
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed ultrafine Ni particles embedded into MOF-74 arrays by partial carbonization for highly efficient hydrogen evolution†
Jing-Bo
Tan
 ,
Jing-Qi
Wu
,
Jia-Wei
Zhao
,
Ling-Jie
Xie
and
Gao-Ren
Li
*
,
Jing-Qi
Wu
,
Jia-Wei
Zhao
,
Ling-Jie
Xie
and
Gao-Ren
Li
*
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: ligaoren@mail.sysu.edu.cn
First published on 23rd June 2020
Abstract
Here, we report the successful fabrication of highly dispersed ultrafine Ni nanoparticles (NPs) embraced into the partially carbonized Ni-MOF-74 (Ni NPs/C@Ni-MOF-74) microrod arrays by the heat treatment of the rod-like Ni-MOF-74 arrays grown on Ni foam. The partial carbonization is an effective strategy to realize the homogeneous dispersion of ultrafine Ni NPs and carbon in the skeleton of rod-like Ni-MOF-74. This can prevent the encapsulated Ni NPs and carbon from aggregation and oxidation, allowing them to serve as highly efficient electrocatalysts. The fabricated Ni NPs/C@Ni-MOF-74 not only provide high specific surface area and high conductivity, but also provide good hydrophilicity and synergetic effects among Ni NPs, carbon and Ni-MOF-74 for the adsorption and activation of H2O molecules. The HER electrocatalytic performance of Ni NPs/C@Ni-MOF-74 arrays can be optimized by the control of the carbonization time and temperature. The optimized Ni NPs/C@Ni-MOF-74/NF shows outstanding HER catalytic performance, with a low overpotential of 131 mV at 10 mA cm−2 and excellent durability, and it is superior to most of the recently reported MOF-based HER electrocatalysts.
Introduction
Owing to the fierce consumption of fossil fuels and increasing serious environmental problems, electrocatalytic water splitting to produce H2 represents an attractive way to store renewable spasmodic energy and supply sustainable clean energy.1,2 To date, the noble metal Pt is still regarded as the state-of-the-art electrocatalyst for the hydrogen evolution reaction (HER).3–5 However, the scarcity and high-cost of Pt lead to the impossibility of large-scale industrial applications. Up to now, the search for non-precious electrocatalysts with high performance for realizing highly efficient HER is still a challenging issue.6–13Numerous studies have demonstrated that a large number of HER catalysts, such as metal–organic frameworks (MOFs),14–17 transition-metal carbides,18–20 chalcogenides,21–25 phosphides and nitrides,26–30 are explored as highly efficient electrocatalysts in basic electrolytes. Especially in recent years, MOFs have attracted more and more interest. As is well known, MOFs are self-assembled by combinations of metal secondary building units and organic ligands, possessing a special porous structure. Benefitting from the unique porous structure of MOFs and highly exposed, homogenously dispersed catalytic active sites inside the frameworks, they can serve as very promising catalysts in the catalytic field, especially for HER.15,31–34 However, the instability in strong acidic/alkaline media and instinctive insulation of MOFs have hindered their development for industrial application of HER.
Recently, various MOF-derived carbon/metal hybrid materials have been widely studied as electrocatalysts for HER.20,24,27,30 However, they have the following drawbacks: (i) the decomposition of MOFs will lead to a decrease in the surface area and diminish the exposure of accessible catalytic active sites; (ii) after carbonization, the hydrophilicity of carbon/metal hybrid materials will become poor compared with MOFs; (iii) after carbonization, the aggregation of catalytic components will hamper the exposure of active sites. To address the above problems, the partial carbonization of MOFs will be an effective strategy to functionalize MOFs with distinctive catalytic components and make them inherit the merits of both MOFs and their derivatives. That is to say, the partial carbonization allows MOFs to controllably decompose to generate metal nanoparticles and limited carbon matrices inside MOFs, instead of only metal-carbon or metal oxides. This perfectly retains the porosity and hydrophilicity of the designed MOFs, and also produces well-distributed nanoscale highly catalytic metal species to promote HER. This self-sacrificial temple method differs from the reported metal@MOFs materials taking advantage of the confined porosity of MOFs via NaBH4 reduction. Moreover, this strategy can contribute to the formation of synergetic effect metal particles and MOFs, which is a crucial factor for highly effective water splitting.
MOF-74 families have drawn much attention in catalysis,35–39 and they show the following impressive advantages: (i) Honeycomb-like channels with a porosity diameter of ∼11 Å, which is accessible for many small molecules, such as H2O, H2, O2, CO2, and NO; (ii) the unsaturated coordination metal units will properly interact with molecules (reactants) by covalent bonding, and also act as catalytic active sites for many catalytic reactions; (iii) metal ions for MOF-74 are diverse and selective, which will easily regulate the MOF catalytic activity. As previously reported, MOF-74 can serve as the catalyst for water disassociation under high temperature with the assistance of a hydroxyl group from the ligand itself.40,41 However, MOF-74 is rarely exploited as an electrocatalyst for HER, except as the precursors to fabricate MOF derivatives. In addition, Ni is regarded as a promising HER electrocatalyst with many merits, such as low-cost, high catalytic activity, and appropriate hydrogen adsorption/desorption energies.8,42 Here, we report the highly dispersed ultrafine Ni nanoparticles (NPs) embraced into partially carbonized Ni-MOF-74 (Ni NPs/C@Ni-MOF-74) microrod arrays were successfully fabricated by the partial carbonization of rod-like Ni-MOF-74 arrays grown on Ni foam (NF), and they can be used as highly effective HER electrocatalysts in alkaline media. Compared with the Ni-MOF-74 arrays, the abundant electroactive sites of Ni NPs, the synergetic effects between Ni NPs and Ni-MOF-74, and the increased conductivity originating from carbon matrices due to the partial carbonization will largely contribute to the remarkable HER performance of the Ni NPs/C@Ni-MOF-74 microrod arrays as shown in Scheme 1.
