removal of Micropollutants and Radioactive Substances (INCT-DATREM), Institute of Chemistry, São Paulo State University, Araraquara, 14800-900, São Paulo, Brazil
cSchool of Arts, Science and Humanities, University of São Paulo, Arlindo Bettio, 1000, 03828-000, São Paulo, SP, Brazil
First published on 30th June 2020
Abstract
The modification of carbon-based materials with electroactive organic compounds can improve the oxygen reduction reaction (ORR) efficiency toward H2O2 electrogeneration. The present work sought to evaluate the electrochemical properties of a Printex L6 carbon matrix modified with 1,4-naphthoquinone (NQE) and its application toward the removal of paracetamol in different advanced oxidation processes. The 1.0% NQE-modified material showed 93% selectivity for H2O2 electrogeneration compared to 85% for the unmodified carbon matrix. Density functional theory calculations helped confirm the NQE action as an H+ donor by improving the ΔG of the reaction from −277.11 kJ mol−1 for PL6C to −986.1 kJ mol−1 for the NQE-modified material. Applied at 75 mA cm−2, the modified gas diffusion electrode presented a 30% increase in H2O2 electrogeneration compared to the unmodified electrode. Paracetamol removal followed pseudo-first-order reaction kinetics in the following order: anodic oxidation with H2O2 electrogeneration (AO-H2O2) < AO-H2O2/UVC < electro-Fenton (EF) < photoelectro-Fenton (PEF). The action of the NQE modifier helped enhance the ORR activity and selectivity for H2O2 electrogeneration, thus making the material suitable for environmental application in wastewater treatment.
Introduction
Over the past decades, the world has witnessed the emergence of several problems stemming from disorderly population growth. A large variety of the new products that are flooding into the market are manufactured by pharmaceutical, agricultural, industrial and food companies. Currently, one can find a wide range of organic compounds in freshwater sources such as rivers and springs; this is attributed to the haphazard discharge of huge volumes of wastewater, which are left either without treatment or treated inefficiently.1 Pharmaceutical compounds specifically require profound attention since they are often disposed of through industrial waste and human excretion.2–4 Paracetamol PRM (acetaminophen, 4-acetylaminophenol) is a drug widely used as an analgesic and antipyretic, and there have been many reports regarding its presence in rivers and wastewater.4–7 With the unrestrained consumption of PRM, studies have shown that 90% of this drug is excreted by humans with no change in its composition. The consequences of the presence of PRM and its metabolites in the aquatic ecosystem are still poorly understood. Its acute toxicity showed variable magnitudes in a study with Danio rerio species (LC50)2 and freshwater arthropods (LD50).8 In tests involving different species of bacteria, ciliates, fish embryos and algae, PRM exhibited highly toxic properties, classified as dangerous.2 In this context, the search for effective and efficient treatment mechanisms capable of ensuring the complete removal of these organic compounds in wastewater and surface water has become a matter of unquestionable relevance.Although several studies have proposed different methods for the removal of contaminants in wastewater, the treatments are commonly incapable of efficiently removing recalcitrant compounds in low concentration.9–11 Among the alternative methods proposed, advanced oxidation processes (AOPs) can complement the traditional treatments and ensure effective/safe removal of pollutants. AOPs are based on the generation of highly reactive oxygen species, such as hydroxyl radicals (˙OH),1,12,13 and are known to promote non-selective oxidation of a wide range of organic molecules. Hydroxyl radicals can be generated using (i) semiconductor–ZnO,14 WO3,15 and TiO2;16 (ii) other precursors, such as ozone (O3)17 and hydrogen peroxide (H2O2);18,19 or (iii) electro-Fenton (EF) reactions where Fe2+ catalyses the decomposition of electrogenerated H2O2 for ˙OH production.20–23 In addition, AOPs can be combined with an irradiation source (ultraviolet or visible) to help enhance the efficiency of the treatment process.
The application of H2O2 in AOPs is particularly interesting in the sense that it can be generated locally in the aqueous matrix; as such, it diminishes the negative effects related to the transport and storage of H2O2 given its corrosive and explosive properties. The H2O2 electrogenerated in aqueous medium using gas diffusion electrodes (GDEs) has led to H2O2 production in high concentration (>100 mg L−1).18,19,24 The electrogeneration of H2O2 typically occurs through the application of carbonaceous materials![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) –usually carbon black–as a cathode in the electrolyte matrix with pressurized O2 gas.18,24–26
–usually carbon black–as a cathode in the electrolyte matrix with pressurized O2 gas.18,24–26
Printex L6 (PL6C) is a carbon black pigment employed as a high efficiency electrocatalytic matrix for H2O2 electrogeneration; its good performance can be attributed to corrosion resistance, large surface area (265 m2 g−1) and oxygenated functional groups (–C–OOH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –C–OH).27 Furthermore, the hydrophobicity of PL6C, associated with the role in the oxygen reduction reaction (ORR), provides high selectivity for H2O228,29 and makes it ideally suitable for the application of O2 gas at the electrolyte/electrode interface in GDE reactions.23,30
O, –C–OH).27 Furthermore, the hydrophobicity of PL6C, associated with the role in the oxygen reduction reaction (ORR), provides high selectivity for H2O228,29 and makes it ideally suitable for the application of O2 gas at the electrolyte/electrode interface in GDE reactions.23,30
The use of the triple interface electrolyte/electrode/gas in acid medium for PL6C in GDE reactions allows the occurrence of the ORR involving 2-electron transfer, leading to the production of H2O2 molecules (eqn (1)).31 However, the reduction process may be followed by other parallel reactions via a pathway involving 4-electron transfer (eqn (2)) or H2O2 electroreduction by 2-electron transfer (eqn (3)). Some reactions may have negative effects on H2O2 production and accumulation; these include the H2O2 disproportional reaction (eqn (4)) and reactions that can compete with the ORR such as the H2 evolution reaction (eqn (5)).21,24,30–34
| O2 + 2H+ + 2e− → H2O2 (E0 = 0.695 V vs. SHE) | (1) |
| O2 + 4H+ + 4e− → 2H2O (E0 = 1.229 V vs. SHE) | (2) |
| H2O2 + 2H+ + 2e− → 2H2O (E0 = 1.76 V vs. SHE) | (3) |
| 2H2O2 → ½O2 + H2O | (4) |
| 2H+ + 2e− → H2 (E0 = 0 V vs. SHE) | (5) |
In order to improve the ORR activity toward H2O2 production, some authors have proposed different techniques for modifying carbon-based materials, including the use of metallic oxides,35–37 metallic complexes,19,32 metallic nanoparticles38,39 and organic compounds.18,26,40–42 These modifications can lead to a decrease of the overpotentials required for the ORR and an improvement of oxygen supply in solution, increasing the H2O2 generation.
The modification of carbon-based electrochemical matrices by quinones has become widely applied because these organic modifiers are ORR facilitators for the 2-electron pathway; this also explains why they are commonly used in the traditional process of hydrogen peroxide generation.25,31Eqn (6) shows the electrochemical reduction of quinones in acidic medium.42–46
| Q + 2H+ + 2e− ⇌ QH2 | (6) |
The technique involving heterogeneous catalysis of cathodic O2 is based on the application of carbon-based materials modified with organic redox compounds as the electrode matrix. The literature on the inorganic–organic interface for ORR catalysis considers the QH2 groups present in the carbon matrix as facilitators of hydrogen peroxide production (eqn (7)).42–46
| QH2 + O2 → H2O2 | (7) |
In this sense, several studies published in the literature have thoroughly investigated the use of quinones as modifiers of carbon matrices for H2O2 production.18,26,47,48 The advantage being that the mechanism for the ORR involves two-electron transfer, favouring H2O2 production. However, the mechanism for H2O2 electrogeneration is still not clearly understood.
Huissoud and Tissot48 tested 2-ethyl-9,10-anthraquinone as a modifier in vitreous carbon; the modification significantly increased the production of H2O2 in catalyst-free electrodes. Valim et al. (2013) showed that the addition of 1% (w/w) tert-butylanthraquinone in a carbon black matrix contributed to an increase in efficiency of 89.6% in H2O2 production compared to 76.6% for unmodified Printex L6 carbon.18 More recently, Moreira et al. (2019) reported the use of 2% anthraquinone-2-carboxylic acid in a carbon matrix (w/w), which led to greater current efficiency in H2O2 electrogeneration compared to the unmodified carbon electrode.26 Although these authors have demonstrated the crucial contribution of quinones as modifiers of the carbonaceous matrix, the mechanism has not been thoroughly clarified.
Moreover, none of these authors used theoretical simulation studies to prove their theories regarding the action mechanism of the organic modifiers. Thus, the novelty of the present work lies in the use of theoretical simulation studies to corroborate the proposed action mechanism of the organic modifier employed in this investigation. Quílez-Bermejo et al. (2019) reported a theoretical study of the ORR involving the use of Vulcan carbon and its N-doped groups, which resulted in the enhancement of selectivity for H2O2.49 However, they did not evaluate the functional groups present in carbon; instead, they demonstrated how a separate organic molecule in the carbon matrix can act as an H+ donor for the ORR.
Considering the application potential of quinones as carbon modifiers, the present work aims to investigate, for the first time, the incorporation of 1,4-naphthoquinone in Printex L6 carbon (PL6C) for the electrogeneration of H2O2 in acid medium in an electrochemical reactor. Taking into account the proposed mechanism of action of these organic molecules, this work will present theoretical simulations that can prove the reaction mechanisms in the ORR. To illustrate the applicability of the methodology, GDEs were studied in the oxidation of paracetamol solution by anodic oxidation (AO), photolysis (UVC), AO-H2O2, AO-H2O2/UVC, electro-Fenton (EF) and photoelectron-Fenton (PEF) processes.
