
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thorough analysis and categorization of bacterial interrupted adenylation domains, including previously unidentified families†
Taylor A.
Lundy‡
,
Shogo
Mori‡
 and
Sylvie
Garneau-Tsodikova
*
and
Sylvie
Garneau-Tsodikova
*
University of Kentucky, Department of Pharmaceutical Sciences, College of Pharmacy, Lexington, KY 40536-0596, USA. E-mail: sylviegtsodikova@uky.edu
First published on 18th August 2020
Abstract
Interrupted adenylation (A) domains are key to the immense structural diversity seen in the nonribosomal peptide (NRP) class of natural products (NPs). Interrupted A domains are A domains that contain within them the catalytic portion of another domain, most commonly a methylation (M) domain. It has been well documented that methylation events occur with extreme specificity on either the backbone (N-) or side chain (O- or S-) of the amino acid (or amino acid-like) building blocks of NRPs. Here, through taxonomic and phylogenetic analyses as well as multiple sequence alignments, we evaluated the similarities and differences between interrupted A domains. We probed their taxonomic distribution amongst bacterial organisms, their evolutionary relatedness, and described conserved motifs of each type of M domain found to be embedded in interrupted A domains. Additionally, we categorized interrupted A domains and the M domains within them into a total of seven distinct families and six different types, respectively. The families of interrupted A domains include two new families, 6 and 7, that possess new architectures. Rather than being interrupted between the previously described a2–a3 or a8–a9 of the ten conserved A domain sequence motifs (a1–a10), family 6 contains an M domain between a6–a7, a previously unknown interruption site. Family 7 demonstrates that di-interrupted A domains exist in Nature, containing an M domain between a2–a3 as well as one between a6–a7, displaying a novel arrangement. These in-depth investigations of amino acid sequences deposited in the NCBI database highlighted the prevalence of interrupted A domains in bacterial organisms, with each family of interrupted A domains having a different taxonomic distribution. They also emphasized the importance of utilizing a broad range of bacteria for NP discovery. Categorization of the families of interrupted A domains and types of M domains allowed for a better understanding of the trends of naturally occurring interrupted A domains, which illuminated patterns and insights on how to harness them for future engineering studies.
Introduction
Historically, natural products (NPs), secondary metabolites made by plants, bacteria, and fungi that are not essential for normal growth and development, have been the foundation for modern medicine, leading to life-saving medications such as vancomycin and paclitaxel.1 These biologically active molecules are produced by these organisms via biosynthetic pathways, a set of proteins that work together to create the final NP, encoded in their genome. With the boom in genomic data available over the last few decades, we are beginning to understand and harness the full potential of these biosynthetic pathways. It is now understood that, even with all the incredible work that has been done, we have barely scratched the surface in terms of understanding the intricacies and nuances of NP production.Nonribosomal peptides (NRPs) are one of the major classes of NPs. They are biosynthesized using amino acid (or amino acid-like) building blocks by nonribosomal peptide synthetase (NRPS) mega-enzymes in an assembly-line fashion. Each NRPS mega-enzyme can be divided into modules, and those modules are further subdivided into domains. It is these domains that are responsible for carrying out individual biosynthetic steps. Each module is comprised, at minimum, of an adenylation (A), a condensation (C), and a thiolation (T) domain. The NPRS cycle2 starts with amino acid activation by the A domain via adenylation. Each A domain is specific for a particular amino acid or a set of structurally similar amino acids. This specificity is dictated by the binding pocket of the A domain, which accommodates the variable side chains of amino acids.3 The T domain must be converted from its inactive (apo) to its active (holo) state by the addition of a 4′-phosphopantetheine (Ppant) prosthetic arm, transferred from coenzyme A (CoA) by a 4′-phosphopantetheinyltransferase.4 This Ppant arm of the T domain is long and flexible, and once the activated amino acid is covalently attached, the T domain transfers the amino acid to subsequent catalytic pockets, such as that of the C domain, where condensation of amino acids takes place. Further modifications of the NRP can be done through the action of auxiliary domains that decorate the NRP substrate with additional chemistry. These auxiliary domains, such as methylation (M), epimerization (E), halogenation (HAL), ketoreduction (KR), and oxygenation/monooxygenation (Ox/MOx) domains, are vital in providing structural complexity and diversity amongst NRPs.5,6
In recent years, A domains have proved to be complex and fascinating, especially with regard to interrupted A domains. Generically, the structure of A domains (Fig. 1A) contains a core N-terminal domain and a small C-terminal subdomain as well as ten conserved sequence motifs (a1–a10).3,7 In order to perform its functions, the core and subdomain cycle through open, closed, and thiolation conformations.8–10 Interrupted A domains are A domains that harbor the catalytic portion of an auxiliary domain within their structures, creating a multifunctional protein that can adenylate and derivatize amino acid substrates. The most common type of interruption observed is an M domain, however there have been reports of KR and Ox/MOx domains within A domains.6,11,12 Recently though, it was shown that KR domains are not true interruptions of A domains, but instead, consist of an intact A domain with a KR domain immediately after followed by a “pseudo-A subdomain”.13 In 2018, the first structure of an interrupted A domain that embeds an M domain was published (Fig. 1B),14 which illuminated two key aspects of interrupted A domains: (i) the overall folding and placement of the core and subdomain of A domains was maintained, allowing them to function normally, and (ii) the M domain contains a Rossmann-like fold, characteristic of class I methyltransferases (Fig. 1B, light green outline). There are five structurally different classes (I–V) of methyltransferases (Fig. 2C), of which class I S-adenosyl-L-methionine (SAM)-dependent methyltransferase is the most common class.15,16
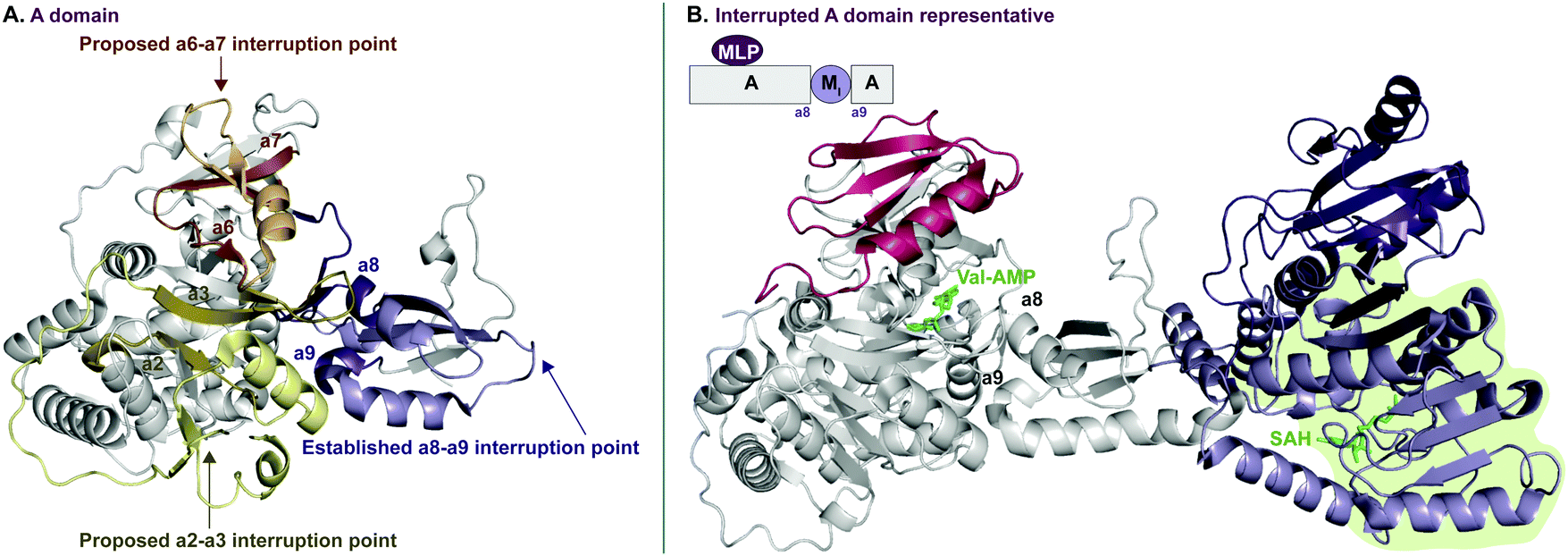 | ||
| Fig. 1 Crystal structure of (A) non-interrupted A domain EntF(A) (PDB ID: 5T3D)8 and (B) interrupted A domain TioS(A8MIA9)4 (PDB ID: 5WMM).14 In the EntF(A) structure (panel A), a2–a3, a6–a7, and a8–a9 conserved sequence motifs of A domains where M domain interruptions are observed/proposed, are colored light yellow, orange, and purple, respectively. In the TioS(A8MIA9)4 structure (panel B), the partner protein, MbtH-like protein (MLP, TioT), of TioS(A8MIA9)4 is colored red. The M domain interrupted between a8–a9 motifs is colored purple where the class I methyltransferase conserved region is light purple and the region for a structural purpose is dark purple. The ligands, L-Val-AMP in the A domain active site and S-adenosylhomocysteine (SAH) in the M domain active site, are colored green. The Rossmann-like fold of the M domain structure is highlighted in light green. | ||
 | ||
| Fig. 2 A representation of (A) the seven families of interrupted A domains, nomenclature used, representative for each of the known families, type of M domain, and the type(s) of methylation they carry out, (B) a schematic showing where in the A domain each type of M domain is found in this manuscript, and (C) the five classes of methyltransferases. Note: aa indicates amino acid. | ||
Interruptions within A domains are known to occur in specific locations. There have been reports of interrupted A domains with embedding M domains between the a2–a317–20 and a8–a914,21,22 regions of A domains. Originally, A domains interrupted between a2–a3 were proposed to be inactive.23,24 The M domains between a8–a9 were originally reported to be located between the A and T domains.25 Additionally, there have been reports of two back-to-back M domains between a8–a9.26–30 Recent work has demonstrated that interrupting M domains can perform backbone N-methylation (Mb) or side chain O- or S-methylation (Ms(O) and Ms(S), respectively).14,17,18,22,26 In order to assess the true prevalence and abundance of these interrupted A domains, we set out to search the National Center for Biotechnology Information (NCBI) database for the purpose of identifying, categorizing, and establishing the distribution of interrupted A domains exclusively amongst bacteria, although interrupted A domains from fungi do exist.31 Through our efforts, we were able to identify seven distinct families (1–7, of which families 2 and 5 were further divided into two (2a and 2b) and three (5a, 5b, and 5c) subfamilies, respectively) of interrupted A domains containing six types of interrupting M domains (I–VI) (Fig. 2A). The families of interrupted A domains were classified based on the position of interruption as well as methylation regiospecificity (N-, O-, or S-). Of these families of interrupted A domains, family 1 is the only one with a known structure.14 Families 1, 2a, 3, and 5a contain representative interrupted A domains that have been biochemically characterized.14,17,18,22,26 Families 2b, 4, 5b, and 5c have the interrupted A domains published in a biosynthetic pathway for a known natural product. Though the interrupted A domain itself in these families have not been studied individually, their substrate and methylation activity were inferred based on elucidation of the pathway and/or creation of knockouts of those proteins.19,20,28–30,32 Families 6 and 7 represent never before identified interrupted A domains’ architectures. Family 6 contains an M domain not between the previously reported a2–a3 or a8–a9, but between a6–a7 of the ten conserved A domain motifs. Family 7 comprises di-interrupted A domains with two M domains, one embedded between a2–a3 and the other between a6–a7. We established nomenclature for each family of interrupted A domains as indicated in Fig. 2A. The subscripts of the A domain represent the interruption point by M domains (e.g., A8MXA9 means that the MX domain is embedded between a8–a9), and the subscripts of M domains depict methylation site of the substrate, where “b” and “s” indicate backbone and side chain methylation, respectively. Specific information about the regiospecificity of side chain methylation is displayed in parentheses (e.g., Ms(O,Ser/Thr) indicates that this M domain catalyzes side chain O-methylation on Ser or Thr). For simplicity, the information about the M domains is replaced with types (I–VI) of M domains in the representative interrupted A domains. The M domain types were categorized based on similarities of amino acid sequences. Each type of M domain is proposed to have specific substrate and interruption point in the A domain (Fig. 2A and B), which is discussed in detail in the “Phylogenetic tree analyses” section in the results and discussion.
