
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topology and porosity control of metal–organic frameworks through linker functionalization†
Jiafei
Lyu
 abc,
Xuan
Zhang
c,
Ken-ichi
Otake
c,
Xingjie
Wang
c,
Peng
Li
c,
Zhanyong
Li
c,
Zhijie
Chen
c,
Yuanyuan
Zhang
c,
Megan C.
Wasson
c,
Ying
Yang
c,
Peng
Bai
ab,
Xianghai
Guo
ab,
Timur
Islamoglu
c and
Omar K.
Farha
*c
abc,
Xuan
Zhang
c,
Ken-ichi
Otake
c,
Xingjie
Wang
c,
Peng
Li
c,
Zhanyong
Li
c,
Zhijie
Chen
c,
Yuanyuan
Zhang
c,
Megan C.
Wasson
c,
Ying
Yang
c,
Peng
Bai
ab,
Xianghai
Guo
ab,
Timur
Islamoglu
c and
Omar K.
Farha
*c
aDepartment of Pharmaceutical Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China
bKey Laboratory of Systems Bioengineering, Ministry of Education, Tianjin University, Tianjin 300350, China
cDepartment of Chemistry and International Institute of Nanotechnology, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: o-farha@northwestern.edu
First published on 9th November 2018
Abstract
Tetratopic organic linkers have been extensively used in Zr-based metal–organic frameworks (MOFs) where diverse topologies have been observed. Achieving meticulous control over the topologies to tune the pore sizes and shapes of the resulting materials, however, remains a great challenge. Herein, by introducing substituents to the backbone of tetratopic linkers to affect the linker conformation, phase-pure Zr-MOFs with different topologies and porosity were successfully obtained under the same synthetic conditions. The conversion of CO2 to valuable cyclic carbonates is a promising route for the mitigation of the greenhouse gas. Owing to the presence of substrate accessible Lewis acidic Zr(IV) sites in the 8-connected Zr6 nodes, the Zr-MOFs in this study have been investigated as heterogenous acid catalysts for CO2 cycloaddition to styrene oxide. The MOFs exhibited drastically different catalytic activities depending on their distinct pore structures. Compared to previously reported MOF materials, a superior catalytic activity was observed with the mesoporous NU-1008, giving an almost 100% conversion under mild conditions.
Introduction
Metal–organic frameworks (MOFs),1–3 a class of crystalline porous materials constructed from metal nodes and organic ligands by coordination bonds, have found a wide range of applications such as gas storage and separation,4–11 catalysis,12–16 magnetism,17,18 drug delivery,19–23 small-molecule recognition24 and chemical sensing.25,26 This functional diversity is largely due to their versatile structural tunability where a variety of components can be combined to generate an almost indefinite number of novel materials. However, elucidating the parameters that dictate the MOF structures, including their topology and porosity, is non-trivial. Considering Zr6 cluster-based MOFs as an example, although linkers of lower connectivity generally favor one particular topology,27–29 more topologies can be accessed from linkers with higher connectivity30,31 due to their tendency to undergo conformational changes, resulting in challenges in predicting MOF structures.32–34Since the first reports of Zr-MOFs with tetratopic linkers,35–37 several topologies have been observed, including ftw,36,38–41csq,35–37,42she,43shp,44scu,32,45flu,27,46,47ith,27sqc,48lvt49 and stp50 which can be attributed to the linker conformation adjustments under different synthetic conditions. This conformational change can be induced by controlling one of the many factors, such as the modulating reagent, concentration, metal salt, temperature and solvent. For example, a csq-net MOF (NU-1000)42,51 is produced when using benzoic acid as the modulator whereas a scu-net MOF (NU-901)32 is obtained using 4-amino-benzoic acid with the same 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) linker and Zr precursor. Using a tetratopic linker with arms of a high degree of rotational freedom, such as tetracarboxyphenylporphyrin (TCPP), multiple topologies have been observed.35–37,43,44,48 A similar phenomenon was also observed when we explored the isoreticular tetracarboxylate Zr-MOF csq-net NU-100X series for enzyme immobilization where MOFs with ftw topology were produced instead when using linkers with longer arms. Therefore, our group concluded that the torsion angle between the planar “backbone” (benzene, pyrene, porphyrin) and the arms (carboxylic acid) had a critical influence on the framework topology. Specifically, if the torsion angle was close to 60°, the csq topology was favored, whereas the ftw topology formed if the torsion angle was close to 0°.52
In addition to using organic linkers with higher rotational freedom, installing functional groups to the organic linkers affords another effective strategy to affect the linker conformation, thereby the material topology. For instance, Yaghi and co-workers successfully synthesized qom-net MOF-177 series with uncommon topologies (pyr and rtl) by introducing functional groups.53 Recently, Zhou and co-workers introduced steric hinderance into the biphenyl-3,3′,5,5′-tetra(phenyl-4-carboxylic acid) (TPCB) linker to affect linker conformation, which subsequently effect the topology of the resulting Zr-tetracarboxylate MOFs. However, accompanying the original structure were mixed phase materials that formed as byproducts, most likely due to the introduction of the bulky substituents.54 In high agreement with experimental results, computational studies compared the energy of MOFs with different substituents to demonstrate the influence of introduced steric hinderance on linker conformation and MOF topology.55 With these design rules and challenges in mind, we set out to tune the steric hindrance of the organic linkers in a stepwise fashion, aiming to achieve topology control in the resulting series of Zr-MOFs.