Results and discussion
Electrochemical study of catalytic materials for the ORR
Materials containing different percentages of NQE incorporated into the PL6C matrix were studied by cyclic voltammetry; the redox profile of the materials can be observed in Fig. 1A. The electrochemical study was performed using N2-saturated electrolyte in order to avoid O2 dissolution and its respective electrochemical reaction. Comparing the voltammetry profile of the PL6C matrix with the materials of different NQE percentages in the carbon matrix, the incorporation of the organic modifier showed a redox profile in the region of 0.0 to 0.2 VAg/AgCl, which is found to be related to the redox reaction of NQE to NQE-H. In the cathodic region, the current observed is related to the reduction of quinone to hydroquinone by the transfer of 2e− and 2H+. The reversibility of the reaction is in accordance with the potential difference of the anodic and cathodic peaks of 40 mV with Q1/Q2 ≈ 1.50 Considering that Q is the charge related to the cathodic (Q1) and anodic (Q2) reaction, the ratio Q1/Q2 ≈ 1 indicates that the charge involved in the cathodic reaction is practically the same as in the reverse reaction. The slight shift in the NQE redox peak in CV analysis is attributed to the change in the electrochemical properties, such as the increase in resistivity of the matrix surface in the presence of organic contents.26,41,42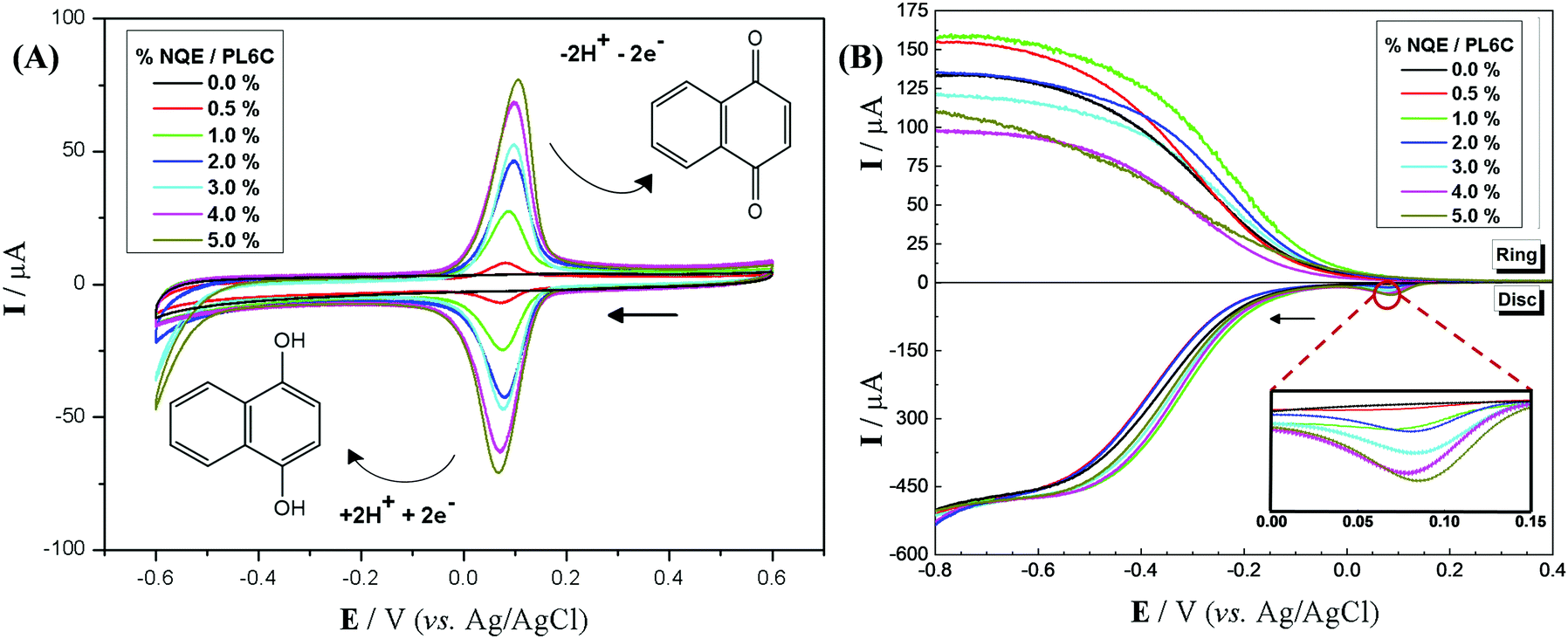 | ||
| Fig. 1 (A) Cyclic voltammetry on an RRDE with different percentages of NQE and PL6C matrix in N2-saturated 0.1 mol L−1 H2SO4 (pH 2), at a scan rate of 50 mV s−1. (B) Comparative linear sweep voltammogram of Printex L6 carbon microlayers and different percentages of NQE on the RRDE electrode in O2-saturated 0.1 mol L−1 H2SO4 (pH 2), at 900 rpm and a 5 mV s−1 scan rate. The inset between 0.0 and 0.15 VAg/AgCl is related to reduced quinone, while the arrows indicate the direction of the scan. | ||
The implication is that NQE-H has the ability to donate electrons and hydrogen for reduction reactions, such as the ORR, and to decrease the activation energy of the reaction or increase its charge transfer, acting as a catalyst. With regard to the capacitive region (−0.1 V < E > 0.2 VAg/AgCl), all the materials investigated here showed that the capacitive current related to the electrical double layer was very close to that of the Printex L6 carbon; this implies that the resistance obtained for the microlayers is very similar, which thus confirms the reproducibility of the methodology. Another point that deserves mentioning is the increase observed in the peak current with the increase in the percentage of NQE in the catalytic matrix; this shows a direct response regarding the quantity of the modifier and the redox peak current, thus validating the method of incorporation adopted.
The O2-saturated electrolyte gives rise to another reaction involving the reduction of O2 to H2O2 or H2O, which is referred to as the oxygen reduction reaction (ORR). Through this system, one is able to evaluate the influence of the organic modifier on the displacement of the ORR potential and the selectivity of the reaction in the generation of H2O2. Here, a hydrodynamic study of the materials in the RRDE was carried out with the aid of an O2-saturated electrolyte. The RRDE exhibited a rotation speed of 900 rpm for all the materials containing different percentages of NQE in the modified PL6C matrix and the unmodified matrix. For the detection of H2O2, the working electrode ring was polarized at +1.0 VAg/AgCl, as proposed by some authors.26,51,52 This overpotential applied promoted the oxidation of H2O2, exhibiting a current proportional to the amount of H2O2 involved.
Fig. 1B illustrates two analyses: (i) hydrodynamic voltammetry of the disk intended for studying the electrochemical activity of the material for the ORR; and (ii) the detection of H2O2 in the ring via the application of chronoamperometry at 1.0 V. The highlighted area of the disk response shows the reduction peak of NQE, which is in the same range of potentials previously observed in the CV analysis. The materials containing different amounts of NQE (in the modified PL6C matrix) showed variations in terms of current efficiency and selectivity for H2O2 electrogeneration (see Table 1). The percentage of H2O2 in the system and the electron number (ne−) involved in the reaction were calculated using eqn (8) and (9) proposed by Paulus et al. (2014).18,51
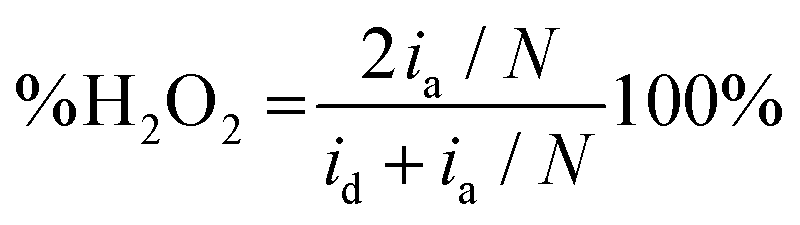 | (8) |
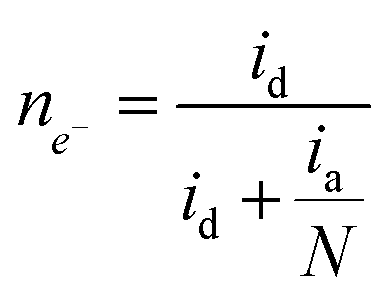 | (9) |
| Material | PL6C matrix | Percentages of NQE/PL6C (w/w) | |||||
|---|---|---|---|---|---|---|---|
| 0.5% | 1.0% | 2.0% | 3.0% | 4.0% | 5.0% | ||
| %H2O2 | 85.3 (1.2) | 91.5 (0.9) | 92.9 (0.8) | 78.9 (2.1) | 84.1 (1.9) | 69.5 (1.5) | 54.5 (1.9) |
| n e− | 2.30 | 2.16 | 2.14 | 2.42 | 2.22 | 2.60 | 2.92 |
Here, id is the current from the ORR disk, ia is the current observed in the ring, and N is the RRDE collection number N = 0.37.
The incorporation of NQE in the PL6C matrix led to an increase in both the electrogeneration of H2O2 and in the reaction potential displacement. Table 1 shows that both 0.5 and 1.0% NQE/PL6C presented great values of selectivity for H2O2 electrogeneration with 91.5 and 92.9% efficiency, respectively. For the modified carbon matrix, a considerable increase in current efficiency of 10% was observed compared to the unmodified carbon matrix.