Results and discussion
Construction of data sets for taxonomic and phylogenetic tree analyses
To bioinformatically study interrupted A domains, we first created data sets of interrupted A domain protein sequences by obtaining information from the NCBI database by protein BLAST (basic local alignment search tool) search on the Genome Workbench software. The BLAST searches were performed using characterized and/or reported interrupted A domains for each family (Fig. 2A) (TioS(A8MIA9)4,14 KtzH(A8MIIA9)422/ThxA2(A8MIIIA9)6,32 TioN(A2MIVA3),17 TtbB(A2MVA3)5,19,20 and ColG(A8MIIMIA9)26/DidJ(A8MIIIMIA9)11,29 for families 1 (A8MbA9), 2a/b (A8Ms(O)A9), 3 (A2Ms(S)A3), 4 (A2MbA3), and 5a/b (A8Ms(O)MbA9), respectively) as query sequences. The newly discovered families 6 (A6Ms(O,arom)A7) and 7 (A2MbA3–6Ms(O,arom)A7) were identified during this process while investigating family 4. The data sets were shaped by setting a threshold of real (true interrupted A domain)/random hits by the “Query Cover” values (the percentage of the query sequence that overlaps with the reference sequence) for the four most common families of interrupted A domains (families 1–4) or by examining top hits individually for the three uncommon families of interrupted A domains (families 5–7). The duplicated NCBI IDs, proteins from unknown (environmental) sources, as well as proteins from other families were removed from each data set. These analyses resulted in sample sizes of n = 536 for family 1, n = 149 for family 2a, n = 79 for family 2b, n = 64 for family 3, n = 188 for family 4, n = 11 for family 5a/b, n = 14 for family 6, and n = 3 for family 7.From these data sets, we found that almost all family 3 (A2Ms(S)A3) and many of family 4 (A2MbA3) are stand-alone (i.e., not a part of an NRPS module) interrupted A domains. Most of the interrupted A domains in the remaining families are paired with other NRPS domains, such as C and T domains. This trend suggests that the A domain's interaction with other domains is maintained when the interruption occurs in the later part of the A domain (between a6–a7 or a8–a9). Such plasticity is best preserved in family 1 (A8MbA9). There are many NPRS proteins that contain multiple family 1 interrupted A domains. The most dramatic display of this is accession number AIW58892.1, which contained six interrupted A domains in a single protein. This feature of family 1 results in many huge proteins that have more than 1 MDa molecular weight. We also found two (WP_084161146.1 and WP_134733373.1) and one (WP_087914619.1) proteins that contained multiple interrupted A domains of families 2a (A8Ms(O,Ser/Thr)A9) and 4 (A2MbA3), respectively. No proteins in these families were found to consist of more than two interrupted A domains. There was one protein (WP_141643221.1) that contained families 2a and 6 (A6Ms(O,arom)A7) interrupted A domains.
Taxonomic tree analyses
To analyze how widespread interrupted A domains are amongst bacteria, we created taxonomic trees of each family of interrupted A domains from the data sets obtained above (Fig. 3 and Fig. S1–S7, Tables S1–S7, ESI†). Family 5c (A8Ms(O,Ser/Thr)A9–10MbA9) was not subjected to taxonomic analysis because it only has one member. Family 7 (A2MbA3–6Ms(O,arom)A7) was not subjected to taxonomic analysis either because all three examples found were in the phylum of FCB (Fibrobacteres, Chlorobi, and Bacteroidetes) group. These taxonomic analyses showed that the interrupted A domains are commonly found in five bacterial phyla: Actinobacteria, Cyanobacteria, Firmicutes, Proteobacteria, and FCB group, where three of these (Actinobacteria, Cyanobacteria, and Firmicutes) and two others (Proteobacteria and FCB group) are broadly categorized as terrabacteria and hydrobacteria, respectively, depending on where they were exposed to environmental pressure during evolution. It is important to note that (i) although there are fungal interrupted A domains, such as a family 1 (A8MbA9) interrupted A domain in the NRPSXY protein in Xylaria sp. BCC1067,31 they were not included in our analyses as the focus of this manuscript is bacterial interrupted A domains, and (ii) there are a few family 4 (A2MbA3) interrupted A domains found in genome sequencing data from sponges (such as XP_028402765 and XP_031561672), which could have originated from symbiotic bacterial species since these genes are found in NCBI under “unplaced genomic scaffolds” of whole genome sequencing data.33 However, since the source of these interrupted A domain genes is not experimentally confirmed to be sponge or bacteria, these data were not included in our analyses. It was found that the distribution of organisms that contain interrupted A domains is significantly diverse and not equal between families of interrupted A domains. For example, the majority (95%) of family 1 interrupted A domains are found in Actinobacteria. The ratios of actinobacterial interrupted A domains are lower in other families, for example, families 2a (A8Ms(O,Ser/Thr)A9), 2b (A8Ms(O,Tyr)A9), 3 (A2Ms(S)A3), 4 (A2MbA3), 5 (A8Ms(O)MbA9), and 6 (A6Ms(O,arom)A7) contain 59%, 61%, 0%, 3%, 0%, and 21%, respectively. In these families of interrupted A domains, ratios of other bacterial phyla are observed at a higher percentage, such as FCB group, Proteobacteria, Cyanobacteria, and Firmicutes. Intriguingly, Firmicutes in family 4 accounts for a large portion (43%) of the organisms (of that 43%, 81% are from the class of Bacilli), although Firmicutes are rare in other interrupted A domain families, there are none in families 1, 2b, and 5, 0.7% in family 2a, 22% in family 3, and 7% in family 6.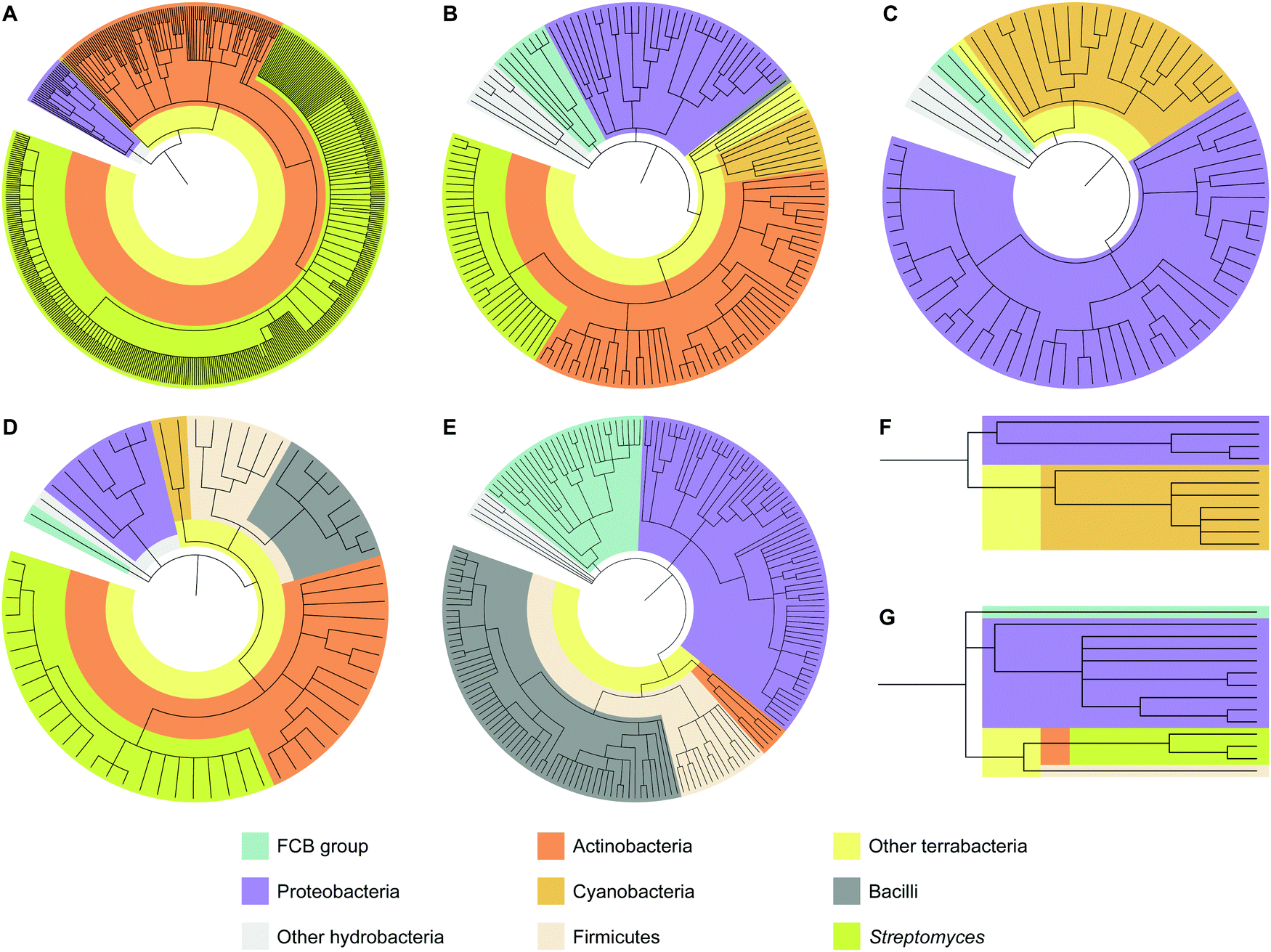 | ||
| Fig. 3 Taxonomic trees of interrupted A domains for (A) family 1 (n = 536), (B) family 2a (n = 149), (C) family 2b (n = 79), (D) family 3 (n = 64), (E) family 4 (n = 188), (F) family 5 (n = 11), and (G) family 6 (n = 14). Phyla of FCB group, Proteobacteria, Actinobacteria, Cyanobacteria, and Firmicutes are colored turquoise, light purple, dark orange, light orange, and light peach, respectively. Other hydrobacteria and terrabacteria are colored light grey and light yellow, respectively. The class of Bacilli and the genus of Streptomyces are colored dark grey and light olive, respectively. The NCBI ID numbers, range of sequences used for these analyses, and organisms are available in Fig. S1–S7 and Tables S1–S7 (ESI†). | ||
Interestingly, the bacteria that contain family 6 (A6Ms(O,arom)A7) interrupted A domains are very diverse and observed in four different phyla: FCB group, Proteobacteria, Actinobacteria, and Firmicutes, even though the sample size of this family is very small (n = 14). These phyla diversity are greater than that observed for family 1 (A8MbA9), which was discovered in only three phyla: Proteobacteria, Cyanobacteria, and Actinobacteria, even though this family contained the largest sample size (n = 536). These taxonomic analyses could also explain why family 1 has been studied and reported the most and why families 2b (A8Ms(O,Tyr)A9) and 4 (A2MbA3) have been described the least in the literature within the four common families (families 1–4), even though the BLAST search results of family 4 are greater in number (n = 188) than that of families 2a (A8Ms(O,Ser/Thr)A9) (n = 149) and 3 (A2Ms(S)A3) (n = 64). Interrupted A domains in families 1, 2a, and 3 are well distributed within Actinobacteria, such as Streptomyces species, which have been extensively studied as NP producers because this genus has a great number of NP gene clusters and is easy to cultivate in standard laboratory settings.34 However, families 2b and 4 are not found in this genus and, thus, have not been well studied.
