DOI:
10.1039/C8NA00156A
(Paper)
Nanoscale Adv., 2019,
1, 565-580
Phase evolution in calcium molybdate nanoparticles as a function of synthesis temperature and its electrochemical effect on energy storage†
Received
21st August 2018
, Accepted 7th October 2018
First published on 8th October 2018
Abstract
The design of a suitable electrode is an essential and fundamental research challenge in the field of electrochemical energy storage because the electronic structures and morphologies determine the surface redox reactions. Calcium molybdate (CaMoO4) was synthesized by a combustion route at 300 °C and 500 °C. We describe new findings on the behaviour of CaMoO4 and evaluate the influence of crystallinity on energy storage performance. A wide range of characterization techniques was used to obtain detailed information about the physical and morphological characteristics of CaMoO4. The characterization results enable the phase evolution as a function of the electrode synthesis temperature to be understood. The crystallinity of the materials was found to increase with increasing temperature but with no second phases observed. Molecular dynamics simulation of electronic structures correlated well with the experimental findings. These results show that to enable faster energy storage and release for a given surface area, amorphous CaMoO4 is required, while larger energy storage can be obtained by using crystalline CaMoO4. CaMoO4 has been evaluated as a cathode material in classical lithium-ion batteries recently. However, determining the surface properties in a sodium-ion system experimentally, combined with computational modelling to understand the results has not been reported. The superior electrochemical properties of crystalline CaMoO4 are attributed to its morphology providing enhanced Na+ ion diffusivity and electron transport. However, the presence of carbon in amorphous CaMoO4 resulted in excellent rate capability, suitable for supercapacitor applications.
Introduction
Global energy supply is currently dominated by fossil fuels. Although wind and solar generated electricity is becoming increasingly available across the world, these provide energy intermittently. Energy storage systems are therefore required for load leveling, allowing energy to be stored when generated and used when needed. The transition to reliable renewable energy sources depends on the availability of storage.1 Rechargeable batteries and supercapacitors are emerging as two important classes of electrochemical energy storage, which can supply energy consistently and allow greater penetration of renewable energy into the electricity grid.1–3 Energy storage capability also has obvious benefits in terms of reducing greenhouse emissions by displacing the need for fossil-fueled spinning reserves. The current generation lithium-ion batteries are used for portable applications in the main due to their high specific capacity and power to weight ratio. For static energy storage, where such properties are less important, new materials must be developed with lower cost and improved long-term performance.
Electrochemical energy storage involves chemical redox reactions at a surface or interface. The major difference between a rechargeable battery and a supercapacitor is the processes that take place at the electrode surfaces.4 In the battery, it is governed by the faradaic process, i.e. electron transfer (reduction/oxidation). In the supercapacitor, it is governed by the non-faradaic process, i.e. the charge stored electrostatically on the electrode surfaces.4 Thus, the interaction between ions in the electrolyte and the surface of an electrode is critically important for both batteries and supercapacitors. To design suitable electrode materials with a specific shape-controlled morphology is a key step in obtaining materials for these applications.5
Electrode materials play a significant role in the development of high-performance energy storage devices. Transition metal oxides (MO2)6 and phosphates (LiMPO4),7 (M refers to transition metal cations), are a well-researched area for over two decades. In particular, manganese di oxide (MnO2)6 and lithium iron phosphate (LiFePO4)7 are promising electrode materials because they are readily available, low cost, environmentally friendly, have a wide electrochemical window, and have rich redox chemistry delivering high theoretical specific capacity. In addition, both these materials function well with both aqueous and non-aqueous electrolytes.8,9 Recently, metal molybdates MMoO4i.e. Mo-containing mixed oxides, have emerged as “hosts” for lithium insertion (as opposed to single metal oxides). These show particular promise for storing renewable energy. Ternary metal oxides have also been reported10,11 to exhibit a wide voltage window, which enhances energy storage performance. Among molybdates, alkaline-earth metal molybdates form an important class of functional materials, which has application in many areas such as luminescent devices, optical fibers, catalysis, and energy storage.12,13 They crystallize with a powellite-type structure, containing large bivalent cations (MMoO4, where M = Ca, Mg or Ba), in which the molybdate ions are loosely connected to M cations and adopt a tetrahedral coordination.12,13 Among the alkaline-earth metal molybdates, CaMoO4 has been widely explored for applications such as luminescent hosts, catalysis, LEDs, and energy storage but all the studies related to energy storage are restricted to lithium-ion batteries.14,15 Despite the merits of using lithium, serious concerns arise with its high and escalating cost and currently limited supply. For this reason, sodium has been considered as an attractive alternative to lithium, due to its low cost and high abundance. In this work, we evaluate CaMoO4 in sodium-based energy storage devices.
Research on molybdate structural frameworks MMoO4 with various metal cations (M = Ni, Mn, and Co) has been reported widely and shown their potential as electrodes in both lithium-ion batteries and supercapacitors.16–18 Among the molybdate frameworks based on metal cations, nickel molybdate (NiMoO4)16 has caused great interest due to its high redox potential, low toxicity and being environmentally benign than its Co counterpart. Cobalt molybdate (CoMoO4)17 is another potential candidate as it exhibits a fast reversible redox reaction but is less electrochemically active than its nickel counterpart. However, alkaline-earth-based molybdate systems are much more important functional materials because of their unique intrinsic properties, which have not yet been explored for sodium-ion supercapacitors and batteries.
Based on the above considerations, CaMoO4 was synthesized at various temperatures. The influence of synthesis temperature on the crystallinity of the phase and the resulting electrochemical performance has been evaluated. To better understand the experimental findings, the electronic structural evolution of CaMoO4 by means of the density of states (DOS) patterns is obtained from the spin-polarized Density Functional Theory (DFT) computations. We report high power capability for the material synthesised at 300 °C and high energy density for that produced at 500 °C. The cycling performance for both materials showed an excellent retention rate. We also evaluated the possible causes of these effects by using electron microscopy and spectroscopy.
Experimental section
Synthesis of calcium molybdate
Calcium molybdate (CaMoO4) was synthesized using a combustion process. All reagents were purchased from Sigma-Aldrich. In a typical synthesis, stoichiometric amounts of calcium nitrate (Ca(NO3)2·4H2O) in the presence of ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and urea (CO(NH2)2) as a fuel were dissolved in de-ionized water (about 50 mL) with magnetic stirring. The pH of the solution was adjusted to 7 by dropwise addition of ammonia solution. In neutral solutions, ammonium molybdate tetrahydrate is present as the monomeric (MoO4)2− ion.19 Subsequently, the solution was evaporated to dryness followed by heating in an oven to 150 °C for 2 h for complete dehydration of the precursors. Once dried, the dehydrated samples were then transferred into a furnace at either 300 °C for 3 h or 500 °C for 4 h. Both reactions produced CaMoO4. However, the lower temperature reaction produced a poorly crystalline phase, while the higher temperature reaction produced a highly crystalline phase.
Brief outline of the computation method
The present computational work has been carried out based on spin-polarized Density Functional Theory (DFT)20,21 as implemented in the Vienna Ab-initio Simulation Package (VASP)22 package. The core and valence electrons are treated separately by employing the Projector-Augmented Wave (PAW) approach23 as embedded in the VASP. The valence electron configurations of Ca, Mo and O are 3s2 3p6 4s2; 4p6 5s2 4d5 and 2s2 2p4, respectively. We have employed the Perdew–Burke–Ernzerhof (PBE)24 type generalized gradient approximation (GGA) as the exchange–correlation functional term in the Kohn–Sham equation. However, it is not able to provide the reliable band gap value for CaMoO4 due to the constraint arising from self-interaction errors, especially, for d-localized electrons. Therefore, we have used the DFT + U (Hubbard U term) method in which the Ueffective value was set as 4.38 eV for Mo, which could significantly improve the accuracy of the band gap values and the corresponding electronic properties of CaMoO4. All the structures are fully optimized using the conjugate-gradient (CG) algorithm and are converged until the total energy differences and the residue Hellmann–Feynman forces are below 0.001 meV and 0.01 eV Å−1, respectively. This enabled us to find the minimum energy configurations of CaMoO4 at 0 K, 300 °C, and 500 °C temperature. The energy convergence has also been tested for an energy cutoff of 500 eV for the considered system and the 3 × 3 × 5 Monkhorst Pack k-points25 have been used throughout the electronic structure calculations. As our investigation of CaMoO4 is not only confined to 0 K, but also to 300 and 500 K, we have performed the molecular dynamics (MD) simulation within the framework of DFT + U. The simulation time for the MD calculation is 14 ps with a time step of 2 fs. After getting the optimized geometries of CaMoO4 at 0 K, 300 °C, and 500 °C, we have determined the projected density of states of the corresponding minimum energy configurations.
Physical characterization of CaMoO4
The phase purity of the as-synthesized sample was characterized by X-ray diffraction (XRD, Philips PANalytical, Bruker D8) analysis using Cu-Kα radiation at an accelerating voltage and current of 40 kV and 30 mA, respectively. Crystallographic identifications and intensities of the samples prepared at 300 °C and 500 °C were accomplished through comparing the experimental XRD patterns with standards from the JCPDS (077-2238) together with the software X'Pert Highscore and Match version 3 for analysis. The morphology of the CaMoO4 material was characterized by Mira VP – Field Emission Scanning Electron Microscope (FESEM). High-magnification imaging of the CaMoO4 material was carried out using scanning transmission electron microscopy (STEM) – imaging in various imaging modes: high angle annular dark field imaging (HAADF), bright field (BF) and secondary electron (SE) on a JEOL ARM 200F TEM operated at 200 kV. STEM specimens were prepared by grinding a small amount of powder in ethanol and dispersing it on a holey carbon film. Brunauer–Emmett–Teller (BET) surface area measurements and porosity analysis were also carried out using a Micromeritics Tristar II surface area and porosity analyzer. For porosity measurements, all samples were degassed at 100 °C overnight prior to analysis. X-ray photoelectron spectra were acquired using a Kratos AXIS Nova instrument (Kratos Analytical Ltd, U.K.) equipped with a monochromated Al Kα radiation source (1486.69 eV) operating at 150 W power (15 kV, 10 mA). The analysis chamber pressure was <1 × 10−9 Torr at the beginning of the analysis. High-resolution spectra acquired for selected photoemissions were recorded at 0.1 eV per step and a pass energy of 20 eV. The analysis area was approx. 300 μm × 700 μm. All spectra are calibrated by setting the main O 1s component position to 530.45 eV.
Electrochemical characterisation of CaMoO4
Aliquots (5 μL) of electrolytes used were aqueous 2 M NaOH and non-aqueous 1 M NaClO4 in ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl carbonate:fluoroethylene carbonate (EC:DMC:FEC) (88:10:2) for all sodium-ion battery and supercapacitor measurements. For the sodium-ion battery, metallic sodium (from Sigma Aldrich) was used as the anode.
:dimethyl carbonate:fluoroethylene carbonate (EC:DMC:FEC) (88:10:2) for all sodium-ion battery and supercapacitor measurements. For the sodium-ion battery, metallic sodium (from Sigma Aldrich) was used as the anode.
For the single electrode tests, a platinum wire of 10 cm length and 1 mm diameter and mercury–mercuric oxide (Hg/HgO) served as the counter and reference electrodes, respectively. CaMoO4 was the working electrode. For electrochemical measurements, the materials were investigated by constructing a working electrode consisting of CaMoO4 (70 wt%), carbon black (20 wt%), and poly-vinylidene fluoride (PVDF) binder (10 wt%) with 0.4 mL of N-methyl-2-pyrrolidine (NMP) to make a slurry. Ingredients were mixed in an agate mortar to produce a uniform paste, which was coated onto a 1 cm2 graphite sheet/stainless steel disc as a current collector. Galvanostatic charge–discharge experiments and cyclic voltammetry measurements were performed using an SP-150 Biologic potentiostat and an Arbin battery testing unit (ARBIN BT-2000) at Helmholtz Institute Ulm (HIU). All the measurements related to the non-aqueous electrolyte involved a sealed Swagelok-type cell which was assembled in a glovebox under an Ar atmosphere and the analysis was then carried out in air in a similar manner to that conducted in the aqueous system but with different electrolytes.
Results and discussion
Physical characterization and computation
(a) X-ray diffraction.
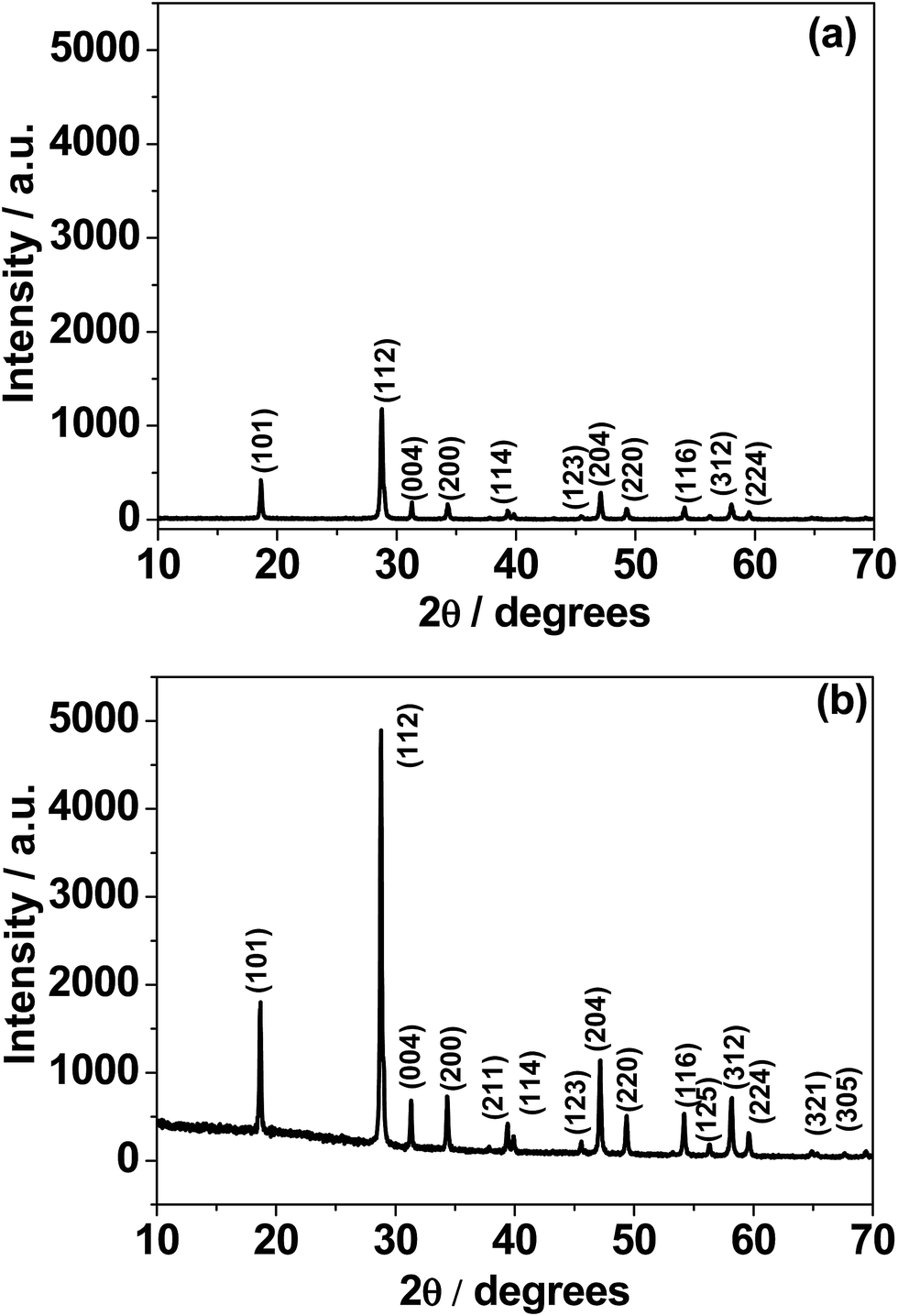
Fig. 1 shows the X-ray diffraction (XRD) patterns of calcium molybdate (CaMoO4) synthesized via a combustion route at temperatures of: (a) 300 °C and (b) 500 °C. All of the observed diffraction peaks were indexed to the family of the powellite structured compounds (JCPDS card no. 077-2238) having a tetragonal space group I41/a with cell constants a = 5.22 Å and c = 11.42 Å. Although several reports15,26 have indexed CaMoO4 to the tetragonal scheelite-type, we did not observe this structure. Fig. S1† shows the baseline corrected XRD patterns. For the 300 °C sample, the intensity of the peaks (in red) is relatively low and the weakest peaks are difficult to discern, implying that the synthesized product is poorly crystalline and/or the crystallite size is very small. The material synthesized at 500 °C shows more intense (in black) and sharper peaks, suggesting that the CaMoO4 has become more crystalline and/or the crystallites are coarser. No second phases were present in either of these XRD patterns indicating that phase-pure calcium molybdate can be produced through a combustion route at synthesis temperatures as low as 300 °C. From the analysis of the XRD data, it seems that the microstructure of the CaMoO4 crystallite/grain size increases with synthesis temperature. Fig. S2a and b† show respective details of the patterns in Fig. 1a and b. The separation between the Bragg reflections (211) and (114) was found to be 0.55 and 0.5 for the 300 and 500 °C materials, respectively. The corresponding (211) to (114) intensity ratios were 68% and 66%, respectively. This estimation may indicate a slight structural deviation from powellite to scheelite at higher temperature. XRD patterns were obtained during in situ heating of the CaMoO4 material from room temperature to 600 °C (Fig. S3†). There were only little changes in peak width or positions, indicating that no changes occur in the phase and crystal structure. An increase in the peak intensities was found to occur on ramping the temperature to 600 °C. This confirmed the results of the ex situ XRD on as-prepared materials (Fig. 1), namely that crystallinity is enhanced at higher temperature.
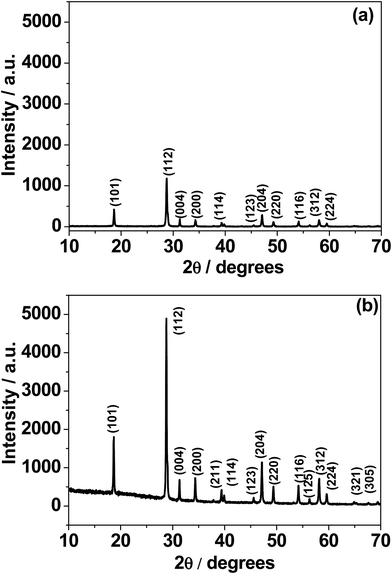 |
| | Fig. 1 X-ray diffraction (XRD) patterns of CaMoO4 synthesized at: (a) 300 and (b) 500 °C. The lower temperature material displayed a much weaker pattern, suggesting that it was not as crystalline as the higher temperature counterpart. | |
(b) Electronic structure of CaMoO4.
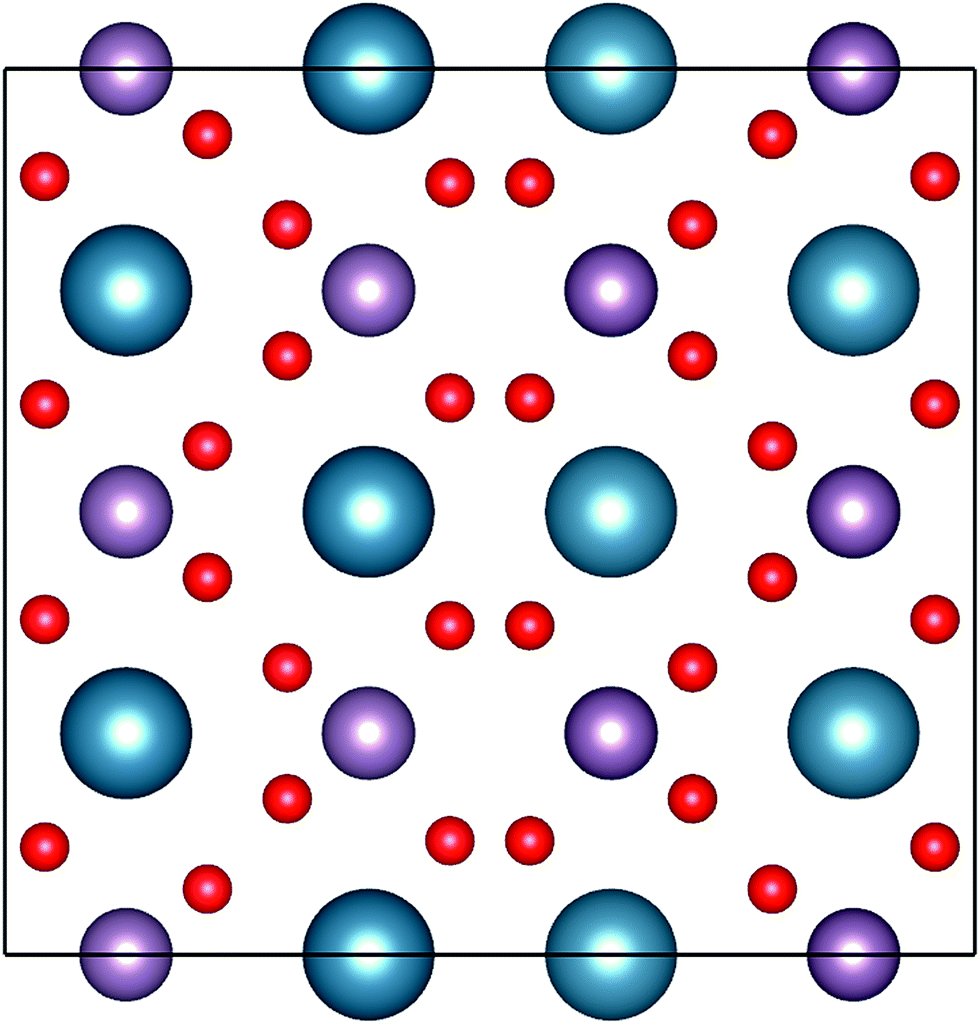
To validate the experimental findings obtained from the XRD, we have performed electronic structure calculations coupled with molecular dynamics (MD) simulation for computations of the structure of CaMoO4. The crystal structure parameters of CaMoO4 computed at temperatures 0 K, 300 °C, and 500 °C temperatures are given in Table 1. The MD simulations, to achieve the minimum energy configuration at 300 °C and 500 °C, were performed using two different approaches in order to bring out the variations in the electronic structure. In the first approach, the respective structures were stabilized at 300 °C and 500 °C, whereas in the second approach, the structures were quenched from a higher temperature of 1725 (arbitrarily chosen) to 300 and 500 °C (denoted as Q300 and Q500) with a cooling rate of 20 °C per step. The increase in volume for the minimum energy configuration of CaMoO4 for the first approach has been found from 1282.625 Å3 to 1283.681 Å3 (300 °C) and 1283.715 Å3 (500 °C). In the case of the second approach, the volume increases to 1290.745 Å3 (Q300) and 1290.513 Å3 (Q500). This shows that, as a function of synthesis temperature and the way it has been relaxed, deviation occurs in the crystal parameter. The optimized crystal structure corresponding to the minimum energy configurations of CaMoO4 is shown in Fig. 2.
Table 1 Lattice parameters and unit cell volume of CaMoO4 computed at different temperatures
| Temp (K) |
a (Å) |
b (Å) |
c (Å) |
α (°) |
β (°) |
γ (°) |
Vol (Å3) |
VBM (eV) |
CBM (eV) |
Bandgap (eV) |
| 0 |
10.54356 |
10.54357 |
11.53786 |
89.9 |
89.9 |
90 |
1282.625 |
−0.451 |
2.515 |
2.966 |
| 300 |
10.54446 |
10.54596 |
11.54375 |
89.9 |
90 |
89.9 |
1283.681 |
−0.512 |
2.562 |
3.074 |
| Q300 |
10.55743 |
10.55798 |
11.57981 |
90.01 |
90.01 |
90.01 |
1290.745 |
−0.449 |
2.539 |
2.988 |
| 500 |
10.54666 |
10.54346 |
11.54439 |
89.9 |
90 |
90 |
1283.715 |
−0.454 |
2.535 |
2.989 |
| Q500 |
10.55579 |
10.55737 |
11.5802 |
90 |
90 |
90.01 |
1290.513 |
−0.438 |
2.519 |
2.966 |
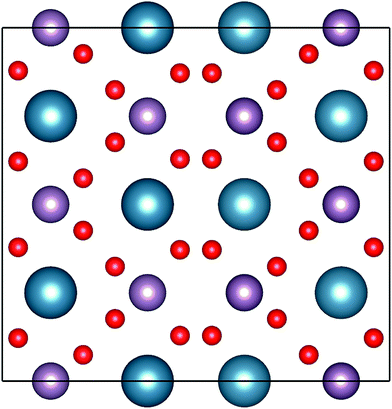 |
| | Fig. 2 Representation of the powellite-type crystal structures of CaMoO4. The atoms with blue, pink and red indicate Ca, Mo and O, respectively. | |
The electronic structure calculations have been performed in order to determine the projected density of states (PDOS) of CaMoO4 at 0 K, 300 °C, and 500 °C (Fig. 3a–c and e). The PDOS of CaMoO4 at Q300 and Q500 are displayed in Fig. 3d and f. The calculated band-gap width increased by 0.1 eV (Fig. 3a and c) of CaMoO4 on going from 0 K to 300 °C and 0.023 eV (Fig. 3a and e) while heating up to 500 °C. Overall, the band gap of CaMoO4 stabilized around 3.0 eV. In the case of the second modelling approach, following quenching the system from 1750 °C with a rate of 20 °C per step, no significant variation in the band gap (Fig. 3d and f) was observed. This suggests that the change in the band gap is sensitive to the synthesis temperature, supported by the XRD data (Fig. 1). The modelled results show a shift in the conduction band minima (CBM) and valence band maxima (VBM) in Fig. 3 as a result of increasing the temperature. In the case of 0 K to 300 °C, the VBM shifted from −0.451 eV to −0.512 eV, and the CBM shifted from 2.515 eV to 2.562 eV. Increasing the temperature to 500 °C, the VBM shift was minimal while the shift in the CBM has been observed from 2.515 eV to 2.535 eV. In the case of the second approach, the shifts (Table 1) in the VBM and CBM are not significant.
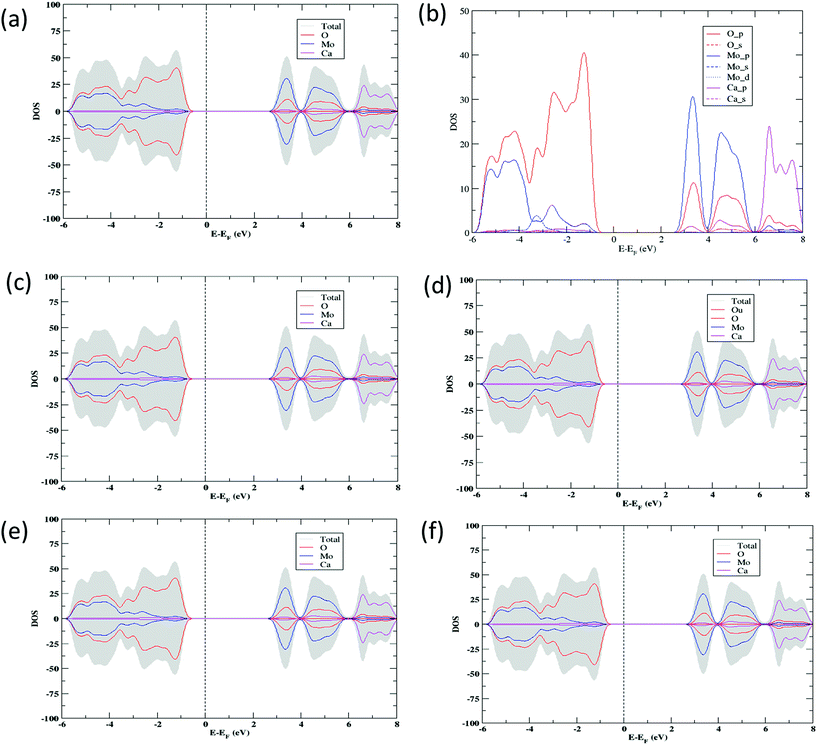 |
| | Fig. 3 Total DOS and PDOS for CaMoO4 computed in a range of temperatures (a) 0 K, (c) 300 °C (d) Q300, (e) 500 °C, and (f) Q500 using the DFT + U exchange correlation functional. The prefix Q signifies a model quenched from high temperature and then allowed to relax. The vertical dotted line represents the Fermi level set at 0 eV; right and left sides represent the conduction and valence bands, respectively. (b) Representation of atomic orbital PDOS for CaMoO4 at 0 K. (c, d) and (e, f) indicate the electronic structure of CaMoO4 at 300 and 500 °C with two different approaches (unquenched or quenched), respectively. | |
From the projected DOS of CaMoO4 at 0 K, 300 °C, and 500 °C (Fig. 3b), it has been found that the conduction band is formed predominantly by the Mo 4p states while the valence band is composed of mainly the O 2p states near the Fermi region. The contribution of Ca to the PDOS appears to be relatively smaller in both the bands, indicating that CaMoO4 is a less conducting material. The hybridization between Mo-4d and O-2p orbitals is observed in a similar fashion for CaMoO4 at 0 K, 300 °C, and 500 °C. The electronic structures of CaMoO4 obtained at various temperature are the consequence of the inherent structural reformation. The computed results summarize the values of band-gap widths and positions of the local levels and these provide some new insights into CaMoO4 as a function of temperature.
(c) X-ray photoelectron spectroscopy.
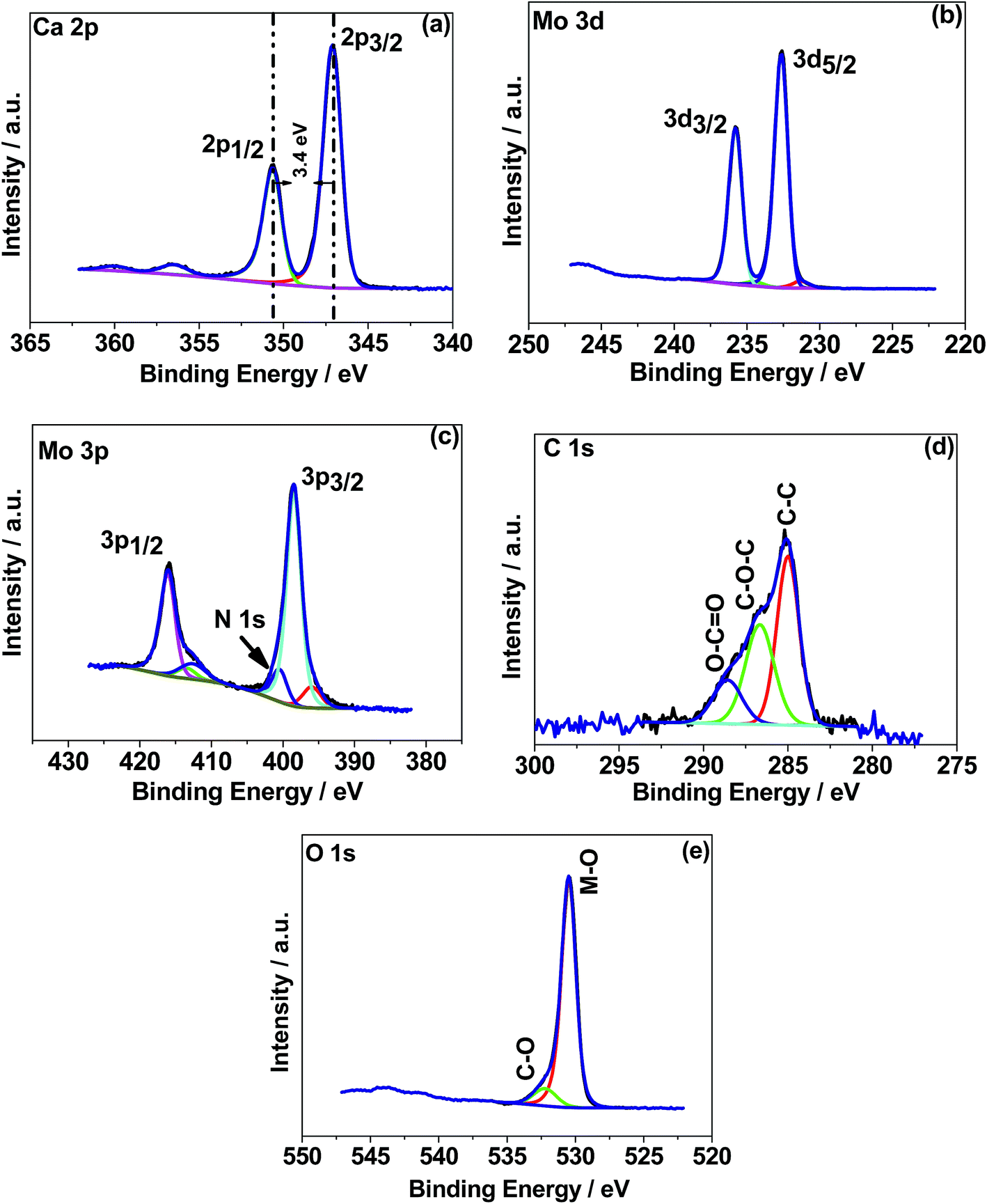
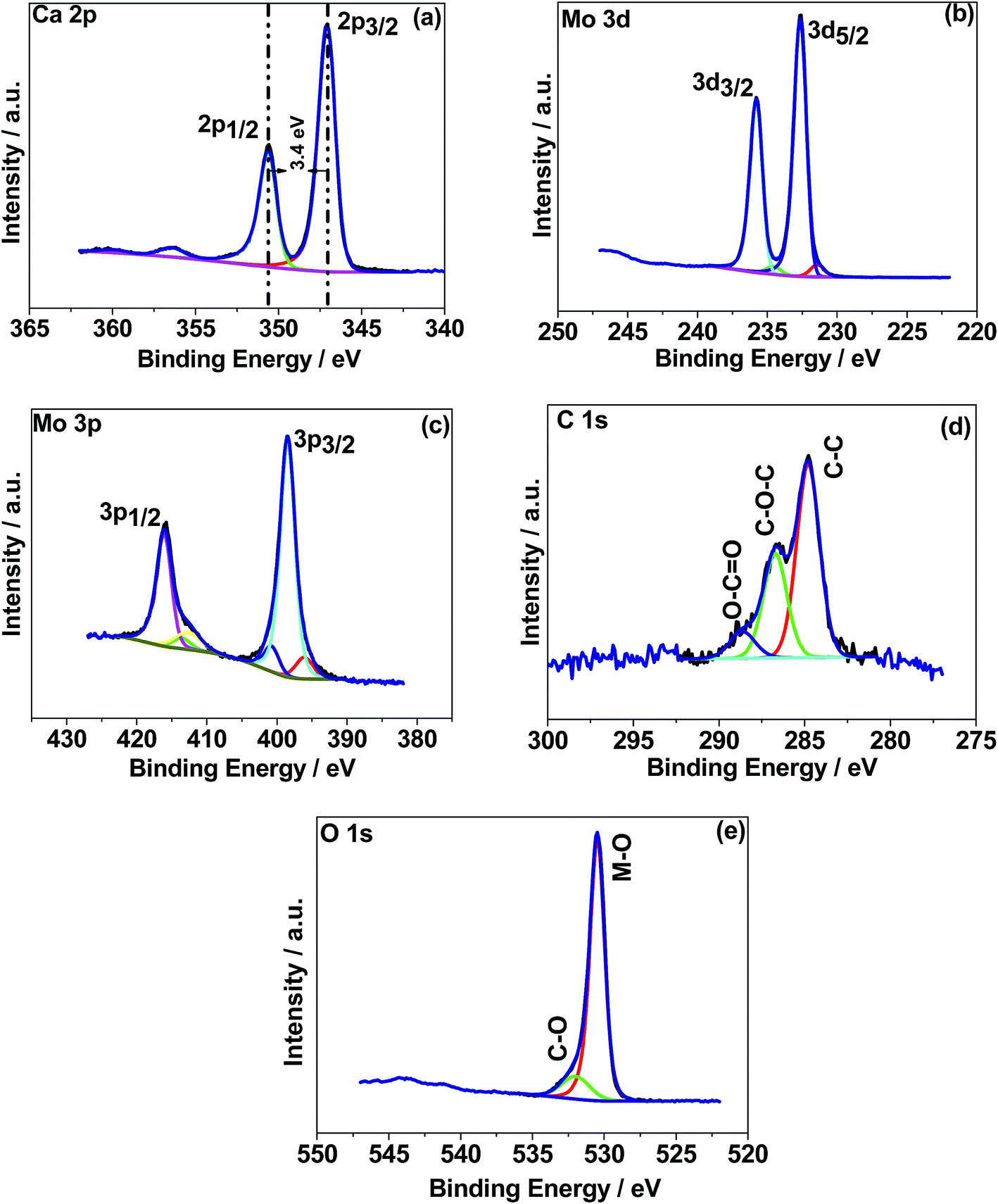
X-ray photoelectron spectroscopy (XPS) was used to determine the chemical (oxidation) states of the elements in the CaMoO4 samples synthesized at 300 °C and 500 °C. The XPS spectra were processed using a Shirley background correction followed by peak fitting with Gaussian–Lorentzian line shapes (Fig. 4 and 5). The binding energy values for all the cations are in accordance with the reported values in the literature.27–29 The Ca 2p spectrum in Fig. 4a exhibits peaks at 347.2 eV and 350.6 eV which can be attributed to Ca 2p3/2 and Ca 2p1/2, respectively. The separation of these peaks at 3.4 eV indicates Ca in the 2+ oxidation state.22 For the material produced at 500 °C the peak positions of Ca 2p (Fig. 5a) are unchanged but their intensity is varied to a small extent. Fig. 4b and c show Mo 3d and 3p spectra. Mo 3d spectra were fitted with two doublets, having spin–orbit components Mo 3d5/2 and Mo 3d3/2 at 232.6 eV and 235.7 eV. These are associated with a single oxidation state (Mo6+) in molybdenum oxides.27–29 The fitted Mo6+ doublet is slightly asymmetric in shape, again being characteristic of the 6+ oxidation state.27 The Mo 3p doublet comprising 3p3/2 and 3p1/2 at 398.4 eV and 415.9 eV obscures the presence of an N 1s peak. A possible fit to this region is shown in Fig. 4c, using a main Mo 3p doublet for Mo6+ and a single peak to account for a loss feature that exists at the low binding energy of the Mo 3p1/2 peak. The remaining area is then fitted with a single N 1s peak set to the 400 eV position. Very similar behaviour was observed in Fig. 5b and c for the 500 °C sample, except for the intensity variation. The correlations of Ca and Mo oxidation states with binding energies are in accordance with the literature value, suggesting the formation of the CaMoO4 structure as a function of temperature. Fig. 4d presents the deconvolution of the C 1s spectrum exhibiting three characteristic peaks at 284.6–285 eV, 286.3–286.7 eV, and 288.6 eV that can be assigned to C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.30 The C–C is likely due to adventitious carbon contamination, and C–O arises from the residual urea as a fuel during the combustion synthesis and the C–NH2 functional group would be assigned to the CO peak.30 Similar behaviour was observed for the 500 °C material relating to C–C and C–O while about 3 at% of carbon (CO) is less in the 500 °C material. This suggests residual carbon from the decomposed urea, which is present on the surface of the 300 °C material. This is reduced in the 500 °C material due to oxidation. Fig. 4e shows the deconvoluted O 1s spectra exhibiting two peaks: the main peak around 530 eV is associated with the metal oxide, and the peak at 532 eV is attributed to the carbon–oxygen species.30 Overall, the XPS study confirms the formation of CaMoO4 for the samples synthesized at two different temperatures 300 °C and 500 °C with a relatively higher intensity and low carbon content observed for the sample synthesized at a higher temperature. The effect of crystallinity and particle size may have no effect on the oxidation state and therefore no changes are observed in the binding energies.
O, respectively.30 The C–C is likely due to adventitious carbon contamination, and C–O arises from the residual urea as a fuel during the combustion synthesis and the C–NH2 functional group would be assigned to the CO peak.30 Similar behaviour was observed for the 500 °C material relating to C–C and C–O while about 3 at% of carbon (CO) is less in the 500 °C material. This suggests residual carbon from the decomposed urea, which is present on the surface of the 300 °C material. This is reduced in the 500 °C material due to oxidation. Fig. 4e shows the deconvoluted O 1s spectra exhibiting two peaks: the main peak around 530 eV is associated with the metal oxide, and the peak at 532 eV is attributed to the carbon–oxygen species.30 Overall, the XPS study confirms the formation of CaMoO4 for the samples synthesized at two different temperatures 300 °C and 500 °C with a relatively higher intensity and low carbon content observed for the sample synthesized at a higher temperature. The effect of crystallinity and particle size may have no effect on the oxidation state and therefore no changes are observed in the binding energies.
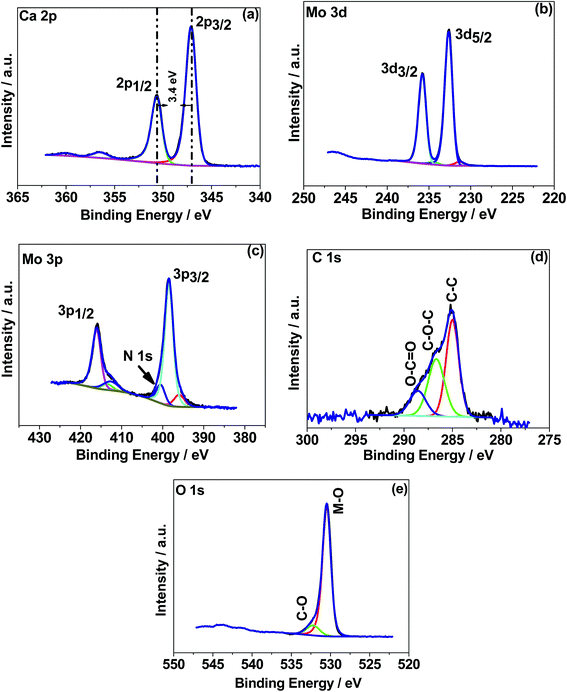 |
| | Fig. 4 X-ray photoelectron spectroscopy (XPS) spectra of CaMoO4 synthesized at 300 °C. High resolution spectra for regions (a) Ca 1s, (b) Mo 3d, (c) Mo 3p, (d) C 1s, and (e) O 1s. | |
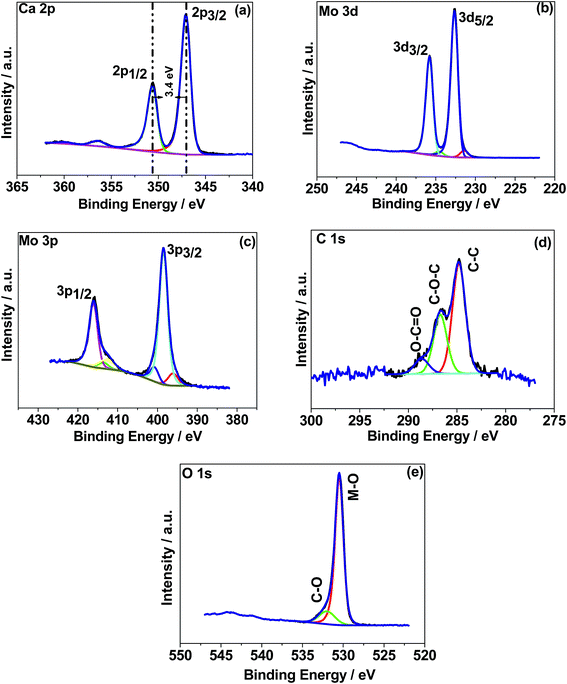 |
| | Fig. 5 X-ray photoelectron spectroscopy (XPS) spectra of CaMoO4 synthesized at 500 °C. High resolution spectra for regions (a) Ca 1s, (b) Mo 3d, (c) Mo 3p, (d) C 1s, and (e) O 1s. | |
(d) Field emission SEM and transmission electron microscopy.
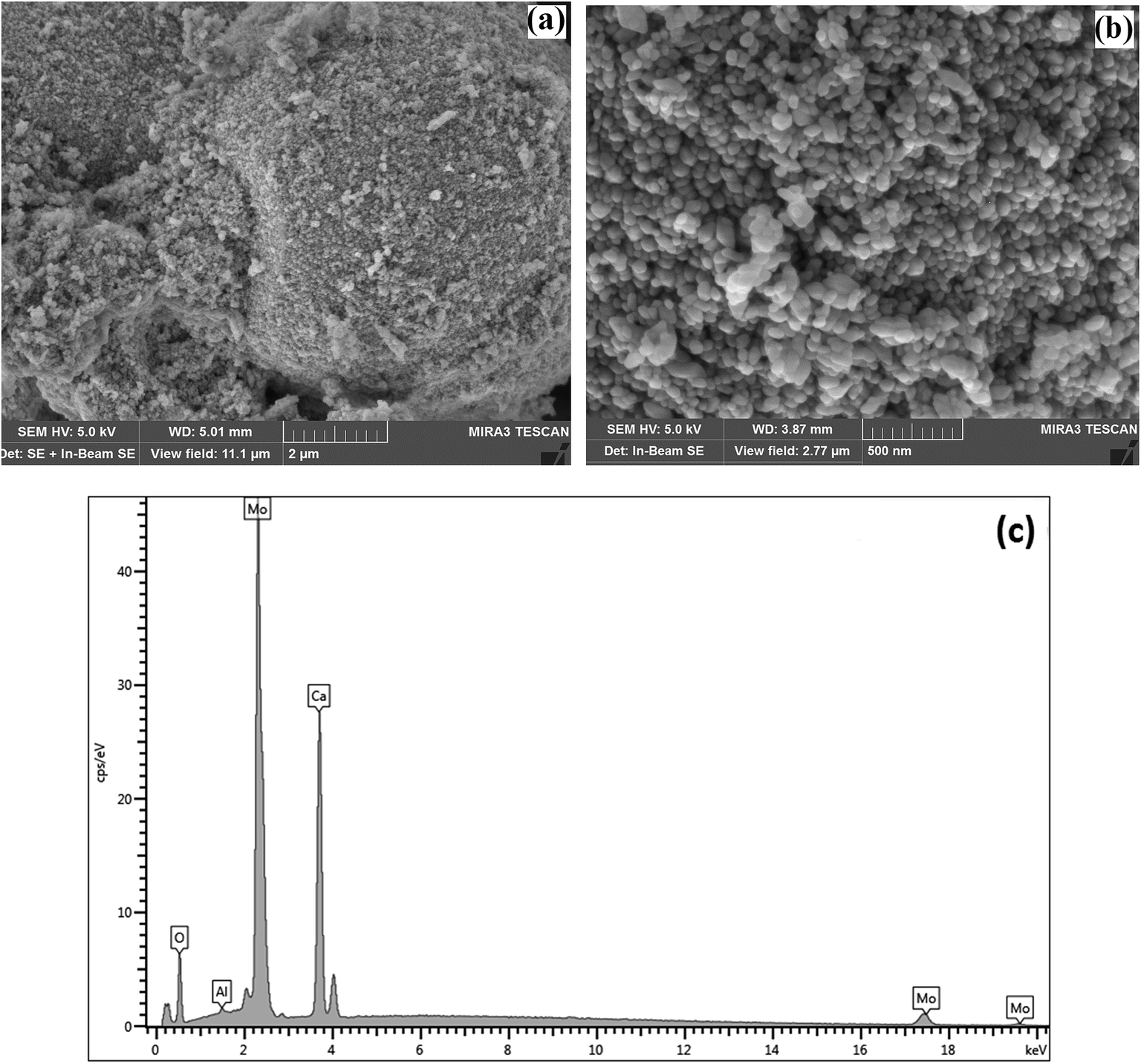
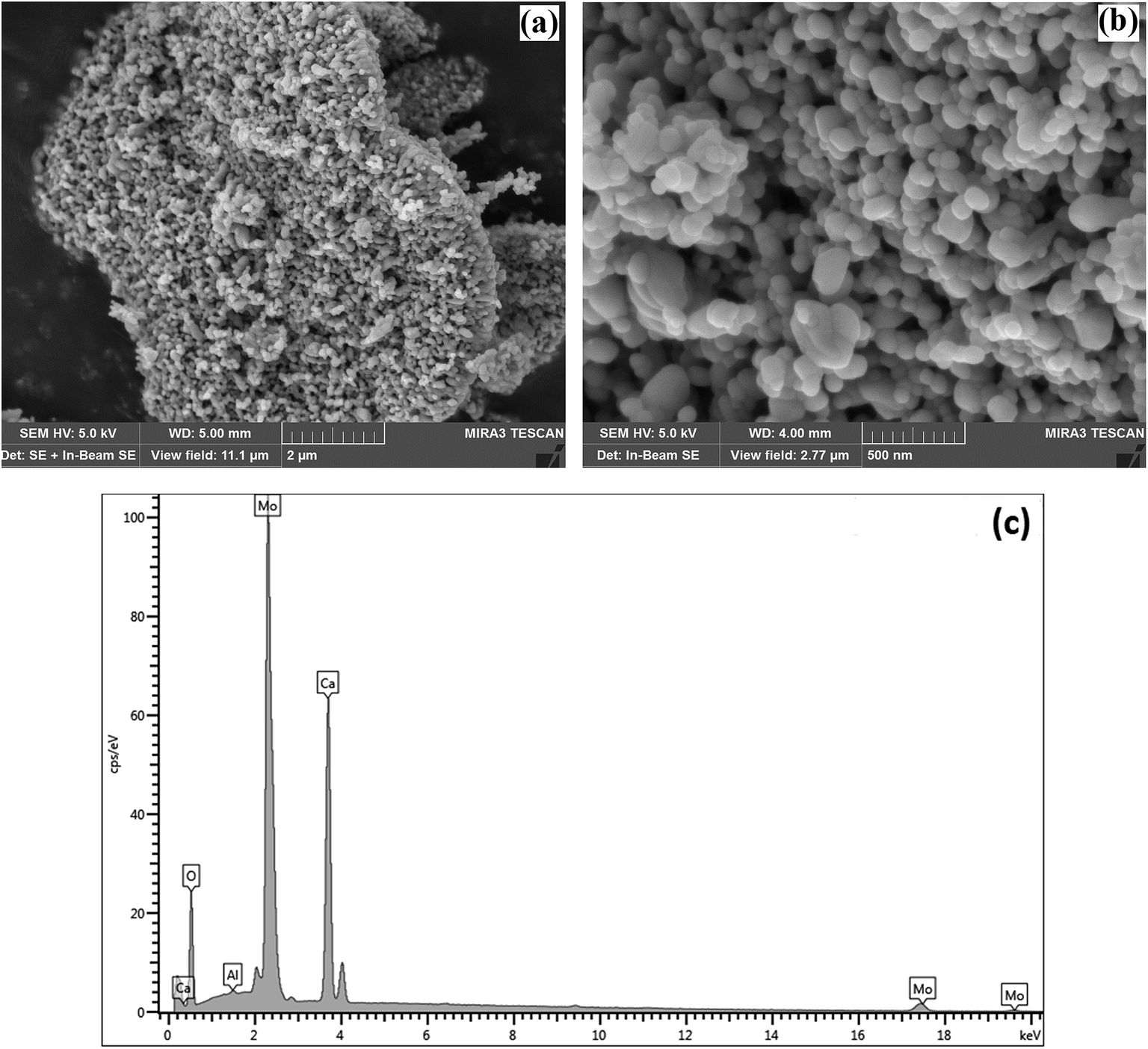
SEM was used to characterise the surface shape and size of the CaMoO4 powder synthesized at 300 °C (Fig. 6) and 500 °C (Fig. 7). The 300 °C material comprised densely agglomerated particles (Fig. 6a). These were roughly spherical being 120–100 nm in diameter (Fig. 6b). The EDS analysis (Fig. 6c) was consistent with pure CaMoO4, with a trace amount of Al arising from the support stub, upon which the specimen is mounted. The microstructure of the material produced at 500 °C was quite different. The grains (Fig. 7a) are fused together and irregular in shape. The agglomerates appear to be less dense, with larger open pores and the size of the individual particles is much coarser (50–250 nm) (Fig. 7b) than the lower temperature material. EDS analysis (Fig. 7c) again indicated high purity. This indicates that the coalescence of the smaller particles occurs at higher temperature. In turn, this produces a coarser grained, more porous microstructure. XRD patterns show that this coarsening is not associated with any change in the crystal structure of CaMoO4.
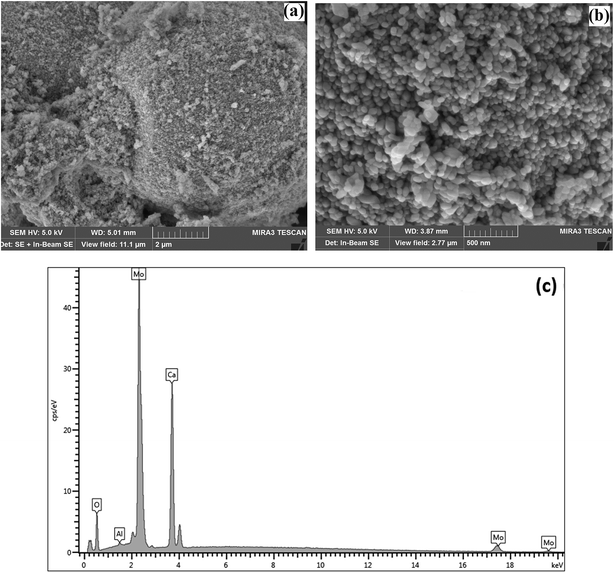 |
| | Fig. 6 SEM secondary electron images of the CaMoO4 synthesised at 300 °C showing: (a) a low magnification view of dense agglomerates of small spherical particles; (b) details of (a) showing the rounded particles to be 120–100 nm in diameter with fine scale pores between them; (c) EDS analysis showing the elemental composition of Ca, Mo, and O. | |
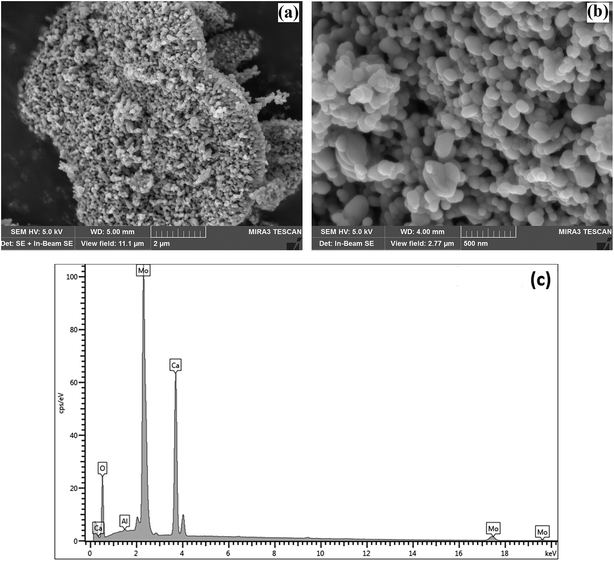 |
| | Fig. 7 (a) SEM secondary electron images of the CaMoO4 synthesised at 500 °C showing: (a) a low magnification view of dense agglomerates of irregular-shaped particles; (b) details of (a) showing the rounded and elongated particles to be 50–250 nm in diameter with coarser scale pores between them; (c) EDS analysis showing the elemental composition of Ca, Mo, and O. | |
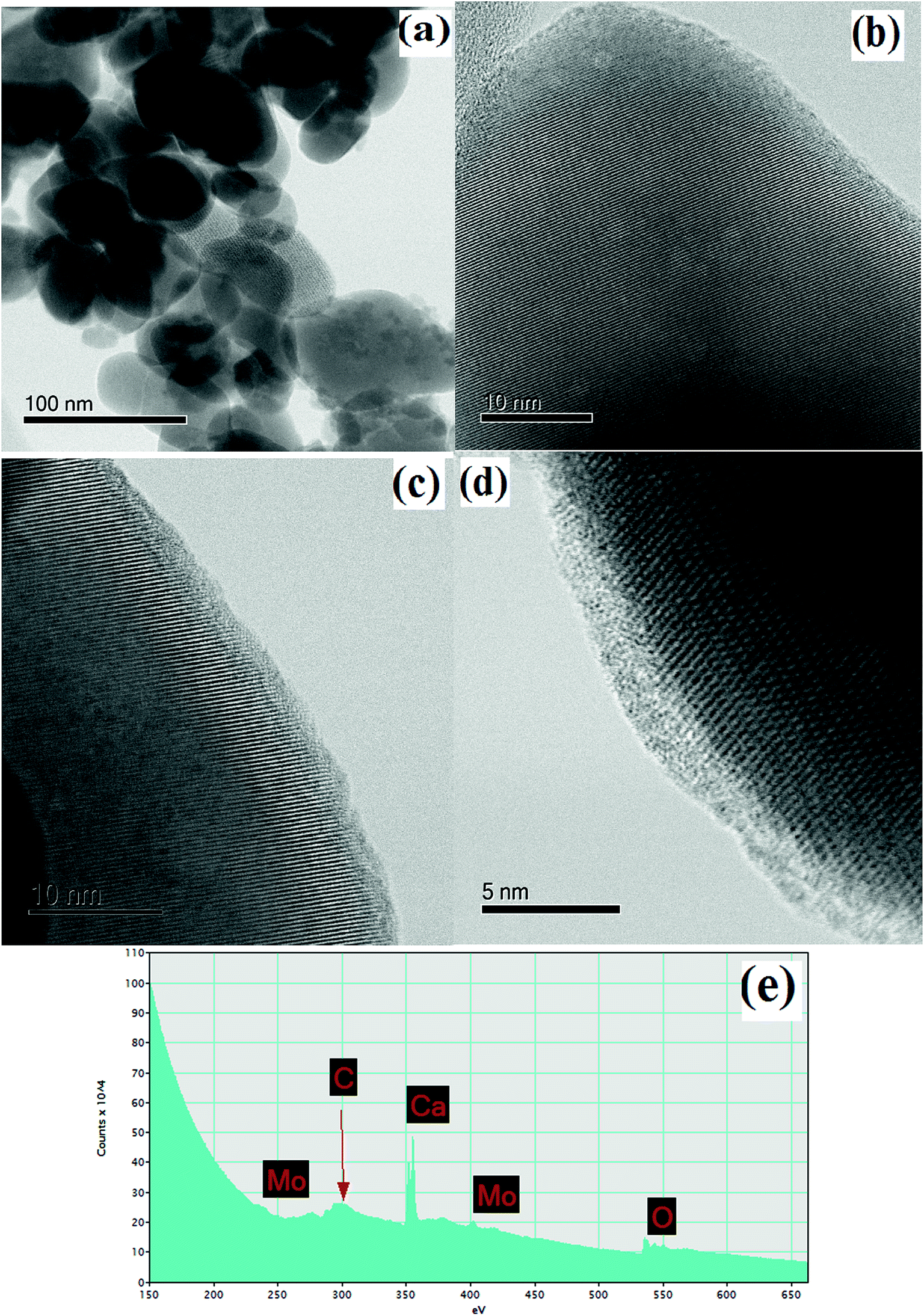
To study the materials more in detail, scanning transmission electron microscopy (STEM) imaging has been employed. High angle annular dark field (HAADF) and bright field (BF) images of the CaMoO4 synthesized at 300 °C and 500 °C are shown in Fig. 8 and 9. Fig. 8a shows a bright field image of a cluster of spheroidal particles 20–100 nm in diameter. The particles are tightly packed with small void spaces between them. Fig. 8b shows details of one of the particles. The bright regions within the crystal are voids 2–5 nm in diameter. These are most likely densification voids which form by coalescence of vacancies as the particles grow during the synthesis. Atomic resolution images (Fig. 8c and d) confirm that the particles are crystalline. Aside from the voids, defects were not evident. Some particles (Fig. 8c) showed lattice fringes almost up to the crystal edge (bounded by a very thin <2 nm amorphous layer), while others (Fig. 8d) clearly showed a thicker (2–3 nm) amorphous surface layer. This was identified as being carbon using electron energy loss spectroscopy (Fig. 8e). This carbon is likely residual carbon from incomplete removal of the urea during combustion synthesis at a low temperature (300 °C).
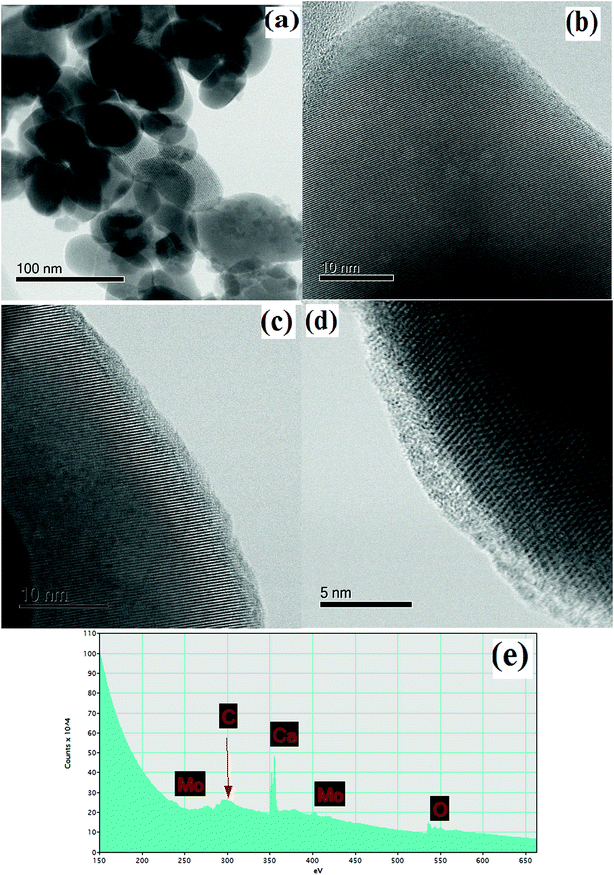 |
| | Fig. 8 STEM images/spectra of CaMoO4 synthesized at 300 °C showing: (a) a bright field image of the rounded particles with small pores between them; (b) details of a particle showing a dispersion of fine scale spherical porosity (bright regions within the crystal) 2–5 nm in diameter; (c) and (d) atomic resolution bright field images showing the presence of (c) a thin (<2 nm) and (d) thicker (2–3 nm) surface amorphous layer on the otherwise crystalline particles and the amorphous layer was identified as being carbon; (e) EELS spectrum of (d) showing a distinct carbon peak. | |
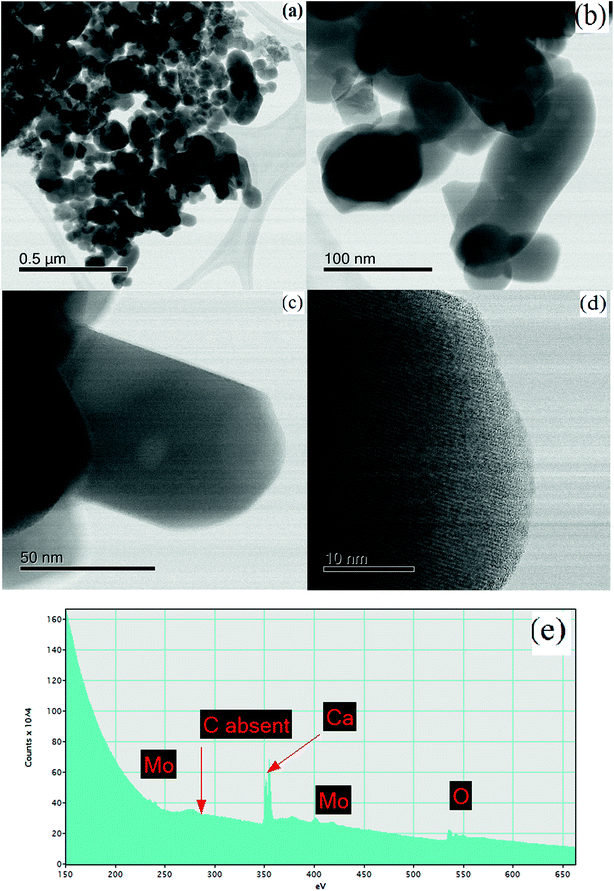 |
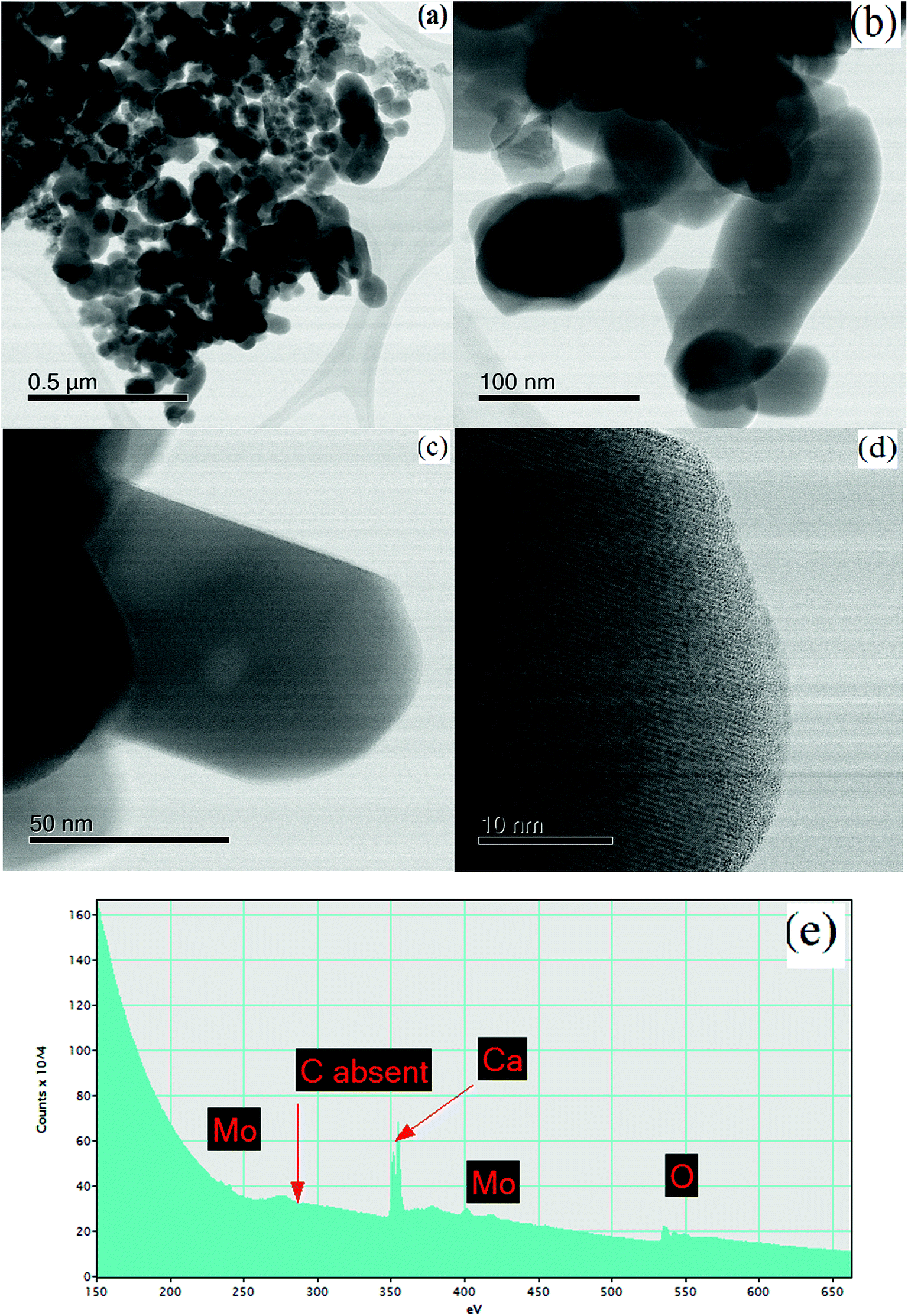
| | Fig. 9 STEM images of CaMoO4 synthesized at 500 °C showing (a and b) a bright field image highlighting the irregular shapes and coarser particle size (50–250 nm), relative to the 300 °C material; (c) details of a rod-like crystal showing a single large void (bright region) approximately 10 nm in diameter; (d) an atomic resolution bright field image showing the lattice fringes to extend to the crystal edge, with no amorphous phase present; (e) EELS spectrum of (e) showing the absence of carbon. | |
For the material synthesised at 500 °C, the particles were both coarser and more irregular (Fig. 9a and b) than at lower temperature (Fig. 8). The particle size was in the 50–250 nm range with some particles becoming cylindrical. Within individual crystals, a small number of relatively large (10 nm) pores could be seen (Fig. 9c). Comparison with the lower temperature material (Fig. 8b) showed that the effect of increasing the combustion temperature from 300 to 500 °C caused the voids to increase in size, but decrease in number. At higher temperature, voids could shrink by emitting vacancies. Any vacancies reaching the surface of the crystal would be eliminated. Within the bulk of the crystal, thermally activated vacancy migration would result in void coalescence and coarsening. The result would be a less strained/defective structure, which would account for the improved crystallinity observed in XRD for the higher temperature material (Fig. 1, S2 and S3†). High-resolution imaging (Fig. 9d) showed no surface amorphous layers of carbon, suggesting complete removal of carbon at this higher temperature. This was confirmed by electron energy loss spectroscopy (EELS) (Fig. 9e). Note the Mo M edges form a broad hump, the peak of which occurs just to the left of where the C K edge is marked (283 eV). A lower carbon content for this higher temperature material was also found in XPS.
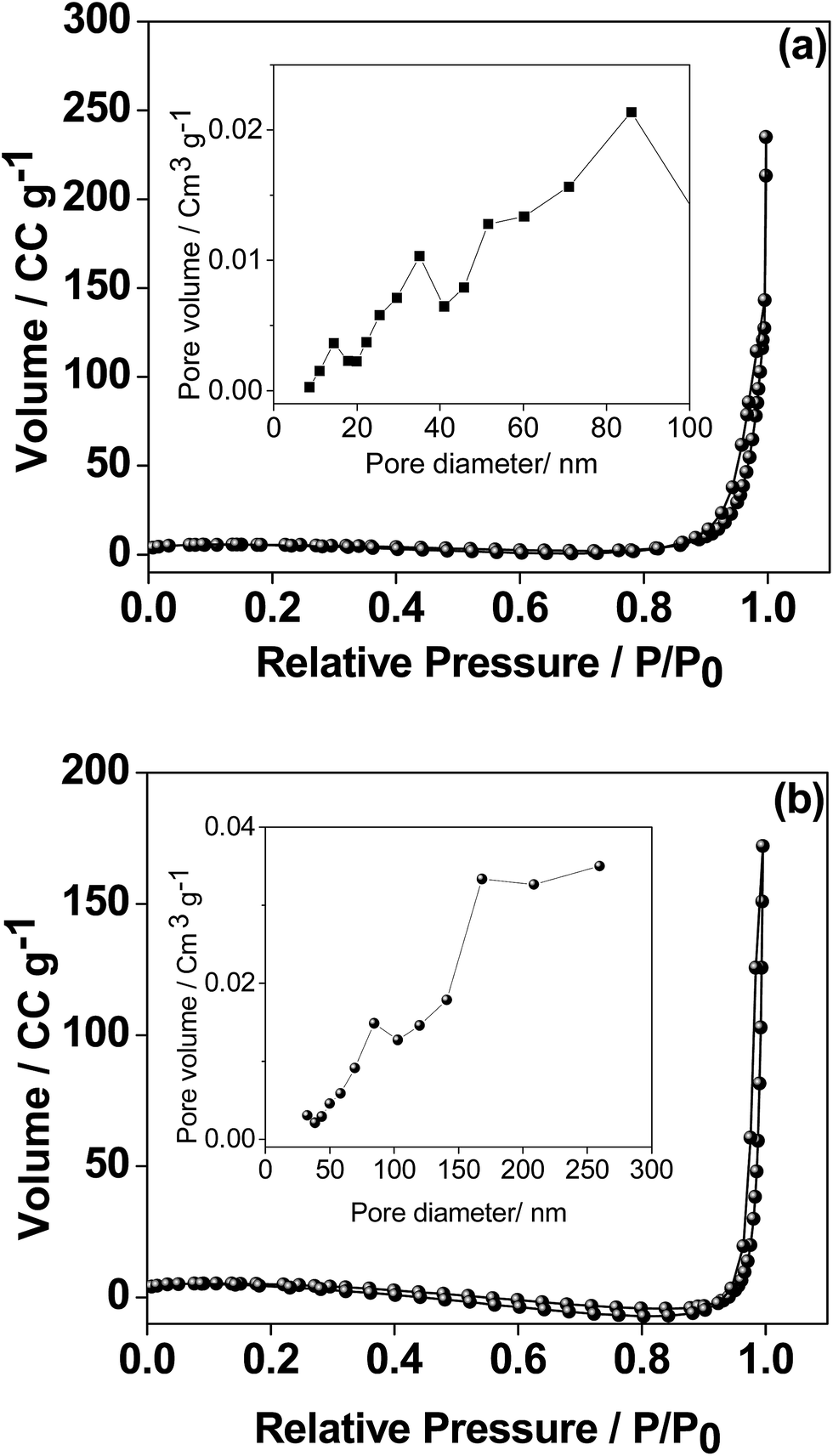
(e) Surface area analysis.
As the electrochemical performance of the material is heavily influenced by the surface area, the porosity of the CaMoO4 materials was measured by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) analyses and the results are shown in Fig. 10. The 300 °C material possesses well-developed porosity, reflected by the N2 sorption isotherm of type III.31 The BET surface area was 20.6 m2 g−1 compared with 12.2 m2 g−1 for the 500 °C material. The proportion of small pores (<20 nm) is higher (Fig. 10a inset) for the sample synthesized at 300 °C while the proportion of large pores (>20 nm) is higher (Fig. 10b inset) for the 500 °C material. The coarsening of the particles and a larger pore structure in the higher temperature material were also evident in the microscopy results (Fig. 6–9).
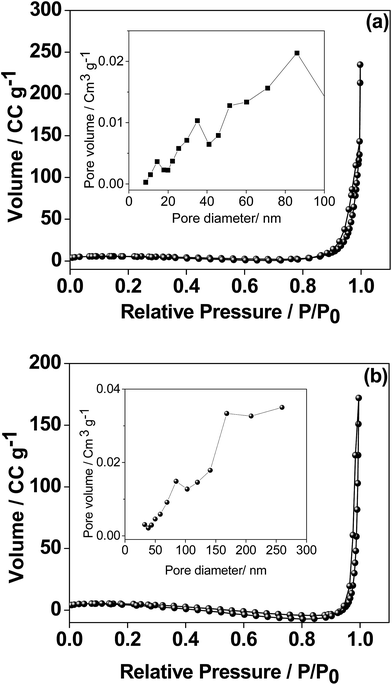 |
| | Fig. 10 Nitrogen adsorption and desorption isotherms and inset BJH adsorption pore size distribution of CaMoO4 synthesized at (a) 300 and (b) 500 °C. The isotherms for the two are quite similar while the pore size distribution is quite different – note different pore diameter scaling in the insets. | |
Electrochemical characterization
(a) Single electrode configuration.
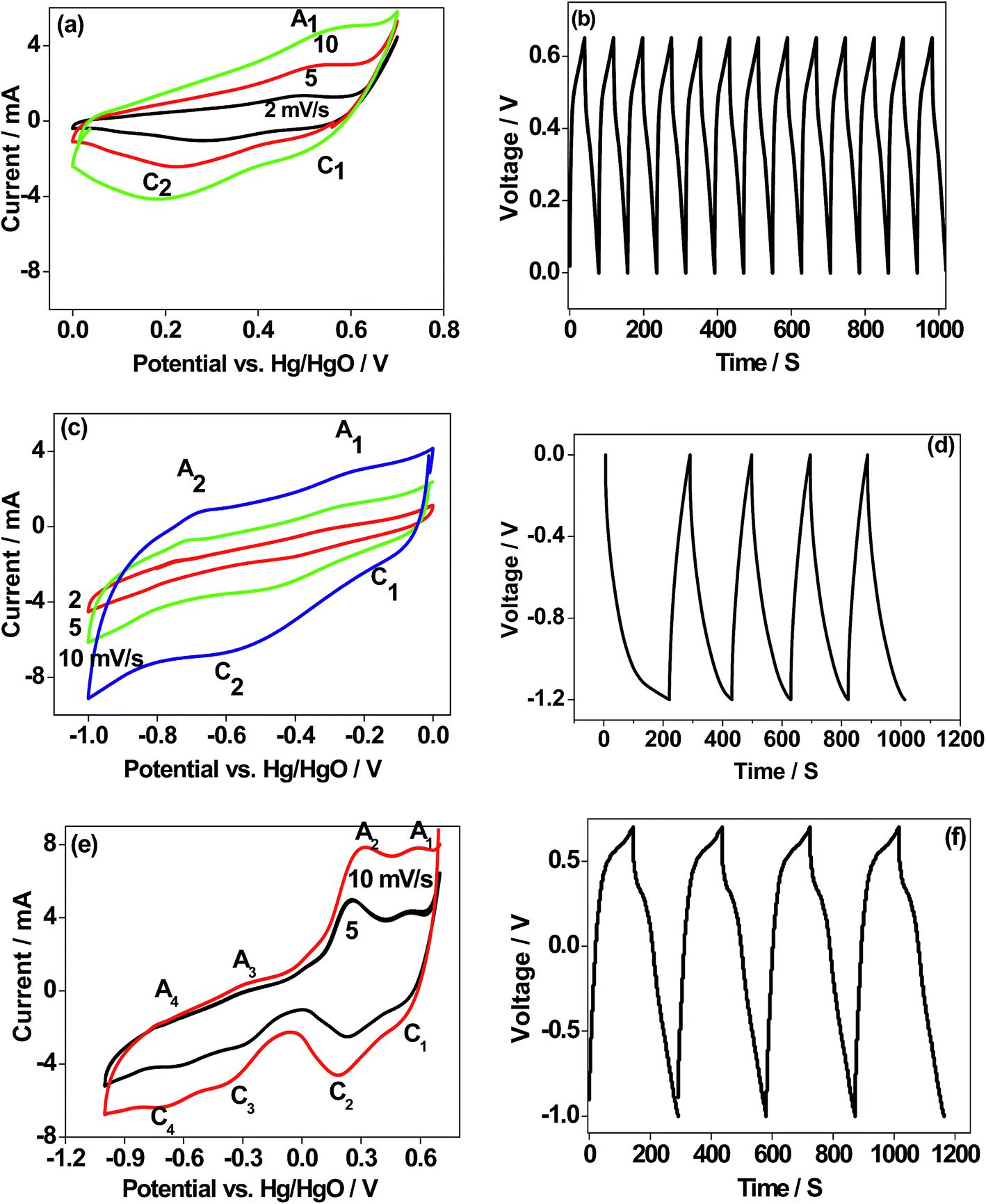
There are few reports14,15 on the use of CaMoO4 as an electrode material for Li-ion batteries. There are no reports on its use in sodium-ion batteries. Nor there has been any emphasis placed on optimising the synthesis protocol and applications such as in batteries and capacitors. The electrochemical analysis shown here provides an insight into the behaviour of CaMoO4 in various potential regions and its suitability as an electrode to store energy. To assess the potential viability of CaMoO4, single electrode studies in potentiostatic and galvanostatic modes have been carried out in a positive region (0.6 V), negative region (−1.0 V), and the entire voltage region (1.6 V) for the 300 °C material. Potentials are reported with reference to the Hg/HgO standard electrode. The cyclic voltammograms (CV) for the 300 °C material at various scan rates are shown in Fig. 11a, c and e. The scan was initiated at 0 V going in the forward (anodic) direction to +0.6 V and then reversing back to the starting potential. As can be seen in Fig. 11a, CV profiles at different sweep rates invariably consist of two reduction peaks (C1 and C2) and one oxidation peak (A1) suggesting that the CaMoO4 electrode undergoes only one oxidation process and the product formed during the oxidation undergoes two separate reductions. Moreover, with an increasing sweep rate, the area under the redox curves is proportional to the sweep rate indicating excellent reversibility of the electrodes.10,11 When the CaMoO4 electrode is introduced into the alkaline electrolyte with a pH of over 8.0, it is oxidized to form Ca(OH)3− (eqn (1)) prior to any electrochemical measurements. It is reported that MoO42− in the pH range of 8 will not exhibit any redox peaks.19,32,33| | | CaMoO4 + H2O + OH− → Ca(OH)3− | (1) |
(the tetrahedral MoO42− ion dominates only in concentrated NaOH solution19).
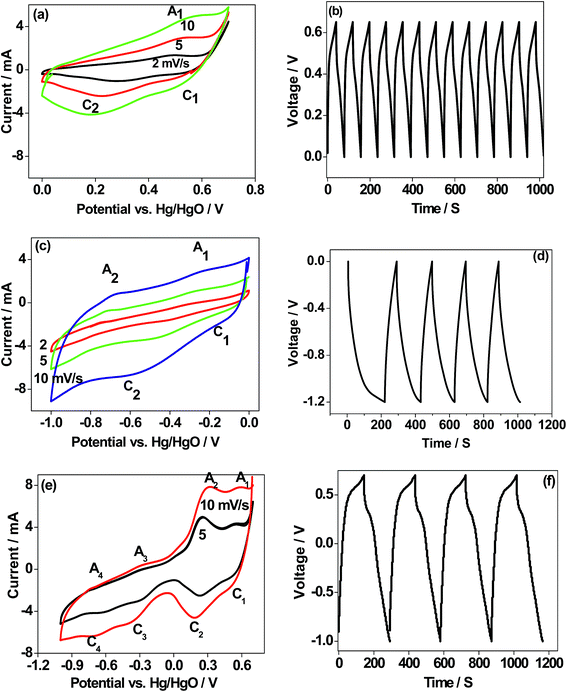 |
| | Fig. 11 (a, c and e) Cyclic voltammetry (CV), and (b, d and f) charge–discharge profiles (single electrode study/three-electrode configuration) of CaMoO4 (300 °C) in different potential regions. The electrode is cycled in the (a and b) positive 0.6 V window, (c and d) negative −1.0 V window, and (e and f) whole region 1.6 V window vs. Hg/HgO, respectively. Sweep rates are indicated in the respective figures. Consecutive charge–discharge cycles at 1 A g−1 are shown for 1000 seconds. d and f show a longer time to charge and discharge. | |
During the anodic scan, Ca(OH)3− is oxidized to form an intermediate product of Ca3O4. On the reverse sweep, Ca3O4 undergoes a change in the reduction state of the Ca atom in CaMoO4 governed by faradaic reactions. The reactions correspond to the redox peaks with several intermediate products as per the following eqn (2)–(4)
| | | 3[Ca(OH)3]− ↔ Ca3O4 + 4H2O + OH− + 2e− (peak A1) | (2) |
| | | Ca3O4 + H2O + OH− ↔ 3CaOOH + e− (peak C1) | (3) |
| | | CaOOH + OH− → CaO‖2OH− (‖ indicates double layer) (peak C2) | (4) |
The electrochemical behaviour of CaMoO4 in Fig. 11a exhibits a quasi-reversible faradaic process. This involves electron transfer in the Ca redox couple, with the ability of OH− to be reversibly intercalated into Ca3O4 for improved charge storage (forming CaOOH) which is attributed to pseudocapacitance.4 However, the peak C2 (eqn (4)) intensity arises from the formation of calcium oxide (CaO) and adsorption of ions on the surface of the active sites of the nanoparticles. This corresponds to a non-faradaic process, which is not reversible during the oxidation process but contributes to the capacitance. The above studies indicate that the reduction of Ca3O4 is reversible while the further reduction of CaOOH forms a mixture of CaO and adsorbed OH− ions, which is irreversible. This behaviour is also substantiated by the corresponding charge–discharge curve (Fig. 11b), which is a quasi-triangular curve. Hence, the observed specific capacitance of 20 F g−1 is contributed by both the faradaic (eqn (2) and (3)) and non-faradaic reactions (eqn (4)). To examine this material for its suitability as a negative electrode, the scan was initiated at 0 V going in the reverse (cathodic) direction to −1.0 V and then reversing it back to the starting potential. Hydrogen reactions may occur in the far negative region, so the potential was limited to −1.0 V. As can be seen in Fig. 11c, the process is fully reversible with a larger area under the curve indicating that the storage capability is higher than that observed in the positive region (Fig. 11a). The proportional increase in the current at a higher sweep rate indicates excellent rate capability of this material, a desirable feature for supercapacitors. The reactions in the negative region correspond to the redox peaks as per the following eqn (5) and (6)
| | | Ca(OH)3− + 4OH− ↔ CaO42− + 4H2O + 3e− peak A1/C1 | (5) |
| | | Ca(OH)2 + OH− ↔ Ca(OH)3 + e− peak A2/C2 | (6) |
The CV profile of CaMoO4 in Fig. 11c exhibits a quasi-rectangular shape indicating a capacitive like behaviour with an observed specific capacitance of 59 F g−1 while the charge–discharge curves are triangular as shown in Fig. 11d. These results show that the electrochemical reactions are strongly dependent on the chosen potential region. The material was then examined over a larger voltage window. The scan was initiated at 0.6 V going in the cathodic direction to −1.0 V and then reversing it to the starting potential, spanning a voltage window of 1.6 V. A typical characteristic curve is shown in Fig. 11e. It can be seen that CaMoO4 behaves as a battery electrode in the positive region (0 to 0.6 V) while in the negative region (0 to −1.0 V) it behaves as a capacitor electrode, resulting in 80 F g−1. At a higher scan rate of 10 mV s−1, a lower capacitance of 70 F g−1 is obtained because the faradaic reaction in the positive region is limited by slower ion diffusion. Identical behaviour is observed in the corresponding charge–discharge cycles, implying a plateau-like curve in the positive region and a triangular-like curve in the negative region with a specific capacitance of 80 F g−1. On the other hand, the electrochemical properties of the sample prepared at 500 °C (Fig. S4a and b†) under identical conditions showed a similar behaviour but lower electrochemical activity and the available specific capacitance is half the value of that of the sample prepared at 300 °C resulting in 40 F g−1. Therefore, CaMoO4 prepared at 300 °C with enhanced storage properties behaves as a hybrid material, which is suitable to act as both the anode and cathode. However, the available storage is three times larger in the negative region. This is in contrast to CoMoO4, MgMoO4 and NiMoO4 electrodes tested for supercapacitors in aqueous 2 M NaOH solutions, for which the potential window is limited to a maximum of 0.7 V.16–18 The versatile nature of the Ca cation, which can operate over a wide voltage range, is a key factor contributing to its excellent performance. The reported capacitance for metal molybdates such as MnMoO4 and CoMoO4 is around 100 F g−1, which is comparable to the current value of 80 F g−1 with an energy density of 102 W h kg−1.34
(b) Two-electrode configuration.
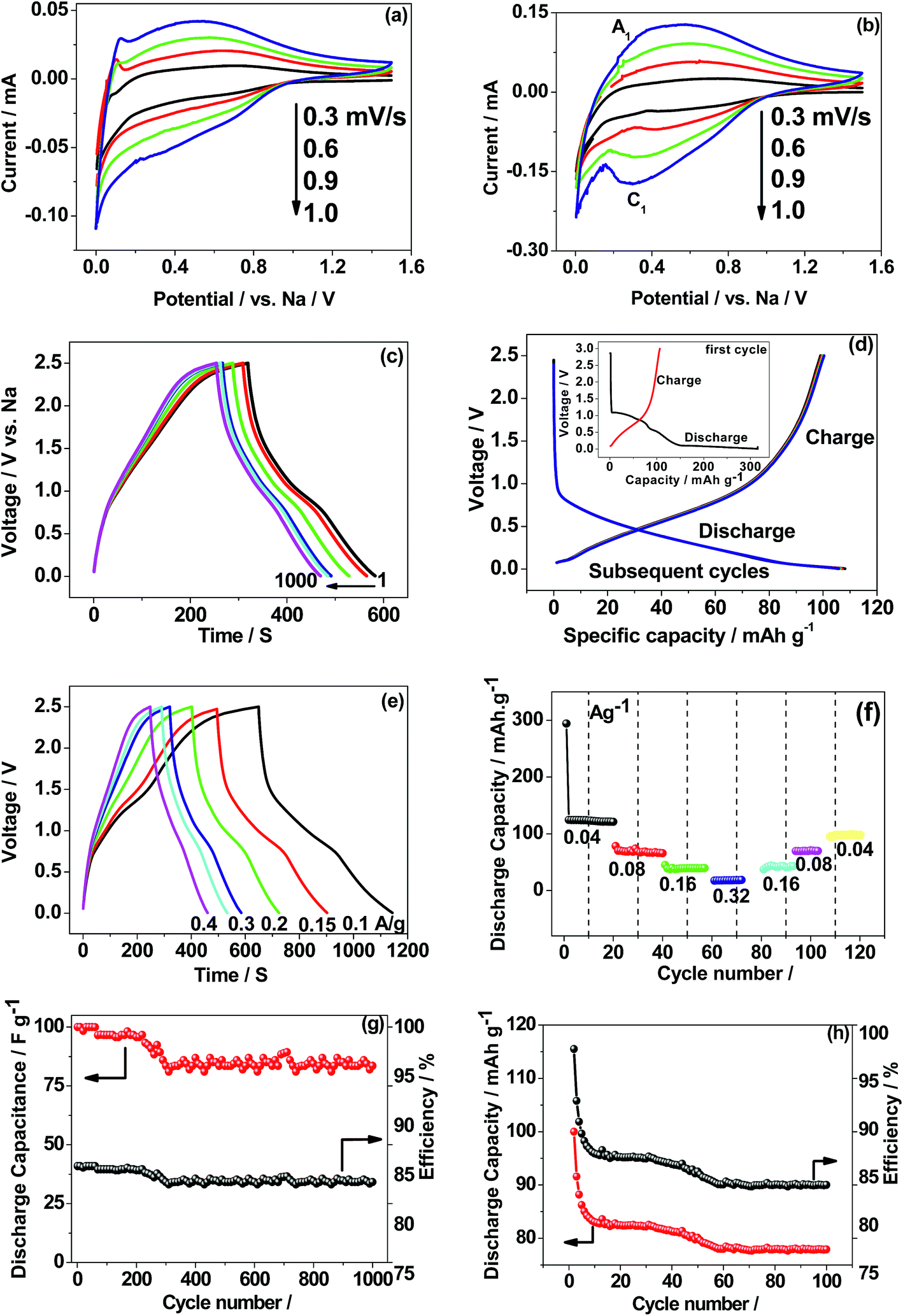
Based on the above facts as a single electrode, CaMoO4 was fabricated in a two-cell configuration (full-cell), coupled with metallic sodium in a non-aqueous electrolyte to facilitate its use in storage applications as an anode material. Reported potentials are with reference to the metallic sodium electrode. The electrochemical results for 300 °C and 500 °C are shown in Fig. 12. The CV profile of CaMoO4 was recorded in the voltage range of 0.005–2.5 V vs. Na at different sweep rates ranging from 0.3–1 mV s−1. Interestingly, the CV profile of the 300 °C material (Fig. 12a) differs in shape from that of the 500 °C sample (Fig. 12b). The sweep was initiated in the cathodic direction and then reversed back anodically to 2.5 V. The characteristics of the curves for the 500 °C material (Fig. 12b) show a reduction peak (C1 at 0.3 V) and a corresponding oxidation peak (A1 at 0.5 V) while the curve for the 300 °C material (Fig. 12a) shows no obvious peak and has a quasi-rectangular curve, essentially at higher sweep rates, as opposed to Fig. 12b that shows redox behaviour. Fig. 12a indicates capacitive-like behaviour based on surface Na+ ion adsorption/desorption (eqn (7)). The lower temperature material has a larger surface area which can facilitate this process| | | CaMoO4 + NaClO4 ↔ CaMoO4‖Na+ (‖ indicates double layer) | (7) |
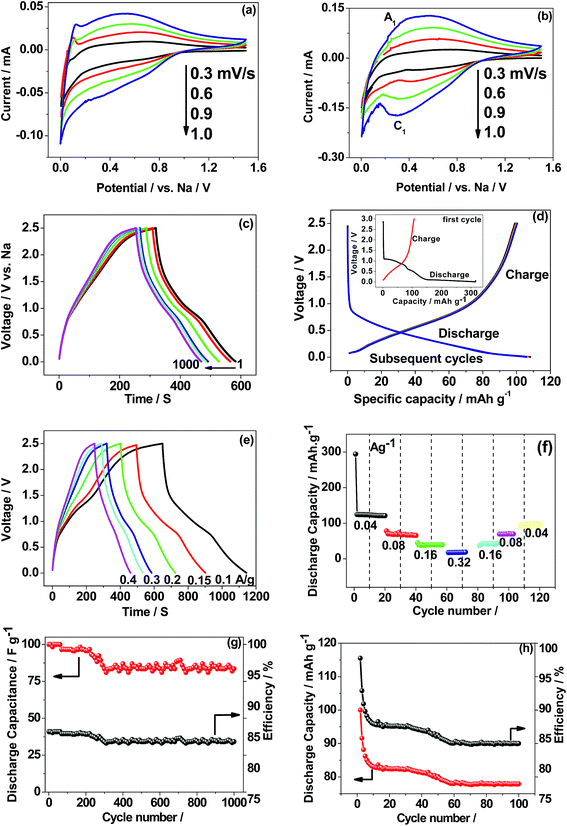 |
| | Fig. 12 First cyclic voltammetry (CV) curves (two-electrode configuration) of CaMoO4vs. metallic sodium in non-aqueous 1 M NaClO4 in EC:DMC:FEC (88:10:2) electrolyte synthesized at (a) 300 and (b) 500 °C; (c) charge–discharge curves of CaMoO4 300 °C exhibiting triangular-like curves at a C/5 rate suitable for supercapacitors; (d) 500 °C shows plateau-like curves at a C/10 rate suitable for battery applications. Rate capability and cycling stability of CaMoO4 synthesized at (e and g) 300 and (f and h) 500 °C. | |
The charge/discharge performance at a C/5 rate (corresponding to a full charge or discharge of the theoretical capacity of CaMoO4 in 5 h) of the 300 °C material (Fig. 12c) showed a triangular-shaped curve. It is asymmetrical in shape but shows an excellent retention of 85% after 1000 cycles. The initial capacitance was 40 F g−1 and after 1000 cycles, it reduced to 34 F g−1. The charge–discharge at the C/10 rate for the 500 °C material (Fig. 12d) shows a relatively flat charge/discharge profile, which is a typical battery-type behaviour. The 2nd charge/discharge capacity was calculated to be 115 mA h g−1, which is 35% of the value obtained in the first cycle. This is due to the formation of a solid electrolyte interface (SEI) layer (shown in the Fig. 12d inset). The redox reaction which corresponds to the charge and discharge curve is shown in eqn (8).
| | | CaMoO4 + Na+ + 2e− ↔ CaO + NaMoO3 | (8) |
It is clear that the 300 and 500 °C materials exhibit different electrochemical behaviour. The rate capability and cycling stability of both materials as a function of current density were tested (Fig. 12e–h). At a current rate of 0.1, 0.15, 0.2, 0.3 and 0.4 A g−1, the specific capacitances for the supercapacitor (Fig. 12e) are 35, 34, 31, 25 and 24 F g−1, respectively. The corresponding energy density values are 110, 105, 96, 78 and 75 W h kg−1 at a current rate of 0.1, 0.15, 0.2, 0.3 and 0.4 A g−1, respectively. In the case of the 500 °C material (Fig. 12f), the specific capacity of the battery at current rates of 0.04, 0.08, 0.16, and 0.32 was found to be 130, 80, 40, and 20 mA h g−1, respectively. CaMoO4 (Fig. 12g) outperforms the higher temperature material by displaying both good capacitance and stable cycling. The low temperature material morphology and higher surface area may contribute to this. Its initial capacitance (40 F g−1) initially decreases to 35 F g−1 but thereafter, it maintained an excellent stability with a retention of 83% capacitance.
In the case of the 500 °C material, the cycling stability is shown to be stable (Fig. 12h) after the initial few cycles, with a reversible capacity of 80 mA h g−1 after 100 cycles. The initial rapid drop in capacity is attributed to the formation of a SEI layer during the initial sodiation that resists the flow of ions through the electrode. After a few cycles it eventually settled down, then allowing the access to the active material. However, apart from stable cycling, all other performance characteristics are inferior to those of the 300 °C material. The reversible capacity is considerably lower than 200 mA h g−1 reported for as-synthesized CaMoO4 in lithium-ion batteries by Sharma et al.,14 Culver et al.,15 Liang et al.,35 and You et al.36 The key effects of synthesis temperature were to produce a larger surface area, smaller particle size, more densely packed morphology and more defective crystal structure in CaMoO4 synthesised at 300 °C compared with 500 °C. Collectively, these influence the electrochemical reactions, which can be either faradaic or non-faradaic.
Conclusions
The electrochemical properties of CaMoO4 synthesized at different temperatures (300 °C and 500 °C) have been assessed for sodium battery/supercapacitor applications. The synthesis temperature exerts an influence on its behaviour. CaMoO4 is found to be a hybrid material, which makes it suitable to work in a wide potential window. The relationship between the degree of particle crystallinity, morphology and their influence on electrochemical reactions and the operating voltage window have been analyzed and discussed. The materials' structural parameters are dependent on the synthesis temperature and so optimisation of that temperature is of key importance in producing high performance materials for energy storage. Molybdenum ions are active in both aqueous and non-aqueous electrolytes but they do not exhibit any reactions in a basic aqueous electrolyte. The key to improving upon the performance of this material lies in further optimising materials synthesis. These results shed new insights on the use of alkaline earth molybdates in aqueous and non-aqueous solutions and highlight their distinct electrochemical properties and their interplay with material synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Karlsruhe Institute of Technology (KIT) is acknowledged for providing a visiting fellowship and to accommodate (M. M) at HIU and carry out the electrochemical work. The STEM work was carried out using JEOL ARM200F instrument equipment funded by the Australian Research Council (ARC) – Linkage, Infrastructure, Equipment and Facilities (LIEF) grant LE120100104, located at the UOW Electron Microscopy Centre. This work was performed in part at the Australian National Fabrication Facility (ANFF), a company established under the National Collaborative Research Infrastructure Strategy, through the La Trobe University Centre for Materials and Surface Science. Authors AB, SC and RA wish to acknowledge Carl Tryggers Stifelse for Vetenskaplig Forskning (CTS), Swedish Research Council (VR) and Erasmus Mundus (EMINTE) for the doctoral fellowship. SNIC and HPC2N are also acknowledged for providing computing time.
References
- A. Evans, V. Stezov and T. J. Evans, Assessment of utility energy storage options for increased renewable energy penetration, Renewable Sustainable Energy Rev., 2012, 16, 4141–4147 CrossRef.
- H. Ibrahim, A. Ilinca and J. Perron, Energy storage systems – characteristics and comparisons, Renewable Sustainable Energy Rev., 2008, 12, 1221–1250 CrossRef CAS.
- J. Cho, S. Jeong and Y. Kim, Commercial and research battery technologies for electrical energy storage applications, Prog. Energy Combust. Sci., 2015, 48, 84–101 CrossRef.
- M. Winter and R. J. Brodd, What are Batteries, Fuel Cells, and Supercapacitors?, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- F. Wang, X. Wang, Z. Chang, Y. Zhu, L. Fu, X. Liu and Y. Wu, Electrode materials with tailored facets for electrochemical energy storage, Nanoscale Horiz., 2016, 1, 272–289 RSC.
- J. F. Whitacre, T. Wiley, S. Shanbhag, Y. Wenzhuo, A. Mohamed, S. E. Chun, E. Weber, D. Blackwood, E. Lynch-Bell, J. Gulakowski, C. Smith and D. Humphreys, An aqueous electrolyte, sodium ion functional, large format energy storage device for stationary applications, J. Power Sources, 2012, 213, 255–264 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, Phospho-olivines as positive electrode materials for rechargeable lithium batteries, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- M. Minakshi, Lithium intercalation into amorphous FePO4 cathode in aqueous solutions, Electrochim. Acta, 2010, 55, 9174–9178 CrossRef CAS.
- S. Li and C. A. Wang, Design and synthesis of hierarchically porous MnO2/carbon hybrids for high performance electrochemical capacitors, J. Colloid Interface Sci., 2015, 438, 61–67 CrossRef CAS PubMed.
- T. Watcharatharapong, M. Minakshi Sundaram, S. Chakraborty, D. Li, G. M. Shafiullah, R. D. Aughterson and R. Ahuja, Effect of transition metal cations on stability enhancement for molybdate-based hybrid supercapacitor, ACS Appl. Mater. Interfaces, 2017, 9, 17977–17991 CrossRef CAS PubMed.
- M. Minakshi, T. Watcharatharapong, S. Chakraborty and R. Ahuja, A combined theoretical and experimental approach of a new ternary metal oxide in molybdate composite for hybrid energy storage capacitors, APL Mater., 2018, 6, 047701 CrossRef.
- E. Wierzbicka, A. Malinowska, K. Wieteska, W. Wierzchowski, M. Lefeld-Sosnowska, M. Swirkowicz, T. Lukasiewicz and C. Paulmann, Characterization crystal lattice defects in calcium molybdate single crystals (CaMoO4) by means of X-ray diffraction topography, X-Ray Spectrom., 2015, 44, 351–355 CrossRef CAS.
- S. Vidya, S. Solomon and J. K. Thomas, Synthesis, sintering and optical properties of CaMoO4: A promising scheelite LTCC and photoluminescent material, Phys. Status Solidi A, 2012, 209, 1067–1074 CrossRef CAS.
- N. Sharma, K. M. Shaju, G. V. Subba Rao, B. V. R. Chowdari, Z. L. Dong and T. J. White, Carbon-coated nanophase CaMoO4 as anode material for Li ion batteries, Chem. Mater., 2004, 16, 504–512 CrossRef CAS.
- S. P. Culver, F. A. Rabuffeti, S. Zhou, M. Mecklenburg, Y. Song, B. C. Melot and R. L. Brutchey, Low-temperature synthesis of AMoO4 (A = Ca, Sr, Ba) Scheelite nanocrystals, Chem. Mater., 2013, 25, 4129–4134 CrossRef CAS.
- Z. Zhang, Y. Liu, Z. Huang, L. Ren, X. Qi, X. Wei and J. Zhong, Facile hydrothermal synthesis of NiMoO4@CoMoO4 hierarchical nanospheres for supercapacitor applications, Phys. Chem. Chem. Phys., 2015, 17, 20795–20804 RSC.
- J. Candler, T. Elmore, B. K. Gupta, L. Dong, S. Palchoudhury and R. K. Gupta, New insight into high-temperature driven morphology reliant CoMoO4 flexible supercapacitors, New J. Chem., 2015, 39, 6108–6116 RSC.
- X. Hu, W. Zhang, X. Liu, Y. Mei and Y. Huang, Nanostructured Mo-based electrode materials for electrochemical energy storage, Chem. Soc. Rev., 2015, 44, 2376–2404 RSC.
- T. P. Dadze, G. A. Kashirtseva, M. P. Novikov and A. V. Plyasunov, Solubility of calcium molybdate in aqueous solutions at 573 K and thermodynamics of monomer hydrolysis of Mo(VI) at elevated temperatures, Monatsh. Chem., 2018, 149, 261–282 CrossRef CAS.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- W. Janbua, T. Bongkarn, W. Vittayakorn and N. Vittayakorn, Direct synthesis and growth mechanism of metal molybdate (AMoO4; A = Ca and Ba) fine particles via the Mechanochemical method, Ceram. Int., 2017, 43, S443–S445 CrossRef.
- W. Grunert, A. Y. Stakheev, R. Feldhaus, L. Anders, E. S. Shpiro and K. M. Minachev, Analysis of Mo (3d) XPS spectra of supported Mo catalysts: An alternative approach, J. Phys. Chem., 1991, 95, 1323–1328 CrossRef.
- S. K. Gupta, M. Sahu, P. S. Ghosh, D. Tyagi, M. K. Saxena and R. M. Kadam, Energy transfer dynamics and luminescence properties of Eu3+ in CaMoO4 and SrMoO4, Dalton Trans., 2015, 44, 18957–18969 RSC.
- J. Baltrusaitis, B. M. Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Generalized molybdenum oxide surface chemical state XPS determination via informed amorphous sample model, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
-
A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST X-ray Photoelectron Spectroscopy Database v4.1, 2012, http://srdata.nist.gov/xps/ Search PubMed.
- G. Aranovich and M. Donohue, Analysis of Adsorption Isotherms: Lattice theory predictions, classifications of isotherms for gas-solid equilibria, and similarities in gas and liquid adsorption behaviour, J. Colloid Interface Sci., 1998, 200, 273–290 CrossRef CAS.
- C. V. Krishnan, M. Garnett, B. Hsiao and B. Chu, Electrochemical measurements of isopolyoxomolybdates: 1. pH dependent behaviour of sodium molybdate, Int. J. Electrochem. Sci., 2007, 2, 29–51 CAS.
- W. Wang, Y. Hu, J. Goebl, Z. Lu, L. Zhen and Y. Yin, Shape- and size-controlled synthesis of calcium molybdate – doughnut shaped microstructures, J. Phys. Chem. C, 2009, 113, 16414–16423 CrossRef CAS.
- L. Q. Mai, F. Yang, Y.-L. Zhao, X. Xu, L. Xu and Y.-Z. Luo, Hierarchical MnMoO4/CoMoO4 heterostructured nanowires with enhanced supercapacitor performance, Nat. Commun., 2011, 2, 381 CrossRef PubMed.
- Y. Liang, X. Han, Z. Yi, W. Tang, L. Zhou, J. Sun, S. Yang and Y. Zhou, Synthesis, characterization and lithium-intercalation properties of rod-like CaMoO4 nanocrystals, J. Solid State Electrochem., 2007, 11, 1127–1131 CrossRef CAS.
- J. You, L. Xin, X. Yu, X. Zhou and Y. Liu, Synthesis of homogenous CaMoO4 microspheres with nanopits for high-caapcity anode material fin Li-ion battery, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 271 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00156a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
David R. G.
Mitchell
c,
Christian
Baur
*ab,
David R. G.
Mitchell
c,
Christian
Baur