Both results are in agreement with the hypothesis suggested regarding the catalytic activity of the modifier in the ORR consisting of lower reaction activation energy and higher selectivity for H2O2 electrogeneration. Table S1 (see the ESI†) presents a comparative analysis of the performance of similar cathodic materials reported in the literature for H2O2 production. The modification of carbon-based materials with organic redox compounds has been shown to increase the reaction activity by ∼400 mV and can lead to more than 90% selectivity for H2O2.40,47,53 Two studies that deserve mentioning here are the studies reported by Valim et al. (2013) and Rocha et al. (2020), which obtained 95 and 89% selectivity after modifying the carbon matrix with 1.0% tert-butyl-anthraquinone and 1.0% 1,2-dihydroxyanthraquinone, respectively.18,40 In the present work, the modification of PL6C with 1.0% NQE resulted in higher ORR activity, shifting the onset reaction potential to ∼60 mV compared to the unmodified carbon matrix, in addition to increasing the selectivity for H2O2 production to ∼93%. Based on these results, one can say that 1,4-naphthoquinone is one of the organic modifiers with the greatest ORR electrochemical catalysis for H2O2 electrogeneration.
The catalytic activity of quinone modifiers involving an electrochemical–chemical mechanism has been previously reported in the literature.18,26 In this mechanism, quinone is electrochemically hydrogenated, and, in a subsequent process, O2 chemically reacts with hydroquinone, receiving 2H+ and paving the way toward the formation of H2O2. However, the present study shows the first reaction of NQE, where the compound undergoes reduction to NQE-H at the peak observed at 0.1 VAg/AgCl with the presence of a subsequent reduction current related to the ORR at more negative potentials. Based on this observation, the action mechanism of this quinone can be defined as purely electrochemical. This process occurs through the action of the organic compound (quinone) as an H+ donor for the electrochemical reaction of O2. The adsorbed O2, which is originally required to react with H+ supplied by the acid medium, reacts more easily with the H+ donor species. The inset of Fig. 1B shows an overpotential region between 0.0 and 0.2 VAg/AgCl; here, one can observe the presence of the peak related to the reduction of quinone (NQE) to hydroquinone (NQE-H). This can also be found in the cyclic voltammetry data. Thus, the whole O2 reduction reaction process is seen to be influenced by NQE-H (hydrogenated species), which can be considered a hydrogen donor for the adsorbed O2.
This electrochemical reaction favors the release of H+ species for O2 reduction on the surface of the PL6C matrix by the chemical oxidation reaction of hydroquinone, leading to the regeneration of the original quinone. Fig. 2 shows that the presence of H+ donor species provokes ORR catalysis in the sense that the donating species directly supplies H+ species on the electrode surface and diminishes the dependence of the reaction on the H+ coming from the diffusion layer.
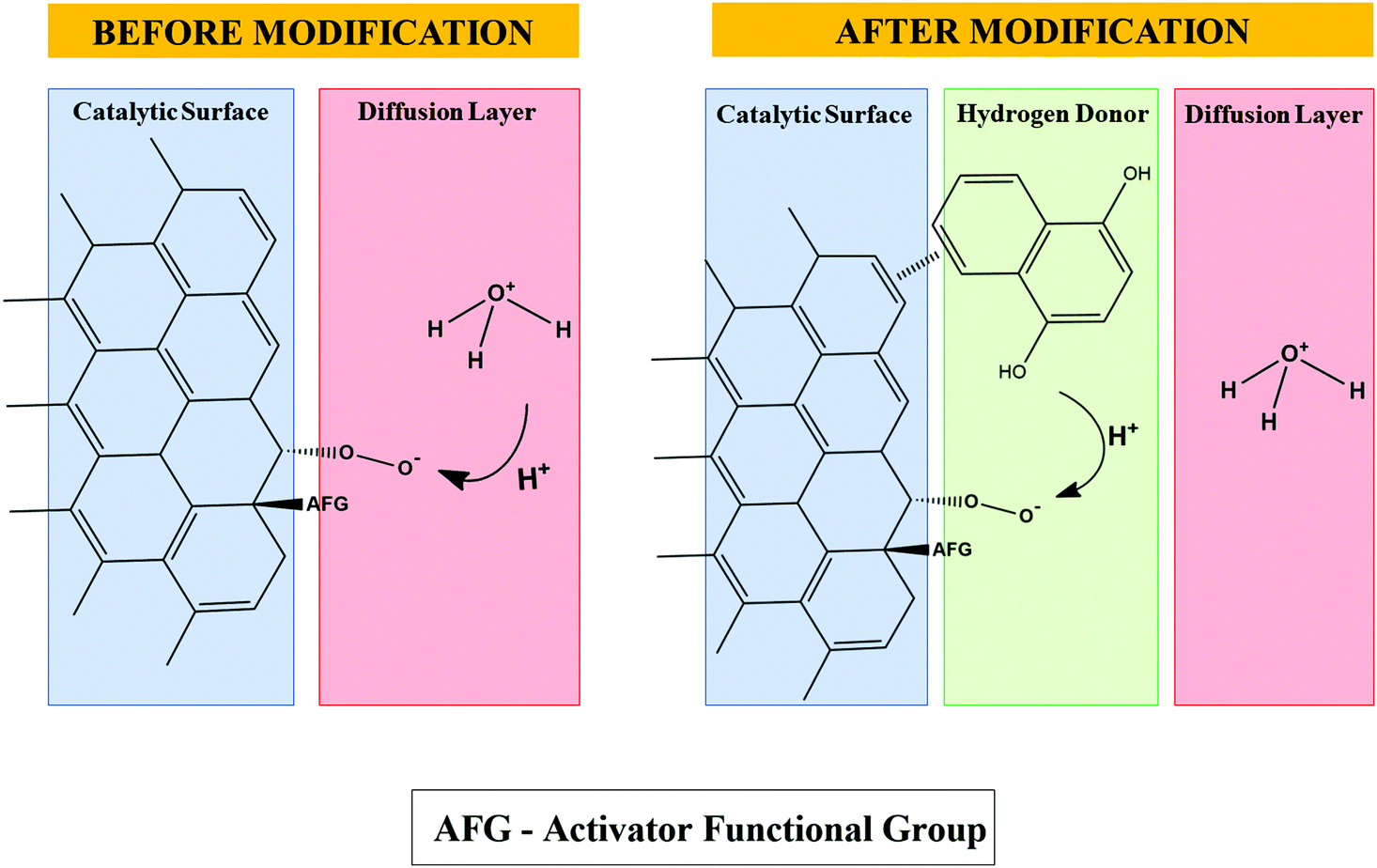 | ||
| Fig. 2 Proposed scheme for the unmodified carbon surface and incorporation of the organic compound 1,4-naphthoquinone, which acts as an H+ donor for the ORR. | ||
To confirm the previous hypothesis, computational studies related to the free energy (ΔG) of the reaction were performed comparing the bare PL6C matrix and its modification with H+ donor species. Essentially, the first stage of the theoretical study involved choosing the most suitable functional and basis set for the system under investigation through a comparison of the experimental and theoretical dipole moment of 1,4-naphthoquinone. Based on the results obtained, the method considered as the most reliable was the one involving CAM-B3LYP and 6-311G(2d,2p), since the theoretical dipole moment was found to be the closest to the experimental data (the values obtained for the other basis sets and the functional B3LYP can be seen in Table S2, ESI†).
Following the validation of the computational methodology, the systems containing Printex L6 (with the –COOH group) with the hydronium ion (structure 1), hydroquinone (structure 2) and the hydronium ion in the presence of hydroquinone (structure 3) were simulated in Gaussian09 with CAM-B3LYP/6-311G (2d, 2p), where the optimized geometries and vibrational frequencies in aqueous medium (CPCM) were calculated, and dispersion corrections were also accounted for. The values of ε0 + Gcorr and, consequently, the ΔG values obtained based on the simulations conducted for structures 1, 2 and 3 are shown in Table 2.
| Structure 1 (Printex 6L matrix + hydronium ion) | ||
| Reagent | ε 0 + Gcorr | ΔG |
| −4335716.4 kJ mol−1 |
−277.4 kJ mol−1 | |
| Product | ε 0 + Gcorr | |
| −4335993.8 kJ mol−1 |
||
| Structure 2 (Printex 6L matrix + hydroquinone) | ||
| Reagent | ε 0 + Gcorr | ΔG |
| −5539711.0 kJ mol−1 |
−986.1 kJ mol−1 | |
| Product | ε 0 + Gcorr | |
| −5540697.1 kJ mol−1 |
||
| Structure 3 (Printex 6L matrix + hydroquinone + hydronium ion) | ||
| Reagent | ε 0 + Gcorr | ΔG |
| −5743123.2 kJ mol−1 |
−341.5 kJ mol−1 | |
| Product | ε 0 + Gcorr | |
| −5743464.7 kJ mol−1 |
||
From Table 2, one can observe that the reaction involving structure 2 (Printex 6L + hydroquinone) exhibits greater stability, which is attributed to the value of ΔG (−986.1 kJ mol−1); this shows that the hydrogen bond between the hydroxide group of hydroquinone and the O2 adsorbed in the carbon matrix is more stable in this configuration. The data related to structure 3 shows that the hydrogen bond with the hydronium ion in the presence of hydroquinone is more stable than the interaction without the presence of the organic molecule (structure 1), but it is still less stable than the interaction involving the hydroquinone hydroxide group (structure 2). Hence, one can conclude that the theoretical values corroborate the experimental data. This can be explained by the fact that the ΔG values are lower when the hydrogen bond occurs with hydroquinone, which implies that the efficiency of the electrogeneration of H2O2 is increased under this configuration.