Phylogenetic tree analyses
To better understand the diversity of each family of interrupted A domains, phylogenetic trees were constructed by utilizing the data sets obtained above (Fig. 4 and Fig. S6–S12, Tables S1–S7, ESI†). The phylogenetic trees show that the interrupted A domains in the same bacterial phylum are relatively homologous to each other. This suggests that interrupted A domain genes tend to be transferred within the same phylum and/or that the species are further diversified after obtaining these genes. However, there are some interrupted A domains, which are in the same phylum, significantly separated on the phylogenetic tree. Examples include Proteobacteria in family 1 (A8MbA9), FCB group/Proteobacteria in family 2a (A8Ms(O,Ser/Thr)A9), and FCB group in family 4 (A2MbA3). This indicates that the above inter-phyla evolutionally events happened multiple times while exchanging biosynthetic genes, including those for interrupted A domains.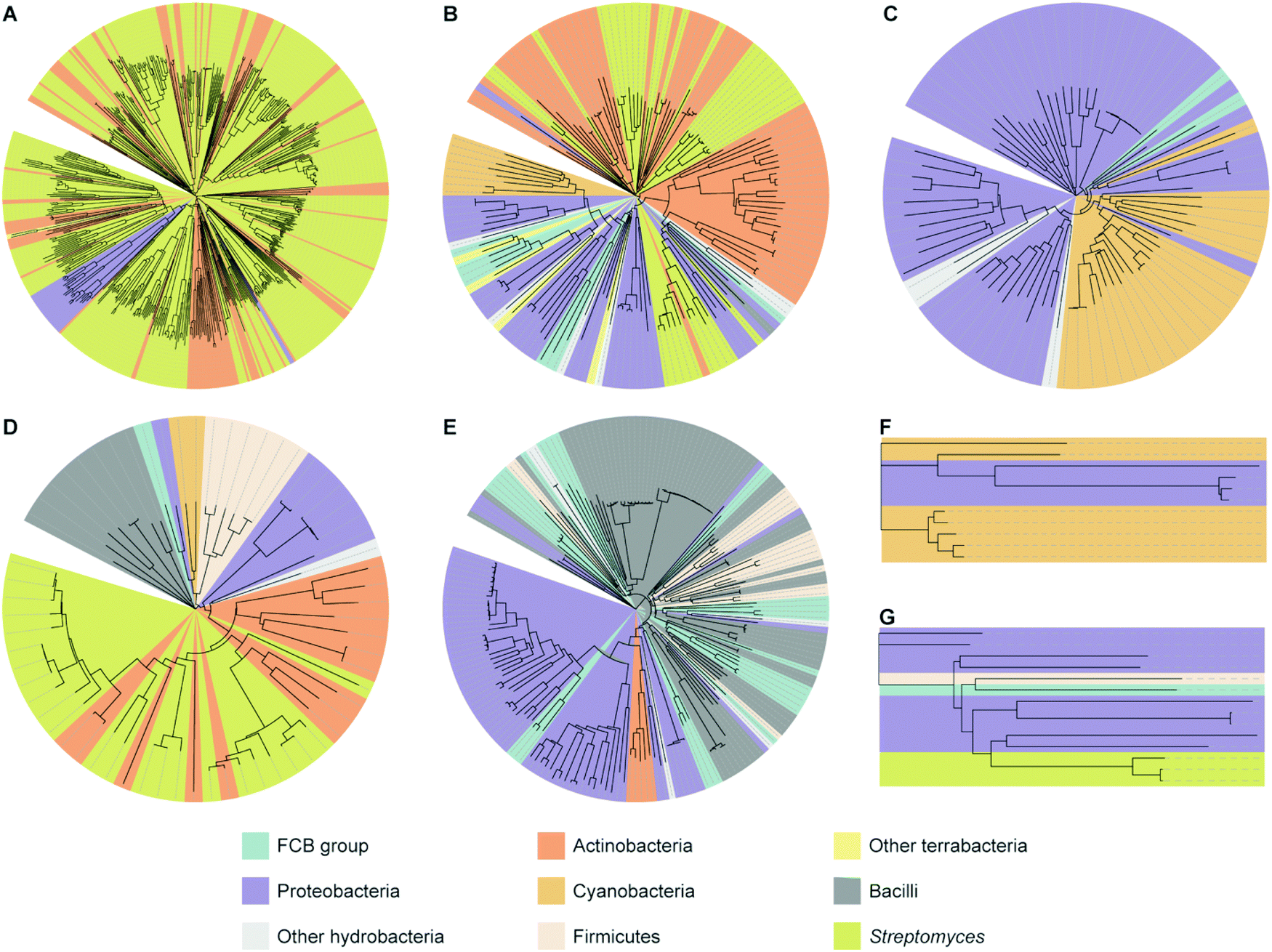 | ||
| Fig. 4 Phylogenetic trees of interrupted A domains for (A) family 1 (n = 536), (B) family 2a (n = 149), (C) family 2b (n = 79), (D) family 3 (n = 64), (E) family 4 (n = 188), (F) family 5 (n = 11), and (G) family 6 (n = 14). Phyla of FCB group, Proteobacteria, Actinobacteria, Cyanobacteria, and Firmicutes are colored turquoise, light purple, dark orange, light orange, and light peach, respectively. Other hydrobacteria and terrabacteria are colored light grey and light yellow, respectively. The class of Bacilli and the genus of Streptomyces are colored dark grey and light olive, respectively. The NCBI ID numbers, range of sequences used for these analyses, and organisms are available in Fig. S6–S12 and Tables S1–S7 (ESI†). | ||
Based on phylogenetic tree analyses, pairs of interrupted A domains that reside on the same proteins can be categorized into two subgroups, which could have evolved by two distinct pathways. In the first subgroup, a pair of interrupted A domains in a single protein is highly homologous (sitting next to or very close to each other on the phylogenetic tree) (burgundy balloons in Fig. S8, S9, and S12, ESI†). This suggests that the origin of these interrupted A domains is identical and copied one from the other, or that genes coding interrupted A domains were duplicated during a gene transfer event. Interrupted A domains of the first subgroup were found in families 1 (A8MbA9), 2a (A8Ms(O,Ser/Thr)A9), and 4 (A2MbA3). In the second subgroup, interrupted A domains that lie on the same protein are heterologous (significantly separated on the phylogenetic tree), implying that interrupted A domains in this subgroup were derived from independent sources. Interrupted A domains of the second subgroup were found exclusively in family 1 (orange balloons in Fig. S8, ESI†). These two subgroups of interrupted A domain pairs are likely the result of different evolutionary pathways.
In an effort to better understand the similarities and differences in the M domains found embedded within A domains, we expanded the phylogenetic analysis to compare M domains from the seven different families of interrupted A domains. The sequences of the M domains from each family of interrupted A domains were extracted (using the M domain regions specified by the multiple sequence alignments; Fig. S13–S21, ESI†) and aligned to construct a phylogenetic tree (Fig. 5). This phylogenetic tree revealed six distinct clusters of M domains, which we designated as types I–VI (Fig. 2A). We assigned type I M domain to the Mb domains of families 1 (A8MbA9) and 5 (A8Ms(O)MbA9). It was revealed that there are two distinct types of Ms domains in families 2a/b (A8Ms(O)A9) and 5a/b depending on substrate specificity (Ser/Thr vs. Tyr). We assigned type II to the Ms(O,Ser/Thr,a8–a9) domain of families 2a and 5a, and type III to the Ms(O,Tyr,a8–a9) domain of families 2b and 5b. The M domains from families 3 (A2Ms(S)A3) and 4 (A2MbA3) also formed their own clusters, which led us to assign type IV to the Ms(S) domain of family 3 and type V to the Mb domain of family 4. We found that the M domains between a6–a7 in the newly identified families 6 (A6Ms(O,arom)A7) and 7 (A2MbA3–6Ms(O,arom)A7) also formed their own cluster. We appointed this M domain type VI. The phylogenetic analysis of the M domains also depicted the relatedness between the different M domain types. For example, Mb domains of families 5 and 7 di-interrupted A domains were found very close to or within the clusters of those of families 1 and 4, respectively (which are types I and V M domains, respectively), which strongly suggests that Mb domains of families 5 and 7 were derived from those of families 1 and 4. The novel family 7 interrupted A domains are di-interrupted ones with a2–a3 interruption of type V and a6–a7 interruption of type VI, thus, are strongly suggested to be derived from combinations of families 4 and 6.
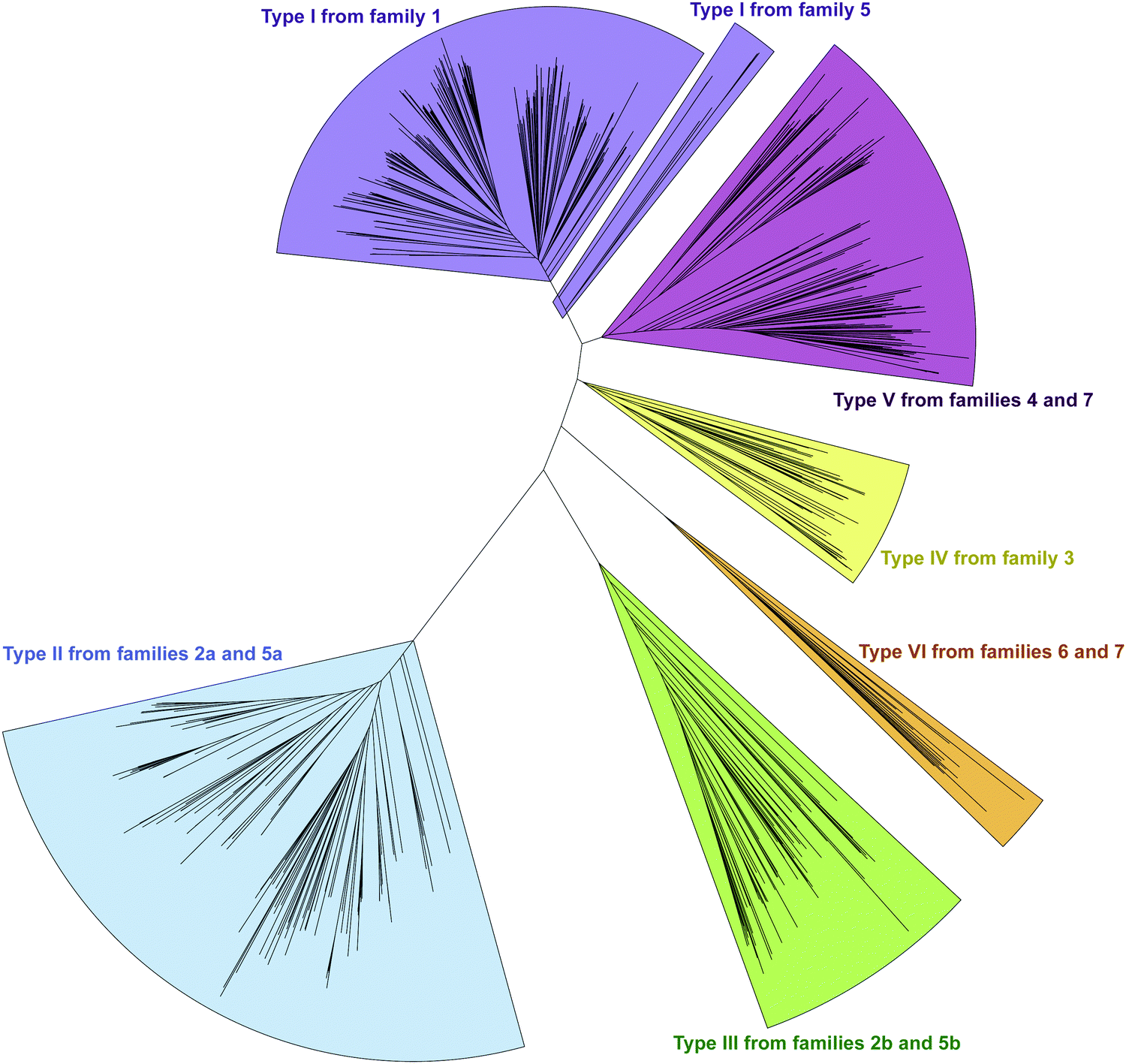 | ||
| Fig. 5 Phylogenetic tree of M domains embedded in interrupted A domains. The M domains used in this analysis were extracted from interrupted A domains family 1 (n = 499), family 2a (n = 148), family 2b (n = 79), family 3 (n = 63), family 4 (n = 188), family 5 (n = 11 for each M domain), family 6 (n = 14), and family 7 (n = 3 for each M domain). Clusters for Mb(a8–a9) (type I), Ms(O,Ser/Thr,a8–a9) (type II), Ms(O,Tyr,a8–a9) (type III), Ms(S,a2–a3) (type IV), Mb(a2–a3) (type V), and Ms(O,arom,a6–a7) (type VI) domains are colored light purple, light blue, light green, light yellow, light pink, and light orange, respectively. | ||
