
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated photoanode based on non-critical raw materials for robust solar water splitting†
Drialys
Cardenas-Morcoso
a,
Miguel
García-Tecedor
 a,
Tsvetelina
Merdzhanova
b,
Vladimir
Smirnov
b,
Friedhelm
Finger
b,
Bernhard
Kaiser
c,
Wolfram
Jaegermann
c and
Sixto
Gimenez
*a
a,
Tsvetelina
Merdzhanova
b,
Vladimir
Smirnov
b,
Friedhelm
Finger
b,
Bernhard
Kaiser
c,
Wolfram
Jaegermann
c and
Sixto
Gimenez
*a
aInstitute of Advanced Materials (INAM), Universitat Jaume I, Avenida de Vicent Sos Baynat, s/n, 12006 Castelló de la Plana, Castellón, Spain. E-mail: sjulia@uji.es
bIEK-5 Photovoltaik, Forschungszentrum Jülich, Jülich, 52425, Germany
cInstitute of Materials Science, TU Darmstadt, Darmstadt, 64287, Germany
First published on 6th July 2020
Abstract
Herein, we have developed an integrated photoanode for solar water splitting based on an “Earth-abundant” Ni–Fe based electrocatalyst combined with a versatile multijunction Si-based photovoltaic device, designed in such a way to allow a direct coupling with the electrocatalyst with minimal losses. The water oxidation catalyst was prepared by electrochemical deposition of iron on a nickel foil, followed by thermal annealing, leading to the formation of NiO, α-Fe2O3, and NiFe2O4 phases. Detailed structural and surface characterization revealed the effect of the addition of different Fe contents and the subsequent implications on the electrocatalytic performance. The optimized integrated photoanode delivered a maximum photocurrent density of 6.2 mA cm−2 at 0 V applied bias, which corresponds to a 7.7% of Solar-To-Hydrogen conversion efficiency, which remained stable for more than 20 hours. These results pave the way towards large-scale, efficient and low-cost solar energy conversion solutions based on non-critical raw materials.
Introduction
The development of sustainable, fossil-free strategies to synthesize fuels and added-value chemicals has raised enormous interest in recent years, in order to provide reliable energy vectors as well as the feedstocks needed for the chemical industry at a global scale.1–3 One of the most promising alternatives involves the use of renewable electricity (wind, solar, hydropower, etc.) to power electrochemical conversion processes, which convert abundant molecules (e.g., water, carbon dioxide, and nitrogen) into higher-value products (e.g., hydrogen, hydrocarbons, oxygenates, and ammonia). In all these processes, water oxidation stands out as the preferred reaction to provide the protons and electrons needed for the target reduction reactions, although this process is considered a kinetic bottleneck and consequently, the development of electrocatalytic materials that effectively oxidise water is essential for improving the efficiency of the overall electrochemical conversion process.4Currently, the most efficient water oxidation catalysts are based on scarce and excessively expensive materials as iridium and ruthenium oxides, IrO2 and RuO2.5,6 Consequently, an intensive search for catalytic materials based on earth-abundant elements and low-cost synthetic processes has been carried out in order to find sustainable and cost-effective alternatives to minimize the use of these critical raw materials. Among them, Ni-based catalysts constitute one of the best alternatives due to their high electrocatalytic activity and stability under alkaline conditions, as a consequence of their high electrical conductivity and corrosion resistance.7,8 Indeed, large scale commercial alkaline electrolyzers, preferentially use nickel-based anodes.9,10 Furthermore, several Oxygen Evolution Reaction (OER) electrocatalysts, which combine Ni with other transition metals exhibit low overpotentials and high stability under alkaline conditions.11,12 Specifically, Ni–Fe alloys have been reported as synergistic catalysts towards OER, significantly more active compared to the individual Ni or Fe components.13 In this context, the beneficial interaction between Fe and Ni towards water oxidation was firstly observed by the unintentional incorporation of Fe into the structure of the Ni-based materials during electrochemical testing.14 Once this effect was reported, several studies focused on understanding the catalytic role of Fe ions incorporated by several methods, particularly on Ni/Fe oxides and oxyhydroxides,15–19 becoming a controversial topic among the scientific community in the recent years. At present, there are two different views about the role of Fe ions on Ni-based catalysts: some authors claim that Fe atoms provide true active sites for catalysis of the OER,15 while others support that Fe ions synergistically enhance the catalytic activity of Ni.14,19,20
A further step towards the exploitation of efficient water oxidation catalysts (WOC) involves their integration in more complex energy conversion devices fed by renewable electricity, in order to minimize the balance of system components, which is essential for large scale applications.21 The paradigmatic example of robust and cost-effective ‘bias-free’ photoelectrochemical water splitting devices resulting from the combination of different WOC with thin-film photo-absorbers, stands out as a promising strategy for conversion of solar energy into chemical energy, stored in solar fuels or added-value chemicals.22,23 This approach is based on an adapted photovoltaic device providing the electricity input as photogenerated charge carries, which are transferred to the electrocatalyst, to drive the water oxidation reaction. Indeed, similar configurations can be employed for other more complex electrochemical reactions leading to the production of added-value chemicals such as hydrogen peroxide (H2O2), hypochlorous acid (HClO), persulfates (H2S2O8), IO4−, Ce4+, hydrocarbons and oxygenates.3,24 Consequently, several approaches of photo- and electrocatalyst/photovoltaic devices combinations have been designed and tested, leading to higher Solar-To-Fuel (STF) efficiencies, compared to their photoelectrochemical (PEC) counterparts based on photoactive electrodes, where the photovoltage is generated at the semiconductor–liquid junction (SCLJ).25 As a relevant example, an unprecedented 17.5% STH efficiency has been recently reported for self-driven solar water-splitting, using a tandem PV-photocathode system.26
From the technological point of view, compared to wired systems, the photovoltaic–electrocatalyst (PV–EC) integrated architectures where both components are intimately connected, offer a compact and less complex design for the realization of an “artificial leaf” for practical solar fuel production.27,28 The highest reported efficiencies in PV–EC systems have been achieved with scarce and expensive materials as GaInP and GaAs as photo-absorbers, and noble metal-based oxides as IrOx or RuOx for the electrocatalyst.1,28,29 However, the use of critical raw materials jeopardizes the future technological deployment of this technology and consequently, the use of Earth-abundant and low-cost materials is imperative for further development of these systems. In this context, silicon thin films, particularly as multijunction architectures, are at the forefront of photovoltaic technologies with application in the production of solar hydrogen.22,30–32 In particular, record Solar-To-Hydrogen (STH) efficiencies of 10% using crystalline silicon photovoltaic27 and 14% with silicon heterojunction cells,33 have been reported. More recently, some of us developed triple and quadruple junction solar cells based on amorphous (a-Si:H) and microcrystalline (μc-Si:H) silicon thin films, providing higher STH efficiencies, up to 9.5%, with an integrated photocathode using the a-Si:H/a-Si:H/μc-Si:H triple junction as photo-absorber.34 One of the major advantages of these systems consists of the versatility to power any electrochemical reaction, since the delivered photovoltage can be conveniently tuned by adjusting the solar cell layers stack, providing higher flexibility to choose the electrocatalytic systems depending on the overpotential requirements.31,32 On the other hand, stability issues remain a key parameter when considering up-scaling of integrated PV–EC devices, which in several cases is limited to a few hours.35 As solid–liquid junction type devices, integrated PV–EC electrodes require tailored strategies to prevent performance losses, mostly induced by the partial dissolution of the photovoltaic component layers. Those strategies include the encapsulation of the electrode and the use of conductive materials acting as a barrier between the PV component and the electrocatalyst.13,20 On the other hand, PV–EC integration as photoanodes is a rarely explored application of multijunction silicon thin-film solar cells. The main reason is the conventional architecture of the Si solar cells, where the hole selective contact is placed at the bottom of the device, limiting the connection of the electrocatalyst to the external wiring.