To reinforce the theoretical analysis regarding the hydrogen bond in the system, the QTAIM methodology was employed. The parameter considered for conducting this analysis was the Laplacian of the electron density (∇2ρ) of BCPs (Bond Critical Points) of the hydrogen bonds; here, it is important to note that the values of this parameter are positive, and when they are bigger, the hydrogen bonds are likely to be stronger. The results obtained from the QTAIM analysis can be found in the ESI† in Fig. S1 and Table S3.
Based on Fig. S1 (ESI†), one can check the representations of the hydronium ion and hydroquinone (circled in blue), NACPs (Nuclear Attractor Critical Points), BCPs (Bond Critical Points; green points between atoms) and RCPs (Ring Critical Points; red points), and the possible intermolecular interactions (trace). The representation of the BCP for the hydrogen bond in structure 1 can be found between atoms H67 and O68; in the case of structure 2, this representation can be found between H85 and O79. The values for the BCPs are presented in Table S3 (ESI†). Looking at the values, one can see that the BCP value for the hydrogen bond in structure 2 is more positive (0.16 a.u.); this points to the presence of a stronger hydrogen bond. For structure 3, the QTAIM analysis for the hydrogen bond (H87–O67) shows that this hydrogen bond is relatively weaker. Thus, the theoretical analysis confirms that hydroquinone increases the effectiveness of H+ donation for the electrogeneration of H2O2.
However, an increase in the amount (in percentage terms) of the modifier resulted in a decrease in its catalytic effect in terms of H2O2 electrogeneration. This can be attributed to the larger amounts of organic molecules on the surface of the PL6C matrix, which induce changes in the O2 molecule adsorption in the ORR active sites. As shown in Fig. 2, the NQE molecules can change the steric hindrance of the carbon surface, and this can affect the interaction of O2 with its adsorption sites. Considering that there is an increase in steric impediment by the overlapping of the O2 adsorption sites, this is expected to diminish the efficiency of H2O2 electrogeneration. Other effects of increasing the amounts of organic compounds in the catalytic matrix include the increase in the hydrophobicity of the matrix and the decrease of its wettability. This can be well understood by observing the analysis involving the contact angle for both the unmodified and modified matrices, which can be found in the ESI† (see Fig. S2).
The addition of organic compounds in the carbon matrix leads to changes in its wettability. According to Moreira et al. (2019),26 an increase in the amount of organic compound in the carbon matrix causes an increase in the matrix hydrophilicity and, consequently, a decrease in its contact angle. However, a non-carbon organic material presents the greatest contact angle among the materials, since organic materials do not interact well with water. Thus, the present work shows that an increase in the amounts of organic compounds leads to a decrease in the wettability of the matrix. This effect is seen to be directly related to the decrease in the efficiency of H2O2 electrogeneration, once it is found to affect the interaction of O2 with the matrix surface (Fig. S2, ESI†). Lower NQE values of 0.5 and 1.0% exerted a relatively smaller influence on the contact angle, reaching values of 39.4 and 50.2°, respectively, compared to the contact angle of 37.5° observed for the unmodified carbon matrix. Higher amounts of NQE in the carbon matrix led to a dramatic increase in the contact angle, where values of 67.1 and 85.2° were recorded for the carbon matrix modified with 3.0 and 5.0% NQE, respectively.
Considering the aforementioned results obtained in the study, the modification of the PL6C matrix with 1.0% NQE was chosen as the optimal condition for promoting an improvement in the catalytic efficiency of the material for H2O2 electrogeneration. For a preliminary study of the ORR, the use of conventional 2D electrodes yielded a good electrochemical response in terms of efficiency and selectivity. However, these electrodes are found to present some limitations with regard to the ORR; the limitations include the low solubility of O2 in aqueous solution, in addition to the need for the transport of O2 from the bulk solution to the electrode. GDEs are porous electrodes that have advantages over conventional electrodes due to the fact that they allow the flow of gas through their structure and help make O2 directly available on the electrode surface. To conduct large scale experiments with GDEs, the area was increased from 0.25 cm2 to 20 cm2 while the solution volume was increased from 150 to 450 mL; this enabled ex situ quantitative analysis of H2O2 electrogeneration by UV-vis spectroscopy.
Quantitative, kinetics and energetic study using a GDE
The analysis of H2O2 electrogeneration was carried out by chronopotentiometry, with applied current densities ranging from 10 to 150 mA cm−2 on a 20 cm2 GDE. The data obtained are presented in terms of H2O2 concentration as a function of time (Fig. 3A and B).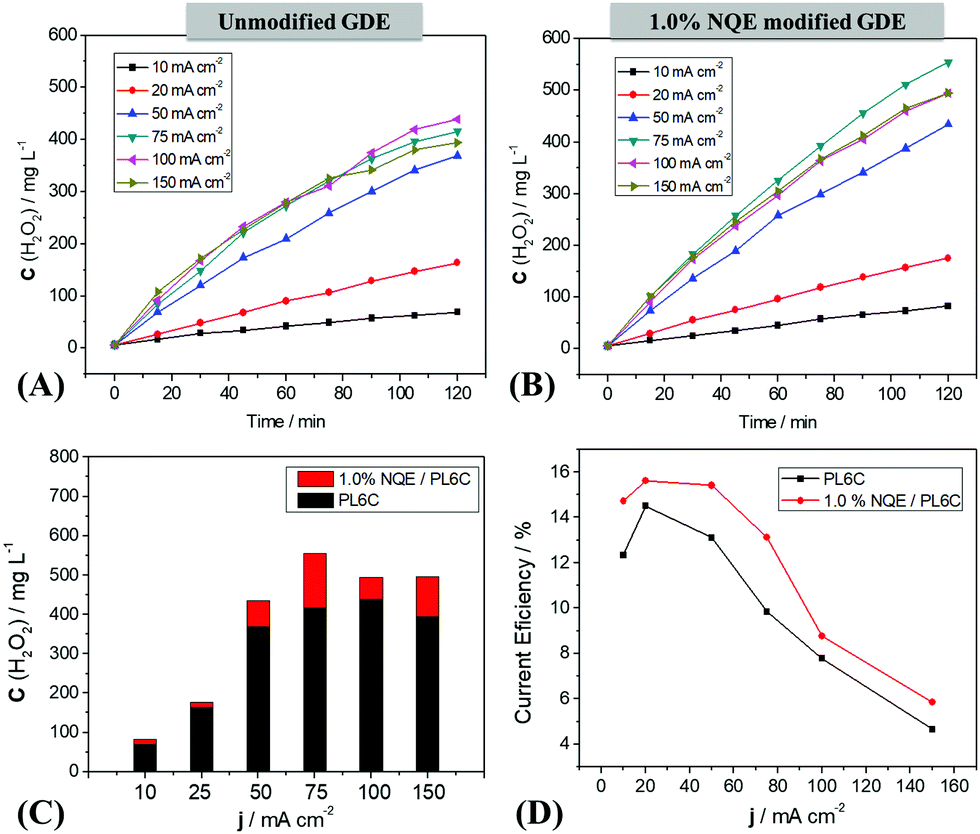 | ||
| Fig. 3 (A) Electrogenerated H2O2 concentration at different current densities on the unmodified GDE and (B) on the 1.0% NQE modified GDE; (C) comparison of the maximum concentration obtained in 120 minutes of electrosynthesis; and (D) current efficiency of all the densities investigated in 0.1 mol L−1 K2SO4 electrolyte, pH = 2.5 (H2SO4) under 0.2 bar O2 pressure. | ||
The unmodified carbon electrode showed an improvement in H2O2 electrogeneration with the increase of the applied current density (Fig. 3A); the optimal condition for the reaction was obtained at 100 mA cm−2, with 420 mg L−1 of electrogenerated H2O2. For the 1.0% NQE modified electrode, an increase was also observed in H2O2 electrogeneration with the current density. Nevertheless, this modified electrode presented a shift in the optimum condition to 75 mA cm−2 (Fig. 3B) and an increase in the amount of accumulated H2O2 compared to the unmodified electrode. In addition, as shown in Fig. 3C (a comparison is made regarding the maximum H2O2 concentration after 120 min at each current density), the modification of the GDE catalytic mass contributed to an increase in H2O2 generation at all current densities. The concentration of H2O2 was found to decrease for current densities above these optimum values; this is attributed to the energy efficiency of the system (Fig. 3D). At all the current densities applied, the modified GDE exhibited an increase in the system efficiency for the electrogeneration of H2O2 compared to the unmodified GDE; this provides support for the proposed mechanism involving the use of NQE as a hydrogen donor for the reaction. For higher current values (100–150 mA cm−2), the current efficiency declined significantly for both modified and unmodified electrodes, which may have been lost to the parallel reactions in the ORR process. In order to study the consequences of PL6C modification with NQE, the influence of NQE on the kinetic velocity was also investigated. The results showed a pattern of behavior typically similar to those obtained in other organic modification studies, where an increase was observed in the final concentration of H2O2 as well as a decrease in energy consumption.18,19,26 Moreira et al. (2019) reported an increase in H2O2 electrogeneration by the modification of PL6C with quinones and azo compounds without any changes in the optimal current density.26 Apart from an increase in efficiency for H2O2, the incorporation of NQE in the carbon matrix displaced the optimal current density to 75 mA cm−2, indicating lower energy consumption for the reaction.