Results and discussion
Introduction of steric hinderance
In addition to the 1,2,4,5-tetrakis(4-carboxyphenyl)benzene (TCPB) linker (Fig. 1, L1), two more linkers were designed. A nitro group (Fig. 1, L2) and two bromo groups (Fig. 1, L3) were installed on the central benzene respectively to introduce steric hindrance to affect the rotational freedom of the peripheral benzoate arms and in turn to control the topology of the resulting MOFs. To ensure consistency and to eliminate variability from factors such as solvent, temperature and modulator on the topology of the resulting MOFs, the syntheses with the three TCPB-based linkers were carried out under identical solvothermal conditions (ESI†).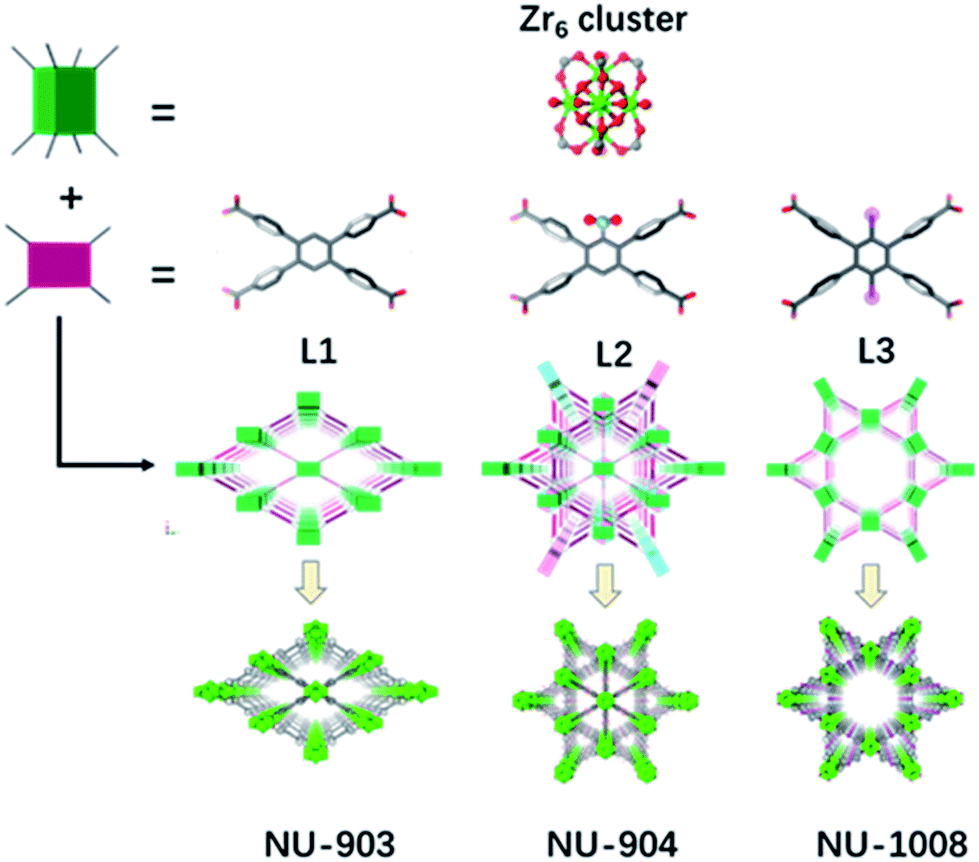 | ||
| Fig. 1 Schematic representation of the construction of NU-903, NU-904 and NU-1008. Atom colour scheme: C, grey; N, blue; O, red; Br, pink; Zr, green polyhedra. H atoms are omitted for clarity. | ||
Topology and porosity diversity of the as-synthesized MOFs
As previously reported by Stock and co-workers,45 Zr6 nodes and the L1 linker form NU-903 with an scu topology with characteristic diamond-shaped channels along the c axis (Fig. 1). NU-903 is isoreticular to TBAPy-based NU-901![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 and TCPP-based NU-902.56
32 and TCPP-based NU-902.56
The crystals of the resulting MOF with linker L2 are oval in shape (Fig. 3b). Single crystal X-ray diffraction analysis (Table S1, ESI†) revealed that NU-904 crystallized in the P2/m space group (a = 19.64 (1) Å, b = 12.63 (1) Å, c = 19.635 (4) Å and β = 119.994° (2) at 200 K) with the formula as Zr6(μ-O)4(μ-OH)4(HCOO)1.5(H2O)2.5(OH)2.5(L2)2. The 3D structure consists of 8-connected Zr6 nodes and mononitro-substituted TCPB linkers, yielding a rare scu topology.32 The single crystal structure of NU-904 is characteristic of a reticular-merohedral twin structure in which three orientations stack together along the b axis, twisting 60° from each other (Fig. 2). The reticular twins of three scu-net components gave rise to the overall 6-fold symmetry in the structure. Topologically, the average structure of the threefold twinned NU-904 is based on the highly connected 4,12-c shp net.44,57 Interestingly, the threefold twins of ordered 4,8-c scu structure led to a twinned 4,12-c rare shp structure (Fig. S17, ESI†). To the best of our knowledge, the inherent correlation between these two topological nets has not been realized until this work. As a result, the average structure is observed as triangular-shaped instead of the expected diamond-shaped channels in NU-903 (Fig. 2).