Interestingly, the Ms(O) domains of families 2 (A8Ms(O)A9) and 5 (A8Ms(O)MbA9) could be divided into two distinct clusters (M domains from subfamilies 2a/b and 5a/b as discussed above) based on their amino acid sequences depicted by the phylogenetic tree of the M domains (Fig. 5). One cluster is comprised of Ms(O) domains of those members homologous to KtzH(A8MIIA9)4 (family 2a) and two members (out of a total of 11) of back-to-back interrupted A domains (ColG(A8MIIMIA9) and an uncharacterized protein RKH86437.1) (family 5a). However, the Ms(O) domains homologous to ThxA2(A8MIIIA9)6 (family 2b) and nine other members of back-to-back interrupted A domains (family 5b) formed an independent cluster, reflecting an independently unique type (III) of O-methyltransferase. While both types (II and III) of M domains are predicted O-methyltransferases, the difference between them can be attributed to their substrate specificity. Type II likely carries out O-methylation of L-Ser/L-Thr, whereas type III likely carries out O-methylation of L-Tyr (or related non canonical amino acids). We reached this conclusion by a combination of substrate predictions, alignments, and known substrates of characterized interrupted A domains or those with the published NP biosynthetic pathway. The representative for type II M domains, KtzH(A8MIIA9)4,22 has the same L-Ser substrate as ColG(A8MIIMIA9), the representative for family 5a.26 The conserved motifs for type II M domain can only be found in the two members of family 5a, whereas type III M domains have a different set of M domain motifs (Fig. 7, 9, Fig. S14, S15, S18, and Table 1). For type III from family 2b interrupted A domain ThxA2(A8MIIIA9)6 as well as two of the nine members of family 5b, DidJ(A8MIIIMIA9) and VatN(A8MIIIMIA9), the NP biosynthetic pathways are published,28,29,32 which indicate their substrate to be L-Tyr. Additionally, the substrates of several members of family 2b and all nine members of family 5b were predicted (using exclusively the A domain portion of the amino acid sequence, without the M domain portion, designated by the alignments) by the website Non-Ribosomal Peptide Synthase Substrate Predictor (NRPSsp)35 to be mostly L-Tyr or L-Phe (with high values of prediction-conditioned fall out (higher probability of errors)), which indicates that their substrates are likely L-Tyr or its analogues. The same reasoning and analyses were also performed for families 6 (A6Ms(O,arom)A7) and 7 (A2MbA3–6Ms(O,arom)A7). The M domains of family 6 formed their own cluster in the phylogenetic analysis of the M domains, indicating that it is not the same as types I–V M domains. However, since there is no known representative or corresponding NP, we relied on substrate predictions by NRPSsp, conserved M domain motifs, and NCBI's “Identify Conserved Domains” function. We predicted the substrates of the A domains and found that most of their substrates were L-Phe with high values of prediction-conditioned fallout. Additionally, aside from the SAM binding motif (present in all class I methyltransferases), there were no conserved motifs from any other types I–V M domains. When predicted with the NCBI's “Identify Conserved Domain” function, type VI M domains had three of five conserved domains (amino acid regions that dictate specific family of enzymes) identified as O-methyltransferases related to those involved in ubiquinone biosynthesis (Table S8, ESI†). These predictions suggest that the substrates for type VI M domains from families 6 and 7 are hydroxylated aromatic molecules, likely derived from other biosynthetic pathways, such as polyketide synthases (PKSs). In fact, when looking at other genes surrounding family 6 or 7 interrupted A domains, there are genes that encode PKSs and/or oxidoreductases in many cases. Therefore, we hypothesize that type VI M domains are a type of Ms(O) domain, which act on hydroxylated aromatic substrates.
| Type of M domain | Nomenclature | Core | Previously called/published | Conserved sequence | # of amino acids per M domain |
|---|---|---|---|---|---|
| I | Mb(a8–a9) | mb(a8–a9)i | Motif 125 | (D/L)Fx(GWxS)(S/N)Y | 390 ± 21 |
| mb(a8–a9)ii | Motif 225/Motif I36 | δ(L/x)E(I/L)GxGxG* | |||
| mb(a8–a9)iii | Motif II/Y36 | x(Y/I)(W/x)(G/A)(T/I)DxS | |||
| mb(a8–a9)iv | Motif 325/Motif IV36 | Dx(V/I)δδ(N/S)S(V/I)δQYFPxxxYL | |||
| mb(a8–a9)v | Motif 425 | (E/D)xELδδ(D/A/S)Px(F/W/L)F | |||
| mb(a8–a9)vi | Motif 525 | NE(L/M)x(K/R/Q)(F/Y/H)RY | |||
| II | Ms(O,Ser/Thr,a8–a9) | ms(O,Ser/Thr,a8–a9)i | N/A | EIFxxxxYxxxG | 290 ± 13 |
| ms(O,Ser/Thr,a8–a9)ii | Motif I36 | (V/I)(F/I/V)DVGx(N/H)xG(L/M)F(S/T)L* | |||
| ms(O,Ser/Thr,a8–a9)iii | N/A | EPδP(E/P)xxxxx(R/A/E)xN | |||
| ms(O,Ser/Thr,a8–a9)iv | N/A | FT(Y/F)YPxx(S/T)x(L/M)SG | |||
| ms(O,Ser/Thr,a8–a9)v | N/A | (I/V)DLLKδD(V/A)ExxE | |||
| ms(O,Ser/Thr,a8–a9)vi | N/A | WxxIxQδxxEVH | |||
| III | Ms(O,Tyr,a8–a9) | ms(O,Tyr,a8–a9)i | Motif I36 | V(V/L)(E/D)(I/V)GxGxxA* | 350 ± 10 |
| ms(O,Tyr,a8–a9)ii | N/A | GDδxxδx(L/I)PEx(A/V)(D/E)xC(V/I)SEI(V/I/F)GxI | |||
| ms(O,Tyr,a8–a9)iii | N/A | (I/V)FxxxxxxFDLR | |||
| ms(O,Tyr,a8–a9)iv | N/A | WLPV(F/Y)(F/L)Pδ | |||
| IV | Ms(S,a2–a3) | ms(S,a2–a3)i | N/A | VLEδGxGxG* | 260 ± 25 |
| ms(S,a2–a3)ii | N/A | Dδ(V/I)δ(L/I)AS(T/V/A)δQF(F/L)PxxxY(L/T) | |||
| ms(S,a2–a3)iii | N/A | x(E/V)LxxR(Y/F)D | |||
| V | Mb(a2–a3) | mb(a2–a3)i | Motif 125 | xxGGWx(S/N)x(Y/F) | 270 ± 6 |
| mb(a2–a3)ii | Motif 225/Motif I36 | (V/I)LE(I/L)Gx(A/S/G)xG* | |||
| mb(a2–a3)iii | Motif II/Y36 | xY(V/Y/L)(G/A)(T/V/I)Dδ(S/T/A) | |||
| mb(a2–a3)iv | Motif 325/Motif IV36 | Dx(V/I)(V/I)δNSV(V/I)(Q/E)xFxGx(N/G)Y(L/F) | |||
| mb(a2–a3)vi | Motif 425 | (N/F/P/S)(E/D)Lxx(F/Y)x(F/Y) | |||
| VI | Ms(O,arom,a6–a7) | ms(O,arom,a6–a7)i | N/A | G(V/A/T)(D/E)(F/I/L)GxGxG* | 300 ± 4 |
| ms(O,arom,a6–a7)ii | N/A | GxxxxGδ(D/E)xxPxx(V/I) | |||
| ms(O,arom,a6–a7)iii | N/A | DF(A/V)δ(S/T)x(L/M)xLD(R/Q) | |||
| ms(O,arom,a6–a7)iv | N/A | G(R/K)F(A/S)(I/L)(Q/G)TδLP |
Interestingly, type IV Ms(S,a2–a3) domains are highly related to type V Mb(a2–a3) domains. This was also implied by a BLAST search using the family 3 interrupted A domain TioN(A2MIVA3) as a query sequence, which had significant numbers of hits that overlapped with another BLAST search using family 4 TtbB(A2MVA3)5. However, these overlapped hits were apparently all members of family 3. Such high degree of overlaps in BLAST searches were not observed for any of the other common interrupted A domains.