The electrogeneration of H2O2 in acid medium without separation of the anode and cathode presents some different types of parallel reactions (reactions (1)–(5)) which influence the reaction kinetics. According to some authors,47,53,54 in the systems that employ a GDE as the working electrode, the global reaction kinetics is of pseudo-zero order, where the first 60 min period presents a linear increase of the H2O2 concentration. Under the pseudo-zero order condition, the angular coefficient of H2O2 concentration vs. time is proportional to the apparent velocity constant (kapp) and reaction velocity (V). In Table S4 (ESI†), the values obtained display an increasing pattern of reaction kinetics for the unmodified GDE, where a value of 4.6 mg L s−1 was recorded at the optimal density condition of 100 mA cm−2. The incorporation of NQE in the carbon matrix resulted in an increase in H2O2 electrogeneration and, consequently, led to the higher reaction kinetics observed for the modified electrode under all the current densities investigated. Thus, at a current density of 75 mA cm−2, the modified GDE presented a higher amount of electrogenerated H2O2 (555 mg L−1) and higher reaction kinetics (5.3 mg L−1 min−1) compared to the unmodified GDE (which presented an amount of 418 mg L−1 electrogenerated H2O2 and reaction kinetics of 4.5 mg L−1 min−1). This result was obtained by the addition of a hydrogen-donating material (NQE) on the electrode surface, which facilitated the reaction of H+ and increased the electrogeneration rate of H2O2.
The value of the energy consumption (EC) represents the amount of energy consumed under each applied current density. Clearly, it is conceivably important to investigate the effects of carbon modification and its influence on the reduction of the EC. Eqn (10) was used to calculate the amount of energy consumption in H2O2 electrogeneration systems:55
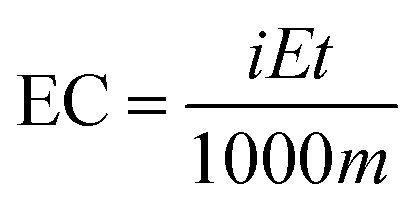 | (10) |
The modified GDE system (with 1.0% NQE) exhibited lower energy consumption and higher H2O2 electrogeneration than the unmodified GDE system under all the current densities investigated. This shows that the modification of the PL6C matrix with NQE yielded higher energy for the oxygen reduction reaction process, and, consequently, required less energy for the electrogeneration of H2O2 from O2. A gradual increase in the applied current density was also found to promote an increase in the energy consumption of the system, with a current density of 150 mA cm−2 presenting the highest energy consumption of 500 kW h kg−1. The conditions that yielded optimum values for both H2O2 electrogeneration and energy consumption were current densities of 75 and 100 mA cm−2. In the case of the unmodified GDE, the variation of the applied current density from 75 to 100 mA cm−2 led to a 6% increase (small) in the concentration of H2O2 generated with a more than 50% increase in the energy consumption of the system (Fig. 4). Based on the comparative analysis of the current densities applied (j values), the best ratio of C(H2O2) to EC for both the modified and unmodified electrodes was obtained at a current density of 75 mA cm−2.
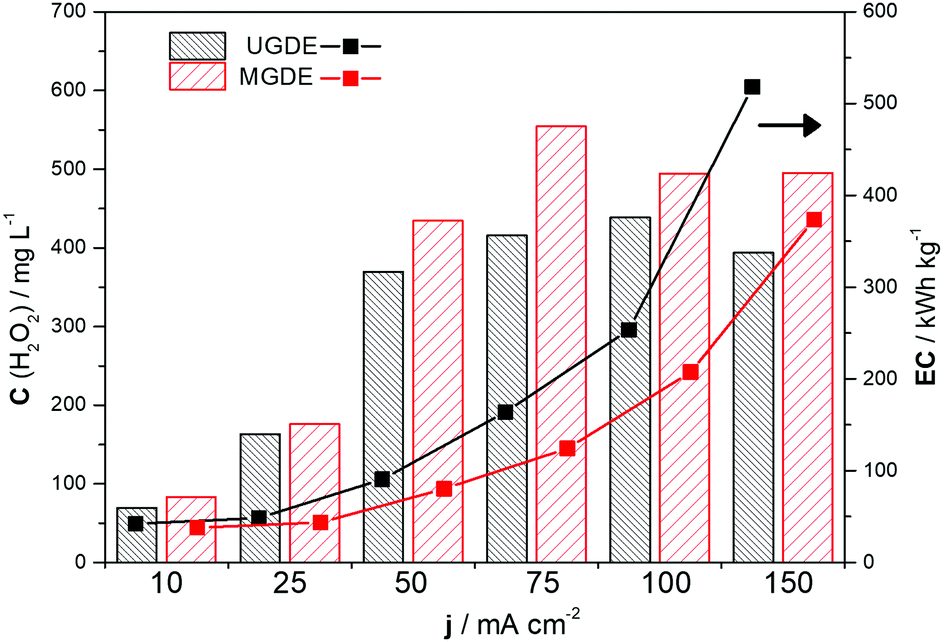 | ||
Fig. 4 Comparison of the maximum concentration of H2O2 electrogenerated in 120 min on the  unmodified GDE (UGDE) and unmodified GDE (UGDE) and  modified GDE (MGDE) with the respective energy consumption for the –■– UGDE and modified GDE (MGDE) with the respective energy consumption for the –■– UGDE and  MGDE, calculated based on eqn (10). MGDE, calculated based on eqn (10). | ||
The tests conducted involving the electrogeneration and accumulation of H2O2 on both the unmodified GDE and 1% NQE-modified GDE showed that the modification of the GDE with NQE contributed to an increase in the amount of H2O2 generated with the application of a lower current density. Thus, the modified electrode was used to carry out paracetamol (PRM) degradation tests under the best current density condition (j = 75 mA cm−2) in order to evaluate the homogeneous ˙OH catalysis processes based on advanced electrochemical oxidation processes (AEOPs).
Paracetamol degradation under optimized H2O2 electrosynthesis conditions
Paracetamol (PRM) degradation was performed (with the aid of an electrochemical cell) by the chronopotentiometry technique using a fixed current of −1.5 A (j = 75 mA cm−2) for 3 h with a 0.1 mol L−1 solution of K2SO4 (pH = 2.0) containing 80 mg L−1 of PRM. The monitoring of the degradation process was performed by HPLC measurements. Aliquots were removed from the solution over the 3 h degradation period and the degradation of the PRM concentration was monitored at a retention time of 5.8 min.The degradation study was performed considering the relative concentration of the compound according to the degradation time, given by C/C0 (cf.Fig. 5A), where C is the PRM concentration at a given time, and C0 stands for the initial concentration of PRM. C/C0 varies from 1 (0% PRM removal) to 0 (100% PRM removal). Six different types of experiments were performed aiming at evaluating the best degradation condition. Apart from the removal of PRM, the reduction of the total organic carbon (TOC) of the system was also investigated (based on the % mineralization for each process – see Fig. 5C). The energy consumption of the system was assessed based on the data on the TOC mineralization, applied current (i), and cell potential.