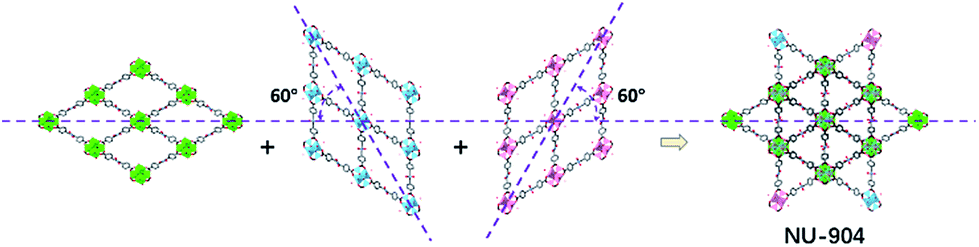 | ||
| Fig. 2 The reticular twin structure of NU-904. | ||
The MOF synthesis with linker L3 yields hexagonal rod-shaped crystals (Fig. 3b). Single crystal X-ray diffraction analysis (Table S1, ESI†) revealed that NU-1008 crystallized in the hexagonal P6/mmm space group with the chemical formula as Zr6(μ-O)4(μ-OH)4(HCOO)(H2O)3(OH)3(L3)2; the 3D structure consists of 8-connected Zr6 nodes and dibromo-substituted TCPB linkers in a csq topology. Isoreticular to NU-1000,58 there are two types of channels along the c axis, a 1 nm wide triangular channel and a 3 nm hexagonal channel (Fig. 1). The incorporation of the dibromo groups does not compromise the porosity of the material because the bromo groups reside in the window connecting the hierarchically triangular micropores and hexagonal mesopores. Therefore, the pore size distribution of NU-1008 is similar to NU-1000.
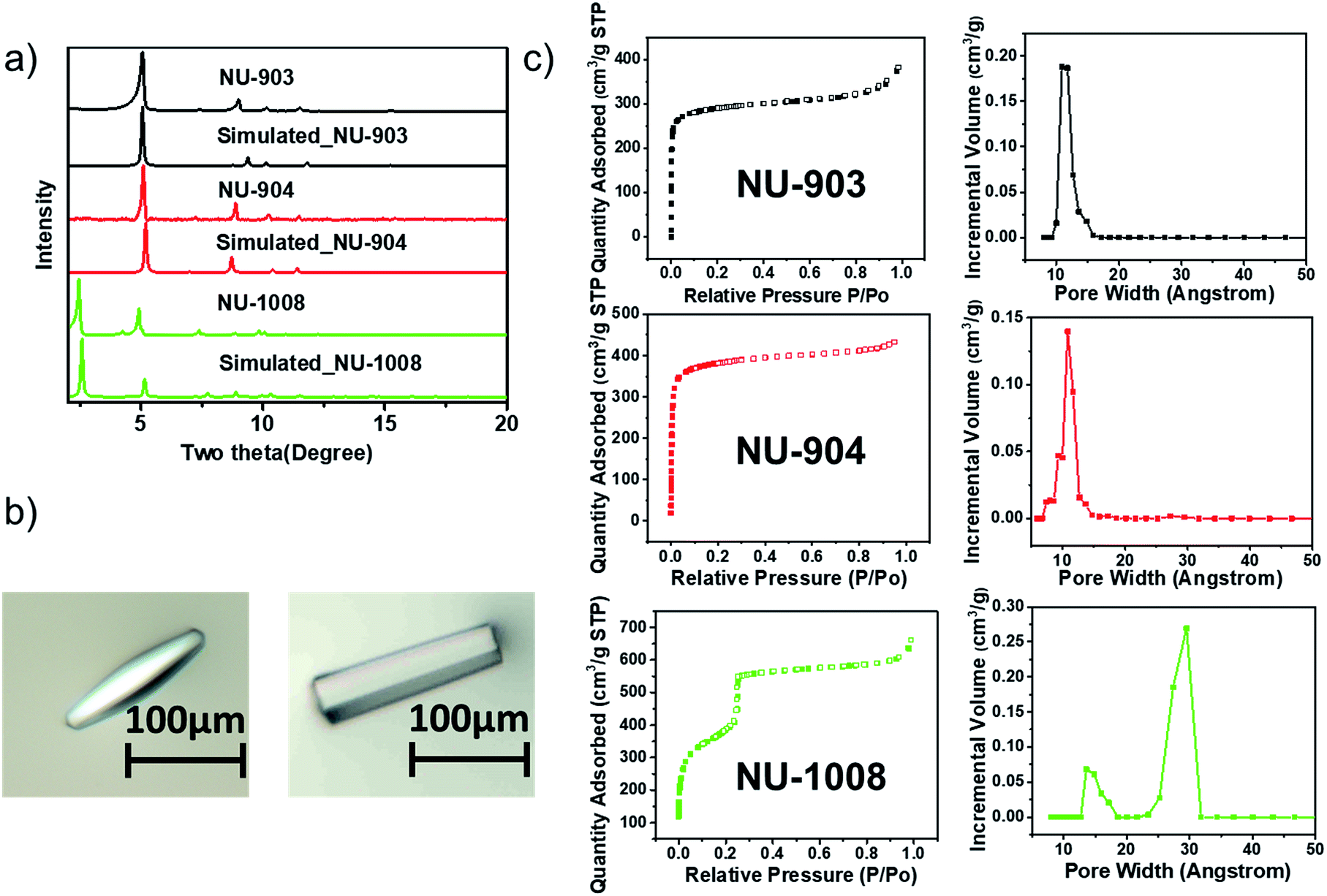 | ||
| Fig. 3 Characterization of the three MOFs. (a) PXRD patterns; (b) optical images of the single crystals of NU-904 (left) and NU-1008 (right), the scale bars in the images are 100 μm; (c) N2 sorption isotherms and DFT pore size distribution of NU-903, NU-904 and NU-1008. | ||
Characterization of as-synthesized MOFs
Due to the 8-connected Zr6 nodes present in all three MOFs, 1H NMR was employed to identify the coordinated species on the remaining accessible sites of the node. Upon digestion of the MOFs with a dilute NaOD solution, a peak was observed for all three materials around 8.37 ppm that is characteristic of formate ligand. Quantifying the amounts of formate relative to the organic linker in the three samples gives ca. 2, 2 and 3 formate groups per Zr6 node in NU-903 (Fig. S4, ESI†), NU-904 (Fig. S5, ESI†) and NU-1008 (Fig. S6, ESI†), respectively. Diffuse reflectance infrared