In the present study, we have designed and fabricated an integrated PV–EC photoanode from an Earth-abundant water oxidation electrocatalyst and a multijunction Silicon thin-film solar cell. A mixed Ni–Fe oxide electrocatalyst was prepared by a simple method based on direct electrochemical deposition of Fe on the surface of a Ni foil, followed by thermal annealing. The resulting electrocatalyst shows enhanced performance compared to the reference nickel oxide, decreasing the overpotential by more than 50 mV. Furthermore, we have explored the integration of the electrocatalyst on two different PV devices such as thin-film silicon triple-junction solar cell and a solar module based on two tandem junction solar cells connected in series. The PV–EC integration was possible due to the flexible design of the photovoltaic component, were both front and back contacts are placed at the top side of the device. The optimized configuration provided bias-free water splitting with a 7.7% STH efficiency, stable for more than 20 hours.
Experimental
Preparation of Ni–Fe based electrocatalyst
Prior to deposition, a nickel sheet (99.2% purity, from Metall Jobst, Germany) used as substrate was cut into 1.5 × 1.5 cm2 samples, which were first cleaned by ultrasonication in acetone (AnalaR NORMAPUR) for 30 min, followed by rinsing with deionized water (Millipore), and then in 3 M HCl solution prepared from HCl (25%, Emsure) and deionized water. Finally, the samples were rinsed with deionized water and ethanol (GPR Rectapur, 99.5% denaturated with 1% MEK) and dried with compressed air. The overall process for the synthesis of the electrocatalyst is showed in ESI,† Fig. SI1. Fe incorporation on the Ni samples was carried out by electrodeposition from a 20 mM FeCl2·4H2O (Emsure) solution using dimethyl sulfoxide, (DMSO) as solvent, by applying a constant potential of −2 V vs. Ag/AgCl with a Gamry Instruments Reference 600 potentiostat, in a three-electrode cell configuration. An Ag/AgCl (3 M KCl) electrode and a Pt mesh were used as reference and counter electrode, respectively. The total deposited charge was varied as 2, 5, 12, 25 and 36 mC cm−2, on a geometrical area of 0.5 cm−2, defined by an O-ring sealing aperture. After Fe deposition, the samples were rinsed with ethanol to remove the solvent and dried with compressed air. Finally, the substrates were annealed at 450 °C for 1 h in air atmosphere, with a heating rate of 2 °C min−1, to promote the conversion of Fe0 to Fe3+.Morphological characterization and chemical composition of the Ni–Fe electrocatalyst
Field Emission Scanning Electron Microscopy (SEM) performed with a JSM-7000F JEOL FEG-SEM system (Tokyo, Japan) equipped with an INCA 400 Oxford EDS analyzer (Oxford, U.K.) and operating at 15 kV, was used for the morphological characterization of the samples. Their crystalline structure was assessed by X-ray diffraction (XRD) collected on a Rigaku Miniflex 600, (Rigaku Corporation, Tokyo, Japan) with copper Kα radiation (λ = 1.5418 Å) operating at a grazing incidence of 1°, at a scan rate of 3° min−1. The chemical composition of the resulting electrocatalytic electrodes was investigated by Confocal Multi-Spectral Imaging (CMSI) Raman Spectroscopy and X-ray Photoelectron Spectroscopy (XPS). CMSI Raman spectroscopy was carried out with a WiTec apyron system, equipped with a 300 mm focal length UHTS 300 spectrometer system. Measurements were performed with an excitation wavelength of 532.165 nm and 24.737 mW laser power. Raman imaging was obtained from the simultaneous treatment of Raman spectra recorded every 0.5 μm in a selected area (28 × 30 μm2) of the sample surface. The software transforms the multispectral maps into images, showing the heterogeneity of the surface composition. The data were analyzed with the WiTec software Project FIVE. XPS measurements were performed in the Daisy-Fun laboratory, at a pressure of 5 × 10−10 mbar using a Specs Phoibos 150 setup.36 A monochromatized Al Kα line of 1486.64 eV was used as X-ray excitation source. Survey measurements were obtained with a pass energy of 20 eV, while all detailed spectra were measured with a pass energy of 10 eV. The obtained data were evaluated using the CasaXPS software package.Electrochemical characterization of the electrocatalysts
Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were performed with a Gamry Instruments Reference 600 potentiostat, at 10 mV s−1 scan rate, in a three-electrode configuration cell, using a Pt mesh as counter electrode, and an Ag/AgCl (3 M KCl) electrode as reference. A 1 M KOH solution at pH 13.6 was used as electrolyte. The potentials were referred to the Reversible Hydrogen Electrode (RHE) through the Nernst equation: VRHE = V(Ag/AgCl) + V0(Ag/AgCl) + 0.059·pH. Measurements in two-electrode configurations were also performed for further calculations in combination with the photovoltaic devices. The electrode/electrolyte contact area was defined as 0.5 cm2 by an O-ring sealing aperture. The turnover frequency (TOF) was calculated as: TOF = jgeo/nq, where jgeo is the geometrical current density at an overpotential of 350 mV (selected for comparison), n is the number of electrons transferred during the reaction (for water oxidation, n = 4), and q is the integrated area under the cathodic redox wave, divided by the scan rate. The O2 evolution at the electrocatalyst surface was determined by gas chromatography measurements using a sealed cell coupled to an Agilent Micro-GC gas chromatograph, during a chronoamperometric measurement at 1.6 V vs. RHE, in 1 M KOH solution. The faradaic efficiency (FE) was estimated through the relation: FE (%) = O2(exp)/O2(theo), were O2(exp) is the amount of evolved O2 in mol, monitored every 5 min, and O2(theo) is the theoretical O2 evolution calculated with the Faraday's law: n (mol) = jO2t/nF, where jO2 is the current density recorded in the chronoamperometry measurement, t is the time in seconds, n is the number of electrons transferred in the reaction and F is the Faraday constant, 96485.33 C mol−1.Preparation and characterization of Si-based multijunction photovoltaic devices
In this work two types of PV devices were investigated in terms of their potential application as integrated photoanodes for solar water splitting. The first device is a solar cell based on a-Si:H/a-Si:H/μc-SiH triple junction prepared on fluorine-doped tin oxide (F:SnO2) coated glass substrates that serves as a front contact. The (p–i–n) a-Si:H top and middle sub-cells and (p–i–n) μc-Si:H bottom sub-cell were deposited by a Plasma Enhanced Chemical Vapor Deposition (PECVD) technique at an excitation frequency of 13.56 MHz and substrate temperature of about 180 °C. The intrinsic absorber layers were prepared with a mixture of silane (SiH4) and hydrogen (H2) gases. The n- and p-type layers, were prepared using phosphine (PH3), trimethylborate (TMB) and methane (CH4) gases, added to the silane–hydrogen mixture. A zinc oxide/silver (ZnO:Al/Ag) reflecting layer was sputtered as a back contact. The size of the solar cell is defined from the metal contact area of 1 cm2. The laser scribing process was used to prepare the solar cell in a design where the front and back contacts were placed outside of the ‘photo-active area’, at the top sides of the device, which allows a direct coupling with the electrocatalyst. The second device is a solar module consisting of two a-Si:H/μc-Si:H tandem solar cells connected in series via laser scribing. A detailed description of the laser scribing process is given elsewhere.37 In order to simplify the reference to the PV devices along the text, the a-Si:H/a-Si:H/μc-Si:H triple-junction solar cell and the module of two a-Si:H/μc-Si:H tandem cells connected in series will be referred as “PV-1” and “PV-2”, respectively. The solar cells and modules were characterized by current–voltage measurements at standard test conditions (100 mW cm−2, 25 °C) using a double source (Class A) AM 1.5G sun simulator. The spectral response measurements to determine the external quantum efficiency (EQE), were conducted using a monochromator at a wavelength range between 300 nm and 1100 nm.Photoelectrochemical characterization of the integrated PV–EC photoanode