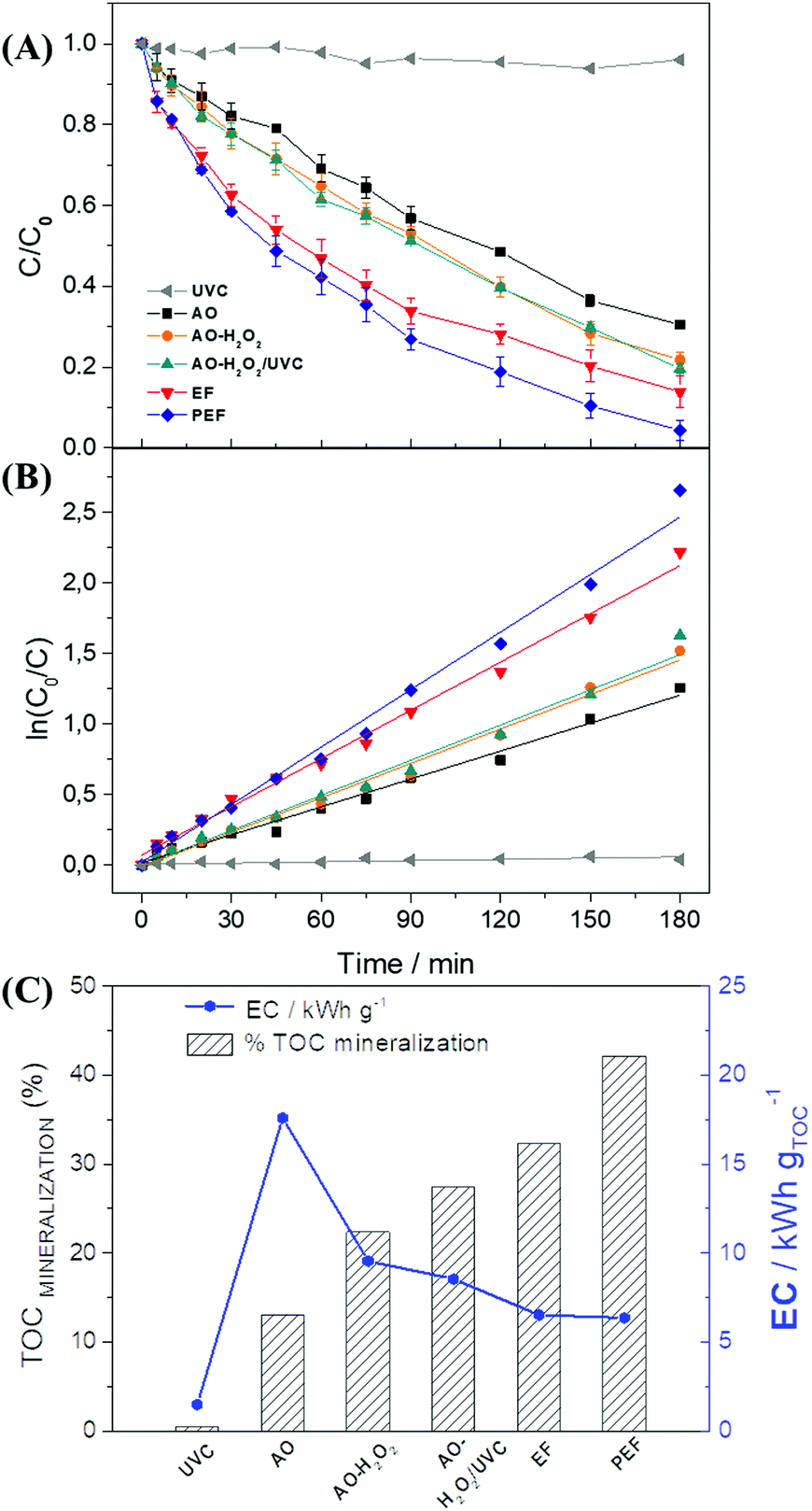 | ||
| Fig. 5 (A) Effect of different AEOPs on PRM degradation in 180 min of experiment at j = 75 mA cm−2 using the 1.0% NQE modified GDE with 0.1 mol L−1 K2SO4 under pH 2 and 0.2 bar of O2 flow; (B) analysis of the pseudo-first order kinetics; and (C) comparison between the TOC mineralization and energy consumption in each system. | ||
Fig. 5A illustrates the difference between the degradation processes employed in the study using the modified electrode, and Fig. 5B shows a comparison of kinetic constant curves for each process. The PRM degradation curve for direct photolysis with UVC light showed a negligible decrease in PRM concentration (4%), in addition to a low kinetic constant of 0.4 min−1 (R2 = 0.99). The monochromatic intensity emitted by the UV lamp exhibits low energy for paracetamol degradation, and, while this may lead the molecule to an excited state, it does not oxidize it.
It is worth noting that three processes exhibit very similar relative concentration values: the anodic oxidation (AO), AO-H2O2, and AO-H2O2/UVC processes. The processes involving the formation of H2O2 have a better degradation efficiency, and they are characterized by the combination of direct and indirect processes; however, the electrogenerated H2O2 tends to exhibit low oxidizing power. The direct process comes from the anodic contribution, while the indirect process is associated with the action of H2O2 molecules. The degradation of PRM involving the application of the AO-H2O2/UVC technique presented results found to be very similar to those of the AO-H2O2 technique, which does not involve the use of UV light; this can be attributed to the low efficiency of monochromatic ultraviolet radiation in the formation of ˙OH, which is evident by the similar kinetic constants observed for both techniques (8.1 × 10−3 min−1 for AO-H2O2 and 8.3 × 10−3 min−1 for AO-H2O2/UVC). Kinetic constant values related to the other experiments can be found in the ESI† (Table S5).
Electro-Fenton processes (EF) employ Fe2+ as a catalyst in order to obtain ˙OH (see eqn (11)); as a result, these processes show relatively higher PRM removal (∼97%) after 180 min of treatment, and an increase in degradation kinetics (11.4 × 10−3 min−1) when compared to processes that do not employ this catalyst. In the EF process, the recovery of Fe2+via the reduction of Fe3+ at the cathode (see eqn (12)) makes the process viable, in addition to enhancing the efficiency over time.
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (11) |
| Fe3+ e− → Fe2+ (E0 = 0.77 vs. SHE) | (12) |
Usually, the degradation processes of organic compounds give rise to by-products, such as carboxylic acids, which form complexes with Fe3+ species. This complexation makes the metallic ions unavailable in the system and decreases the efficiency of the Fenton process.22,56 Unlike the previous tests involving the use of UVC radiation, which did not play any effective role either in direct photolysis or in the catalysis of H2O2 to ˙OH, in the PEF process ultraviolet light played an effective role in the release of iron complexes. When UVC light is applied, one notices the action of this source of radiation in the decarboxylation of iron complexes (see eqn (13)), which leads to a rise in the availability of the catalyst and the promotion of synergy in the removal of PRM. The application of the PEF process resulted in nearly 99.5% degradation of PRM in 180 min of treatment and generated the highest kinetic constant value (kapp = 16.4 × 10−3 min−1) among the processes investigated in this work.
| [Fe(OOCR)]2+ + hν → Fe2+ + CO2 + R | (13) |
The mineralization of organic compounds was evaluated based on the results obtained from TOC analysis. It is worth noting that the organic load of the solution under investigation is directly related to both the PRM concentration and the concentration of its degradation byproducts, which may or may not be eliminated during the experiment. The mineralization process can also be used to estimate the energy consumption (EC) of the electrochemical degradation system.32Fig. 5C shows the comparison between the mineralization percentage and the EC in kW h gTOC−1 under each of the degradation processes investigated. One will notice the presence of Fe2+ as a catalyst in the mechanism involving the derivation of ˙OH from H2O2; clearly, the use of the catalyst enhances the efficiency of the processes when it comes to the mineralization of organic compounds, as can be observed in the case of the EF (32%) and PEF (39%) processes after 180 minutes of experiment. Furthermore, the higher the mineralization efficiency, the lower the EC of the process.
Another point that deserves mentioning here is that even though the amount of energy consumed by the UVC lamp employed in the PEF process is roughly similar to that consumed under the EF process, the PEF process exhibited a relatively higher TOC mineralization percentage (∼42%) compared to the EF process (∼32%) after 180 min of experiment. Considering that the application of the PEF process yielded the highest rate of mineralization and the lowest energy consumption, an exhaustive 360 min degradation process was conducted based on this technique.
Fig. S3 (ESI†) shows the 3D-chromatograms for PRM in a 6 h PEF degradation experiment using the 1.0% NQE modified GDE. At the end of 180 min of experiment, the characteristic peak of PRM was no longer detected, and all the PRM molecules were found to have been completely eliminated in the degradation process. In addition, at the end of 6 h of experiment, no chromatographic peaks related to possible by-products were observed for the 1.0% NQE modified electrode. Furthermore, the TOC values obtained exhibited sequential increases in the percentage of organic compound removal, with 42.1, 58.2 and 79.3% removal rates after 3, 4 and 6 h of experiment, respectively.
The incorporation of a hydrogen donor on the carbon matrix surface resulted in an increase in in situ H2O2 generation, which, in turn, contributed toward good PRM degradation with low energy consumption and high reaction kinetic velocity. Another point worth considering is that the greater availability of H2O2 led to an increase in the removal rate of PRM and a high percentage of mineralization as shown by TOC and 3D chromatogram results. Considering the results obtained in this study, one can conclude that the treatment technique proposed here, involving the modification of the catalytic matrix PL6C with 1.0% 1,4-naphthoquinone for the decontamination of water using a contaminant model molecule (PRM), promotes greater efficiency of the process with relatively lower costs.