fourier transform spectroscopy (DRIFTS) also confirms the presence of formate groups with the observed C–H stretching at 2745 cm−1. In addition, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1658 and 1373 cm−1) and C–Br stretching (712 cm−1) in the DRIFTS spectra of NU-904 and NU-1008 confirmed the presence of L2 and L3, respectively (Fig. S10–S12, ESI†).
O stretching (1658 and 1373 cm−1) and C–Br stretching (712 cm−1) in the DRIFTS spectra of NU-904 and NU-1008 confirmed the presence of L2 and L3, respectively (Fig. S10–S12, ESI†).
The phase purity of the bulk materials was confirmed by PXRD (Fig. 3a). The particle morphology of NU-904 is oval-shaped and NU-1008 is hexagonal rod-shaped (Fig. 3b), as evidenced by the SEM images (Fig. S13, ESI†). Nitrogen sorption isotherms of three different MOFs were measured at 77 K; the type I isotherms of NU-903 and NU-904 are indicative of microporous structure of the MOFs, while the type IV isotherm of NU-1008 indicates the presence of both micropores and mesopores in the material. The BET areas were calculated to be 1140, 1410 and 1400 m2 g−1 for NU-903, NU-904 and NU-1008, respectively. DFT pore size distributions reveal micropores of 11 Å for NU-903, 10 Å for NU-904 and hierarchical micropores of 11 Å and mesopores of 29 Å for NU-1008 (Fig. 3c). The total pore volumes were 0.515, 0.613 and 0.819 cm3 g−1 for NU-903, NU-904 and NU-1008, respectively. The larger gravimetric pore volume and the type IV isotherm of NU-1008 is again consistent with a mesoporous structure. Thermogravimetric analysis (TGA) under air showed no sign of mass loss up to 400 °C (Fig. S7–S9, ESI†), demonstrating the high thermal stability of the three MOFs. In addition, all three materials showed good stability under acidic condition with treatment of 0.5 M HCl aqueous solution for 10 hours (Fig. S14, ESI†).
