
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A biomimetic approach towards phorone sesterterpenoids†
Harry J.
Shirley
 and
Christopher D.
Bray
*
and
Christopher D.
Bray
*
The School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.bray@qmul.ac.uk
First published on 10th July 2019
Abstract
We report an investigation towards a unified total synthesis of the Korean sponge derived sesterterpenoids, phorones A (1) and B (2), via a biomimetic strategy. This work has established a new synthetic strategy to the parent ansellane sesterterpenoid skeleton with unanticipated diversion to a biogenetically related pathway.
Introduction
Phorones A (1) and B (2) are isoprenoid natural products and constitutional isomers which bear the unprecedented tetracyclic phorane carbon skeleton. Phorone A (1) was extracted from a sample of the marine sponge Phorbas sp. collected off the coast of Korea, and reported in 2012 by Nam, Kang and their co-workers.1 More recently, the isolation of phorone B (2), which exhibits anti-cancer activity, was reported in 2015 by Woo et al.2 from the Korean marine sponge Clathria gombawuiensis. While isolated from two distinct genera of sponge, the phorones are likely derived from a common biogenesis related to that of a large family of bioactive sesterterpenoids: encompassing the phorbaketals (6),3 alotaketals (7)4 and ansellones (8, 9)5 (Scheme 1) as well as the gombaspiroketals,6 phorbasones7 and isophorbasones1 whose isolations have spawned multiple total synthesis efforts over recent years.8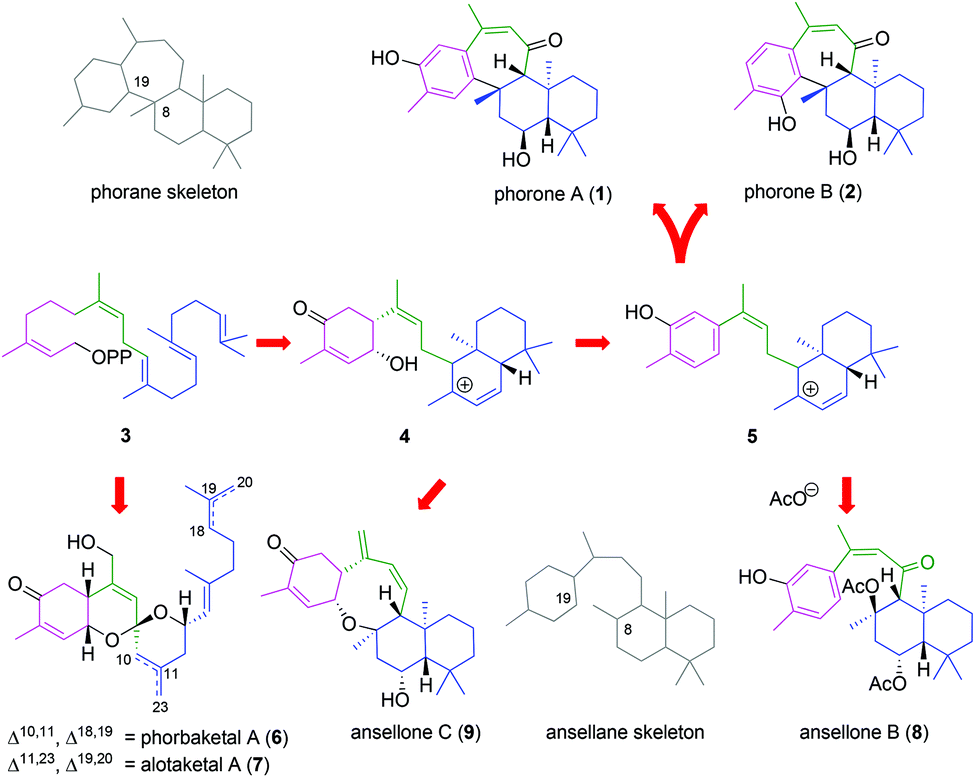 | ||
| Scheme 1 Proposed biogeneses of phorones and ansellones (OPP = pyrophosphate). | ||
A key proposed biogenetic step in the formation of the phorane skeleton from geranylfarnesyl pyrophosphate (3) involves intramolecular cyclization of the ansellane tertiary carbocation 5via electrophilic aromatic substitution of the tethered phenol to give para- and ortho-adducts, as 1 and 2 respectively. Of note is the resulting stereochemical relationship between the seven-membered ring and drimane bicycle, since cis-annulation has not previously been reported. The isolation and structural assignments of ansellones B (8) and C (9) further signifies trapping of the putative tertiary carbocation with either a γ-hydroxycyclohexenone or exogenous acetate. The parent ansellane skeleton is devoid of the C8-19 bridge and so alternative nomenclature for the phorane skeleton is 8β-19-cycloansellane.
In the present work we have investigated a biomimetic synthesis of the phorone isoprenoids in order to elucidate key events in their biogenesis, confirm their assigned relative stereochemistry and assist future structure–activity relationship studies.
Results and discussion
In light of their proposed biogeneses, our retrosynthetic analysis of phorones A (1) and B (2) (Scheme 2) began with disconnection of the key C8–C19 bond bridging the decalin and aromatic systems.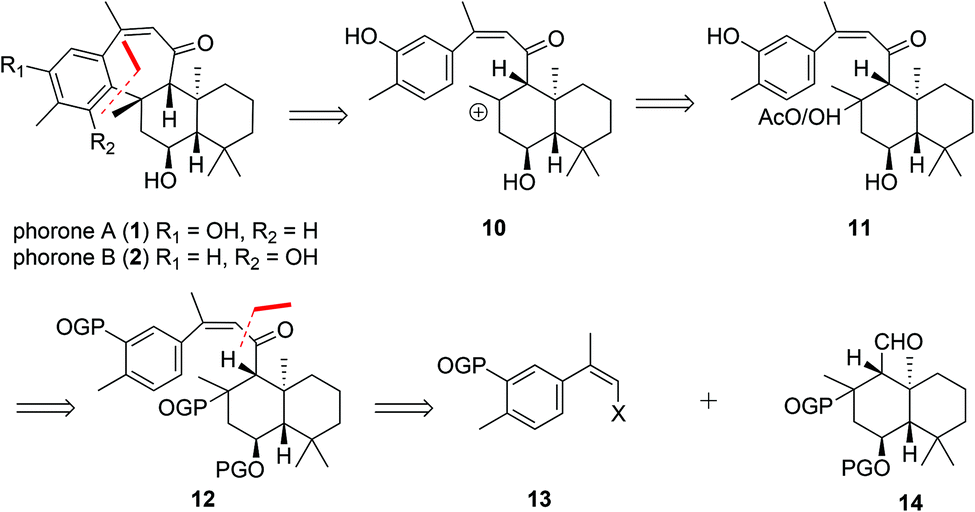 | ||
| Scheme 2 Retrosynthetic analysis of phorones A (1) and B (2). | ||
This gave synthon 10 which would enable trapping of the tertiary carbocation via electrophilic aromatic substitution of the phenol at the ortho- or para-positions to give 1 or 2 respectively. We anticipated that a mixture of regioisomers may form from this step. The formation of such a carbocation intermediate 10 could be achieved by treatment of a tertiary alcohol or acetate 11 with Lewis- or Brønsted acid. Construction of the ansellane skeleton 12 was planned to proceed via coupling of the (Z)-haloalkene 13 with the drimane aldehyde 14 by way of a halogen-metal exchange/electrophile trapping process.9 The (Z)-haloalkene 13 would in turn be accessed by halo-olefination of a suitable acetophenone.
The synthesis of a drimane aldehyde such as 14 commenced by treatment of commercially available β-ionone with NaH in the presence of dimethylcarbonate to give the ester 15, which could be accessed in >20 g. Photochemically-mediated cyclisation of the ester 15 gave enone 16 (Scheme 3).10
 | ||
| Scheme 3 Synthesis of drimane aldehyde 21. | ||
Enone 16 was then readily converted to the diol 18 in three steps by reduction with LiAlH4, allylic alcohol oxidation and then nucleophilic addition using an excess MeLi.9 Diol 18 was then oxidised with m-CPBA to give the epoxide 19 as a single diastereomer. The stereochemistry of the epoxide 19 was confirmed by X-ray crystallographic analysis (see ESI†). The observed non-chelation controlled diastereofacial selectivity anti to the pendant tertiary alcohol may be a consequence of unfavourable steric interactions between the axial methyl groups and the peracid. Following this, the epoxide 19 was reductively ring-opened using Red-Al in refluxing PhMe, to allow regiospecific formation of the desired 1,3-diol in quantitative yield (1,2-diol product was not observed) to give the triol 20. The primary alcohol of triol 20 was selectively oxidised, in preference to the secondary and tertiary alcohols, using PhI(OAc)2 with catalytic TEMPO, to the aldehyde 21. The structure of the aldehyde 21 was confirmed by X-ray crystallographic analysis (Fig. 1, see ESI†) revealing the syn-relationship between the tertiary alchohol and the potentially epimerizable aldehyde, with the remaining secondary alcohol on the opposite face of the decalin system.
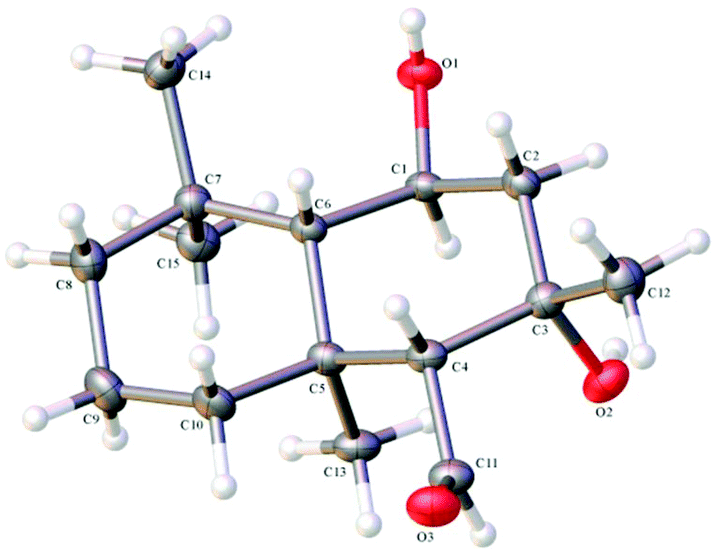 | ||
| Fig. 1 ORTEP image of aldehyde 21. | ||
Following 8 synthetic steps without the need for protecting groups, silylation of the secondary and tertiary alcohols using standard silyl chloride and base combinations returned starting material only, perhaps reflective of a sterically demanding environment. However, rapid trimethylsilylation of both alcohols was achieved using TMSOTf to give the aldehyde 22 (cf.14) which was used to probe the envisaged coupling with the appropriate vinyl metal species.
The synthesis of a (Z)-haloalkene fragment such as 13 began with aromatic nitration of commercially purchased 4-methylacetophenone using HNO3/H2SO4 to nitro compound 23 and hydrogenolysis of the nitro group to an aniline intermediate, before diazotisation and in situ hydrolysis finally gave phenol 24.11 O-Protection of 24 as the tert-butyldimethlsilyl ether using TBSCl in the presence of imidazole gave the siloxyacetophenone 25 (Scheme 4). There are scarcely any methods for the halo-olefination of acetophenones. Indeed, the use of standard halogenated Wittig and Horner–Wadsworth–Emmons reagents in combination with a range of bases was initially investigated but this resulted in either the return of starting materials or the observed formation of only traces of suspected alkenes. Of the few known methods for such a transformation we investigated a three-step process firstly reported by Barluenga12 and later modified by Ando.13 Treatment of acetophenone 25 with dibromomethyl lithium, formed through deprotonation of CH2Br2 with LiNi-Pr2, facilitated conversion to alcohol 26. The tertiary alcohol of 26 was then protected as the trimethylsilyl ether using TMSCl in the presence of imidazole, to give gem-dibromide 27. Finally, treatment of 27 with n-BuLi gave an isomeric mixture of bromoalkenes (Z) and (E)-28 in a 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 ratio respectively. These apolar bromoalkenes were separated by careful column chromatography on standard silica gel, although this essential separation (which could not be achieved using basic alumina or other non-acidic stationary phases) resulted in apparent loss of large amounts of material through degradation during elution. The stereochemistry of each isolated alkene isomer was determined by NOESY experiments (see ESI†).
:1.0 ratio respectively. These apolar bromoalkenes were separated by careful column chromatography on standard silica gel, although this essential separation (which could not be achieved using basic alumina or other non-acidic stationary phases) resulted in apparent loss of large amounts of material through degradation during elution. The stereochemistry of each isolated alkene isomer was determined by NOESY experiments (see ESI†).
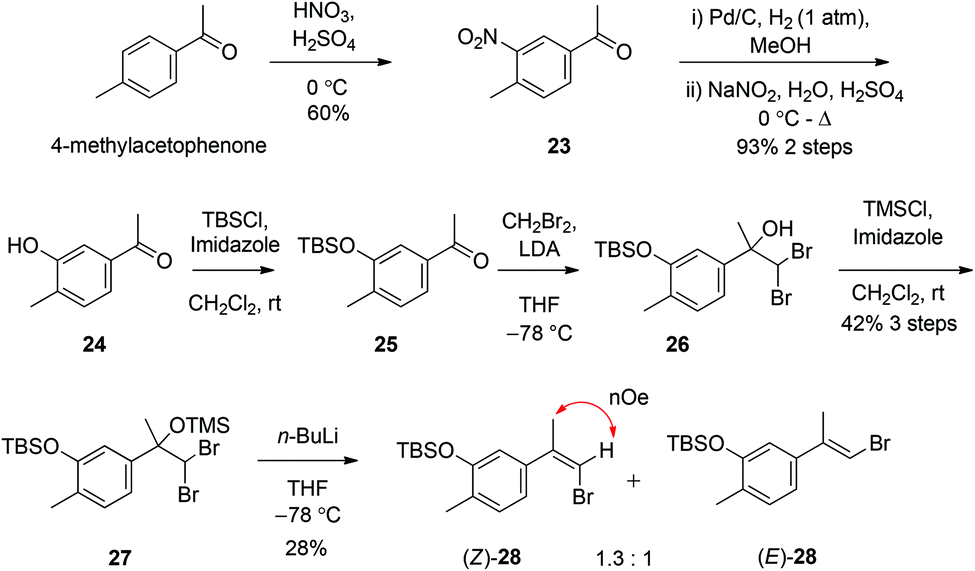 | ||
| Scheme 4 Synthesis of (Z)-bromoalkene 28. | ||
Having achieved the synthesis of drimane aldehyde 22 and (Z)-bromoalkene 28, work was focused upon their convergence (Scheme 5). Treatment of (Z)-28 with t-BuLi at −78 °C caused bromine–lithium exchange to form an organolithium species, to which drimane aldehyde 22 was added. The ansellane alcohol 29 was isolated as a single diastereomer. The stereochemistry of the secondary alcohol was not determined as it would be oxidised to a ketone in the next step. Retention of the crucial Z-alkene was confirmed through a NOESY experiment (see ESI†). This point denotes a new chemical synthesis of the novel ansellane skeleton. Following storage at room temp for 72 hours we observed that alcohol 29 degraded to a complex mixture. Isolation of the major degradation product was achieved and after thorough spectroscopic analysis identified as ansellane diene 30, bearing a 1,3-diene motif. This seemingly labile transformation involved elimination of the secondary alcohol. It was suggested that this process may reflect a sterically demanding environment of the alcohol, whose elimination is thermodynamically favoured, furthermore with extended π-conjugation. It was interesting to observe a similar 1,3-diene moiety in the biosynthetically related ansellone C (9) (which was reported five months after our synthesis of 30). We hypothesise that the 1,3-diene moiety may be acquired in nature through an analogous deoxygenative elimination process.
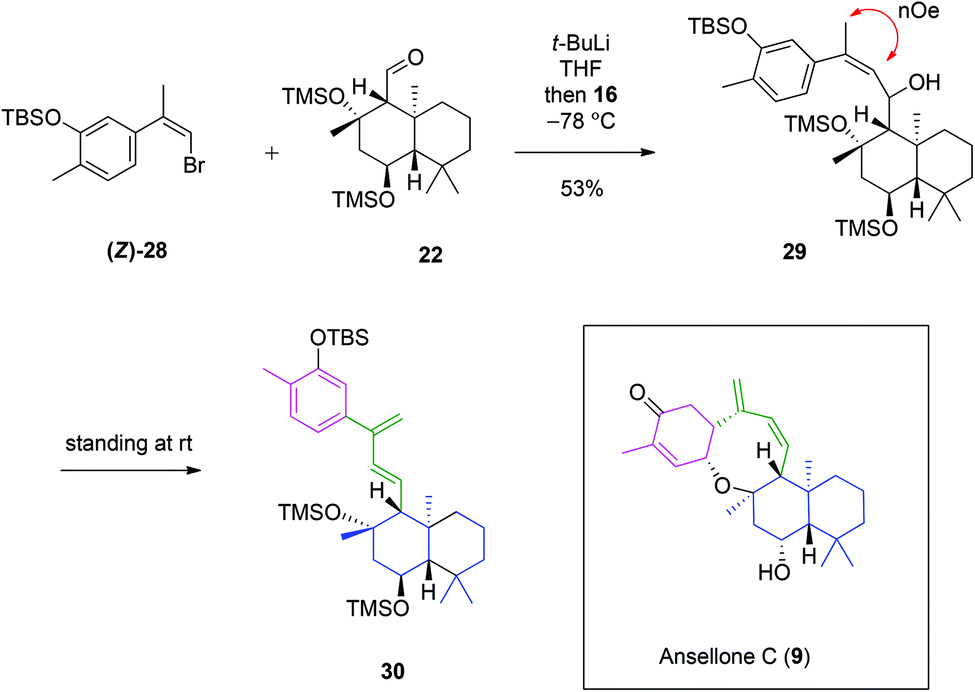 | ||
| Scheme 5 Synthesis of ansellane alcohol 29 and diene 30. | ||
After this set-back, storage of the ansellane alcohol in a −30 °C freezer and using within 3/4 days allowed a short interval for probing of oxidation of the allylic alcohol 29 to enone 31. To our surprise the oxidation of the secondary alcohol could not be achieved using any conventional methods (Dess-Martin's periodinane, MnO2, IBX and PhI(OAc)2/TEMPO), returning starting material only. It was found that Swern oxidation assisted degradation to a mixture of products of a virtually identical composition to that previously observed upon storage of 29 at room temperature (Scheme 6).
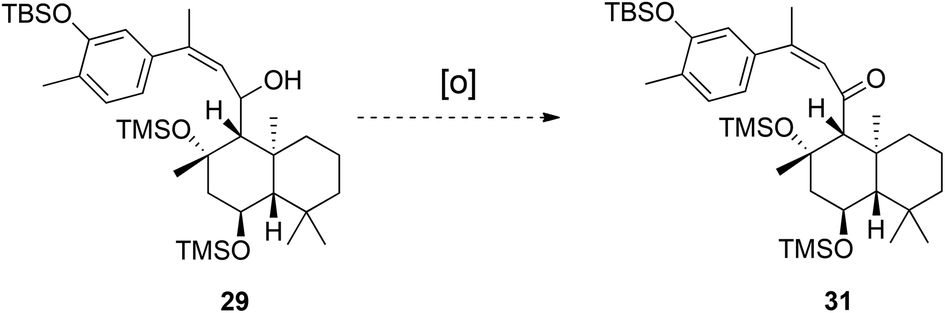 | ||
| Scheme 6 Attempted oxidation of allylic alcohol 29. | ||
Conclusions
These synthetic accomplishments leading to ansellane alcohol 29 and ansellane diene 30 provide novel chemical syntheses of the important ansellane sesterterpenoid skeleton. Key steps include diastereofacial selective epoxide installation to 19 and subsequent reductive epoxide opening to 20, before t-BuLi-mediated addition of (Z)-bromoalkene 28 to aldehyde 22. This work will provide valuable guidance for future total synthesis endeavours of phorone, ansellone and related natural products, in which we anticipate sustained interest. The labile transformation of ansellane alcohol 29 into ansellane diene 30 has shed some light on the biogenesis of this family of sesterterpenoids, and may provide guidance to related future synthetic endeavours.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank Queen Mary University of London and the Engineering & Physical Sciences Research Council for a DTA studentship (EP/J50029X/1) (HJS). We thank the Engineering & Physical Sciences Research Council UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data (performed by Dr. Peter Horton).14Notes and references
- W. Wang, Y. Lee, T. G. Lee, B. Mun, A. G. Giri, J. Lee, H. Kim, D. Hahn, I. Yang, J. Chin, H. Choi, S. Nam and H. Kang, Org. Lett., 2012, 14, 4486–4489 CrossRef CAS PubMed.
- J. Woo, C. Kim, C. Ahn, D. Oh, K. Oh and J. Shin, J. Nat. Prod., 2015, 78, 218–224 CrossRef CAS.
- (a) J. Rho, B. Hwang, C. J. Sim, S. Joung, H. Lee and H. Kim, Org. Lett., 2009, 11, 5590–5593 CrossRef CAS PubMed; (b) M. R. Byun, A. R. Kim, J. Hwang, M. K. Sung, Y. K. Lee, B. S. Hwang, J. Rho, E. S. Hwang and J. Hong, FEBS Lett., 2012, 586, 1086–1092 CrossRef CAS; (c) W. Wang, Y. Lee, R. M. Venkat, Y. Park, J. Lee, H. Kim, D. Hahn, J. Chin, M. Ekins, S. J. Nam and H. Kang, J. Nat. Prod., 2013, 76, 170–177 CrossRef CAS; (d) Y. Lee, W. Wang, H. Kim, A. G. Won, D. Hahn, K. R. Baek, J. Lee, I. Yang, J. Lee, H. Choi and S. J. Nam, Bioorg. Med. Chem. Lett., 2014, 24, 4095–4098 CrossRef CAS PubMed.
- (a) R. Forestieri, C. E. Merchant, N. J. De Voogd, T. Matainaho, T. J. Kieffer and R. J. Andersen, Org. Lett., 2009, 11, 5166 CrossRef CAS PubMed; (b) J. Arias, A. S. Alberts, P. Brindle, F. X. Claret, T. Smeal, M. Karin, J. Feramisco and M. Montminy, Nature, 1994, 370, 226–229 CrossRef CAS PubMed.
- (a) J. Daoust, A. Fontana, C. E. Merchant, N. J. De Voogd, B. O. Patrick, T. J. Kieffer and R. J. Andersen, Org. Lett., 2010, 12, 3208–3211 CrossRef CAS PubMed; (b) J. Daoust, M. Chen, M. Wang, D. E. Williams, M. A. G. Chavez, A. Wang, C. E. Merchant, A. Fontana, T. J. Kieffer and R. J. Andersen, J. Org. Chem., 2013, 78, 8267–8273 CrossRef CAS; (c) J. Daoust, PhD thesis, The University of British Columbia, 2005.
- J. Woo, C. Kim, S. Kim, H. Kim, D. Oh, K. Oh and J. Shin, Org. Lett., 2014, 16, 2826–2829 CrossRef CAS PubMed.
- J. R. Rho, B. S. Hwang, S. Joung, M. R. Byun, J. Hong and Y. Lee, Org. Lett., 2011, 13, 884–887 CrossRef CAS.
- For a review of syntheses: (a) Y. Chen, J. Zhao, S. Li and J. Xu, Nat. Prod. Rep., 2019, 36, 263–288 RSC. For synthetic efforts towards the ansellones: (b) W. Zhang, H. Yao, J. Yu and Z. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4787–4791 CrossRef CAS PubMed. Phorbaketals: (c) J. Hubert, D. Furkert and M. A. Brimble, J. Org. Chem., 2015, 80, 2715–2723 CrossRef CAS; (d) J. Hubert, D. Furkert and M. A. Brimble, J. Org. Chem., 2015, 80, 2231–2239 CrossRef CAS; (e) H. J. Shirley and C. D. Bray, Eur. J. Org. Chem., 2016, 1504–1507 CrossRef CAS. For alotaketal A: (f) J. Huang, J. R. Yang, J. Zhang and J. Yang, Org. Biomol. Chem., 2013, 11, 3212–3222 RSC; (g) J. Huang, J. R. Yang, J. Zhang and J. Yang, J. Am. Chem. Soc., 2012, 134, 8806–8809 CrossRef CAS; (h) M. Xuan, I. Paterson and S. M. Dalby, Org. Lett., 2012, 14, 5492–5495 CrossRef CAS. For a review on whole family: (i) H. J. Shirley, M. Jamieson, M. A. Brimble and C. D. Bray, Nat. Prod. Rep., 2018, 35, 210–219 RSC.
- W. Gao, K. Sakaguchi, S. Iseo and Y. Ohfune, Tetrahedron Lett., 1996, 37, 7071–7074 CrossRef CAS.
- J. White, R. Skeean and G. Trammell, J. Org. Chem., 1985, 50, 1939–1948 CrossRef CAS.
- (a) L. Montiel, G. L. Zepeda and J. Tamariz, Helv. Chim. Acta, 2010, 93, 1261–1273 CrossRef CAS; (b) A. P. Phadnis, B. Nanda, S. A. Patwardhan and A. S. Gupta, Indian J. Chem., Sect B, 1984, 23, 1098–1102 Search PubMed.
- J. Barluenga, J. L. Fernandez-Simon, J. M. Concellon and M. Yus, J. Chem. Soc., Perkin Trans. 1, 1989, 691–694 RSC.
- K. Ando, M. Kitamura, K. Miura and K. Narasaka, Org. Lett., 2004, 6, 2461–2463 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043793 and 1043794. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob00745h |
| This journal is © The Royal Society of Chemistry 2019 |