Conclusions
The preliminary electrochemical study using an RRDE showed that the modification of the PL6C matrix with an organic compound (NQE) with potential H+ donation characteristics can increase the efficiency of H2O2 electrogeneration. The modification of the carbon matrix with 1.0% NQE resulted in higher current efficiency for the ORR with high selectivity for H2O2; this was subsequently confirmed by large scale analyses using GDE electrodes. In the quantitative analysis conducted, the modified GDE presented a 30% increase in the concentration of electrogenerated H2O2 compared to the unmodified GDE. In addition, the incorporation of an H+ donor molecule into the catalytic surface improved ΔG of the reaction from −277.11 kJ mol−1 for the bare PL6C matrix to −986.1 kJ mol−1 for the NQE-modified PL6C matrix; this led to an increase in the synthesis kinetics and a reduction in the energy consumption of the system. The study of PRM degradation by the PEF process presented the highest removal rate of PRM (∼100%), the highest mineralization percentage (42.1%), and the lowest energy consumption (6.35 kW h gTOC−1) in 180 min of experiment. An exhaustive assay conducted in the study showed a decline in HPLC peaks related to the by-products of degradation, where approximately 80% removal of TOC was obtained after 6 h of experiment. The novelty of this work lies in the fact that it merges theoretical concepts with practical application. The findings of the work show that the incorporation of an electroactive H+ donor species (NQE) onto the surface of the PL6C matrix can improve H2O2 electrogeneration by reducing ΔG of the reaction system. The hydrogen donor species employed in the study was found to enhance the removal of organic compounds present in water with low energy consumption, which thus improves the efficiency of the water treatment process.Experimental section
Reagents and materials
Printex L6 carbon (PL6C) acquired from Evonik do Brasil Ltda was used as a matrix for hydrogen peroxide generation and as a support medium for the 1,4-naphthoquinone (NQE) organic modifier (97% purity) obtained from Sigma-Aldrich. The solutions employed in conducting the experiments included the following: 97.8% sulfuric acid (J. T. Baker), 99.5% isopropyl alcohol (J. T. Baker), 99% potassium sulfate (Synth), iron sulfate heptahydrate (Sigma-Aldrich), 60% w/w poly(tetrafluoroethylene) (PTFE) dispersion (Uniflon), sodium bisulfite P. A. (Nuclear), methanol (UV-IR-HPLC) 99.9% (Panreac) and ultrapure water (Milli-Q, Millipore 18 MΩ cm).NQE modification theoretical study
Initially, calculations of the dipole moment of 1,4-naphthoquinone (isolated and constructed with the aid of the Avogadro 1.1.1 program57) were performed using DFT (Density Functional Theory)58,59 with two functionals, namely B3LYP and CAM-B3LYP,60–62 and Pople, Dunning and def2-TZVPD (Valence triple-zeta polarization with diffuse functions) basis sets, as shown in Table S2 (see the ESI†).63–65 The simulations were carried out considering benzene as a solvent and using the CPCM (Conductor-Like Polarizable Continuum Model) technique,66 while maintaining the experimental conditions.67 After choosing the method and basis set, the next simulations were performed to confirm the experimental data obtained from the ORR study for H2O2 electrogeneration using the amorphous carbon matrix as the ORR catalyst.The carbon structures with the –COOH group (used for simulating Printex 6L) were divided into structures 1 (O2 adsorbed on carbon with the hydronium ion), 2 (O2 adsorbed on carbon with hydroquinone) and 3 (O2 adsorbed on carbon with the hydronium ion in the presence of hydroquinone). The geometry optimization and frequency calculations were performed in Gaussian09, using DFT (CAM-B3LYP/6-311G (2d,2p) – the most suitable functional and basis set for the system obtained previously). The calculation details are displayed in Table S3 (ESI†). Thermochemical analysis was also performed in order to analyze the hydrogen bonds found in structures 1, 2 and 3 (aiming at evaluating the possible formation of H2O2). The value of the free energy (ΔG) is calculated based on eqn (14):68
 | (14) |
The reagents for structures 1, 2 and 3 are related to hydrogen bonds (O2 + hydronium ion for structure 1; O2 + hydroquinone for structure 2; and O2 + hydronium ion in the presence of hydroquinone for structure 3). For the reaction product related to structure 1, the hydronium ion is transformed into one H2O molecule, while with regard to structure 2, hydroquinone is converted to 1,4-naphthoquinone, and in the case of structure 3, the hydronium ion is converted into a water molecule (in the presence of hydroquinone). In addition to the aforementioned simulations, QTAIM (Quantum Theory of Atoms in Molecules)69 analysis of structures 1 and 2 was carried out, using the AIMALL program (Version 17.11.14, TK Gristmill Software: 2017), in order to verify the strength of the hydrogen bonds involved in these species.
Porous microlayer analysis on an RRDE
The organic modifier NQE was incorporated into the PL6C carbon matrix by the impregnation method. A mixture was produced containing 1 mg of the catalytic material (PL6C + NQE) in 20 mL of isopropyl alcohol. The mixture was subjected to mechanical stirring for 20 min; the material was subsequently oven dried at 100 °C for 3 h. Six samples were produced based on the following nominal percentages: 0.5, 1.0, 2.0, 3.0, 4.0 and 5.0% (w/w) of NQE in relation to PL6C. The catalysts were dispersed in ultrapure water, in a ratio of 1 mg of the catalytic material to 1 mL of water, and subjected to an ultrasonic bath (Soni-top 404A) for 20 min. The microlayer was prepared with a 25 μL droplet of the material, and this was deposited on the glassy carbon disc of a rotating ring-disc electrode (RRDE) system and dried under a constant N2 flow of 1.0 mL min−1. The fact that 25 μg of the material was applied on a 0.2475 cm2 disc meant that the microlayer had a loading of 101 μg cm−2.The redox reactions from the PL6C catalytic matrix surface and NQE organic modifier were evaluated by cyclic voltammetry (CV). The CV analysis was performed in acid medium of 0.1 mol L−1 K2SO4 (pH 2.0) in the potential range of 0.6 to −0.6 VAg/AgCl.
The ORR process was investigated in a hydrodynamic system in a three-compartment bench cell, with 0.1 mol L−1 K2SO4 O2-saturated electrolyte. The assays were performed using a potentiostat Metrohm Autolab PGSTAT-302N. The working electrode employed was a Pine 616 model rotating ring-disc electrode (RRDE) with a glassy carbon disc and platinum ring; Ag/AgCl (KCl 3 mol L−1) was used as the reference electrode and a ∼235 mm2 platinum plate as the counter electrode. Linear Sweep Voltammetry (LSV) analysis was performed in the potential range of 0.4 to −0.8 VAg/AgCl on the disc at a 5 mV s−1 scan rate. To monitor the detection of H2O2 on the ring, a constant potential of 1.0 V was applied. The frequency of the working electrode was kept at 900 rpm.
The hydrophobic character of the materials was evaluated based on the analysis of the contact angle using an Attension Theta Flex tensiometer. In this analysis, a 3.0 μL drop of ultrapure water was deposited on the surface of the material microlayers, and the contact angle between the drop and the surface was measured.
Analyses using a modified gas diffusion electrode (GDE)
The catalytic mass of the unmodified GDE was prepared through the application of the unmodified PL6C matrix and a hydrophobic agent. For the development of the catalytic mass of the modified GDE, the PL6C matrix was modified with a hydrophobic agent and 1.0% of the NQE modifier. PTFE was used as the hydrophobic agent, where 20% PTFE was employed relative to the total mass of the electrode. After drying, the electrode was subjected to sintering under high pressure and temperature so as to obtain a thin and solid electrode based on the sintering procedure reported in the literature.18,19,26 The electrode was used as a cathode in a 450 mL bench top cell and was allocated to a gas chamber to allow the flow of gas into the system.Hydrogen peroxide electrogeneration assays were performed by chronopotentiometry using different current densities (10, 25, 50, 75, 100 and 150 mA cm−2) in 0.1 mol L−1 K2SO4 (pH 2–H2SO4) under pressurized O2 gas of 0.2 bar, with temperature control of 20 °C in a thermostatic bath (obtained from Nova Ética). In the electrochemical cell, Ag/AgCl was used as a reference electrode, a 24 cm2 platinum wire as a counter electrode, and a GDE (modified or unmodified) as a working electrode (20 cm2). The electronic system PGSTAT-30 (Metrohm Autolab) potentiostat with booster module (BSTR-10A) was used as an electron source.
The quantification of hydrogen peroxide was performed by ex situ analysis of UV-vis spectroscopy as described in studies published in the literature.18,19,26
Paracetamol degradation analyses
For the study of paracetamol degradation, the following advanced oxidative processes were employed: anodic oxidation (AO), AO-H2O2, AO-H2O2/UVC coupling, electro-Fenton (EF) and photoelectron-Fenton (PEF) performed in a conventional one-compartment cell using 450 mL of solution (supporting electrolyte + standard paracetamol) with a controlled temperature of 20 °C. H2O2 electrogeneration was conducted by applying a current density of 75 mA cm−2 in K2SO4 0.1 mol L−1 (pH 2.0) with 80 mg L−1 initial paracetamol concentration under an O2 pressure of 0.2 bar. To carry out the EF and PEF processes, 0.15 mmol of Fe2+52 was used, and for the UVC radiation processes, the mercury lamp Pen-Ray model 11SC-2.12 with monochrome emission of 254 nm was employed. In all the samples employed in this investigation, sodium bisulfite was used to remove all residual H2O2 and to halt the drug degradation reaction.32,52
Degradation monitoring
The monitoring of the PRM concentration was performed by high performance liquid chromatography (HPLC) using a Shimadzu Prominence model LC-20 AT chromatograph coupled to an SPD-20A UV detector. To conduct the analysis, an analytical curve was constructed based on an external standard (Synth U.S.P. standard) using a concentration range of 0.5–100 ppm of PRM (Fig. S4, ESI†). A Phenomenex Luna C18 column (250 × 4.6 mm, 5 μm) and a Supelcosil C18 pre-column (4 × 3.0 mm i.d.) were used to separate paracetamol from its byproducts. Methanol/ultrapure water in a ratio of 40:60 (v/v) was used as the mobile phase and the drug detection was performed with the aid of a UV detector at 243 nm wavelength. The mineralization process of the organic compounds (paracetamol and its byproducts) was monitored based on the total organic carbon (TOC) of the final samples using a Shimadzu model TOC-VCPN analyzer.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors do acknowledge the financial support provided by the Brazilian research funding agencies, including the Brazilian National Council for Scientific and Technological Development – CNPq (grants no. 465571/2014-0, 301492/2013-1, 302874/2017-8 and 427452/2018-0), São Paulo Research Foundation (FAPESP – grants #2014/50945-4, #2015/14669-5, #2017/10118-0 and #2017/23464-3) and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES – Finance Code 001).References
- V. K. Sharma and M. Feng, J. Hazard. Mater., 2019, 372, 3–16 CrossRef CAS PubMed.
- L. H. M. L. M. Santos, A. N. Araújo, A. Fachini, A. Pena, C. Delerue-Matos and M. C. B. S. M. Montenegro, J. Hazard. Mater., 2010, 175, 45–95 CrossRef CAS PubMed.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O’Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS PubMed.
- M. B. Campanha, A. T. Awan, D. N. R. de Sousa, G. M. Grosseli, A. A. Mozeto and P. S. Fadini, Environ. Sci. Pollut. Res., 2015, 22, 7936–7947 CrossRef CAS PubMed.