In the integrated PV–EC device, the Ni foil used as substrate for catalyst deposition, was mechanically attached to the “p-side” (or “front-contact”) of the photovoltaic component, also acting as protective barrier against corrosion. Kapton tape was used for additional protection of the device against possible contact with the electrolyte during the cell assembly. The contribution of the contact resistance on the device performance was found negligible, as evidenced in the unaltered open circuit potential and extracted photocurrent of the integrated device. Linear sweep voltammetry (LSV) and chronoamperometric measurements were recorded with a Gamry Instruments potentiostat, in a two-electrode configuration cell, with a Pt mesh as counter electrode. A 1 M KOH solution was used as electrolyte. The electrode–electrolyte contact area was defined by an O-ring sealing aperture of 0.5 cm2. Measurements under illumination conditions were carried out using simulated AM 1.5 solar illumination (100 mW cm−2), provided by an Oriel LCS-100 solar simulator.Results and discussion
Ni–Fe based water oxidation electrocatalyst
The Ni–Fe based water oxidation electrocatalyst was prepared by electrodeposition of metallic Fe on previously cleaned Ni foil surface, followed by thermal annealing for the conversion of Fe0 to Fe3+. We previously showed that electrochemical deposition of Fe under specific conditions led to the formation of catalytically active α-Fe2O3 nanoparticles on the surface of a water splitting photoanode.38 Although α-Fe2O3 has been widely studied as a water oxidation photoanode,39–43 it has been suggested that electrodeposited α-Fe2O3 particles provide cooperative electrocatalytic behavior on BiVO4 photoanodes for water oxidation.38 Three different deposition charges of Fe were tested: 5, 12 and 25 mC cm−2, corresponding to 0.49, 1.06 and 6.27 at% of Fe detected by EDS analysis on Ni foil (ESI,† Fig. SI2 and Table SI1). Additionally, 36 mC cm−2 of Fe charge (14.65 at%) was used to prepare α-Fe2O3 thin films, as reported earlier.38,41 After thermal annealing at 450 °C, according to the Fe–O,44 and Ni–O45 phase diagrams, it is expected that phase transition from metallic iron to iron(III) oxide takes place, concomitant to phase transition on the Ni foil surface, due to the formation of nickel(II) oxide above 400 °C.46 Furthermore, the formation of the FeNi3 intermetallic phase is expected at temperatures higher than 200 °C according to the Ni–Fe phase diagram.47 This FeNi3 can react with oxygen according to the reaction 4FeNi3 + 9O2 → 2NiFe2O4 + 10NiO,48 after the subsequent increase of the temperature. A reference Ni foil sample thermally treated in the same conditions was used for comparison with the Fe-containing samples.In order to evaluate the electrocatalytic behavior of the prepared materials for water oxidation, cyclic voltammetry measurements in a 1 M KOH electrolyte were performed. Fig. 1a summarizes the evolution of catalytic performance versus Fe content. This performance is expressed as the overpotential (η, mV) required for water oxidation and the turnover frequency (TOF) at 400 mV overpotential, versus the Fe content in Fig. 1b. The optimal performance was obtained at a Fe content of 1.06 at%, (corresponding to 12 mC cm−2 load) with a reduction of the overpotential of about 50 mV, as compared to the reference Ni foil (after annealing), as showed in Fig. 1b. Moreover, the calculated TOF value, which is related to the amount of oxygen evolved per mole of the catalyst per second, also shows to be strongly dependent on the Fe content, reaching the highest value (0.5 s−1) at 1.06 at%. On the other hand, it is accepted that Ni-based electrocatalysts need an electrochemical activation process (several cyclic voltammetry scans) to maximize their catalytic activity towards water oxidation, since water from the electrolyte percolates within the structure of the catalyst introducing Fe impurities and increasing the exposed surface area.14 Consequently, electrochemical activation was carried out on both reference and optimal Ni–Fe sample for comparison. The cyclic voltammograms before and after electrochemical activation (50 cycles), recorded at a scan rate of 10 mV s−1, are represented in Fig. 1c. The observed anodic and cathodic peaks, A1 and C1 respectively, are associated to the α-Ni(OH)2/γ-NiOOH transformation, while the secondary A2 and C2 peaks are related to the β-Ni(OH)2/β-NiOOH conversion.13,49,50 The complete set of cyclic voltammograms is showed as ESI,† Fig. SI3. Furthermore, the faradaic efficiency for oxygen evolution was determined (blue diamonds) on the optimal electrocatalyst under operation from gas chromatography measurements (red circles) and catalytic current, (black line), see Fig. 1d. The obtained values increased with time, reaching the 100% efficiency after 100 min of operation.
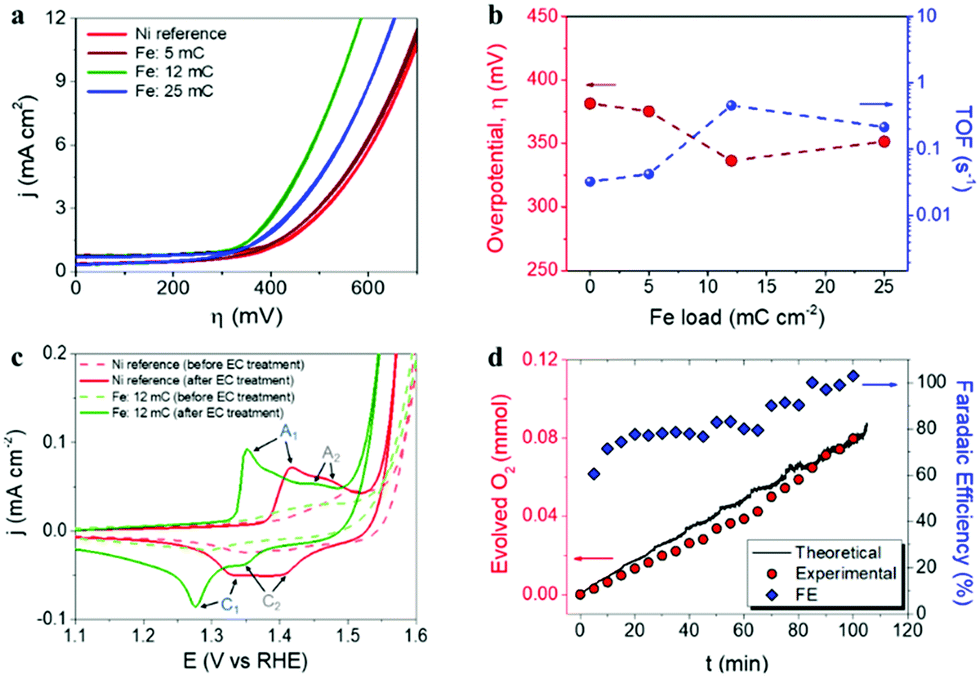 | ||
| Fig. 1 (a) Cyclic voltammetry measurements, recorded with a scan rate of 10 mV s−1, for the different samples prepared with different electrodeposited Fe charges. (b) Overpotential (η) for the water oxidation onset and turn-over frequency (TOF), calculated at an overpotential of 400 mV, as a function of the Fe content on the Ni-based electrocatalysts. (c) Effect of electrochemical activation on reference Ni foil thermally treated and optimal Ni–Fe electrocatalyst before (dashed lines) and after (solid lines) electrochemical activation (50 cycles). (d) Oxygen evolution measurement and faradaic efficiency (FE), performed with the optimal Ni–Fe catalyst (Fe load: 12 mC cm−2 corresponding to 1.06 at% Fe content), after activation at 1.6 V vs. RHE. | ||
To understand the observed electrocatalytic performance, we have combined structural and compositional characterization tools, to determine the structure and composition of the Ni–Fe electrocatalyst. First, morphological and structural characterization of the as-prepared samples was carried by SEM and XRD. Fig. 2a shows the top-view of the reference Ni foil used as substrate, after thermal annealing. 12 mC cm−2 electrodeposited Fe charges led to distributed clusters of particles (3–5 μm) at the surface (Fig. 2b and c). It is expected that this dispersion will increase the surface density of catalytic active sites and subsequently, the electrocatalytic performance. EDS analysis at different locations reveals that Fe is mainly confined at these dispersed clusters, as shown in Fig. 2c (see ESI,† Fig. SI4 for the EDS spectra). Then, it is expected that Ni–Fe active phases are formed in the vicinity of such clusters, providing a higher density of active sites for water oxidation. A detailed compilation of SEM micrographs at all the different Fe contents tested is showed as ESI,† Fig. SI2. From those images, the electrodeposited Fe charge clearly had a significant impact on the morphology of the samples. Furthermore, the highest Fe content (14.65 at%), led to the formation of an iron oxide film covering the Ni foil surface, as showed in ESI,† Fig. SI2d.