Topology and porosity control through introduction of steric effect
To investigate the influence of linker conformation on topology control, we performed a detailed analysis of linker conformation in the three crystal structures. In the scu-net NU-903 structure,45 the TCPB linker with no substituents adopts a C2h symmetry with the C2 axis and the perpendicular σh depicted in Fig. 4a. In the MOF structure, the two adjacent phenyl arms in the upper side rotate away from each other, whereas the lower ones rotate toward each other (Fig. 4a). In NU-904, the introduction of the mononitro group lowers the symmetry of the linker to C2 but the conformation of the peripheral phenyl arms was found to be similar to the non-substituted TCPB linker in NU-903 (Fig. 4b). As a result, each of the twinning portions that comprise this structure has the same scu topology as NU-903, even though the average structure of NU-904 has 1D triangular channels instead of the diamond-shaped channels with a typical scu topology. Notably, the mononitro group on the central benzene rotates ∼32° to fit in the structure. In NU-1008 with dibromo-substituted TCPB, the linker adopts a C2v symmetry due to the rotation of the upper and lower pair of phenyl arms toward each other (Fig. 4c) which directs the framework to csq topology over the scu-net MOF. In addition, to accommodate the dibromo groups in NU-1008, the dihedral angle between the arm benzene and central benzene in the csq-net NU-1008 is closer to 90° than the dihedral angle observed in scu-net NU-903 and NU-904 (Table S3, ESI†). Additionally, the angle between the arms (117°) in NU-1008 is larger than in scu-net NU-903 (115°) and NU-904 (108.5°).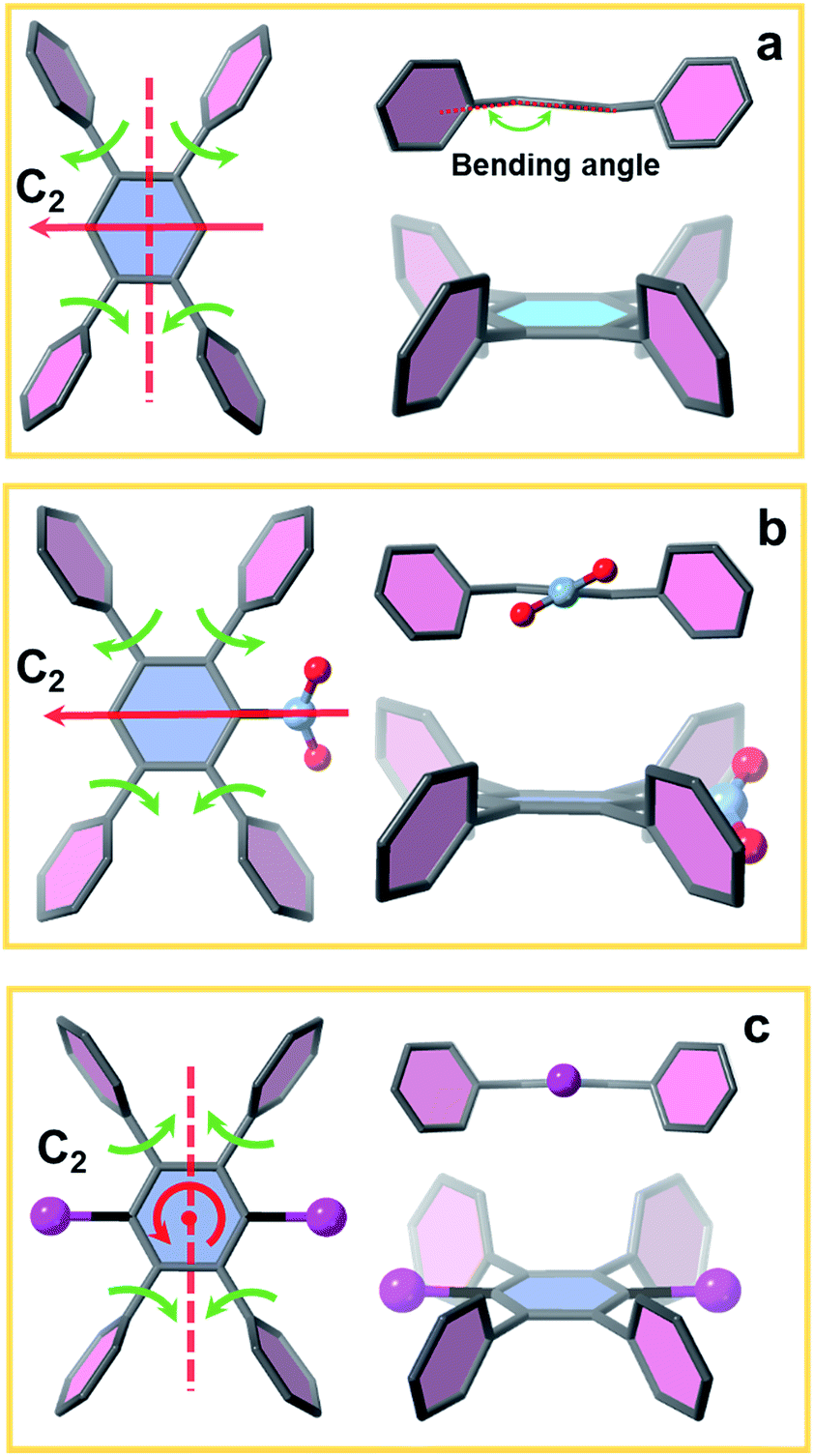 | ||
| Fig. 4 Linker conformation in (a) NU-903, (b) NU-904 and (c) NU-1008. | ||
The 8-connected Zr6 cluster in the three MOFs can be regarded as a cubic shaped node and the linker as a rectangular plane. In NU-903, the non-substituted TCPB linker with C2h symmetry can link the clusters in the same orientation (Fig. 1), giving the scu topology. In contrast, the Zr6 clusters in NU-1008 undergo an orientation change to conform to the C2v symmetry of dibromo TCPB linker, and form a different topology, csq. The topology change confirmed our hypothesis that the steric effect induced by linker functionalization can achieve the topology control of MOF materials.
Catalytic performance for CO2 fixation under mild conditions
CO2 is a well-established greenhouse gas that originates from the carbon footprint of human activities.59 In order to mitigate the adverse effect of CO2 to the environment, considerable progress has been made in its capture and storage in recent years,60,61 and promising strategies for the consumption of CO2 have received much attention. Rational utilization of CO2 is of great significance for the deceleration of global warming and the development of sustainable energy. Chemical fixation of CO2 with epoxides through a facile cycloaddition process, catalysed by acidic sites, is an attractive route to convert this greenhouse gas into highly demanded cyclic organic carbonates.62Owing to the presence of substrate accessible Lewis acidic sites, MOFs have been previously explored to catalyse the cycloaddition of CO2 and epoxides.63,64 However, energy-demanding reaction conditions such as elevated temperatures and pressures are generally required for the efficient conversion of CO2 into cyclic carbonates.65 Inspired by these reports, CO2 fixation into styrene oxide was used as a model reaction to test the catalytic performance of MOFs reported in this study under mild conditions.
NU-903, NU-904 and NU-1008 were investigated for CO2 fixation into styrene oxide under room temperature with 1 bar of CO2. The styrene oxide (0.2 mmol), tetrabutylammonium bromide (6.5 mg, 0.02 mmol) pre-dissolved in 400 μL of acetonitrile and MOF material (4.0 mol%, 0.002 mmol) were added to an autoclave batch reactor, which had previously been dried for 6 h at 80 °C. The autoclave reactor was evacuated, purged with CO2, and then placed under a constant pressure of CO2 under 5 bar for 15 min to allow the system to equilibrate. The reaction was carried out at room temperature for 24 hours after the pressure was reduced to 1 bar of gauge pressure. After the reaction, the catalyst was separated by centrifugation and a small aliquot of the supernatant reaction mixture was taken to be analyzed by 1H NMR to calculate the conversion (Fig. 5).
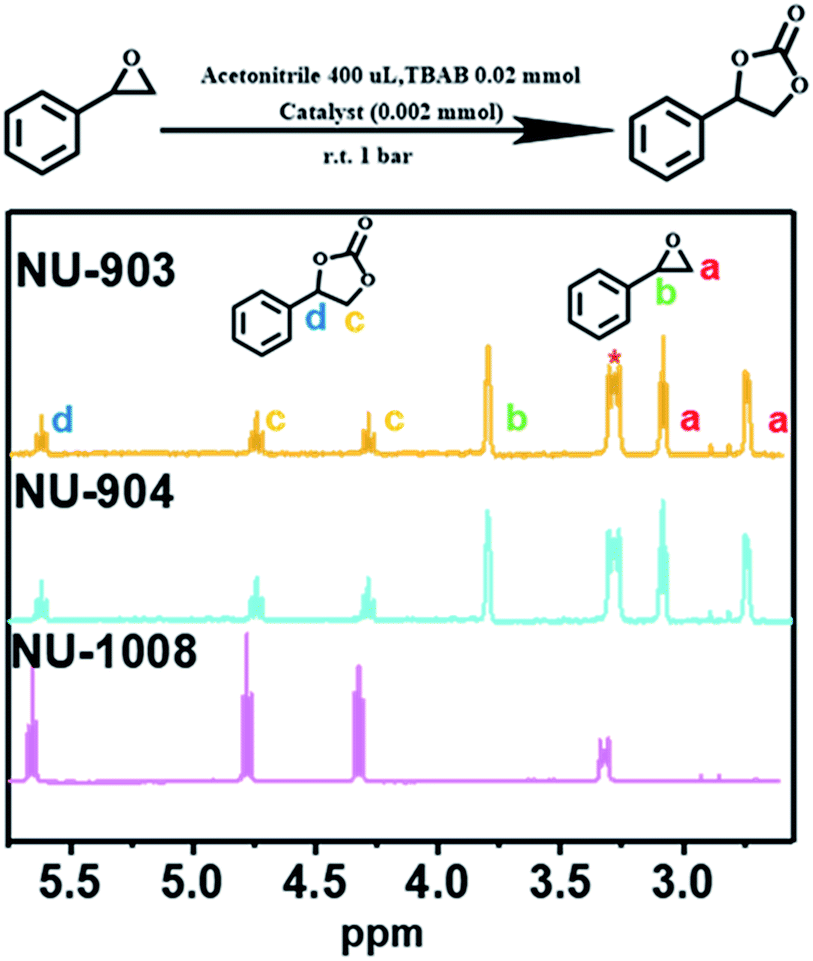 | ||
| Fig. 5 Proton NMR spectra of CO2 fixation into styrene oxide catalyzed by NU-903, NU-904 and NU-1008 (1 bar, room temperature, 24 h; CDCl3 as deuterium solvent, 500 MHz. The peak with asterisk refers to the tetrabutylammonium bromide). | ||
A control experiment without any MOF catalyst exhibited negligible conversion of styrene oxide. However, vastly different CO2 fixation activities were observed within the three MOF catalysts utilized in this report. Remarkably, full conversion of styrene oxide was observed with NU-1008 after 24 hours under mild conditions (room temperature, 1 bar of CO2). Previous reports using MOF catalysts for this reaction required elevated temperatures (∼120 °C), pressures (10–20 bar) or longer reaction time (up to 56 hours) to afford similar or lower substrate conversions (Table S2, ESI†).62 Thus, NU-1008 stands out as a more environmentally friendly solid acid catalyst for the chemical fixation of CO2, compared to others reported.
In comparison, NU-903 and NU-904 showed much lower conversion (Fig. 6a), despite the fact that they had similar CO2 adsorption performance to NU-1008 (Fig. S15, ESI†). This significant difference is likely attributed to the aforementioned narrower pores and smaller pore volumes in NU-903 and NU-904 (around 10 Å, 0.50 cm3 g−1) compared to the mesoporous channels in NU-1008 (around 30 Å, 0.82 cm3 g−1). The microporous NU-903 and NU-904 most likely limited the diffusion of the sizeable substrate and product (Fig. 6b) and resulted in the lower conversion. The turnover numbers (TONs) in 24 hours were calculated to be 20.3, 22.8 and 99.4 for NU-903, NU-904 and NU-1008, respectively.
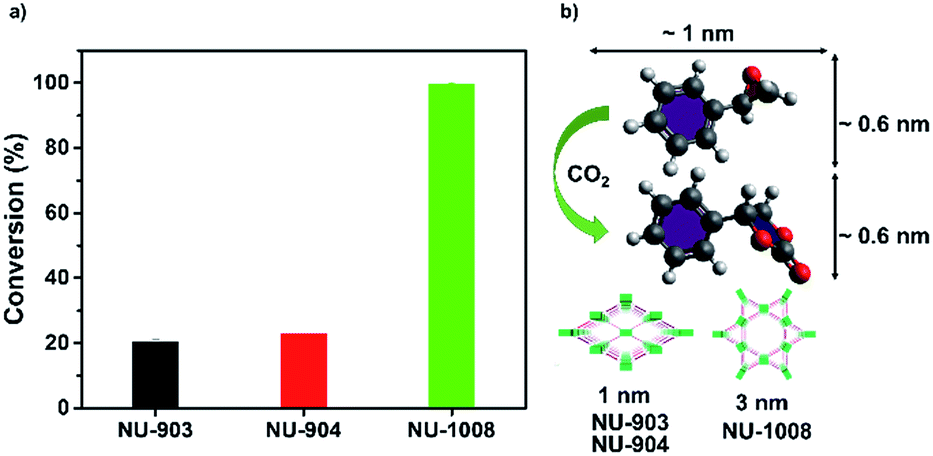 | ||
| Fig. 6 CO2 fixation conversion on NU-903, NU-904 and NU-1008. (a) Catalytic capacities; (b) size of substrate, product and pore size of NU-903, NU-904 and NU-1008. | ||
Conclusions
Three different Zr-MOFs were constructed by the steric control of the conformations of substituted tetratopic carboxylate linkers. Compared to NU-903 with the scu topology, phase pure csq-net NU-1008 supports our initial hypothesis that introducing functionality into the organic linker can influence the linker conformation and direct the topology of targeted MOFs. Consequently, some mechanistic insights regarding MOF synthesis can be inferred in efforts to make MOF topology control possible with the strategic design of organic linkers. Interestingly, with nitro groups, threefold twinning is observed in scu-net NU-904, which yields an average structure of shp-net with triangular microchannels while maintaining the unsaturated metal sites for promising catalytic applications. Significantly different CO2 fixation catalytic activities were observed due to the diverse pore structures. Among them, the mesoporous NU-1008 is found to be a highly active MOF catalyst which displays complete CO2 fixation into styrene oxide in less than 24 hours under room temperature and 1 bar of CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
O. K. F. gratefully acknowledges support from the Defense Threat Reduction Agency (HDTRA1-18-1-0003) for the catalysis study and ARO-STTR (W911SR18C0031) for the linkers and the MOFs syntheses. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. This work made use of the IMSERC at Northwestern University, which has received support from the NSF (CHE-1048773 and DMR-0521267); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). J. L. thanks the China Scholarship Council (CSC) for the financial support.Notes and references
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed.
- A. I. Cooper and M. J. Rosseinsky, Nat. Chem., 2009, 1, 26 CrossRef CAS PubMed.
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- D.-X. Xue, A. J. Cairns, Y. Belmabkhout, L. Wojtas, Y. Liu, M. H. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135, 7660–7667 CrossRef CAS PubMed.
- J.-J. Zheng, S. Kusaka, R. Matsuda, S. Kitagawa and S. Sakaki, J. Phys. Chem. C, 2017, 121, 19129–19139 CrossRef CAS.
- E. J. Cussen, J. B. Claridge, M. J. Rosseinsky and C. J. Kepert, J. Am. Chem. Soc., 2002, 124, 9574–9581 CrossRef CAS PubMed.
- L. Li, R.-B. Lin, R. Krishna, X. Wang, B. Li, H. Wu, J. Li, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 7733–7736 CrossRef CAS PubMed.
- A. J. Rieth and M. Dincă, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef CAS PubMed.
- X. Zhang, Z. Huang, M. Ferrandon, D. Yang, L. Robison, P. Li, T. C. Wang, M. Delferro and O. K. Farha, Nature Catalysis, 2018, 1, 356–362 CrossRef.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- C. R. Kim, T. Uemura and S. Kitagawa, Chem. Soc. Rev., 2016, 45, 3828–3845 RSC.
- H. D. Park, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2018, 140, 10669–10672 CrossRef CAS PubMed.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- D. Aulakh, J. B. Pyser, X. Zhang, A. A. Yakovenko, K. R. Dunbar and M. Wriedt, J. Am. Chem. Soc., 2015, 137, 9254–9257 CrossRef CAS PubMed.
- X. Zhang, M. R. Saber, A. P. Prosvirin, J. H. Reibenspies, L. Sun, M. Ballesteros-Rivas, H. Zhao and K. R. Dunbar, Inorg. Chem. Front., 2015, 2, 904–911 RSC.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
- R. Freund, U. Lächelt, T. Gruber, B. Rühle and S. Wuttke, ACS Nano, 2018, 12, 2094–2105 CrossRef CAS PubMed.
- S. Wuttke, M. Lismont, A. Escudero, B. Rungtaweevoranit and W. J. Parak, Biomaterials, 2017, 123, 172–183 CrossRef CAS PubMed.
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- X. Zhang, Q. Hu, T. Xia, J. Zhang, Y. Yang, Y. Cui, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 32259–32265 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- J. Lippke, B. Brosent, T. von Zons, E. Virmani, S. Lilienthal, T. Preuße, M. Hülsmann, A. M. Schneider, S. Wuttke, P. Behrens and A. Godt, Inorg. Chem., 2017, 56, 748–761 CrossRef CAS PubMed.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673 CrossRef CAS PubMed.
- Z. Chen, Ł. J. Weseliński, K. Adil, Y. Belmabkhout, A. Shkurenko, H. Jiang, P. M. Bhatt, V. Guillerm, E. Dauzon, D.-X. Xue, M. O'Keeffe and M. Eddaoudi, J. Am. Chem. Soc., 2017, 139, 3265–3274 CrossRef CAS PubMed.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS.
- H. Noh, C.-W. Kung, T. Islamoglu, A. W. Peters, Y. Liao, P. Li, S. J. Garibay, X. Zhang, M. R. DeStefano, J. T. Hupp and O. K. Farha, Chem. Mater., 2018, 30, 2193–2197 CrossRef CAS.
- J. Ma, L. D. Tran and A. J. Matzger, Cryst. Growth Des., 2016, 16, 4148–4153 CrossRef CAS.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- Y. Chen, T. Hoang and S. Ma, Inorg. Chem., 2012, 51, 12600–12602 CrossRef CAS PubMed.
- S. B. Kalidindi, S. Nayak, M. E. Briggs, S. Jansat, A. P. Katsoulidis, G. J. Miller, J. E. Warren, D. Antypov, F. Corà, B. Slater, M. R. Prestly, C. Martí-Gastaldo and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2015, 54, 221–226 CrossRef CAS PubMed.
- T. C. Wang, W. Bury, D. A. Gómez-Gualdrón, N. A. Vermeulen, J. E. Mondloch, P. Deria, K. Zhang, P. Z. Moghadam, A. A. Sarjeant, R. Q. Snurr, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 3585–3591 CrossRef CAS PubMed.
- D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661–12667 CrossRef CAS PubMed.
- Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235–2238 CrossRef CAS PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, Z. Wei, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714–17717 CrossRef CAS PubMed.
- M. Lammert, H. Reinsch, C. A. Murray, M. T. Wharmby, H. Terraschke and N. Stock, Dalton Trans., 2016, 45, 18822 RSC.
- S. Wang, J. Wang, W. Cheng, X. Yang, Z. Zhang, Y. Xu, H. Liu, Y. Wu and M. Fang, Dalton Trans., 2015, 44, 8049–8061 RSC.
- M. Zhang, Y.-P. Chen, M. Bosch, T. Gentle, K. Wang, D. Feng, Z. U. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 815–818 CrossRef CAS PubMed.
- H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, J. Lyu, K. I. Otake, X. Wang, L. R. Redfern, C. D. Malliakas, Z. Li, T. Islamoglu, B. Wang and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 11179–11183 CrossRef CAS PubMed.
- P. T. K. Nguyen, H. T. D. Nguyen, H. N. Nguyen, C. A. Trickett, Q. T. Ton, E. Gutiérrez-Puebla, M. A. Monge, K. E. Cordova and F. Gándara, ACS Appl. Mater. Interfaces, 2018, 10, 733–744 CrossRef CAS PubMed.
- T. Islamoglu, K.-i. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibay and O. K. Farha, CrystEngComm, 2018, 20, 5913–5918 RSC.
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson, D. A. Gómez-Gualdrón, F. M. Cetin, J. Jagiello, A. M. Asiri, J. F. Stoddart and O. K. Farha, Chemistry, 2018, 4, 1022–1034 CrossRef CAS.
- Y.-B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H.-C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef CAS PubMed.
- W.-G. Liu and D. Truhlar, Chem. Mater., 2017, 29, 8073–8081 CrossRef CAS.
- P. Deria, D. A. Gómez-Gualdrón, I. Hod, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14449–14457 CrossRef CAS PubMed.
- R. G. AbdulHalim, P. M. Bhatt, Y. Belmabkhout, A. Shkurenko, K. Adil, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2017, 139, 10715–10722 CrossRef CAS PubMed.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175–180 CrossRef CAS.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Proton NMR spectra of organic ligands and MOF materials, crystallographic data, TGA curves, DRIFTS spectra and SEM images are included in the ESI. CCDC [1854453 and 1855836]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04220a |
| This journal is © The Royal Society of Chemistry 2019 |