- C. Gómez-Canela, V. Pueyo, C. Barata, S. Lacorte and R. M. Marcé, Sci. Total Environ, 2019, 666, 57–67 CrossRef PubMed.
- D. Vogna, R. Marotta, A. Napolitano and M. D’Ischia, J. Org. Chem., 2002, 67, 6143–6151 CrossRef CAS PubMed.
- A. Macías-García, J. García-Sanz-Calcedo, J. P. Carrasco-Amador and R. Segura-Cruz, Sustainability, 2019, 11, 1–11 CrossRef.
- A. Sokół, K. Borowska and J. Karpińska, Polish J. Environ. Stud., 2017, 26, 293–302 CrossRef.
- J. Wang, Z. Wang, C. L. Z. Vieira, J. M. Wolfson, G. Pingtian and S. Huang, Ultrason. Sonochem., 2019, 55, 273–278 CrossRef CAS PubMed.
- W. Zhao, I. W. Chen and F. Huang, Nano Today, 2019, 27, 11–27 CrossRef CAS.
- P. M. Gore, M. Naebe, X. Wang and B. Kandasubramanian, Chem. Eng. J., 2019, 374, 437–470 CrossRef CAS.
- D. Kanakaraju, B. D. Glass and M. Oelgemöller, J. Environ. Manage., 2018, 219, 189–207 CrossRef CAS PubMed.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- J. S. Souza, W. M. Carvalho, F. L. Souza, C. Ponce-De-Leon, D. V. Bavykin and W. A. Alves, J. Mater. Chem. A, 2016, 4, 944–952 RSC.
- A. S. Martins, P. J. M. Cordeiro-Junior, L. Nunez and M. R. de V. Lanza, Electrocatalysis, 2017, 8, 115–121 CrossRef CAS.
- A. A. S. Martins, L. Nunez and M. R. V. Lanza, J. Electroanal. Chem, 2017, 802, 33–39 CrossRef CAS.
- G. G. Bessegato, J. C. Cardoso, B. F. da Silva and M. V. B. Zanoni, Appl. Catal., B, 2016, 180, 161–168 CrossRef CAS.
- R. B. Valim, R. M. Reis, P. S. Castro, A. S. Lima, R. S. Rocha, M. Bertotti and M. R. V. Lanza, Carbon, 2013, 61, 236–244 CrossRef CAS.
- F. L. Silva, R. M. Reis, W. R. P. Barros, R. S. Rocha and M. R. V. Lanza, J. Electroanal. Chem., 2014, 722–723, 32–37 CrossRef CAS.
- A. S. Martins, V. M. Vasconcelos, T. C. R. Ferreira, E. R. Pereira-Filho and M. R. V. Lanza, J. Adv. Oxid. Technol., 2015, 18, 9–14 CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- I. Sirés, E. Brillas, M. A. Oturan, M. A. Rodrigo and M. Panizza, Environ. Sci. Pollut. Res., 2014, 21, 8336–8367 CrossRef PubMed.
- W. R. P. Barros, P. C. Franco, J. R. Steter, R. S. Rocha and M. R. V. Lanza, J. Electroanal. Chem., 2014, 722–723, 46–53 CrossRef CAS.
- P. S. Simas, V. S. Antonin, L. S. Parreira, P. Hammer, F. L. Silva, M. S. Kronka, R. B. Valim, M. R. V. Lanza and M. C. Santos, Electrocatalysis, 2017, 8, 311–320 CrossRef CAS.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- J. Moreira, V. Bocalon Lima, L. Athie Goulart and M. R. V. Lanza, Appl. Catal., B, 2019, 248, 95–107 CrossRef CAS.
- P. J. M. Cordeiro-Junior, R. Gonçalves, T. T. Guaraldo, R. da Silva Paiva, E. C. Pereira and M. R. de V. Lanza, Carbon, 2020, 156, 1–9 CrossRef CAS.
- E. C. Paz, L. R. Aveiro, V. S. Pinheiro, F. M. Souza, V. B. Lima, F. L. Silva, P. Hammer, M. R. V. Lanza and M. C. Santos, Appl. Catal., B, 2018, 232, 436–445 CrossRef CAS.
- M. H. M. T. Assumpção, R. F. B. De Souza, D. C. Rascio, J. C. M. Silva, M. L. Calegaro, I. Gaubeur, T. R. L. C. Paixão, P. Hammer, M. R. V. Lanza and M. C. Santos, Carbon, 2011, 49, 2842–2851 CrossRef.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- W. R. P. Barros, M. P. Borges, R. M. Reis, R. S. Rocha, R. Bertazzoli and M. R. V. Lanza, J. Braz. Chem. Soc., 2014, 25, 1673–1680 CAS.
- W. Zhou, J. Gao, Y. Ding, H. Zhao, X. Meng, Y. Wang, K. Kou, Y. Xu, S. Wu and Y. Qin, Chem. Eng. J., 2018, 338, 709–718 CrossRef CAS PubMed.
- W. Zhou, X. Meng, J. Gao and A. N. Alshawabkeh, Chemosphere, 2019, 225, 588–607 CrossRef CAS PubMed.
- J. F. Carneiro, M. J. Paulo, M. Siaj, A. C. Tavares and M. R. V. Lanza, J. Catal., 2015, 332, 51–61 CrossRef CAS.
- J. F. Carneiro, L. C. Trevelin, A. S. Lima, G. N. Meloni, M. Bertotti, P. Hammer, R. Bertazzoli and M. R. V. Lanza, Electrocatalysis, 2017, 8, 189–195 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619–621 CrossRef CAS PubMed.
- E. Pizzutilo, O. Kasian, C. H. Choi, S. Cherevko, G. J. Hutchings, K. J. J. Mayrhofer and S. J. Freakley, Chem. Phys. Lett., 2017, 683, 436–442 CrossRef CAS.
- R. S. Rocha, R. B. Valim, L. C. Trevelin, J. R. Steter, J. F. Carneiro, J. C. Forti, R. Bertazzoli and M. R. V. Lanza, Electrocatalysis, 2020, 11, 338–346 CrossRef CAS.
- K. Tammeveski, K. Kontturi, R. J. Nichols, R. J. Potter and D. J. Schiffrin, J. Electroanal. Chem., 2001, 515, 101–112 CrossRef CAS.
- A. Sarapuu, K. Vaik, D. J. Schiffrin and K. Tammeveski, J. Electroanal. Chem., 2003, 541, 23–29 CrossRef CAS.
- P. S. Guin, S. Das and P. C. Mandal, Int. J. Electrochem., 2011, 2011, 1–22 CrossRef.
- S. M. Golabi, J. B. Raoof, B. Keita, L. Nadjo, M. C. Pham, J. E. Dubois, L. Roullier, E. Waldner and E. Laviron, J. Electroanal. Chem., 1986, 145, 153–164 Search PubMed.
- B. Keita and L. Nadjo, J. Electroanal. Chem., 1983, 145, 431–437 CrossRef CAS.
- A. S. Kumar and P. Swetha, Colloids Surf., A, 2011, 384, 597–604 CrossRef CAS.
- J. C. Forti, R. S. Rocha, M. R. V. Lanza and R. Bertazzoli, J. Electroanal. Chem., 2007, 601, 63–67 CrossRef CAS.
- A. Huissoud and P. Tissot, J. Appl. Electrochem., 1998, 28, 653–657 CrossRef CAS.
- J. Quílez-Bermejo, M. Melle-Franco, E. San-Fabián, E. Morallón and D. Cazorla-Amorós, J. Mater. Chem. A, 2019, 7, 24239–24250 RSC.
- A. J. Bard and Larry R. Faulkner, Electrochemical Methods Fundamentals and Applications, 1985, vol. 126 Search PubMed.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, Res. Gate, 2014, 495, 134–145 Search PubMed.
- W. R. P. Barros, M. P. Borges, J. R. Steter, J. C. Forti, R. S. Rocha and M. R. V. Lanza, J. Electrochem. Soc., 2014, 161, H867–H873 CrossRef CAS.
- J. C. Forti, J. A. Nunes, M. R. V. Lanza and R. Bertazzoli, J. Appl. Electrochem., 2007, 37, 527–532 CrossRef CAS.
- R. M. Reis, A. A. G. F. Beati, R. S. Rocha, M. H. M. T. Assumpção, M. C. Santos, R. Bertazzoli and M. R. V. Lanza, Ind. Eng. Chem. Res., 2012, 51, 649–654 CrossRef.
- W. R. P. Barros, R. M. Reis, R. S. Rocha and M. R. V. Lanza, Electrochim. Acta, 2013, 104, 12–18 CrossRef CAS.
- A. Xu, E. Brillas, W. Han, L. Wang and I. Sirés, Appl. Catal., B, 2019, 259, 118127 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4(1) DOI:10.1186/1758-2946-4-17.
- W. Hohenberg and P. Kohn, Phys. Rev., 1964, 136, 864–871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- W. Chengteh Lee and R. G. P. Yang, Eval. Eng., 2011, 50, 36–39 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133 DOI:10.1063/1.3484283.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- C. C. Meredith, L. Westland and G. F. Wright, J. Am. Chem. Soc., 1957, 79, 2385–2390 CrossRef CAS.
- J. W. Ochterski and D. Ph, Gaussian Inc., Pittsburgh, PA, 2000, 264, 1–19.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00290a |
| This journal is © The Royal Society of Chemistry 2020 |