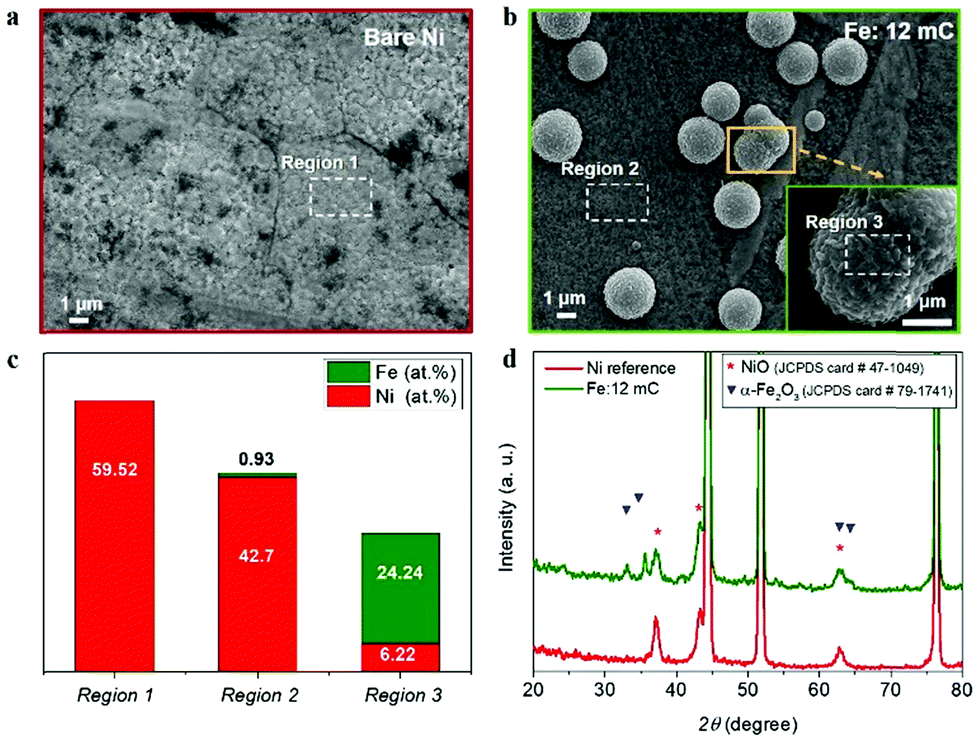 | ||
| Fig. 2 Surface morphological and structural characterization of Ni-based electrocatalyst by SEM and XRD: (a) SEM image of reference Ni substrate after thermal annealing; (b) SEM image of the Ni substrate with optimal Fe content (1.06 at%), showing the clusters formed after Fe loading and thermal annealing; the inset shows a magnification of one of the Ni–Fe clusters. (c) Bars chart with the atomic percentage (at%) of nickel and iron at the different selected regions. (d) X-ray diffractograms of reference Ni substrate, and Ni substrate with optimal Fe content (1.06 at%). The reference peaks of NiO and α-Fe2O3 phases are included for comparison (symbols). | ||
Fig. 2d shows the X-ray diffractograms of the reference Ni substrate after thermal annealing and the Ni substrate with the optimal Fe content (1.06 at%). Both diffractograms show sharp peaks at 2θ values of 44.52°, 51.88° and 76.4° (see ESI,† Fig. SI5), which can be indexed as the (111), (200) and (220) planes of the face-centred cubic (fcc) nickel according to the JCPDS card number 04-0850. Both samples show diffraction peaks at 37.08°, 43.28° and 62.72°, indexed as the (111), (200) and (220) planes of the fcc nickel(II) oxide, NiO, (JCPDS card number 47-1049). Furthermore, the diffractogram of the optimal Fe charge sample shows additional peaks at 33.04°, 35.64°, 63.12° and 64.36° corresponding to the (104), (110), (214) and (300) planes of the monoclinic structure of iron oxide, α-Fe2O3, (JCPDS card number 79-1741). Note that the position of the symbols conveys the relative intensity of the peaks in the reference patterns.
The chemical composition was further investigated through Confocal Multi-Spectral Imaging (CMSI) Raman spectroscopy and X-ray Photoelectron Spectroscopy (XPS). Fig. 3a and b show the chemical mapping and the Raman spectra of a selected area of the optimal Ni–Fe catalyst surface, where three different compositions were detected. The Raman spectrum of the component at the region (i) shows a sharp signal at 550 cm−1, which can be assigned to the Ni–O stretching mode of the nickel(II) oxide, NiO.51,52 The slight shift compared to the characteristic value for this vibrational mode (∼536 cm−1) is attributed to different crystal orientations. The Raman spectrum in the region (ii), which is directly related to the clusters observed in Fig. 2b, shows the characteristic phonon modes at 210 cm−1 (A1g), 270 cm−1 (Eg) and less intense 380 cm−1 (Eg) and 580 cm−1 (Eg), corresponding to the metal-oxide vibrations of hematite, α-Fe2O3.53,54 These observations are in excellent agreement with the SEM and EDS analyses discussed above, where it was found that the deposited Fe is preferably confined at the clusters. Finally, the Raman spectrum corresponding to the region (iii), which is related to the interface between the Fe-rich aggregates and the nickel oxide on the substrate surface, shows the phonon modes at 200 cm−1 (F2g), 290 cm−1 (Eg) and 460 cm−1 (F2g), characteristic of oxygen atom bending in M–O bond at octahedral voids of the spinel NiFe2O4, and the band at 675 cm−1 (A1g), related to a stretching mode of the oxygen atom concerning metal-ion binding in the tetrahedral void, confirming the formation of the mixed oxide NiFe2O4.55,56 The Raman spectrum of the Ni foil sample after the thermal treatment taken as the reference is provided in ESI,† Fig. SI6, showing a principal signal around 536 cm−1, characteristic of the Ni–O stretching mode of the NiO phase.