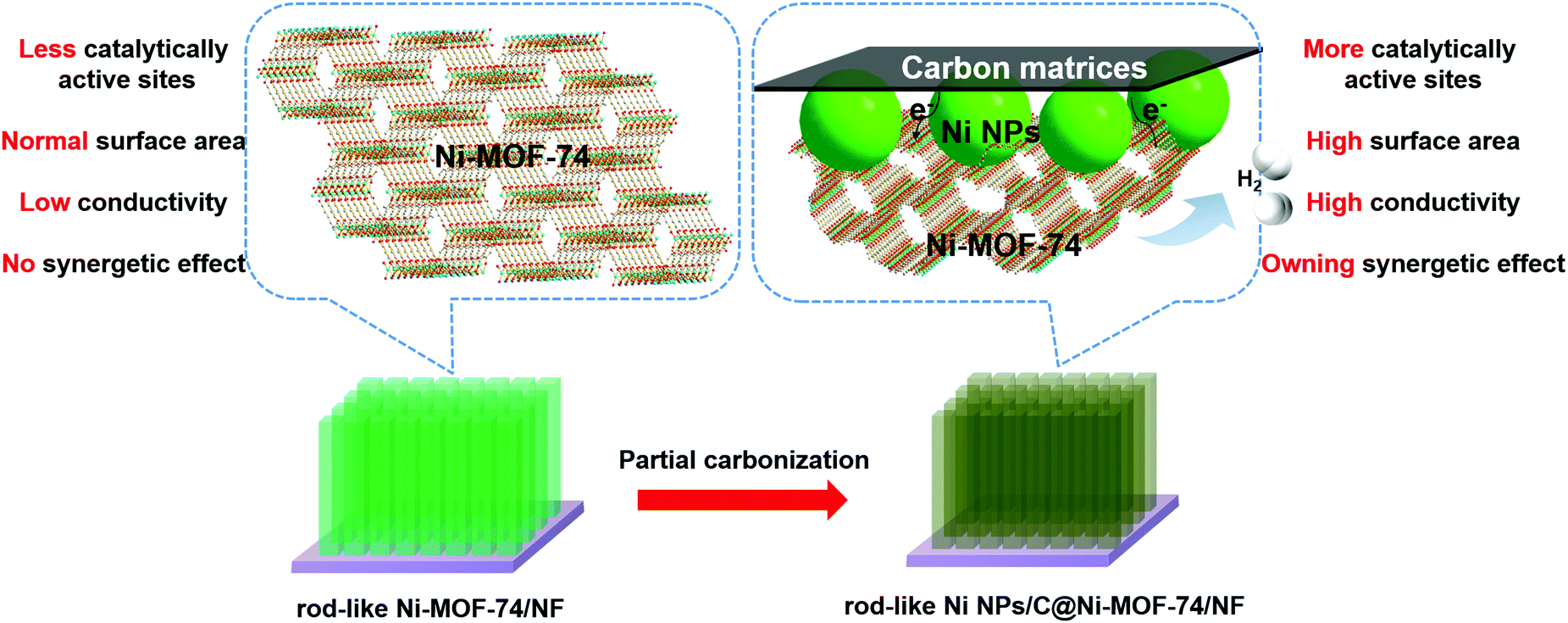 | ||
| Scheme 1 The advantages of Ni NPs/C@Ni-MOF-74/NF obtained from Ni-MOF-74/NF. (Purple substrate: Ni Foam; light green arrays: Ni-MOF-74; dark green arrays: Ni NPs/C@Ni-MOF-74.) | ||
Experimental
Synthesis of materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH:H2O = 1:1:1. After sonication for 30 min, the solution was transferred into a 20 mL stainless-steel autoclave and kept at 120 °C for 48 h. The resulting yellow precipitate was centrifuged and washed thoroughly with DI water and ethanol several times, respectively, and then dried in the oven at 70 °C.
:EtOH:H2O = 1:1:1. After sonication for 30 min, the solution was transferred into a 20 mL stainless-steel autoclave and kept at 120 °C for 48 h. The resulting yellow precipitate was centrifuged and washed thoroughly with DI water and ethanol several times, respectively, and then dried in the oven at 70 °C.
Materials and characterizations
All chemicals were purchased from commercial suppliers and used without further purification (Aladdin Co., Ltd). Raman spectra were characterized by a Laser Micro-Raman Spectrometer Renishaw inVia with 514 nm light. Powder X-ray powder diffraction (PXRD) measurements were performed with a Bruker D8 X-ray diffractometer with Cu Kα radiation at a scanning rate of 1° min−1 with powders collected from the as-prepared catalyst grown onto NF. Scanning electron microscopy (SEM) was performed with a Hitachi SU-8010 scanning electron micro-analyzer with an accelerating voltage of 10 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were conducted using FEI Tecnai G2 F30 field-emission TEM and JEM-ARM200P. All samples were prepared by dropping catalysts uniformly dispersed in ethanol solution onto the carbon-coated copper grids. X-ray photoelectron spectroscopy (XPS) was conducted by an ESCAlab250. Thermal gravity analysis (TGA) was performed by a NETZSCH TG 209 F3 Tarsus under air atmosphere. Brunauer–Emmett–Teller (BET) surface areas were obtained though N2 adsorption–desorption isotherms at 77 K on an ASAP 2460.Electrochemical measurements
The electrocatalytic activity of the samples for HER was studied in 1.0 M KOH (pH = 13.8) solution, and recorded on a CHI760e electrochemical instrument (Shanghai, Changhua Co. Ltd) at ambient temperature. All tests were performed in a three-electrode electrochemical cell with a saturated calomel reference electrode and a carbon rod as the counter electrode. The as-prepared electrodes were used as working electrodes to investigate their electrocatalytic activities toward HER. It was cycled at least 30 times at a scan rate of 50 mV s−1 between −0.9 and −1.4 V vs. Hg/HgCl before data collection. The potential versus saturated calomel electrode was calibrated with respect to the reversible hydrogen electrode (RHE) using the following equation: E(RHE) = E(Hg/HgCl) + 0.242 + pH × 0.0592. The linear sweep voltammogram (LSV) curves were conducted at a scan rate of 5 mV s−1 for HER in 1.0 M KOH solution and shown without iR compensation. Electrochemical impedance spectroscopy (EIS) was performed in potentiostatic mode from 1000 kHz to 10 mHz. To evaluate the effective electrochemical surface areas (ECSAs), CVs were carried out to probe the electrochemical double layer capacitance (Cdl) of all as-prepared samples at the non-faradaic region, and identified from CVs in quiescent solution. By plotting the capacitive current density Δj at 0.58 V vs. RHE against the scan rates, a linear trend was observed. The linear slope, equivalent to twice the double-layer capacitance Cdl, was widely used to represent the ECSA.Results and discussion
The synthetic scheme of Ni NPs/C@Ni-MOF-74/NF is illustrated in Scheme S1 (ESI†). In a typical solvothermal synthesis, nickel chlorides and 2-aminoterephthalic acid with a chemical stoichiometry of 1:2 were dissolved into a mixed solvent with a piece of cleaned bare NF, and were heated up to 120 °C to fabricate rod-like Ni-MOF-74 on NF (here, it is denoted as Ni-MOF-74/NF). Then, all solvent molecules were eliminated thoroughly from the honeycomb-like 1D channel of Ni-MOF-74/NF under vacuum at 150 °C. Finally, the partial carbonization of Ni-MOF-74/NF was carried out under vacuum at 300 °C for 18 h, and the Ni NPs/C@Ni-MOF-74/NF were successfully fabricated. Here, the vacuum environment plays a crucial role in the generation of Ni NPs and carbon matrices, instead of NiO and CO2 since no oxygen exists during the carbonization. In order to achieve the ideal partial carbonization to preserve the MOF frames, a temperature of 300 °C and 18 h are selected in the synthesis procedures.