Sequence analyses and comparisons of the families of interrupted A domains
In order to identify patterns and common trends in the amino acid sequences of these interrupted A domains, we constructed a multiple sequence alignment for each of the seven families of interrupted A domains. The entire sequence alignments can be found in the ESI† (Fig. S13–S21), but here we present shortened versions highlighting important features, such as A and M domain boundaries, conserved sequence motifs, and residues involved or suspected to be involved in substrate or SAM binding (Fig. 6–10). We found, based on conserved regions and the presence of the hallmark SAM binding motif,16 that all types of M domains inserted into A domains fall under the broad umbrella of class I SAM-dependent methyltransferases. For family 1 (A8MbA9) interrupted A domains, this was verified by the structure of the representative of this family, TioS(A8MIA9)4,14 which shows that the type I M domain contains the core Rossmann-like fold consisting of a seven stranded β-sheet sandwiched between three α-helices on each side (Fig. 1B). Overall, we can separate the M domains of interrupted A domains into two broad categories, those that do backbone N-methylation and those that do side chain O- or S-methylation. In the following sections, each type of M domain will be discussed in detail, but for simplicity, we present a summary table of the M domains (Table 1).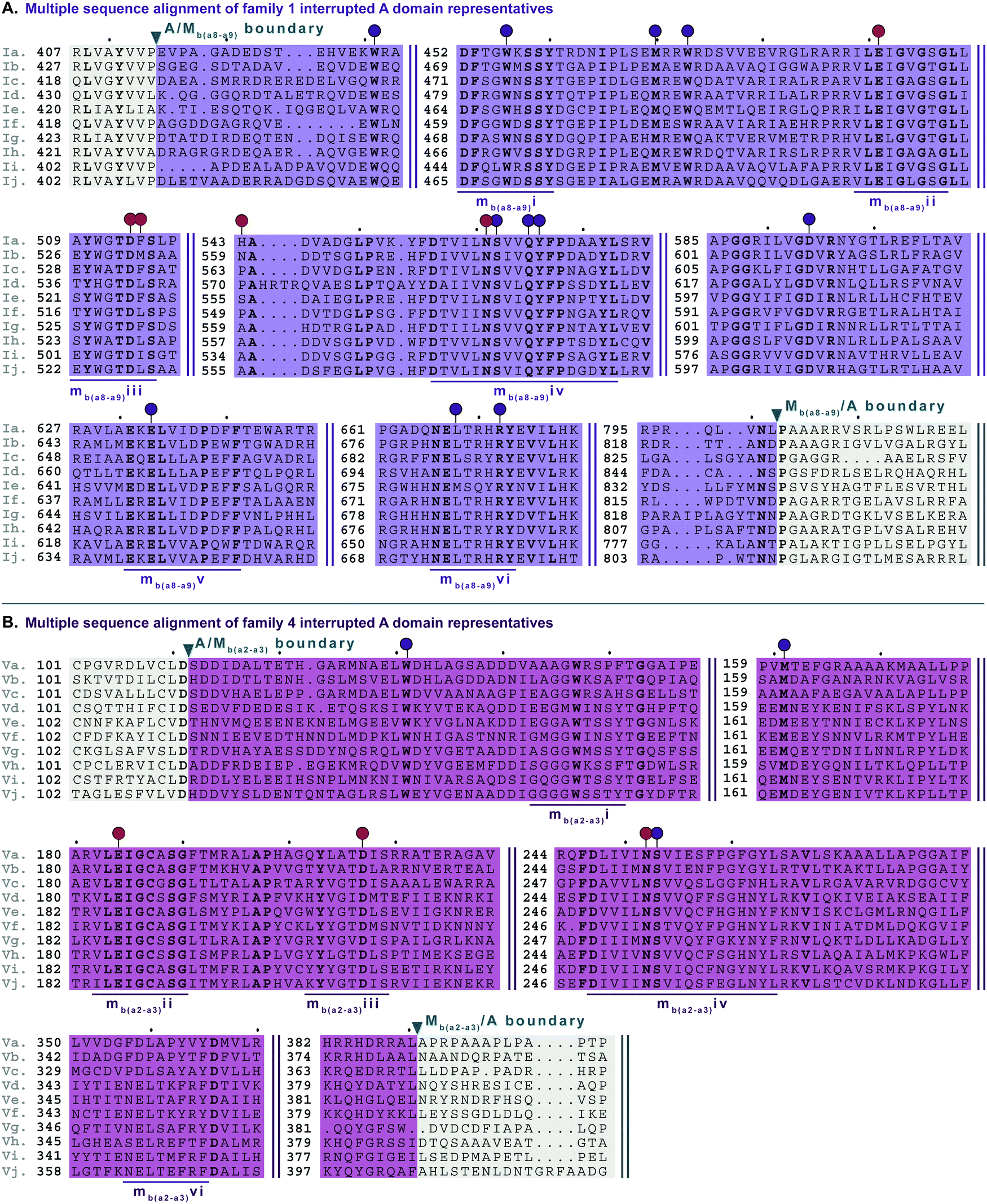 | ||
| Fig. 6 Multiple sequence alignments of (A) family 1 (type I M domain) interrupted A domain representatives as noted in Fig. S1 (ESI†), and (B) family 4 (type V M domain) interrupted A domain representatives as noted in Fig. S5 (ESI†). Types I and V M domains are highlighted in light purple and light pink, respectively. The A domain is highlighted in light grey. The red and dark purple balloons in panel A correspond to residues involved in SAM and amino acid bound Ppant arm binding, respectively, according to the structure of TioS(A8MIA9)4.14 The identical corresponding residues in type V M domain are also indicated in the same way. The conserved M domain motifs for types I and V M domains are underlined in dark purple and dark pink, respectively. The boundaries between the domains are indicated by a triangle. Breaks in the sequences are indicated by two parallel bars. The full sequence alignments and accession numbers are presented in Fig. S13 and S16 (ESI†). | ||
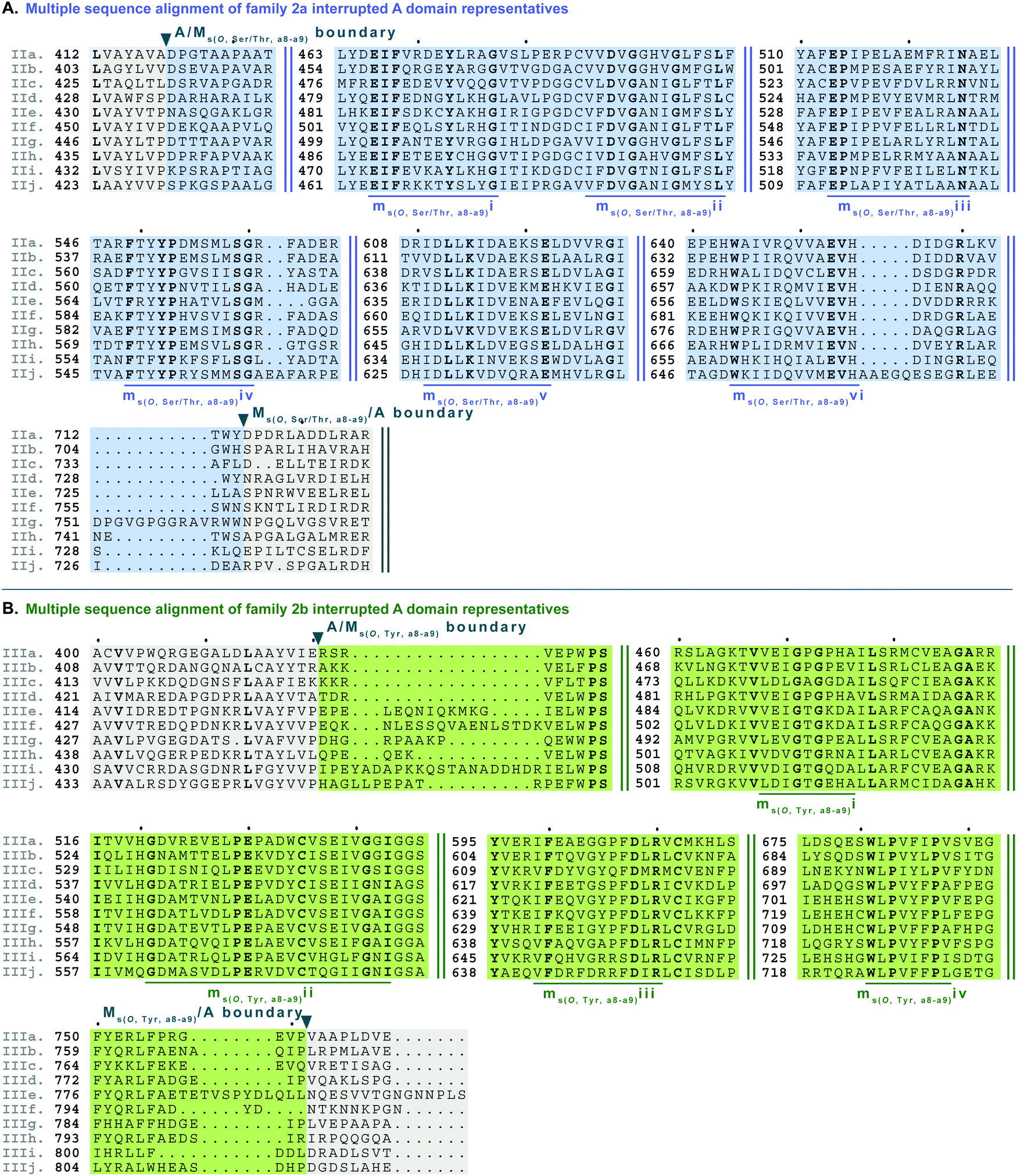 | ||
| Fig. 7 Multiple sequence alignments of (A) family 2a (type II M domain) interrupted A domain representatives as noted in Fig. S2, ESI† and (B) family 2b (type III M domain) interrupted A domain representatives as noted in Fig. S3 (ESI†). Types II and III M domains are highlighted in light blue and light green, respectively. The A domain is highlighted in light grey. The conserved M domain motifs for types II and III M domains are underlined in dark blue and dark green, respectively. The boundaries between the domains are indicated by a triangle. Breaks in the sequences are indicated by two parallel bars. The full sequence alignments and accession numbers are presented in Fig. S14 and S15 (ESI†). | ||
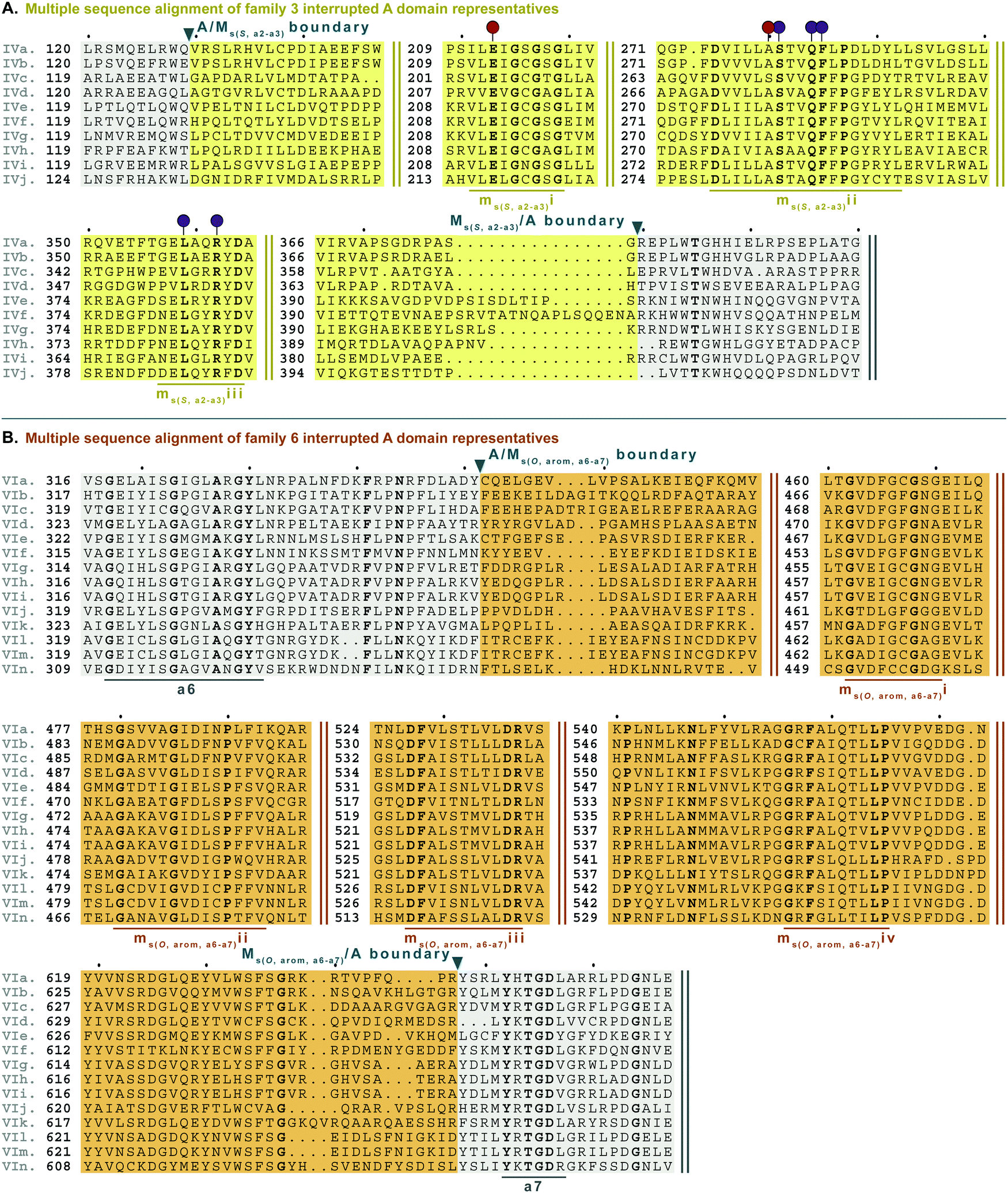 | ||
| Fig. 8 Multiple sequence alignments of (A) family 3 (type IV M domains) interrupted A domain representatives as noted in Fig. S4, ESI† and (B) family 6 (type VI M domains) interrupted A domains. Types IV and VI M domains are highlighted in light yellow and light orange, respectively. The A domain is highlighted in light grey. The red and dark purple balloons in panel A correspond to residues suspected to be involved in SAM and amino acid bound Ppant arm binding, respectively, based on similarities to TioS(A8MIA9)4.14 The conserved M domain motifs for types IV and VI M domains are underlined in dark yellow and dark orange, respectively. The boundaries between the domains are indicated by a triangle. Breaks in the sequences are indicated by two parallel bars. The full sequence alignments and accession numbers are presented in Fig. S16 and S20 (ESI†). | ||
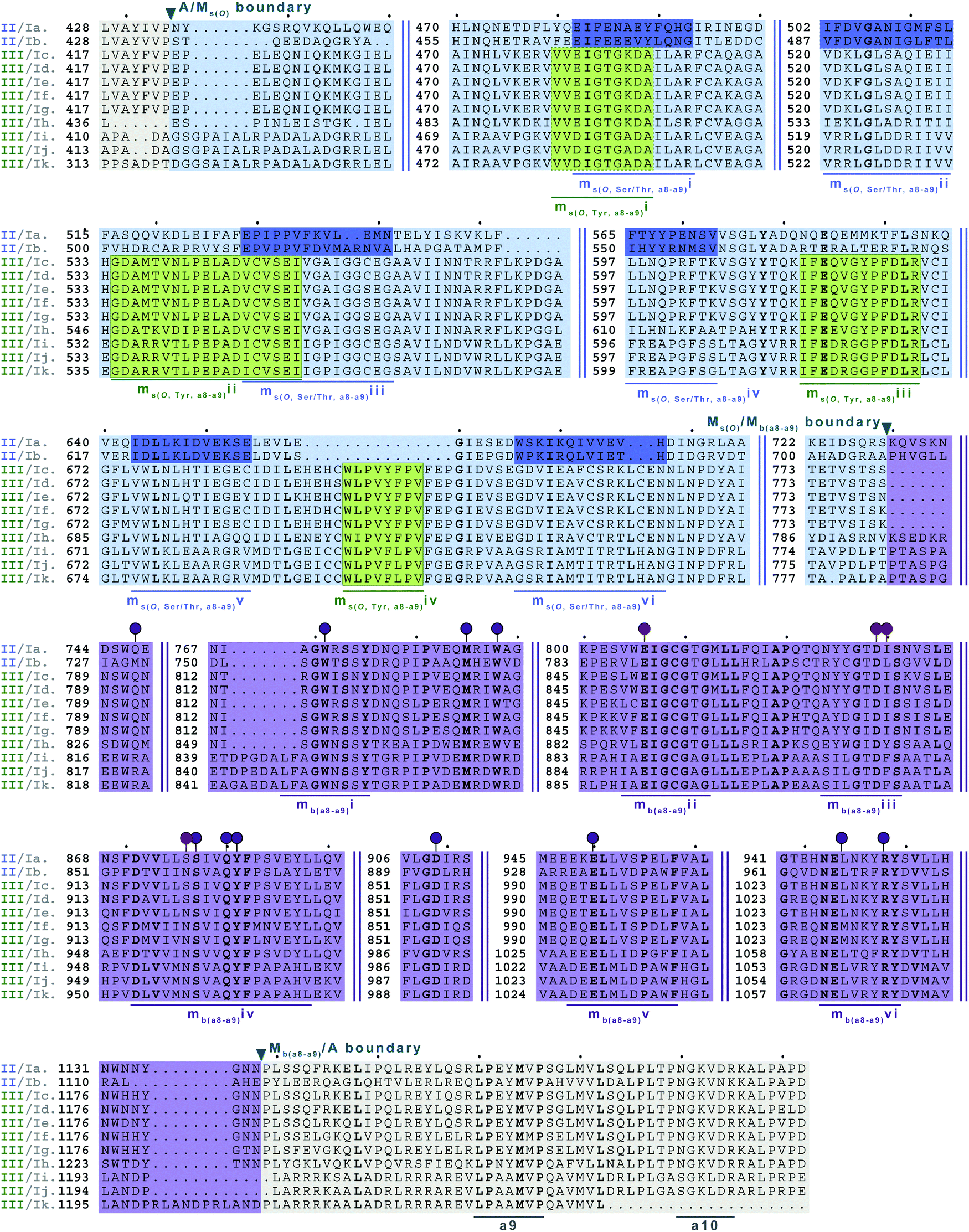 | ||
| Fig. 9 Multiple sequence alignments of families 5a/b interrupted A domains. The A domain portion is highlighted in light grey. The O-methylating M domain is highlighted in light blue. The conserved M domain motifs for type II M domains are boxed and underlined in dark blue. The conserved M domain motifs for type III M domains are boxed in light green and underlined in dark green. Type I M domain is highlighted in purple and the motifs underlined in dark purple. The red and dark purple balloons correspond to residues involved in SAM and amino acid bound Ppant arm binding, respectively, based on their correspondence to the same residues of TioS(A8MIA9)4.14 The boundaries between the domains are indicated by a triangle. Breaks in the sequences are indicated by two parallel bars. The full sequence alignments and accession numbers are presented in Fig. S18 (ESI†). | ||
 | ||
| Fig. 10 Multiple sequence alignments of (A) family 5c interrupted A domain (sequence “a”) alignment with representative type II and type I M domains, KtzH(MII)4 (sequence “b”) and TioS(MI)4 (sequence “c”), respectively, and (B) family 7 interrupted A domains. The A domain is highlighted in light grey and conserved A domain motifs are underlined in dark grey. The MII is highlighted in light blue and the conserved motifs are underlined in dark blue. The MI is highlighted in light purple, and the conserved motifs are underlined in dark purple. The red and dark purple balloons in panel A correspond to residues involved in SAM and amino acid bound Ppant arm binding, respectively, according to the structure of TioS(A8MIA9)4.14 The identical corresponding residues in type V M domain are also indicated in the same way. Type V M domain is highlighted in light pink and the conserved motifs are underlined in dark pink. Type VI M domain portion is highlighted in light orange and the conserved M domain motifs are underlined in dark orange. The boundaries between the domains are indicated by a triangle. Breaks in the sequences are indicated by two parallel bars. The full sequence alignments and accession numbers are presented in Fig. S19 and S21 (ESI†). | ||
Interrupted A domains with a single M domain: backbone N-methylation