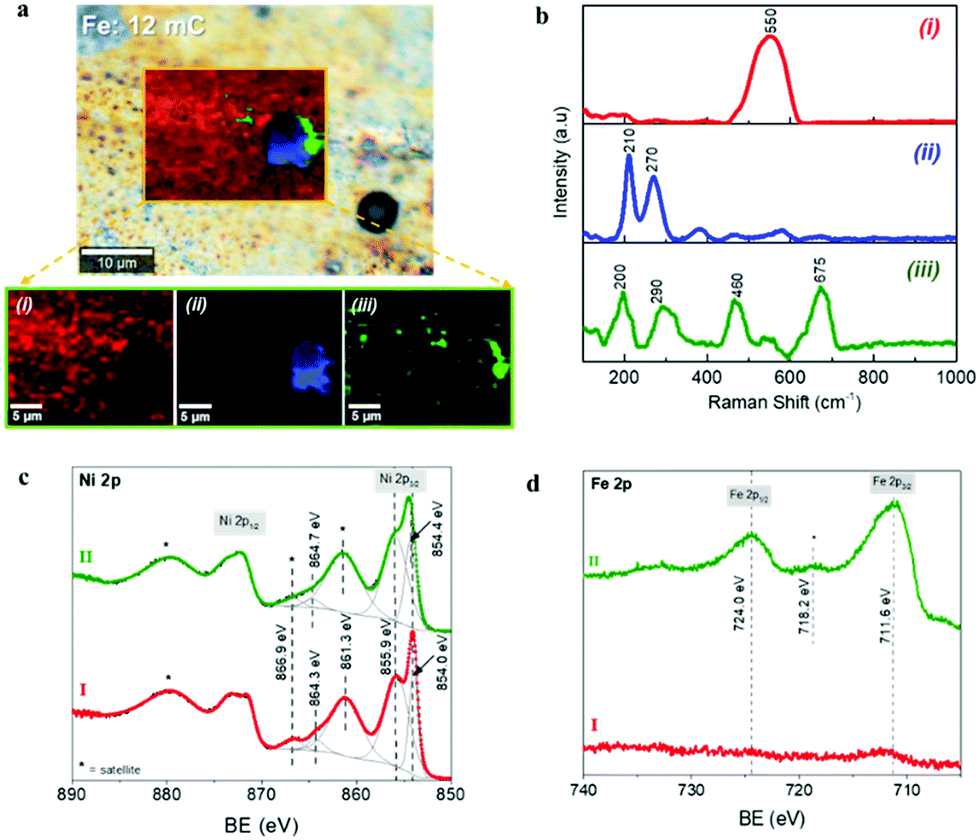 | ||
| Fig. 3 Compositional characterization of Ni-based electrocatalyst by Raman spectroscopy and XPS: (a) optical image of the of the Ni–Fe electrocatalyst with the optimal Fe content (1.06 at%) surface; as an inset, the Confocal Multi-Spectral Imaging (CMSI) maps in a selected area and individual regions from the CMSI mapping, (b) Raman shift spectra corresponding to the different regions from (a.). (c) XPS Ni 2p spectra and (d) XPS Fe 2p spectra of Ni reference (I) and optimal Ni–Fe electrocatalyst (II). | ||
More detailed surface characterization was carried out by XPS analysis. It has been reported that Fe and Ni species with high spin in Ni–Fe phases lead to multiple splitting of the 2p signals, as well as peak asymmetries and overlapping of binding energies. Consequently, the identification of specific chemical states on oxides and hydroxide compounds of such elements is extremely challenging.57,58 The Ni 2p signals of reference and optimal Ni–Fe samples (labeled as I and II respectively) are showed in Fig. 3c and the survey spectra can be found in ESI,† Fig. SI7a. After fitting the Ni 2p3/2 signal on each sample, the peak positions show the presence of Ni2+ as the dominant species at the surface of both materials. Comparing to specific literature related to the identification of Ni and Fe compound through XPS, the Ni 2p spectra obtained here for reference and Fe loaded samples are characteristic fingerprints for NiO and NiFe2O4 respectively.57,58 The Fe 2p spectrum of the optimal Ni–Fe catalyst, depicted in Fig. 3c, shows the characteristic binding energies of Fe3+ species, confirming the successful conversion from metallic Fe during thermal annealing. As expected, no Fe-related signals were found on the reference Ni substrate. The O 2s signal also offers further information about the surface composition (see ESI,† Fig. SI7b). The characteristic peaks related to hydroxides and water (531.2 eV and 532.4 eV respectively) are observed on the reference Ni sample, together with a peak at 529.6 eV, related to oxygen linked to a metal, particularly characteristic of Ni–O. Consequently, we can conclude that nickel(II) oxide, NiO, is the dominant phase at the surface of the reference Ni sample after thermal annealing, in perfect agreement with the SEM, XRD and CMSI analyses. On the other hand, after optimal Fe loading, an intense signal 530.2 eV relates to metal–oxygen bond, characteristic for α-Fe2O3, in excellent agreement with the information provided by XRD and Raman spectroscopy. The Ni–O related signal is still clearly visible but less intense. These results confirm the presence of mixed phases of Ni–Fe compounds in the optimal electrocatalyst, as showed in Fig. 3a. The XPS-VB spectra are shown in ESI,† Fig. SI7c for both samples. The spectrum of the reference Ni substrate also shows a higher energy emission at around 2 eV and a shoulder close to 3.5 eV related to Ni 3d states in NiO.59 However, both features are shifted to higher binding energies at the Ni–Fe sample, while the shoulder is less defined, in good agreement with the features associated to NiFe2O4.60 The position of the Fermi level is closer to the valence band maximum (VBM) for the reference Ni sample compared to the Ni–Fe sample, as shown in ESI,† Fig. SI7c. The effect of the different Fe charges tested on the XPS spectra is illustrated as ESI,† Fig. SI8. The Ni 2p signal reveals that only with the optimal 1.06 at% Fe content, the formation of NiFe2O4 is favored, and the Fe 2p signal clearly reflects the increase of Fe species at the electrode surface with the Fe load, concomitant to the decrease of the Ni signal. Furthermore, compared to the highest Fe content (14.65 at%), (see ESI,† Fig. SI8) the Ni 2p signal is negligible, since the surface is mainly covered by Fe species, (as shown in ESI,† Fig. SI2d) and the density of exposed Ni sites is negligible. All the identified compounds: NiO, α-Fe2O3 and NiFe2O4, constitute efficient water oxidation electrocatalysts, and the best performance has been reported for α-Fe2O3 and NiFe2O4,61,62 which nicely agrees with the functional characterization showed in Fig. 1. NiO and NiFe2O4 have been reported as active water oxidation electrocatalysts,11 after the subsequent CV treatment to develop (Ni, Fe)OOH as the active phase for OER.63 On the other hand, the electrocatalytic activity of α-Fe2O3 towards OER has been reported for electrodeposited α-Fe2O3 nanoparticles.38 Nonetheless, we observe that the activity of the optimal electrocatalyst prepared herein is moderate, compared to the state-of-the-art of Ni–Fe based materials studied as water oxidation catalysts.64 Further optimization, including nanostructuring to increase the surface area and pre/post-synthetic treatments,63,65,66 can be addressed to improve the electrocatalytic activity. However, such optimization is out of the scope of this study, since the final performance of the integrated device, for which the electrocatalyst is aimed, is limited by the photovoltaic component. Consequently, we have focused on the facile modification of the Ni foil used for both contact and protective barrier to the photovoltaic component, to reach a competitive electrocatalytic activity toward water oxidation, and high stability, with the integrated photoanode.
A relevant insight related to the effect of Fe content on the Ni foil surface stems from the redox peaks related to the reversible reaction Ni(OH)2 + OH− ↔ NiOOH + H2O + e−, characteristic of the Ni-based electrocatalysts.67 It has already been reported that the accumulation of active sites, reflected on the size of the redox wave correlates to the catalytic activity.14,68 The sample with 1.06 at% Fe shows both the highest catalytic activity and the largest redox wave (Fig. 1c). Increasing the number of cycles during the electrochemical activation process leads to a significant enhancement of the Ni2+/Ni3+ redox wave (ESI,† Fig. SI2), suggesting the increase of Ni oxo-hydroxide species as actives sites for water oxidation catalysis and hence, a cathodic shift of 50 mV of the onset potential for water oxidation. The presence of Ni oxo-hydroxides was further confirmed by XPS measurements, as shown in ESI,† Fig. SI9.
Both the method to incorporate Fe ions and their concentration play a crucial role on the final electrocatalytic performance of the Ni-based electrocatalysts.19 When Fe is incorporated as an impurity in the host Ni lattice, it improves the activity towards OER by providing active sites or favouring the Ni centres, modifying the local Ni environment. On the other hand, when the Fe content exceeds a certain limit, a less active phase is formed.13,15 Two different effects related to Fe incorporation can be observed in Fig. 1c: the intentional addition of Fe to the catalytic system (red and green lines) and the unintentional incorporation of Fe traces from the electrolyte (dashed and solid lines), both increasing the catalytic activity.
The photovoltaic device
The photovoltaic devices employed in the present study were prepared with a design where both “front” and “back contact” were placed outside the photo-active region, at the top side of the device, as represented in Fig. 4a, allowing both electron and hole extraction to drive either reduction or oxidation reactions. This configuration offers great versatility for the use of photovoltaic devices either as photoanodes or photocathodes in PEC cells. On the other hand, integrated amorphous (a-Si) and microcrystalline (μc-Si) silicon layers provide high versatility for photo-electrocatalytic applications, due to the tunable photovoltage (Open Circuit Potential, OCP), which depends on the specific multijunction Si-based device architecture. Current devices provide OCP values ranging from 1.5 V up to 2.8 V.32,69 A detailed description of multijunction Si architectures for water splitting applications can be found elsewhere.30,33 In the present study, two different multijunction Si cells have been tested as photo-absorber in combination with the Ni–Fe water oxidation electrocatalyst developed: (i) a triple-junction solar cell, a-Si:H/a-Si:H/μc-Si:H, (PV-1) (Fig. 4b) which provides a total 2.15 V OCP and (ii) a solar module with two a-Si:H/μc-Si:H tandem solar cells connected in series (PV-2) (Fig. 4c). In this second device, each cell provides 1.3 V OCP, which is not sufficient to drive the water oxidation reaction, considering the unavoidable thermodynamic and kinetic overpotentials. However, the in-series connection of two a-Si:H/μc-Si:H tandem cells delivers a total of 2.6 V OCP, exceeding the required voltage for water oxidation.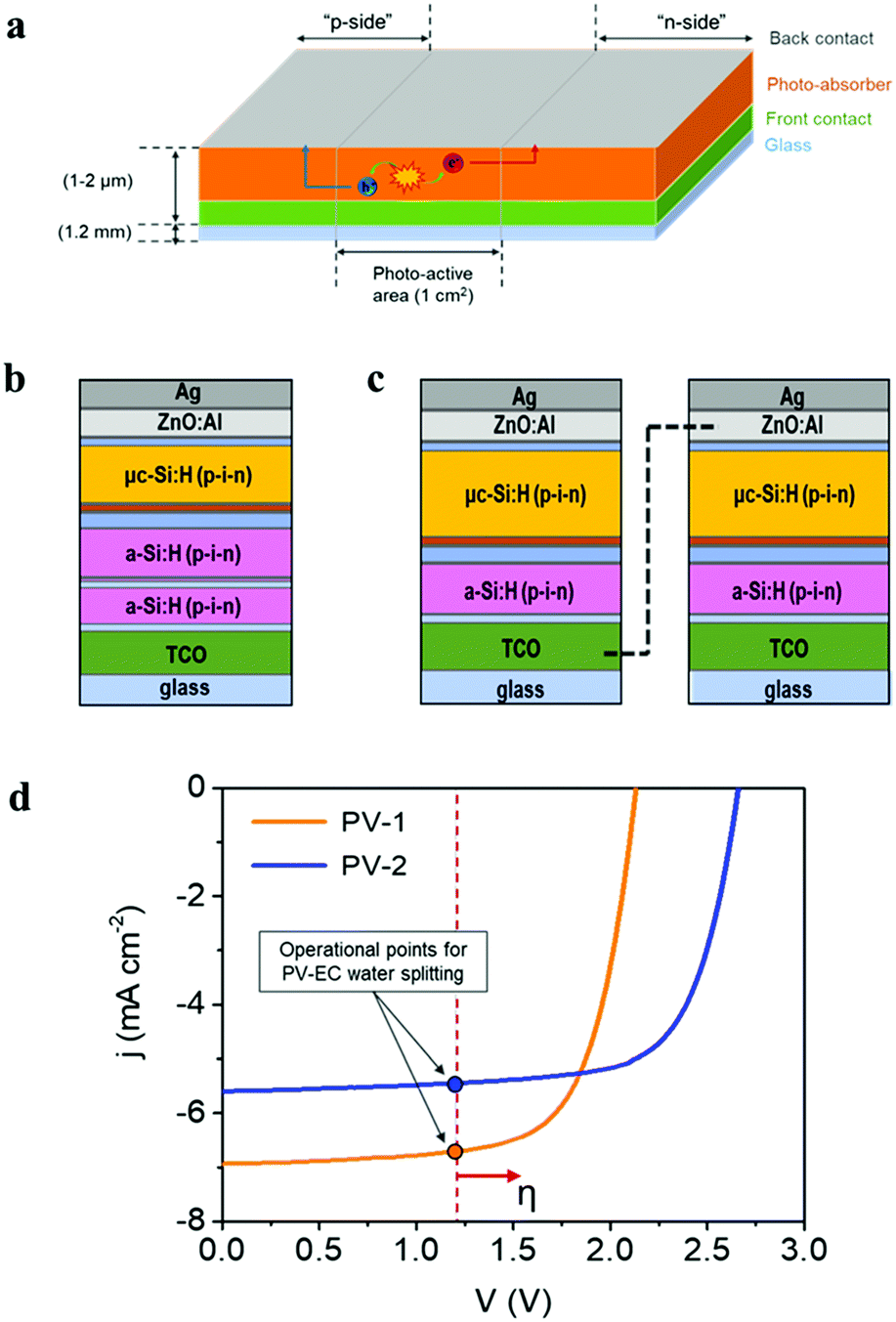 | ||
| Fig. 4 (a) Schematic representation of the photovoltaic device design, where the Ag contacts are outside of the ‘photo-active area’, and serve to transport the photogenerated charge carriers to the electrocatalyst. The photo-active region, with a size of 1 cm2, consists of a Si-based device. The architecture of the two different devices employed: (b) the a-Si:H/a-Si:H/μc-Si:H triple-junction solar cell (PV-1) and (c) the module of two a-Si:H/μc-Si:H tandem cells connected in series (PV-2). (d) Characteristic current–voltage curves of PV-1 and PV-2 devices. The theoretical operational points of the integrated PV–EC water splitting devices are represented (the dashed line indicates the water splitting thermodynamic potential at 1.23 V). The red arrow indicates that the “practical” operating point is shifted due to overpotentials (η). | ||
Fig. 4d shows the characteristic current–voltage curves of the two types of multijunction thin film solar devices described, and the photovoltaic parameters extracted from these curves are summarized in Table 1. The External Quantum Efficiency (EQE), including individual spectra for each sub-cell of the photovoltaic devices is provided as ESI,† Fig. SI10. The theoretical operation point of an integrated PV–EC device for water splitting, at 1.23 V, is also represented in Fig. 4d. Since practical electrochemical water oxidation requires voltages >1.8 V, considering the overpotentials, both configurations can provide a sufficient photovoltage to drive the water oxidation reaction. However, enhanced performance is expected for PV-1 combined with the Ni–Fe electrocatalyst due to its higher photocurrent. Conversely, the higher photovoltage of PV-2 can be more efficiently exploited in photoelectrochemical applications where higher voltages are required, as e.g., CO2 reduction or synthesis of added-value products.3,24
| Device parameters | PV-1 | PV-2 |
|---|---|---|
| η (%) | 10.3 | 10.7 |
| j sc (mA cm−2) | 6.9 | 5.6 |
| V oc (V) | 2.13 | 2.66 |
| FF (%) | 69.6 | 71.1 |
| P mpp (mW) | 10.3 | 10.7 |
The PV–EC integrated photoanode
Fig. 5a schematically represents the combination of the Si photovoltaic device, with the optimal Ni–Fe water oxidation electrocatalyst in the integrated water splitting photoanode. Note that, due to the design of the photovoltaic component (Fig. 4a), it is possible to prepare either a photoanode, placing the catalyst on the “p-side”, or a photocathode, if it is placed on the “n-side”, even when using a Si-based cell with p–i–n configuration, as those employed here. The integrated photoanode and the experimental setup of the photoelectrochemical cell in operation are showed as ESI,† Fig. SI11. The performance of the PV–EC integrated device can be predicted by the estimation of the theoretical operation point, obtained as the intersection of the current–voltage plot of the photovoltaic device with the linear voltammogram of the electrocatalyst in two-electrode configuration,32 as illustrated in Fig. 5b. Hence, the expected STH efficiency, or ηSTH, can be calculated as: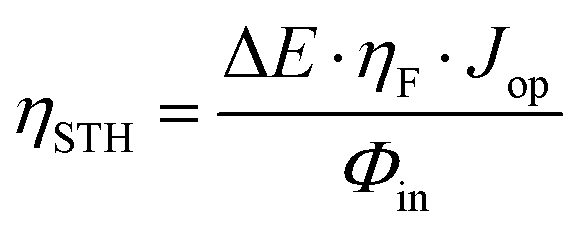 | (1) |
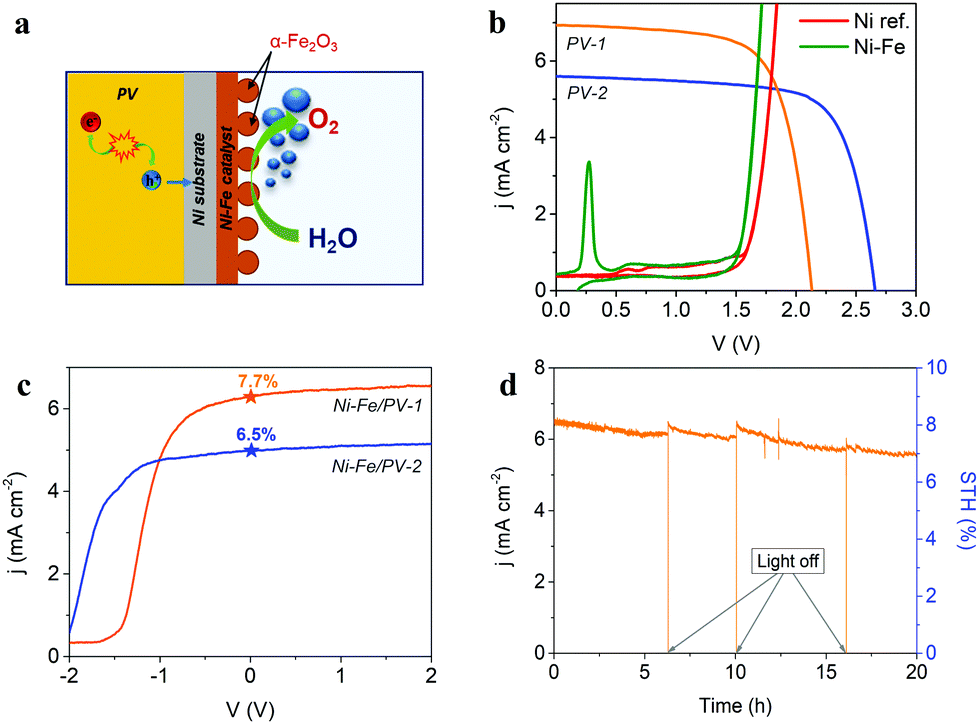 | ||
| Fig. 5 (a) Schematic representation of the general operation of the integrated PV–EC photoanode, obtained by the direct coupling of the Si-based solar device and the Ni–Fe optimized catalyst. (b) Estimation of the theoretical operation point from the characteristic current–voltage curve of the photovoltaic devices and LSV of the reference Ni sample and the optimal electrocatalyst (Ni–Fe), measured in a two-electrode configuration. (c) LSV recorded in two-electrode configuration at 50 mV s−1, of the integrated PV–EC devices as photoanodes and STH efficiencies. (d) Stability test of the integrated photoanode combining the optimal Ni–Fe electrocatalyst and the PV-1 showing promising stability for more than 20 hours under continuous operation. The left y-axis shows the recorded photocurrent density, while the right y-axis represents the calculated STH efficiency. | ||
The measured performance of the integrated photoanodes in a two-electrode configuration cell is showed in Fig. 5c. Both photovoltaic devices PV-1 and PV-2 were tested for comparison. As expected, the higher current density at zero applied bias is obtained for PV-1, despite the detrimental shift of the onset potential compared to the solar module, due to the lower photovoltage. The 7.7% STH efficiency measured for the optimal architecture, nicely agrees with that predicted from Fig. 5b and is comparable to recent reports.28,30,72 As an example, using a NiMo/NiFeOx catalyst system and a-Si:H/a-Si:H/μc-Si:H triple-junction cell in cassette configuration, a 5.1% STH efficiency was reported for a larger area device (AEC = 50.3 cm2; APV = 64 cm2) device.69 Finally, the stability of the best integrated device (resulting from the assemble of the optimal Ni–Fe electrocatalyst and the PV-1 device) was tested by a chronoamperometric test running for 20 h, as shown in Fig. 5d. Ni–Fe mixed electrocatalyst are highly stable and corrosion resistant in alkaline media, particularly at the operating current density of the integrated PV–EC device under study.73 This is evidenced by the long-term stable behaviour of the PV–EC device under operation in Fig. 5d, where there is not a significant decrease of the photocurrent density. SEM and EDS analysis after the long-term stability test, provided as ESI,† Fig. SI13, showed minor changes on both the morphology and the composition on the electrocatalyst surface. The small decrease of the photocurrent density is attributed to the formation of oxygen bubbles at the electrode surface, decreasing the active area and, hence, the extracted current density. Moreover, the initial photocurrent is restored after switching-off the light, and releasing the formed bubbles, showing a stable behavior of the integrated device. It is worth noting, that stability tests on integrated PV–EC electrodes in direct contact with the electrolyte are usually carried out for few minutes, mainly because of the partial dissolution of the photovoltaic component.23,35 Here, since the Ni substrate of the Ni–Fe electrocatalyst acts as a protective barrier, and the photovoltaic component design allows good electrical contact with the electrocatalyst, the device can run under operating conditions without current losses for more than 20 hours.
Conclusions
We have shown that the combination of an optimized Ni–Fe based electrocatalyst (mixed NiO, α-Fe2O3 and NiFe2O4 phases) for water oxidation, with a Si-based multijunction photovoltaic device leads to an efficient and stable integrated photoanode. The concept has been applied here for solar water splitting, although more challenging reactions can be addressed, given the tunability of the accessible photovoltage delivered by the photovoltaic component, and the versatility for driving either oxidation or reduction process, inherent to the device design. The intentional incorporation of Fe by electrodeposition is reflected on the surface morphology of the electrocatalyst, as well as on the development of a high density of active sites for water oxidation during the electrochemical treatment, leading to a 50 mV decreased overpotential compared to the reference Ni substrate. Furthermore, the rational selection of the photovoltaic component is highlighted, targeting the enhancement of the device performance when an optimal electrocatalyst is used. The resulting integrated photoanode shows a competitive 7.7% STH efficiency and promising stability (20 hours test) at zero applied bias. These results demonstrate that PV–EC configurations based on non-critical raw materials constitute a viable pathway for efficient and low-cost solar energy conversion schemes. In addition, compared to externally wired systems, the integrated PV–EC devices open a promising path towards further implementation of monolithic structures for overall water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Union Horizon 2020 project A-LEAF (Grant Agreement No. 732840). The authors would like to thank J. Kirchhoff and G. Schöpe for their support with solar cell fabrication, and C. Zahren for his support in solar cell characterization.References
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- K. Sayama, ACS Energy Lett., 2018, 3, 1093–1101 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 125–131 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- U. De Silva, J. Masud, N. Zhang, Y. Hong, W. P. R. Liyanage, M. Asle Zaeem and M. Nath, J. Mater. Chem. A, 2018, 6, 7608–7622 RSC.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. M. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Ju, Z. Li and Y. Xu, Mater. Res. Bull., 2015, 64, 171–174 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- J. Zaffran, M. B. Stevens, C. D. M. Trang, M. Nagli, M. Shehadeh, S. W. Boettcher and M. Caspary Toroker, Chem. Mater., 2017, 29, 4761–4767 CrossRef CAS.
- V. Fidelsky and M. C. Toroker, Phys. Chem. Chem. Phys., 2017, 19, 7491–7497 RSC.
- F. Song, M. M. Busch, B. Lassalle-Kaiser, C.-S. Hsu, E. Petkucheva, M. Bensimon, H. M. Chen, C. Corminboeuf and X. Hu, ACS Cent. Sci., 2019, 5, 558–568 CrossRef CAS PubMed.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Nat. Commun., 2019, 10, 5208 CrossRef PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC.
- F. Urbain, J. P. Becker, V. Smirnov, J. Ziegler, F. Yang, B. Kaiser, W. Jaegermann, S. Hoch, A. Maljusch, U. Rau and F. Finger, Mater. Sci. Semicond. Process., 2016, 42, 142–146 CrossRef CAS.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5 Search PubMed.
- J. Ronge, T. Bosserez, L. Huguenin, M. Dumortier, S. Haussener and J. A. Martens, Oil Gas Sci. Technol., 2015, 70, 863–876 CrossRef CAS.
- S. K. Karuturi, H. Shen, A. Sharma, F. J. Beck, P. Varadhan, T. Duong, P. R. Narangari, D. Zhang, Y. Wan, J.-H. He, H. H. Tan, C. Jagadish and K. Catchpole, Adv. Energy Mater., 2020, 2000772 CrossRef.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057 CrossRef CAS PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- W. J. Chang, K.-H. Lee, H. Ha, K. Jin, G. Kim, S.-T. Hwang, H.-m. Lee, S.-W. Ahn, W. Yoon, H. Seo, J. S. Hong, Y. K. Go, J.-I. Ha and K. T. Nam, ACS Omega, 2017, 2, 1009–1018 CrossRef CAS PubMed.
- C. S. Tan, K. W. Kemp, M. R. Braun, A. C. Meng, W. Tan, C. E. D. Chidsey, W. Ma, F. Moghadam and P. C. McIntyre, Sustainable Energy Fuels, 2019, 3, 1490–1500 RSC.
- F. Urbain, K. Wilken, V. Smirnov, O. Astakhov, A. Lambertz, J.-P. Becker, U. Rau, J. Ziegler, B. Kaiser, W. Jaegermann and F. Finger, Int. J. Photoenergy, 2014, 10 Search PubMed.
- J. P. Becker, B. Turan, V. Smirnov, K. Welter, F. Urbain, J. Wolff, S. Haas and F. Finger, J. Mater. Chem. A, 2017, 5, 4818–4826 RSC.
- J.-W. Schüttauf, M. A. Modestino, E. Chinello, D. Lambelet, A. Delfino, D. Dominé, A. Faes, M. Despeisse, J. Bailat, D. Psaltis, C. Moser and C. Ballif, J. Electrochem. Soc., 2016, 163, F1177–F1181 CrossRef.
- F. Urbain, V. Smirnov, J.-P. Becker, A. Lambertz, F. Yang, J. Ziegler, B. Kaiser, W. Jaegermann, U. Rau and F. Finger, Energy Environ. Sci., 2016, 9, 145–154 RSC.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- W. Jaegermann, B. Kaiser, J. Ziegler and J. Klett, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, 2016, pp. 199–280 Search PubMed.
- S. Haas, A. Gordijn and H. Stiebig, Prog. Photovoltaics Res. Appl., 2008, 16, 195–203 CrossRef.
- M. N. Shaddad, M. A. Ghanem, A. M. Al-Mayouf, S. Gimenez, J. Bisquert and I. Herraiz-Cardona, ChemSusChem, 2016, 9, 2779–2783 CrossRef CAS PubMed.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- F. S. Hegner, D. Cardenas-Morcoso, S. Giménez, N. López and J. R. Galan-Mascaros, ChemSusChem, 2017, 10, 4552–4560 CrossRef CAS PubMed.
- L. Badia-Bou, E. Mas-Marza, P. Rodenas, E. M. Barea, F. Fabregat-Santiago, S. Gimenez, E. Peris and J. Bisquert, J. Phys. Chem. C, 2013, 117, 3826–3833 CrossRef CAS.
- P. Sharma, J.-W. Jang and J. S. Lee, ChemCatChem, 2019, 11, 157–179 CrossRef CAS.
- H. A. Wriedt, J. Phase Equilib., 1991, 12, 170–200 CrossRef CAS.
- J. P. Neumann, T. Zhong and Y. A. Chang, Bull. Alloy Phase Diagrams, 1984, 5, 141–144 CrossRef CAS.
- Y. Wu, Y. He, T. Wu, T. Chen, W. Weng and H. Wan, Mater. Lett., 2007, 61, 3174–3178 CrossRef CAS.
- L. J. Swartzendruber, V. P. Itkin and C. B. Alcock, J. Phase Equilib., 1992, 13, 585 CrossRef.
- J. Zhu, D. Xiao, J. Li, X. Yang and Y. Wu, Scr. Mater., 2006, 54, 109–113 CrossRef CAS.
- S. R. Mellsop, A. Gardiner, B. Johannessen and A. T. Marshall, Electrochim. Acta, 2015, 168, 356–364 CrossRef CAS.
- M. E. G. Lyons, R. L. Doyle, I. Godwin, M. O'Brien and L. Russell, J. Electrochem. Soc., 2012, 159, H932–H944 CrossRef CAS.
- A. R. Neale, Y. Jin, J. Ouyang, S. Hughes, D. Hesp, V. Dhanak, G. Dearden, S. Edwardson and L. J. Hardwick, J. Power Sources, 2014, 271, 42–47 CrossRef CAS.
- C. Luo, D. Li, W. Wu, Y. Zhang and C. Pan, RSC Adv., 2014, 4, 3090–3095 RSC.
- I. Chourpa, L. Douziech-Eyrolles, L. Ngaboni-Okassa, J.-F. Fouquenet, S. Cohen-Jonathan, M. Soucé, H. Marchais and P. Dubois, Analyst, 2005, 130, 1395–1403 RSC.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- J.-L. Ortiz-Quiñonez, U. Pal and M. S. Villanueva, ACS Omega, 2018, 3, 14986–15001 CrossRef PubMed.
- H. Salazar-Tamayo, K. E. García and C. A. Barrero, J. Magn. Magn. Mater., 2019, 471, 242–249 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- S. A. Krasnikov, A. B. Preobrajenski, T. Chasse and R. Szargan, Thin Solid Films, 2003, 428, 201–205 CrossRef CAS.
- D. Chen, F. Zhang, W. Wang, Y. Yang and G. Qian, Int. J. Hydrogen Energy, 2018, 43, 2121–2129 CrossRef CAS.
- J. Liu, D. Zhu, T. Ling, A. Vasileff and S.-Z. Qiao, Nano Energy, 2017, 40, 264–273 CrossRef CAS.
- G. Liu, K. Wang, X. Gao, D. He and J. Li, Electrochim. Acta, 2016, 211, 871–878 CrossRef CAS.
- L.-A. Stern and X. Hu, Faraday Discuss., 2014, 176, 363–379 RSC.
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS.
- X. Li, X. Hao, Z. Wang, A. Abudula and G. Guan, J. Power Sources, 2017, 347, 193–200 CrossRef CAS.
- M. Yu, G. Moon, E. Bill and H. Tüysüz, ACS Appl. Energy Mater., 2019, 2, 1199–1209 CrossRef CAS.
- M. E. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2018, 3, 1386–1424 Search PubMed.
- S. Corby, M. Garcia-Tecedor, S. Tengeler, C. Steinert, B. Moss, C. A. Mesa, B. Kaiser, W. Jaegermann, L. Francas, S. Gimenez and J. R. Durrant, Sustainable Energy Fuels, 2020 Search PubMed , in press.
- K. Welter, N. Hamzelui, V. Smirnov, J. P. Becker, W. Jaegermann and F. Finger, J. Mater. Chem. A, 2018, 6, 15968–15976 RSC.
- N. Wang, M. Liu, J. Liang, T. Li, H. Tan, B. Liu, Q. Zhang, C. Wei, Y. Zhao and X. Zhang, Catal. Sci. Technol., 2017, 7, 5608–5613 RSC.
- F. Friedhelm, W. Katharina, U. Félix, S. Vladimir, K. Bernhard and J. Wolfram, Z. Phys. Chem., 2019, 20191453, DOI:10.1515/zpch-2019-1453.
- X. Zhou, J. Zhou, G. Huang, R. Fan, S. Ju, Z. Mi and M. Shen, J. Mater. Chem. A, 2018, 6, 20297–20303 RSC.
- J. Mohammed-Ibrahim, J. Power Sources, 2020, 448, 227375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00355g |
| This journal is © The Royal Society of Chemistry 2020 |