Powder X-ray diffraction (PXRD) was conducted to characterize the crystalline phase and purity of the as-prepared materials. As shown in Fig. 1, the diffraction peaks of the prepared Ni-MOF-74 matched well with those of the reported MOF-74,24,36 demonstrating that Ni-MOF-74 were successfully synthesized without any impurities. After the partial carbonization of Ni-MOF-74/NF under vacuum at 300 °C with various remaining times (6–24 h), the different Ni NPs/C@Ni-MOF-74/NF samples were obtained, and their PXRD are also shown in Fig. 1. Only the diffraction peaks of MOF-74 are seen and no other peak is found, indicating that the obtained carbon matrices after the partial carbonization are amorphous and the sizes of the generated Ni NPs are too small to reflect their distinct diffraction peaks.
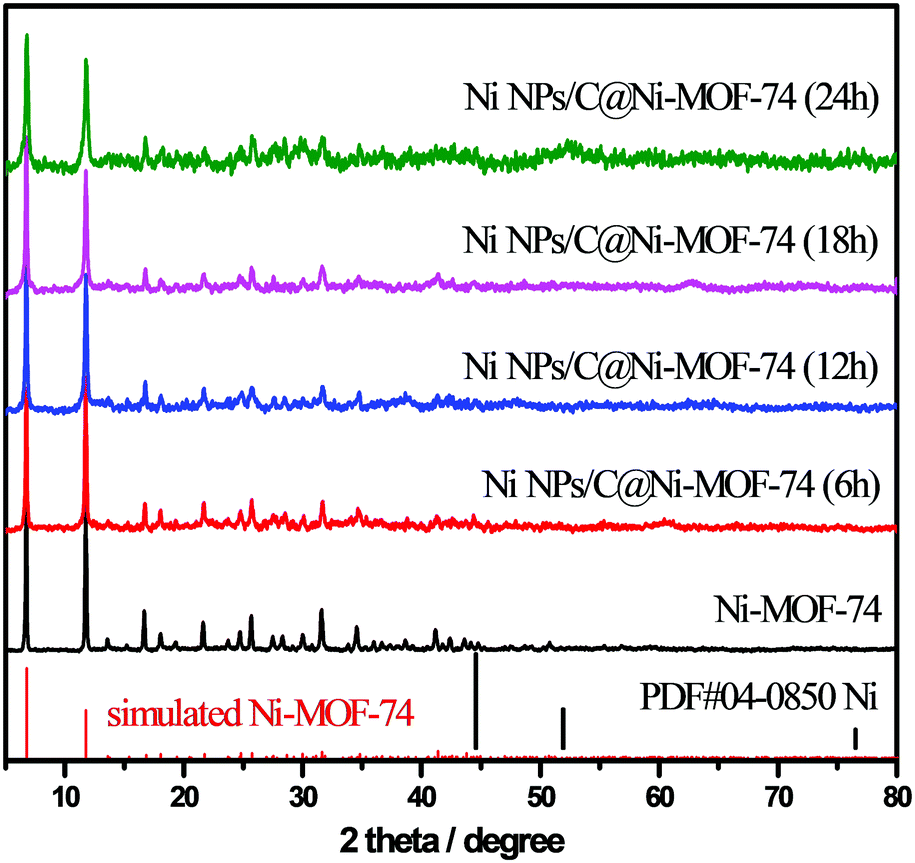 | ||
| Fig. 1 PXRD patterns of Ni-MOF-74 and various Ni NPs/C@Ni-MOF-74 samples, and the simulated XRD patterns of Ni-MOF-74 (red bar) and metal Ni (black bar). | ||
The SEM image in Fig. S1a (ESI†) shows the rod-like Ni-MOF-74 arrays grown on NF with a length of ∼6 μm. In contrast, the branched microrods of Ni-MOF-74 were obtained without the NF substrate, as shown in Fig. S2 (ESI†). To investigate the surface morphologies of Ni NPs/C@Ni-MOF-74/NF, SEM images of various samples with different carbonization times are shown in Fig. S1b–e (ESI†). Intriguingly, after a partial carbonization at 300 °C with remaining time of 6–24 h, the obtained Ni NPs/C@Ni-MOF-74/NF can maintain rod arrays well. TEM and HRTEM images of the Ni NPs/C@Ni-MOF-74/NF(18 h) samples are measured and they show that the ultrafine Ni NPs (∼5 nm) were formed and dispersed homogenously in the Ni-MOF-74 frameworks, as shown in Fig. 2a–c. The distinct lattice spacing of Ni NPs in the sample is about 0.206 nm, as shown in Fig. 2b, and it can be indexed to the (1 1 1) plane of Ni (PDF #04-0850),43 while that of ∼0.212 nm can be attributed to the (−11 1 0) plane of Ni-MOF-74, as shown in Fig. S3b (ESI†).44 The STEM image also illustrates that the generated Ni NPs are uniformly distributed inside Ni-MOF-74, instead of only on the surface of the frameworks, as shown in Fig. 2c. The HAADF-STEM image and elemental mapping images in Fig. 2d–g reveal that the elements of Ni, C, and O are homogenously distributed all over the Ni NPs/C@Ni-MOF-74/NF(18 h), similar to those of Ni-MOF-74, as shown in Fig. S3d–g (ESI†). As we know, the Ni NPs can serve as the electrocatalytic active sites for HER with competent hydrogen adsorption/desorption abilities,8 which will obviously enhance the HER catalytic performance of Ni-MOF-74.
 | ||
| Fig. 2 (a) TEM image; (b) HRTEM image and SAED pattern (inset); (c) STEM image of Ni NPs/C@Ni-MOF-74/NF(18 h); (d) HAADF-STEM image; (e–g) elemental mapping images of Ni NPs/C@Ni-MOF-74/NF(18 h), revealing the homogeneous distributions of C, O, and Ni elements. | ||
Raman spectra of the various Ni NPs/C@Ni-MOF-74/NF were measured to analyse the vibration modes of Ni-MOF-74 to confirm the stability of the MOF skeletons in the sample, as shown in Fig. S4 (ESI†).45 The peak at 576 cm−1 corresponds to the Ni–O stretching mode (ν(Ni–O)) of Ni-MOF-74, and the peaks at 1416 and 1554 cm−1 correspond to the symmetric and asymmetric stretching modes of the carboxylate groups (νas,s(COO−)), respectively. The peak at 820 cm−1 is assigned to the C–H bending of the benzene ring, and the peak at 1489 cm−1 corresponds to the stretching modes of the phenolate groups (ν(C–O)). The stretching modes of the aromatic rings are located at 1275 and 1632 cm−1. Compared with the pristine Ni-MOF-74, it was evidently found from the Raman spectra of all Ni NPs/C@Ni-MOF-74/NF(6–24 h) samples that the skeleton of Ni-MOF-74 can be well maintained after partial carbonization.
X-ray photoelectron spectroscopy (XPS) spectrum was measured to elucidate the elemental states of Ni NPs/C@Ni-MOF-74(18 h). The XPS survey scans spectrum in Fig. 3a shows the presence of Ni, C and O in Ni NPs/C@Ni-MOF-74(18 h). As shown in Fig. 3b, the appearances of the peaks of Ni 2p3/2 (856.11 eV) and Ni 2p1/2 (873.78 eV), and their corresponding satellite peaks (861.04 eV, 879.38 eV), demonstrate the existence of Ni2+ with a high-spin state in Ni-MOF-74.46 Intriguingly, the peak of Ni 2p3/2 at 852.45 eV obviously appears after partial carbonization, and it can be ascribed to metallic Ni,47,48 strongly demonstrating the existence of the metal Ni NPs inside the frameworks, which is consistent with the HRTEM result. In addition, to confirm the existence of carbon matrices in the frameworks, the Ni NPs/C@Ni-MOF-74(18 h) powders were treated with 0.1 M HCl solution. It was found that the carbon matrices were precipitated in the solution as residues after centrifugation, as shown in Fig. S5 (ESI†). In addition, Ni-MOF-74 was decomposed and Ni NPs was dissolved by HCl solution, indicating that the carbon matrices were generated after partial carbonization. XPS survey spectra of Ni NPs/C@Ni-MOF-74(18 h) before and after acid treatment were measured, as shown in Fig. S6 (ESI†), which demonstrates the existence of carbon matrices after the partial carbonization. The carbon matrices in the electrocatalysts not only can enhance the electrical conductivity and lower the instinctive insulating of MOF-74, but also can protect the metallic Ni catalytic sites and stabilize the structure of Ni-MOF-74 during the HER process.
 | ||
| Fig. 3 (a) XPS survey spectrum of Ni NPs/C@Ni-MOF-74(18 h); (b) XPS spectra of Ni 2p of Ni-MOF-74 and Ni NPs/C@Ni-MOF-74(18 h). | ||
N2 absorption isotherms were measured at 77 K and 1 atm to calculate the specific surface area and pore size of the catalysts. As shown in Fig. S7 (ESI†), the Ni NPs/C@Ni-MOF-74(18 h) possesses a higher Brunauer–Emmett–Teller surface area of 1263.13 m2 g−1 than those of Ni-MOF-74 (660.35 m2 g−1), Ni NPs/C@Ni-MOF-74(6 h) (1157.46 m2 g−1), Ni NPs/C@Ni-MOF-74 (12 h) (1233.34 m2 g−1) and Ni NPs/C@Ni-MOF-74 (24 h) (1157.46 m2 g−1). The pore size distributions obtained from their corresponding adsorption isotherms are shown in Fig. S8 (ESI†), which revealed that the pore sizes mainly ranged from 0.8–1.1 nm. In addition, a small part of the mesoporous porosity ranged from 1.68 to 2.39 nm, which is attributed to their microporous nature and mesopores through the partial decomposition of Ni-MOF-74. The highly specific surface area and increased porous volume of Ni NPs/C@Ni-MOF-74(18 h) are very accessible for the electrolyte, and will facilitate the interactions between the Ni active sites and water molecules for HER in alkaline media.
The HER electrocatalytic performances of various Ni NPs/C@Ni-MOF-74/NF catalysts were studied in an aqueous solution of 1.0 M KOH by a three-electrode electrolytic cell, where those catalysts are utilized as working electrodes. As shown in Fig. 4a, the Ni NPs/C@Ni-MOF-74/NF(18 h) affords a low overpotential of only ∼131 mV at the current density of 10 mA cm−2, and it is superior to Ni-MOF-74/NF and other MOF-74-based HER electrocatalysts. The mass ratio of ultrafine Ni NPs in Ni NPs/C@Ni-MOF-74(18 h) was investigated by TGA analysis under air atmosphere, and it is approximately 13.55 wt% as shown in Fig. S9 (ESI†) (the detailed calculations can be seen in the ESI†). As control experiments, the samples adhered on NF or grown on NF were evaluated by LSVs in Fig. S10 (ESI†). The Ni NPs/C@Ni-MOF-74 grown on NF (namely Ni NPs/C@Ni-MOF-74/NF) exhibited much higher catalytic activity than those which were adhered on NF (namely Ni NPs/C@Ni-MOF-74 + NF), indicating that the Ni NPs/C@Ni-MOF-74(18 h) grown on NF can enhance the ability of electron transport between the sample and NF. As shown in Fig. 4a, the Ni NPs/C@Ni-MOF-74/NF(18 h) shows much higher catalytic activity than Ni-MOF-74/NF. This indicates that the partial carbonization can provide much more accessible Ni active sites and carbon matrices, which can obviously increase the conductivity of electrocatalysts for HER. In addition, Fig. 4a shows the HER catalytic activities of various Ni NPs/C@Ni-MOF-74/NF with different carbonization times (6–24 h), and their overpotentials at 10 mA cm−2 are arranged in the following order: η (Ni NPs/C@Ni-MOF-74/NF(18 h)) (131 mV) < η (Ni NPs/C@Ni-MOF-74/NF(24 h)) (143 mV) < η (Ni NPs/C@Ni-MOF-74/NF(12 h)) (165 mV) < η (Ni NPs/C@Ni-MOF-74/NF(6 h)) (183 mV) < η (Ni-MOF-74/NF) (269 mV). Furthermore, the onset overpotential of Ni NPs/C@Ni-MOF-74/NF(18 h) is only 39 mV, which is the lowest onset over-potential in all catalysts (129 mV of Ni-MOF-74/NF, 58 mV of Ni NPs/C@Ni-MOF-74/NF(6 h), 56 mV of Ni NPs/C@Ni-MOF-74/NF(12 h), and 43 mV of Ni NPs/C@Ni-MOF-74/NF(24 h)).
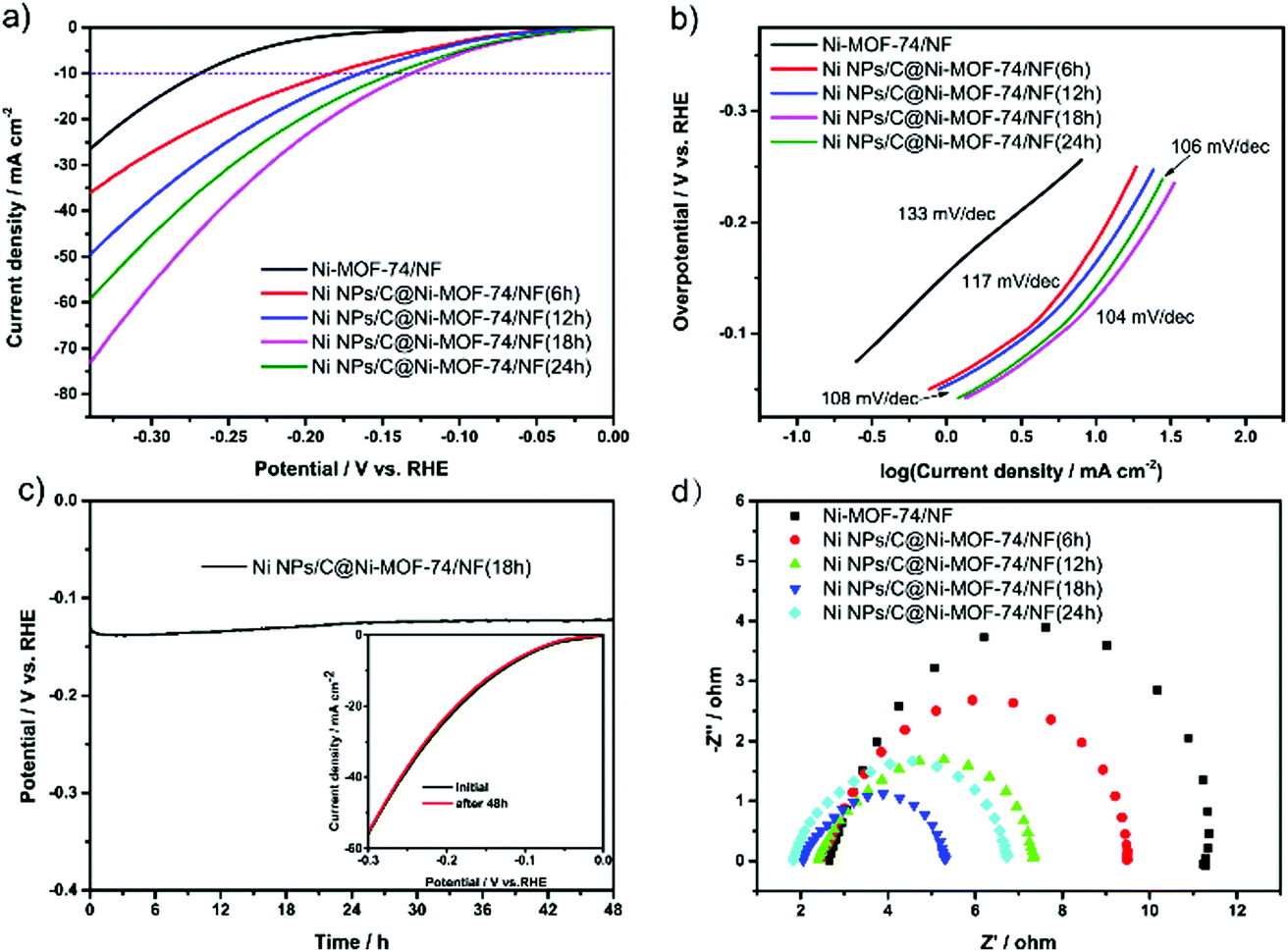 | ||
| Fig. 4 (a) LSVs of Ni-MOF-74/NF and various Ni NPs/C@Ni-MOF-74/NF measured in 1.0 M KOH at 5.0 mV s−1 without iR compensation; (b) Tafel plots obtained from (a); (c) chronopotentiometric curves of Ni NPs/C @Ni-MOF-74/NF(18 h) at a current density of 10 mA cm−2; the inserted image illustrates its LSV curves before and after 48 h; (d) Nyquist plots of various catalysts in 1.0 M KOH solution. | ||
The above results demonstrate that the Ni NPs/C@Ni-MOF-74/NF(18 h) exhibits a remarkable HER electrocatalytic activity in the alkaline solution. The Tafel slopes of various electrocatalysts were calculated to analyse the kinetic activities during the HER process, as shown in Fig. 4b, which shows the Tafel slopes of Ni-MOF-74/NF, Ni NPs/C@Ni-MOF-74/NF(6 h), Ni NPs/C@Ni-MOF-74/NF(12 h), Ni NPs/C@Ni-MOF-74/NF(18 h) and Ni NPs/C@Ni-MOF-74/NF(24 h) are 133, 117, 108, 104 and 106 mV dec−1, respectively. Intriguingly, the Tafel slope of Ni@Ni-MOF-74/NF(18 h) is near the theoretical Tafel value (116 mV dec−1) associated with the Volmer rate-determining step, indicating that the Ni NPs/C@Ni-MOF-74/NF(18 h) undergoes the Volmer–Heyrovsky mechanism for HER in alkaline media.49 The Tafel slopes of these catalysts after partial carbonization are lower than that of Ni-MOF-74/NF, and this reveals a more efficient charge transfer between the catalyst and electrolyte during the HER process. Accordingly, the partial carbonization method represents a powerful strategy to enhance the HER electrocatalytic activity of MOF-based catalysts.
The electrocatalytic stabilities of various electrocatalysts were investigated by chronopotentiometry over a continuous 12 h at 10 mA cm−2 for HER (Fig. 4c and Fig. S12 (ESI†)). As shown in Fig. 4c, it shows that the overpotential of Ni NPs/C@Ni-MOF-74/NF(18 h) increased slightly over −17 mV after a chronopotentiometric test of 48 h, indicating that it possesses excellent long-term durability. The LSV curve after 48 h electrolysis also shows that the current density of Ni NPs/C@Ni-MOF-74/NF(18 h) was nearly retained, as shown in the inset in Fig. 4c, which is consistent with the chronopotentiometry curve. The SEM images and PXRD pattern also demonstrate that the catalysts after the partial carbonization can resist the corrosion of the alkaline electrolyte after 12 h of electrocatalysis (seen in Fig. S13 and S14 (ESI†)). However, the Ni/carbon hybrids obtained by full carbonization of Ni-MOF-74 under vacuum at 400 °C will lead to severe structural collapse.
Compared with the results reported in the literature, Ni NPs/C@Ni-MOF-74/NF(18 h) exhibits the remarkable electro-catalytic performance for HER in alkaline solution among the reported MOF-based electrocatalysts (please see Table S1 (ESI†)). The catalytic performance improvement of Ni NPs/C@Ni-MOF-74/NF(18 h) can be attributed to the following factors: (i) Ni-MOF-74 grown in situ on the conductive substrate NF with excellent adhesion can accelerate the electron transport; (ii) the partial carbonization makes the generation of Ni NPs and carbon matrices without damaging the frame of Ni-MOF-74, and the Ni NPs and carbon matrices will serve as highly catalytic active sites and enhance the conductivity of MOF-based hybrids, respectively; (iii) the synergetic effects between Ni NPs and Ni-MOF-74 will obviously promote HER.
To elucidate the enhancement of the electrocatalytic performance, a series of electrochemical characterizations were conducted on Ni-MOF-74/NF and Ni NPs/C@Ni-MOF-74/NF(18 h). Electrochemical impedance spectra (EIS) measurements were first carried out, and the Nyquist plots revealed that the Ni NPs/C@Ni-MOF-74/NF(18 h) possesses a much smaller charge transfer resistance (Rst) (3.69 Ω) than Ni-MOF-74 (6.98 Ω) and other MOF-based electrocatalysts, as shown in Fig. 4d and Table S2 (ESI†). This can be attributed to the generated carbon matrices endowed with fast electronic transmission capability to Ni NPs/C@Ni-MOF-74/NF(18 h) that will serve as highly conductive MOF-based electrocatalysts for HER. The electro-chemical surface areas (ECSAs) were further evaluated by electrochemical double-layer capacitance (Cdl) measurements of the electrocatalysts, which has been regarded as an efficient method to estimate the number of catalytically active sites of electrocatalysts. As shown in Fig. S15 (ESI†), the CdI value of Ni NPs/C@Ni-MOF-74/NF(18 h) is 5.82 mF cm−2, which is about 20 times higher than that of Ni-MOF-74/NF (0.29 mF cm−2). Moreover, the CdI value of Ni NPs/C@Ni-MOF-74/NF(18 h) is much higher than those of other counterparts, such as 3.61 mF cm−2 of Ni NPs/C@Ni-MOF-74/NF(6 h), 3.62 mF cm−2 of Ni NPs/C@Ni-MOF-74/NF(12 h), and 2.62 mF cm−2 of Ni NPs/C@Ni-MOF-74/NF(24 h). The above results demonstrated that more exposed catalytically active sites were acquired in Ni NPs/C@Ni-MOF-74/NF(18 h) by optimal partial carbonization.
Here, the existence of Ni-MOF-74 in the Ni NPs/C@Ni-MOF-74/NF(18 h) is important for the catalytic performance enhancement, and its positive effects for HER in alkaline media are illustrated as follows. According to the above analyses of the Tafel slope and previous HER studies,49,50 here HER on Ni NPs/C@Ni-MOF-74/NF(18 h) is considered to be the Volmer–Heyrovsky mechanism. As shown in Fig. 5, the HER catalytic process follows three steps: (i) water molecules are first absorbed into 1D channels of Ni-MOF-74, and they will favourably coordinate with the unsaturated coordination centres (Ni2+) of Ni-MOF-74;36,40 (ii) these coordinated water molecules inside the channels of Ni-MOF-74 will be promptly activated, and they will be split easily under the electrocatalysis of Ni NPs to produce the adsorbed H atoms onto the surfaces of Ni NPs (Ni + H2O + e− → Ni–Hads + OH−, where Hads represents the adsorbed H atom on the active site), which is well-known for the strong absorption energy between the Hads and metallic Ni NPs;8 (iii) Finally, the adsorbed H atoms will interact with the absorbed water molecules on Ni-MOF-74 under the catalysis of Ni NPs to generate hydrogen gas (H2 molecules) (Ni–Hads + H2O + e− → Ni + H2 + OH−).51–53 Therefore, the Ni-MOF-74 plays an important role in the adsorption and activation of water molecules during the catalytic process of hydrogen evolution in alkaline media.
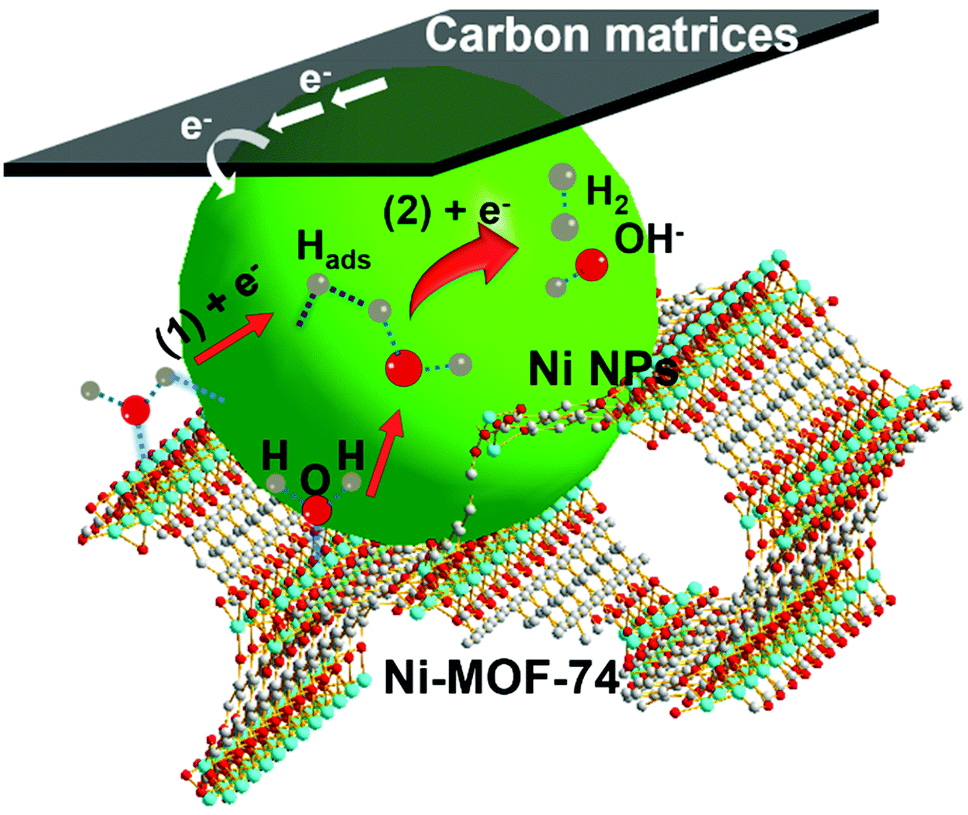 | ||
| Fig. 5 HER catalytic process on Ni NPs/C@Ni-MOF-74/NF(18 h) via steps (1) and (2) in alkaline media. | ||
Conclusions
In summary, we have reported on highly dispersed ultrafine Ni NPs embedded in partially carbonized Ni-MOF-74 (Ni NPs/C@Ni-MOF-74) microrod arrays grown on the conductive NF. The partial carbonization route is an effective strategy to realize the combination of metal NPs, carbon, and MOF frameworks to form highly efficient HER catalysts. N2 absorption isotherms, ECSAs, EIS and XPS studies revealed that the Ni NPs/C@Ni-MOF-74/NF(18 h) possessed a large specific surface area, highly exposed electroactive sites, high conductivity, and the synergetic effects between the ultrafine Ni NPs and Ni-MOF-74 frameworks to promote the adsorption, activation, and dissociation of water molecules. The Ni NPs/C@Ni-MOF-74/NF(18 h) as the catalyst exhibits superior catalytic performance for HER in alkaline media with a low overpotential of 131 mV at 10 mA cm−2 and excellent cycle durability, and it is superior to most of the recently reported MOF-based HER catalysts. This work will provide a new method for the design and synthesis of novel metal NPs/carbon/MOF hybrid electrocatalysts for efficient HER in alkaline media.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (2016YFA0202603), NSFC (91645104), Science and Technology Program of Guangzhou (201704030019), Natural Science Foundation of Guangdong Province (2017A010103007), Guangdong Science and Technology Innovation Leading Talent Fund (2016TX03N187) and the Fundamental Research Funds for the Central Universities (20lgpy84).Notes and references
- (a) K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426 CrossRef CAS PubMed; (b) H. Xu, Z. Shi, Y. Tong and G. Li, Adv. Mater., 2018, 30, 1705442 CrossRef PubMed; (c) J. Feng, H. Xu, S. Ye, G. Ouyang, Y. Tong and G. Li, Angew. Chem., Int. Ed., 2017, 56, 8120 CrossRef CAS PubMed; (d) J. Feng, L. Ding, S. Ye, X. He, H. Xu, Y. Tong and G. Li, Adv. Mater., 2015, 27, 7051 CrossRef CAS PubMed; (e) Q. Hu, G. Li, X. Liu, B. Zhu, G. Li, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, J. Mater. Chem. A, 2019, 7, 461 RSC; (f) X. Cui, S. Lei, A. C. Wang, L. K. Gao, Q. Zhang, Y. K. Yang and Z. Q. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- (a) Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC; (b) A. Wang, X. He, X. Lu, H. Xu, Y. Tong and G. Li, Angew. Chem., Int. Ed., 2015, 54, 3669 CrossRef CAS PubMed; (c) A. Wang, H. Xu, J. Feng, L. Ding, Y. Tong and G. Li, J. Am. Chem. Soc., 2013, 135, 10703 CrossRef CAS PubMed; (d) L. Ding, A. Wang, G. Li, Z. Liu, W. Zhao, C. Su and Y. Tong, J. Am. Chem. Soc., 2012, 134, 5730 CrossRef CAS PubMed; (e) X. Liu, Q. Hu, B. Zhu, G. Li, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Small, 2018, 14, 1802755 CrossRef PubMed; (f) D. Gao, J. Guo, X. Cui, L. Yang, Y. Yang, H. He, P. Xiao and Y. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22420 CrossRef CAS PubMed.
- (a) W. C. Sheng, H. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS; (b) Q. Li, Z. Wang, G. Li, R. Guo, L. Ding and Y. Tong, Nano Lett., 2012, 12, 3803 CrossRef CAS PubMed; (c) A. Wang, H. Xu and G. Li, ACS Energy Lett., 2016, 1, 445 CrossRef CAS; (d) L. Fan, B. Zhu, P. Su and C. He, Nano Energy, 2018, 45, 148 CrossRef CAS.
- (a) J. Guan, C. Li, J. Zhao, Y. Yang, W. Zhou, Y. Wang and G. Li, Appl. Catal., B, 2020, 269, 118600 CrossRef CAS; (b) W. Zhou, J. Zhao, J. Guan, M. Wu and G. Li, ACS Omega, 2019, 4, 20244 CrossRef CAS PubMed; (c) W. Zhou, X. Lu, J. Chen, T. Zhou, P. Liao, M. Wu and G. Li, ACS Appl. Mater. Interfaces, 2018, 10, 38906 CrossRef CAS PubMed.
- (a) I. E. L. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2011, 50, 1476 CrossRef CAS PubMed; (b) X. He, J. Feng, Q. Ren and G. Li, J. Mater. Chem. A, 2018, 6, 19438 RSC; (c) J. Wu, C. He, G. Li and J. Zhang, J. Mater. Chem. A, 2018, 6, 19176 RSC; (d) A. Wang, Y. Dong, M. Li, C. Liang and G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 34954 CrossRef CAS PubMed.
- F. Wang, T. A. Shifa, X. Zhan, Y. Huang, K. Liu, Z. Cheng, C. Jiang and J. He, Nanoscale, 2015, 7, 19764 RSC.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed.
- Z. W. Fang, L. L. Peng, Y. M. Qian, X. Zhang, Y. J. Xie, J. J. Cha and G. H. Yu, J. Am. Chem. Soc., 2018, 140, 5241 CrossRef CAS PubMed.
- F. Li, G. F. Han, H. J. Noh, J. Jeon, I. Ahmad, S. S. Chen, C. Yang, Y. F. Bu, Z. P. Fu, Y. L. Lu and J. B. Baek, Nat. Commun., 2019, 10, 4046 CrossRef PubMed.
- I. Hod, P. Deria, W. Bury, J. E. Mondloch, C. W. Kung, M. So, M. D. Sampson, A. W. Peters, C. P. Kubiak, O. K. Farha and J. T. Hupp, Nat. Commun., 2015, 6, 8304 CrossRef CAS PubMed.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942 CrossRef CAS PubMed.