There are two types of backbone N-methylating M domains, types I and V, contained within A domains. Type I M domain is found in family 1 (A8MbA9), the only family with an accompanying representative structure,14 which yields valuable insight into the residues that are important for activity and substrate binding. Type I M domains are very well conserved amongst interrupted A domains. These M domains contain six previously reported motifs for N-methylating M domains (Fig. 6A, Fig. S13, ESI† and Table 1).25,36 Type V M domains are found in family 4, and its representative, TtbB(A8MVA9)5, was shown to be responsible for N-methylating L-Tyr.20 Although types I and V M domains are in fact very similar in amino acid sequences, there are a few differences worth noting as they yield insight into the difference of interruptions between a2–a3 (MV) and a8–a9 (MI) (Fig. 6). The conserved motifs mbi-iv and vi are present in both types of M domains, however, motif mbv is absent from type V M domain (Fig. 6 and Table 1). The presence of these same motifs, which contain some identical residues determined to be involved in binding of either SAM or the amino acid bound Ppant arm, likely indicates similar structures. Perhaps the most notable difference in these types of M domains is their size. The average number of amino acids in type I M domains is 390 ± 21 amino acids, but it is 270 ± 6 in type V. This difference in length corresponds to the last ∼120 amino acids of type I M domains, which based on the crystal structure, were proposed to be a unique region of the M domain co-opted for structural purposes,14 possibly serving as a bridge between the A and M domains (Fig. 1B and Fig. S13, ESI†). Therefore, we can postulate that this missing region in type V M domains is related to the fact that those are inserted in the A domain between a2–a3 rather than a8–a9 and could be required for proper functionality for family 1 interrupted A domains, but not family 4. Both types I and V M domains appear in other interrupted A domains embedding multiple M domains, families 5a/b and 7, respectively, which is discussed below in the “Interrupted A domains with multiple M domains: backbone N- and side chain O-methylation” section.Interrupted A domains with a single M domain insertion: side chain O- and S-methylation
Side chain methylation can be categorized by type of methylation, O- or S-. Also, O-methylation can be further divided based on the substrate getting methylated: L-Ser/L-Thr (type II) vs.L-Tyr (type III) vs. hydroxylated aromatic residues (type VI). Interestingly, unlike backbone N-methylating types I and V M domains, which are found between a8–a9 and a2–a3, respectively, the M domains that carry out side chain O- or S-methylation, are so far, confined to their specific interruption locations: a2–a3 (type IV), a6–a7 (type VI), or a8–a9 (types II and III). Of the side chain O-methylating M domains, we can see a clear distinction in both the location of interruption, observed between a6–a7 and a8–a9, and the substrate. Given the abundance of O-methylation across all domains of life, it is no surprise that they have been studied in great detail, especially with regard to plant O-methyltransferases.37–41 Categorization of these plant O-methyltransferases is commonly based on phylogenetic relatedness and their substrate.39,41 Thus, we have also divided the interrupting Ms(O) domains by their relatedness (discussed above in the phylogenetic analyses of M domains) and substrate (hypothesized or confirmed in those few cases where it is known or inferred by the NP).Type II M domains are found in family 2a (A8Ms(O,Ser/Thr)A9) (Fig. 7A and Fig. S14, ESI†). The substrate of the representative, KtzH(A8MIIA9)4, was shown biochemically to be L-Ser.22 However, aside from the classic SAM binding motif (ms(O,Ser/Thr,a8–a9)ii), there were no other strongly conserved motifs that match previously published motifs for O-methyltransferase domains,36,41 possibly because most that have been characterized are those that have large aromatic substrates like catechol-O-methyltransferases.42 Therefore, we identified ms(O,Ser/Thr,a8–a9)i and iii-vi as conserved motifs present in all of the representatives from family 2a that were aligned (Fig. 7A, Fig. S14, ESI† and Table 1). It is important to note that the acidic E in ms(O,Ser/Thr,a8–a9)iii is possibly the conserved acidic residue seen at the end of the second β-sheet in the Rossmann-like fold15,16 following the GxGxG motif in class I methyltransferases,16 in which case it would be analogous to the acidic D in motif mb(a8–a9)iii. However, this remains a speculation until a structure for family 2a interrupted A domains is determined. While the SAM binding motif of class I methyltransferases typically contains GxGxG, none of these Gs is universally conserved; substitutions are typically those with small nonpolar replacements such as A.43 However, it is not unprecedented to see larger bulky groups (F or Y) replacing the middle G, especially in O-methyltransferases.44 Therefore, it was unsurprising to find ms(O,Ser/Thr,a8–a9)ii to contain …DVGx(N/H)xG… where the middle G has been replaced with N or H. This change did not impair methylation activity of either KtzH(A8MIIA9)422 nor ColG(A8MIIMIA9).26
Type III M domains, Ms(O,Tyr,a8–a9), are found in family 2b. Like type II M domains, these M domains are found between the a8–a9 conserved motifs of A domains, in almost identical interruption points, and are predicted to O-methylate amino acids. It is worth noting that all of the a8–a9 interruptions have a strikingly similar, though not completely identical (L(V/A/I)(A/G)(Y/F)xxx; where the last x is frequently, but not always a P)26 region of the A/Mb or A/Ms(O) boundary (Fig. 6A, 7, 9, 10A and Fig. S13–S15, S18, S19, ESI†), which corresponds to the last β-sheet before the M domain in TioS(A8MIA9)4.14 The GxGxG of the SAM binding motif is denoted as ms(O,Tyr,a8–a9)i with only GxG present (Fig. 7B), which is known to occur.15 Interestingly, none of these type III M domains contained the third G, but had a conserved A after two amino acids from the middle G. It is possible that there was a single residue insertion here changing an original GxGxA to GxGxxA. However, like the type II M domains, this change to the GxGxG does not appear to impact the M domain activity as there are examples of O-methylated L-Tyr connected to these M domains, indicating they are still functional.32 We suspect, based on NCBI's detection of the SAM binding site, that the conserved acidic residue is the D at the start of motif ms(O,Tyr,a8–a9)ii. However, we cannot be sure until the structure is solved as the acidic residue corresponds to a structural position in class I methyltransferases, the end of the second β-sheet in the Rossmann-like fold.15,16 The remaining motifs (iii and iv) were assigned based on the presence of conserved residues found in the type III M domain of families 2b and 5b. As with type II M domains, with the exception of the SAM binding motif, there were no other strongly conserved motifs that match previously published motifs for O-methyltransferase domains.36,41
Type VI M domains are found in family 6 (A6Ms(O,arom)A7). To our knowledge, this represents the first report of this family of interrupted A domains both in terms of the location of the interruption as well as the type of M domain it contains (Fig. 8B and Fig. S20, ESI†). We were able to identify the conserved GxGxG SAM binding motif of class I methyltransferases and have labeled it ms(O,arom,a6–a7)i. However, there are two sequences where one of the Gs has been swapped for an A or C. As with type II M domains, we surmise based on conservation, similarity, and proximity to ms(O,arom,a6–a7)i, that the conserved acidic E/D of class I methyltransferases is present in ms(O,arom,a6–a7)ii analogous to the D in mb(a8–a9)iii. As described in the “Phylogenetic analyses” section, we suspect that the substrate of type VI M domains is some type of hydroxylated aromatic molecule, resembling the substrate 2-polyprenyl-3-methyl-5-hydroxy-1,4-benzoquinone of UbiG, a methyltransferase with which it shares conserved regions (Table S8, ESI†). However, since it was shown that interrupted A domains must first activate the substrate, load it onto the T domain, then methylate it,21 how this occurs in families 6 and 7 remains to be answered.
M domains that carry out S-methylation (type IV) are found in family 3 (A2Ms(S)A3) interrupted A domains (Fig. 8A and Fig. S16, ESI†). Intriguingly, the conserved motifs ms(S,a2–a3)i, ii, and iii, although not identical, bear a striking resemblance to mb(a8–a9)/(a2–a3)ii, iv, and vi, respectively. This is in part expected since ms(S,a2–a3)i is the SAM binding motif found in all class I methyltransferases. Unexpected is the resemblance of ms(S,a2–a3)ii to mb(a8–a9)/(a2–a3)iv, but there is one key difference. In TioS(A8MIA9)4, the first N of …(N/S)S(V/I)δQY… is involved in SAM binding (indicated by the red balloon in Fig. 6A and also seen in type V M domains, red balloon in Fig. 6B), however in type IV M domains, this critical N is instead a highly conserved A (also marked with a red balloon in Fig. 8A). The third motif, ms(S,a2–a3)iii is similar to mb(a8–a9)/(a2–a3)vi, however between the conserved L and R (indicated by the purple balloons in Fig. 6), there are three residues in types I/V M domains, but only two residues in type IV M domains (Fig. 8A). It is generally accepted that N-, O-, and S-methylations by class I methyltransferases all occur via an SN2 reaction where the orientation of the methyl acceptor allows the lone pair of the nucleophile to point toward the electrophilic methyl group of SAM.15,45 Therefore, it is plausible that a slight change in the substrate or SAM binding position could flip the specificity from N- to S-, which could be the route type IV M domains took, given their sequence and size similarities and subtle differences to type V M domains. This is also supported by the phylogenetic relatedness of types IV and V M domains (Fig. 5). A similar architecture of these M domains would also explain why we find both N- and S-methylation domains between a2–a3, but do not find Ms(O) in that location. The a2–a3 insertion point (Fig. 1A) could be less forgiving than the loop observed between a8–a9 (Fig. 1A), which accommodates Ms(O), Mb, and two back-to-back M domains.
Interrupted A domains with multiple M domains: backbone N- and side chain O-methylation
In order to incorporate both backbone N-methylation and side chain O-methylation on a single amino acid in NRPs, Nature has devised two families of interrupted A domains: (i) two back-to-back M domains within a single interruption site between a8–a9 (family 5), and (ii) a di-interrupted A domain with interruptions between both a2–a3 and a6–a7 (family 7). Family 5 interrupted A domains can be further subdivided into three subfamilies, 5a–5c. Families 5a and 5b both contain an O-methylating M domain followed immediately by an N-methylating M domain (type I) (Fig. 9 and Fig. S18, ESI†). The difference between families 5a and 5b depends on the type of O-methyltating M domain. Family 5a contains a type II (Ms(O,Ser/Thr,a8–a9)) M domain, and the representative of this family is ColG(A8MIIMIA9), which has been shown to N,O-dimethylate L-Ser.26 Family 5b contains a type III (Ms(O,Tyr,a8–a9)) M domain, and the representatives of this family are DidJ(A8MIIIMIA9)29 and VatN(A8MIIIMIA9),28 as predicted based on an N,O-dimethylated L-Tyr in their corresponding NPs. These Ms(O) and Mb domains contain the same motifs and boundaries seen in their corresponding single M domain interrupted A domains counterparts (Fig. 6A, 7, and Fig. S13–S15, ESI†), indicating there were no special modifications to the M domains required to accommodate the back-to-back interruptions in families 5a and 5b. However, there is another unique family of interrupted A domains related to family 5a, family 5c, that to our knowledge only has one known member, FrsG(A8MIIA9-A10MIA9)8.30 Based on the NP of this biosynthetic pathway, the substrate of FrsG(A8MIIA9-A10MIA9)8 is L-Thr. This interrupted A domain is unique because, while it has both Ms(O,Ser/Thr,a8–a9) and Mb(a8–a9) domains between a8–a9, like family 5a, instead of the Mb(a8–a9) immediately following Ms(O,Ser/Thr,a8–a9), there is what appears to be a spacer between the two M domains. This spacer comprises what looks like the end of an A domain complete with a9 and a10 motifs, but instead of going to a T domain, there is an Mb(a8–a9), which then returns to the end of the A domain, where another set of a9 and a10 motifs is present (Fig. 10A and Fig. S19, ESI†). Aside from the spacer between the M domains observed in family 5c, they are otherwise very similar to those in family 5a. The same M domain motifs for types I and II M domains are present (Fig. 6A, 7A, 10A, and Fig. S13, S14, S19, ESI†). However, instead of having an Ms(O)/Mb(a8–a9) boundary (Fig. 9 and Fig. S18, ESI†) of families 5a/b, there is an Ms(O,Ser/Thr,a8–a9)/A boundary resembling that observed in family 2a (Fig. 7A and Fig. S14, ESI†). The A/Ms(O,Ser/Thr,a8–a9) and A/Mb(a8–a9) boundaries mirror those observed in families 2a and 1 (Fig. 6A and 7A), respectively. It is important to note that in all family 5 interrupted A domains the O-methylating M domain comes first followed by the N-methylating M domain. This is in stark contrast to the arrangement of domains seen in family 7 (A2MbA3–6Ms(O,arom)A7) di-interrupted A domains (Fig. 10B and Fig. S21, ESI†). These di-interrupted A domains contain an Ms(O) and Mb domains, just as the family 5, but instead of the M domains occurring back-to-back within the same interruption site, they occur in separate locations in the A domain. The N-methylating M domain in family 7 di-interrupted A domain is a type V M domain and is in fact, essentially the same in terms of conserved motifs and arrangement as that seen in family 4 (A2MbA3) (Fig. 6B and Fig. S17, ESI†). The same holds true for the second M domain, which is a type VI M domain between a6–a7, as observed in family 6 (Fig. 8B and Fig. S20, ESI†). These two interruptions in family 7 di-interrupted A domains are so similar to their mono-interrupted counterparts that it is easy to envision a situation where family 7 emerged from combining families 4 and 6. However, this remains speculative until families 6 and 7 are experimentally characterized. It is interesting that these interrupted A domains with multiple M domains only contained Ms(O) and Mb, but no Ms(S) domains. We could envision that an interrupted A domain of this nature, based on the trends we have observed here, would contain an Ms(S,a2–a3), as naturally found paired with an Mb(a8–a9) interruption. This di-interruption is functionally possible as we showed this conformation can exist artificially,46 although we have yet to discover a natural di-interrupted A domain like this.Conclusion
In summary, we have systematically analyzed and characterized seven distinct families of interrupted A domains along with the six types of M domains that interrupt them. Each family displays a unique taxonomic distribution amongst bacteria, reinforcing the importance of exploring different phyla beyond Streptomyces sp. in the search for structurally peculiar NPs. Although each type of M domain belongs to the broad class I methyltransferase, it is clear that these six types of interrupting M domains display their own unique signature motifs that allow us to make predictions as to their substrates and methylation activity. All six M domains form separate clusters on a phylogenetic tree, indicating that different evolutionary pathways have resulted in the formation of distinct families of interrupted A domains. These insights will help facilitate future engineering of interrupted A domains. Previous work has shown the malleability of interrupted A domains, in that they can have their M domains exchanged,47 be created from scratch,48 and be created to possess unnatural domain arrangements (e.g., a2–a3 and a8–a9 di-interrupted A domains).46 Most significantly, we have identified two new naturally occurring families of interrupted A domains, containing unique architecture and insertion sites. Family 6 interrupted A domains contain an M domain between a6–a7, a site that was previously unknown to withstand insertions. Family 7 interrupted A domains contain two separate interruption sites, between a2–a3 and a6–a7, representing the first examples of naturally occurring di-interruptions. These new interrupted A domains warrant substantial investigation of not only their activity, but also their corresponding NPs. The families of interrupted A domains and types of M domains we reported here may cover all common SN2 methylations that derivatize building blocks of NRPs. Methylation can play an essential role in bioactivity and bioavailability of NPs.49–51 Since there are not many examples of stand-alone methyltransferases in NRP biosyntheses, one could speculate that interrupted A domains are the most efficient way to regiospecifically methylate amino acid-like building blocks in NRPs. Therefore, it is important to further investigate and better understand interrupted A domains functionally and structurally for combinatorial biosynthesis of methylated NRPs.Author contributions
T. A. L., S. M., and S. G.-T. designed the study. T. A. L. and S. M. had equal contributions in performing the analyses and writing of the manuscript and Electronic Supplementary Information (ESI). T. A. L., S. M., and S. G.-T. wrote the manuscript and ESI and prepared all figures.Abbreviations
| A | Adenylation |
| AMP | Adenosine monophosphate |
| BLAST | Basic local alignment search tool |
| C | Condensation |
| CoA | Coenzyme A |
| E | Epimerization |
| FCB | Fibrobacteres, Chlorobi, and Bacteroidetes |
| HAL | Halogenation |
| KR | Ketoreduction |
| M | Methylation |
| MLP | MbtH-like protein |
| MOx | Monooxygenation |
| NCBI | National Center for Biotechnology Information |
| NP | Natural product |
| NRP | Nonribosomal peptide |
| NRPS | Nonribosomal peptide synthetase |
| Ox | Oxygenation |
| PKS | Polyketide synthase |
| Ppant | 4′-Phosphopantetheine |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosyl-L-methionine |
| SN2 | Nucleophilic substitution |
| T | Thiolation |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a National Science Foundation (NSF) CAREER Award MCB-1149427 (to S. G.-T.) and by startup funds from the University of Kentucky College of Pharmacy (to S. G.-T.). T. A. L. was in part supported by a 2019–2020 Pharmaceutical Sciences Excellence in Graduate Achievement Fellowship from the College of Pharmacy at the University of Kentucky as well as a 2019–2020 Pre-doctoral Fellowship in Pharmaceutical Sciences from the American Foundation of Pharmaceutical Education (AFPE).References
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the 30 years from 1981 to 2010, J. Nat. Prod., 2012, 75(3), 311–335 CrossRef CAS PubMed.
- C. T. Walsh, Insights into the chemical logic and enzymatic machinery of NRPS assembly lines, Nat. Prod. Rep., 2016, 33(2), 127–135 RSC.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases, Chem. Biol., 1999, 6(8), 493–505 CrossRef CAS PubMed.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, A new enzyme superfamily - The phosphopantetheinyl transferases, Chem. Biol., 1996, 3(11), 923–936 CrossRef CAS.
- C. T. Walsh, H. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey, L. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines, Curr. Opin. Chem. Biol., 2001, 5(5), 525–534 CrossRef CAS.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Interrupted adenylation domains: Unique bifunctional enzymes involved in nonribosomal peptide biosynthesis, Nat. Prod. Rep., 2015, 32(5), 641–653 RSC.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S, EMBO J., 1997, 16(14), 4174–4183 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Structures of two distinct conformations of holo-non-ribosomal peptide synthetases, Nature, 2016, 529(7585), 235–238 CrossRef CAS PubMed.
- M. Strieker, A. Tanovic and M. A. Marahiel, Nonribosomal peptide synthetases: Structures and dynamics, Curr. Opin. Struct. Biol., 2010, 20(2), 234–240 CrossRef CAS PubMed.
- J. M. Reimer, M. N. Aloise, P. M. Harrison and T. M. Schmeing, Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase, Nature, 2016, 529(7585), 239–242 CrossRef CAS PubMed.
- N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, Characterization of the cereulide NRPS alpha-hydroxy acid specifying modules: Activation of alpha-keto acids and chiral reduction on the assembly line, J. Am. Chem. Soc., 2006, 128(33), 10698–10699 CrossRef CAS PubMed.