- S. Peng, F. Gong, L. Li, D. Yu, D. Ji, T. Zhang, Z. Hu, Z. Zhang, S. Chou, Y. Du and S. Ramakrishna, J. Am. Chem. Soc., 2018, 140, 13644 CrossRef CAS PubMed.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058 CrossRef CAS PubMed.
- H. Feng, Z. Xu, L. Ren, C. Liu, J. Zhuang, Z. Hu, X. Xu, J. Chen, J. Wang, W. Hao, Y. Du and S. X. Dou, ACS Catal., 2018, 8, 4288 CrossRef CAS.
- Z. P. Shi, Y. X. Wang, H. L. Lin, H. B. Zhang, M. K. Shen, S. H. Xie, Y. H. Zhang, Q. S. Gao and Y. Tang, J. Mater. Chem. A, 2016, 4, 6006 RSC.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329 CrossRef CAS PubMed.
- G. Yilmaz, K. M. Yam, C. Zhang, H. J. Fan and G. W. Ho, Adv. Mater., 2017, 29, 1606814 CrossRef PubMed.
- W. H. He, R. Ifraemov, A. Raslin and I. Hod, Adv. Funct. Mater., 2018, 28, 1707244 CrossRef.
- K. Jayaramulu, J. Masa, O. Tomanec, D. Peeters, V. Ranc, A. Schneemann, R. Zboril, W. Schuhmann and R. A. Fischer, Adv. Funct. Mater., 2017, 27, 1700451 CrossRef.
- R. Miao, B. Dutta, S. Sahoo, J. He, W. Zhong, S. A. Cetegen, T. Jiang, S. P. Alpay and S. L. Suib, J. Am. Chem. Soc., 2017, 139, 13604 CrossRef CAS PubMed.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186 CrossRef CAS PubMed.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610 CrossRef CAS PubMed.
- C. Yilmaz, C. F. Tan, Y. F. Lim and C. W. Ho, Adv. Energy Mater., 2019, 9, 1802983 CrossRef.
- R. Wang, X. Y. Dong, J. Du, J. Y. Zhao and S. Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef PubMed.
- L. Gu, J. Chen, T. Zhou, X. Lu and G. Li, Nanoscale, 2020, 12, 11201–11208 RSC.
- X. Lu, P. Liao, J. Wang, J. Wu, X. W. Chen, C. T. He, J. P. Zhang, G. R. Li and X. M. Chen, J. Am. Chem. Soc., 2016, 138, 8336 CrossRef CAS PubMed.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C. T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. X. Wang, J. Q. Zhang, A. M. Khattak, N. A. Khan, Z. X. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- X. H. Zhao, B. Pattengale, D. H. Fan, Z. H. Zou, Y. Q. Zhao, J. Du, J. E. Huang and C. L. Xu, ACS Energy Lett., 2018, 3, 2520 CrossRef CAS.
- W. J. Phang, W. R. Lee, K. Yoo, D. W. Ryu, B. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2014, 53, 8383 CrossRef CAS PubMed.
- J. Xing, K. Guo, Z. Zou, M. Cai, J. Du and C. Xu, Chem. Commun., 2018, 54, 7046 RSC.
- S. H. Ahn and A. Manthiram, J. Mater. Chem. A, 2017, 5, 2496 RSC.
- C. Zhang, J. Xiao, X. L. Lv, L. H. Qian, S. L. Yuan, S. Wang and P. X. Lei, J. Mater. Chem. A, 2016, 4, 16516 RSC.
- K. Tan, S. Zuluaga, Q. H. Gong, P. Canepa, H. Wang, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2014, 26, 6886 CrossRef CAS.
- S. Zuluaga, E. M. A. Fuentes-Fernandez, K. Tan, J. Li, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. A, 2016, 4, 11524 RSC.
- M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- (a) A. Wang, C. Liang, X. Lu, Y. Tong and G. Li, J. Mater. Chem. A, 2016, 4, 1923 RSC; (b) S. Ye, J. Feng, A. Wang, H. Xu and G. Li, J. Mater. Chem. A, 2015, 3, 23201 RSC; (c) X. Lu, A. Wang, H. Xu, X. He, Y. Tong and G. Li, J. Mater. Chem. A, 2015, 3, 16560 RSC.
- (a) H. Xu, L. Ding, C. Liang, Y. Tong and G. Li, NPG Asia Mater., 2013, 5, e69 CrossRef CAS; (b) L. Ding, A. Wang, Y. Ou, Q. Li, R. Guo, W. Zhao, Y. Tong and G. Li, Sci. Rep., 2013, 3, 1181 CrossRef PubMed; (c) G. Li, D. Qu, C. Yao, F. Zheng, Q. Bu, C. Dawa and Y. Tong, J. Phys. Chem. C, 2007, 111, 6678 CrossRef CAS.
- (a) G. Li, C. Dawa, Q. Bu, Z. Ke, H. Hong, F. Zhen, C. Yao, G. Liu and Y. Tong, J. Phys. Chem. C, 2007, 111, 1919 CrossRef CAS; (b) G. Li, C. Su and Y. Tong, J. Phys. Chem. C, 2008, 112, 2927 CrossRef CAS; (c) G. Li, C. Dawa, Q. Bu, F. Zheng, C. Yao, P. Liu and Y. Tong., Electrochem. Commun., 2007, 9, 863 CrossRef CAS.
- M. Mukoyoshi, H. Kobayashi, K. Kusada, M. Hayashi, T. Yamada, M. Maesato, J. M. Taylor, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Matsumura and H. Kitagawa, Chem. Commun., 2015, 51, 12463 RSC.
- P. D. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Chem. Commun., 2006, 959 RSC.
- I. Strauss, A. Mundstock, D. Hinrichs, R. Himstedt, A. Knebel, C. Reinhardt, D. Dorfs and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 7434 CrossRef CAS PubMed.
- J. B. Tan, P. Sahoo, J. W. Wang, Y. W. Hu, Z. M. Zhang and T. B. Lu, Inorg. Chem. Front., 2018, 5, 310 RSC.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- N. Yao, P. Li, Z. Zhou, Y. Zhao, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2019, 9, 1902449 CrossRef CAS.
- P. T. Wang, X. Zhang, J. Zhang, S. Wan, S. J. Guo, G. Lu, J. L. Yao and X. Q. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, J. Am. Chem. Soc., 2016, 138, 14546 CrossRef CAS PubMed.
- M. I. Abdullah, A. Hameed, N. Zhang and M. M. Ma, ChemElectroChem, 2019, 6, 2100 CrossRef CAS.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00253d |
| This journal is © The Royal Society of Chemistry 2020 |