- B. Silakowski, H. U. Schairer, H. Ehret, B. Kunze, S. Weinig, G. Nordsiek, P. Brandt, H. Blocker, G. Hofle, S. Beyer and R. Muller, New lessons for combinatorial biosynthesis from myxobacteria. The myxothiazol biosynthetic gene cluster of Stigmatella aurantiaca DW4/3-1, J. Biol. Chem., 1999, 274(52), 37391–37399 CrossRef CAS PubMed.
- D. A. Alonzo, C. Chiche-Lapierre, M. J. Tarry, J. Wang and T. M. Schmeing, Structural basis of keto acid utilization in nonribosomal depsipeptide synthesis, Nat. Chem. Biol., 2020, 16(5), 493–496 CrossRef CAS PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Structural basis for backbone N-methylation by an interrupted adenylation domain, Nat. Chem. Biol., 2018, 14(5), 428–430 CrossRef CAS PubMed.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Many paths to methyltransfer: A chronicle of convergence, Trends Biochem. Sci., 2003, 28(6), 329–335 CrossRef CAS.
- J. L. Martin and F. M. McMillan, SAM (dependent) I AM: The S-adenosylmethionine-dependent methyltransferase fold, Curr. Opin. Struct. Biol., 2002, 12(6), 783–793 CrossRef CAS PubMed.
- A. H. Al-Mestarihi, G. Villamizar, J. Fernandez, O. E. Zolova, F. Lombó and S. Garneau-Tsodikova, Adenylation and S-methylation of cysteine by the bifunctional enzyme TioN in thiocoraline biosynthesis, J. Am. Chem. Soc., 2014, 136(49), 17350–17354 CrossRef CAS PubMed.
- T. Huang, Y. Duan, Y. Zou, Z. Deng and S. Lin, NRPS protein MarQ catalyzes flexible adenylation and specific S-methylation, ACS Chem. Biol., 2018, 13(9), 2387–2391 CrossRef CAS PubMed.
- A. C. Ross, Y. Xu, L. Lu, R. D. Kersten, Z. Shao, A. M. Al-Suwailem, P. C. Dorrestein, P. Y. Qian and B. S. Moore, Biosynthetic multitasking facilitates thalassospiramide structural diversity in marine bacteria, J. Am. Chem. Soc., 2013, 135(3), 1155–1162 CrossRef CAS PubMed.
- J. J. Zhang, X. Tang, T. Huan, A. C. Ross and B. S. Moore, Pass-back chain extension expands multimodular assembly line biosynthesis, Nat. Chem. Biol., 2020, 16(1), 42–49 CrossRef CAS PubMed.
- S. Mori, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Deciphering Nature's intricate way of N,S-dimethylating L-cysteine: Sequential action of two bifunctional adenylation domains, Biochemistry, 2017, 56(46), 6087–6097 CrossRef CAS PubMed.
- O. E. Zolova and S. Garneau-Tsodikova, KtzJ-dependent serine activation and O-methylation by KtzH for kutznerides biosynthesis, J. Antibiot., 2014, 67(1), 59–64 CrossRef CAS PubMed.
- F. Lombó, A. Velasco, A. Castro, F. de la Calle, A. F. Brana, J. M. Sanchez-Puelles, C. Mendez and J. A. Salas, Deciphering the biosynthesis pathway of the antitumor thiocoraline from a marine actinomycete and its expression in two Streptomyces species, ChemBioChem, 2006, 7(2), 366–376 CrossRef PubMed.
- M. A. Marahiel, T. Stachelhaus and H. D. Mootz, Modular peptide synthetases involved in nonribosomal peptide synthesis, Chem. Rev., 1997, 97(7), 2651–2674 CrossRef CAS PubMed.
- L. Rouhiainen, L. Paulin, S. Suomalainen, H. Hyytiainen, W. Buikema, R. Haselkorn and K. Sivonen, Genes encoding synthetases of cyclic depsipeptides, anabaenopeptilides, in Anabaena strain 90, Mol. Microbiol., 2000, 37(1), 156–167 CrossRef CAS PubMed.
- T. A. Lundy, S. Mori, N. Thamban Chandrika and S. Garneau-Tsodikova, Characterization of a unique interrupted adenylation domain that can catalyze three reactions, ACS Chem. Biol., 2020, 15(1), 282–289 CrossRef CAS PubMed.
- K. Kleigrewe, J. Almaliti, I. Y. Tian, R. B. Kinnel, A. Korobeynikov, E. A. Monroe, B. M. Duggan, V. Di Marzo, D. H. Sherman, P. C. Dorrestein, L. Gerwick and W. H. Gerwick, Combining mass spectrometric metabolic profiling with genomic analysis: A powerful approach for discovering natural products from Cyanobacteria, J. Nat. Prod., 2015, 78(7), 1671–1682 CrossRef CAS PubMed.
- N. A. Moss, G. Seiler, T. F. Leao, G. Castro-Falcon, L. Gerwick, C. C. Hughes and W. H. Gerwick, Nature's combinatorial biosynthesis produces vatiamides A-F, Angew. Chem., 2019, 58(27), 9027–9031 CrossRef CAS PubMed.
- Y. Xu, R. D. Kersten, S. J. Nam, L. Lu, A. M. Al-Suwailem, H. Zheng, W. Fenical, P. C. Dorrestein, B. S. Moore and P. Y. Qian, Bacterial biosynthesis and maturation of the didemnin anti-cancer agents, J. Am. Chem. Soc., 2012, 134(20), 8625–8632 CrossRef CAS PubMed.
- A. Carlier, L. Fehr, M. Pinto-Carbo, T. Schaberle, R. Reher, S. Dessein, G. Konig and L. Eberl, The genome analysis of Candidatus Burkholderia crenata reveals that secondary metabolism may be a key function of the Ardisia crenata leaf nodule symbiosis, Environ. Microbiol., 2016, 18(8), 2507–2522 CrossRef CAS PubMed.
- J. Jirakkakul, J. Punya, S. Pongpattanakitshote, P. Paungmoung, N. Vorapreeda, A. Tachaleat, C. Klomnara, M. Tanticharoen and S. Cheevadhanarak, Identification of the nonribosomal peptide synthetase gene responsible for bassianolide synthesis in wood-decaying fungus Xylaria sp. BCC1067, Microbiology, 2008, 154(Pt 4), 995–1006 CrossRef CAS PubMed.
- E. Oueis, T. Klefisch, N. Zaburannyi, R. Garcia, A. Plaza and R. Muller, Two biosynthetic pathways in Jahnella thaxteri for thaxteramides, distinct types of lipopeptides, Org. Lett., 2019, 21(14), 5407–5412 CrossRef CAS PubMed.
- G. M. Konig, S. Kehraus, S. F. Seibert, A. Abdel-Lateff and D. Muller, Natural products from marine organisms and their associated microbes, ChemBioChem, 2006, 7(2), 229–238 CrossRef PubMed.
- R. E. Procopio, I. R. Silva, M. K. Martins, J. L. Azevedo and J. M. Araujo, Antibiotics produced by Streptomyces. Braz, J. Infect. Dis., 2012, 16(5), 466–471 Search PubMed.
- C. Prieto, C. Garcia-Estrada, D. Lorenzana and J. F. Martin, NRPSsp: Non-ribosomal peptide synthase substrate predictor, Bioinformatics, 2012, 28(3), 426–427 CrossRef CAS PubMed.
- M. Z. Ansari, J. Sharma, R. S. Gokhale and D. Mohanty, In silico analysis of methyltransferase domains involved in biosynthesis of secondary metabolites, BMC Bioinf., 2008, 9, 454 CrossRef PubMed.
- N. Lavid, J. Wang, M. Shalit, I. Guterman, E. Bar, T. Beuerle, N. Menda, S. Shafir, D. Zamir, Z. Adam, A. Vainstein, D. Weiss, E. Pichersky and E. Lewinsohn, O-methyltransferases involved in the biosynthesis of volatile phenolic derivatives in rose petals, Plant Physiol., 2002, 129(4), 1899–1907 CrossRef CAS PubMed.
- J. Wang and E. Pichersky, Identification of specific residues involved in substrate discrimination in two plant O-methyltransferases, Arch. Biochem. Biophys., 1999, 368(1), 172–180 CrossRef CAS PubMed.
- K. C. Lam, R. K. Ibrahim, B. Behdad and S. Dayanandan, Structure, function, and evolution of plant O-methyltransferases, Genome, 2007, 50(11), 1001–1013 CrossRef CAS PubMed.
- C. Zubieta, X. Z. He, R. A. Dixon and J. P. Noel, Structures of two natural product methyltransferases reveal the basis for substrate specificity in plant O-methyltransferases, Nat. Struct. Biol., 2001, 8(3), 271–279 CrossRef CAS PubMed.
- R. K. Ibrahim, A. Bruneau and B. Bantignies, Plant O-methyltransferases: Molecular analysis, common signature and classification, Plant Mol. Biol., 1998, 36(1), 1–10 CrossRef CAS PubMed.
- J. Siegrist, J. Netzer, S. Mordhorst, L. Karst, S. Gerhardt, O. Einsle, M. Richter and J. N. Andexer, Functional and structural characterisation of a bacterial O-methyltransferase and factors determining regioselectivity, FEBS Lett., 2017, 591(2), 312–321 CrossRef CAS PubMed.
- P. Z. Kozbial and A. R. Mushegian, Natural history of S-adenosylmethionine-binding proteins, BMC Struct. Biol., 2005, 5, 19 CrossRef PubMed.
- X. Hou, Y. Wang, Z. Zhou, S. Bao, Y. Lin and W. Gong, Crystal structure of SAM-dependent O-methyltransferase from pathogenic bacterium Leptospira interrogans, J. Struct. Biol., 2007, 159(3), 523–528 CrossRef CAS PubMed.
- R. W. Woodard, M. D. Tsai, H. G. Floss, P. A. Crooks and J. K. Coward, Stereochemical course of the transmethylation catalyzed by catechol O-methyltransferase, J. Biol. Chem., 1980, 255(19), 9124–9127 CAS.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Probing the limits of interrupted adenylation domains by engineering a trifunctional enzyme capable of adenylation, N-, and S-methylation, Org. Biomol. Chem., 2019, 17(5), 1169–1175 RSC.
- S. K. Shrestha and S. Garneau-Tsodikova, Expanding substrate promiscuity by engineering a novel adenylating-methylating NRPS bifunctional enzyme, ChemBioChem, 2016, 17(14), 1328–1332 CrossRef CAS PubMed.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Engineering bifunctional enzymes capable of adenylating and selectively methylating the side chain or core of amino acids, ACS Synth. Biol., 2018, 7(2), 399–404 CrossRef CAS PubMed.
- H. B. Park, Y. J. Kim, J. S. Park, H. O. Yang, K. R. Lee and H. C. Kwon, Glionitrin B, a cancer invasion inhibitory diketopiperazine produced by microbial coculture, J. Nat. Prod., 2011, 74(10), 2309–2312 CrossRef CAS.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, N-methylation of peptides: A new perspective in medicinal chemistry, Acc. Chem. Res., 2008, 41(10), 1331–1342 CrossRef CAS.
- J. Chatterjee, F. Rechenmacher and H. Kessler, N-methylation of peptides and proteins: An important element for modulating biological functions, Angew. Chem., 2013, 52(1), 254–269 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the construction of the data sets used for the taxonomic and phylogenetic trees, multiple sequence alignments and boundary identification of interrupted A domains, and identification of M domain conserved domain motifs and assignment of M domain types. Detailed information of all families 1–6 (Tables S1–S7), and conserved regions of M domain types (Table S8). Taxonomic tree of families 1–4 interrupted A domain (Fig. S1–S5), taxonomic and phylogenetic trees of families 5a, 5b, and 6 (Fig. S6 and S7), phylogenetic tree of families 1–4 (Fig. S8–S12) and full-length multiple sequence alignment of families 1–7 interrupted A domains (Fig. S13–S21). See DOI: 10.1039/d0cb00092b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |