
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencepH-responsive targeted gold nanoparticles for in vivo photoacoustic imaging of tumor microenvironments†
Shiying
Li‡
b,
Kwok-Ho
Lui‡
b,
Tik-Hung
Tsoi
b,
Wai-Sum
Lo
 b,
Xin
Li
b,
Xuesen
Hu
b,
William
Chi-Shing Tai
b,
Clara
Hiu-Ling Hung
c,
Yan-Juan
Gu
*b and
Wing-Tak
Wong
*ab
b,
Xin
Li
b,
Xuesen
Hu
b,
William
Chi-Shing Tai
b,
Clara
Hiu-Ling Hung
c,
Yan-Juan
Gu
*b and
Wing-Tak
Wong
*ab
aThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, 518057, China
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: w.t.wong@polyu.edu.hk
cUniversity Research Facility in Life Science, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China
First published on 13th December 2018
Abstract
The acidic microenvironment of tumor tissues has been proven to be a major characteristic for differentiation from normal tissues, thereby providing a desirable target for both disease diagnosis and functional imaging. We herein introduce a way to endow gold nanoparticles with aggregation behaviour induced by pH tuning. The nanoparticle surface was modified with two thiol conjugate molecules, which could smartly stabilize it at the pH of blood and normal tissues but induce aggregation in response to the acidic extracellular pH in tumor. The surface conjugate molecule composition effect was studied systematically, and at the optimal surface conjugate molecule composition, a pH-responsive active tumor-targeting c(RGDyk)-MHDA/LSC@AuNP nanoprobe was successfully obtained and showed a significantly enhanced contrast effect for both in vitro and in vivo photoacoustic (PA) imaging. Intravenous administration of our nanoprobe to U87MG tumor-bearing nude mice showed PA imaging contrasts almost 3-fold higher than those for the blocking group. Quantitative biodistribution data revealed that 9.7 μg g−1 of nanoprobe accumulated in the U87MG tumor 4 h post-injection. These findings might provide an effective strategy for developing new classes of intelligent and biocompatible contrast agents with a high efficiency for PA imaging and PA imaging-guided cancer therapy.
Introduction
Photoacoustic (PA) imaging is a novel noninvasive and nonionizing imaging modality for preclinical in vivo imaging that has recently attracted significant interest.1–3 It combines the advantages of optical and acoustic imaging and provides deep tissue penetration (up to 5 cm), an excellent spatial resolution (as low as 5 μm) and high sensitivity due to the much weaker scattering of ultrasonic signals in tissues than optical signals. The prevailing application is in cancer research, where PA imaging has been explored for primary tumor detection and molecular characterization, therapeutic monitoring, and identification and assessment of metastatic lymph nodes.4 Since few endogenous chromophores like haemoglobin and melanin can generate PA signals, synthetic exogenous chromophores are often used for enhancing the signal-to-noise ratio of PA imaging.5 To date, various agents such as organic dyes,6–8 metallic nanoparticles,9–11 carbon nanotubes,12 semiconducting macromolecules and polymers13,14 have been studied for use in PA imaging.As one class of strong optical-absorption nanomaterial, gold nanostructures have been widely explored as PA contrast agents due to their ease of synthesis, good biocompatibility and high absorption in the near infrared (NIR) range.15 Several gold nanostructures with a controllable size and uniform shape, such as gold nanorods,16,17 nanocages,18–21 nanostars22,23 and aggregates of nanoparticles24,25 have been used for PA imaging within the NIR window. The key advantage of gold nanostructures for PA imaging signal generation is that they essentially act as a macroscopic dipole, leading to an extinction coefficient several orders of magnitude higher than that of small organic dyes.26 Plasmonic gold nanoparticles are used for tumor imaging as they could intrinsically accumulate within tumors due to the Enhanced Permeability and Retention (EPR) effect; furthermore, various studies have revealed that RGD peptides possess the ability to enhance receptor-mediated binding sensitivity.27,28 Thus, RGD-modified nanoparticles could be a promising platform for elevating the binding sensitivity towards tumor cells. As representative anisotropic nanogeometries, Au-nanorods and nanocages have shown great promise as PA contrast agents because they can absorb NIR light. However, they generally have a relatively large size (50 nm), which limits efficient endocytosis in cancerous cells and results in some unfavourable in vivo behaviours.5,29 Moreover, their ability to maintain a high signal-to-noise ratio for differentiating healthy and diseased tissues in clinical applications is still a major obstacle for these nanomaterials because the signal is always “on”. Hence, smart contrast agents which elicit a significant signal change, such as a shift in the peak absorption wavelength in response to a biological behaviour, are more attractive for minimizing the influence from the background signal of endogenous chromophores for accurate detection with high sensitivity.30 Small gold nanoparticles (AuNPs) possess a longer residence time in blood and a shorter biological life-time than their larger counterpart as reported by Cheng et al.31 However, spherical AuNPs with a size smaller than 50 nm show poor NIR absorption which limits their utility for in vivo therapeutic applications such as PA imaging and plasmonic photothermal therapy (PTT). Fortunately, the optical absorption wavelength of small AuNPs can be tuned into the NIR optical window to minimize any unwanted endogenous absorption and maximize the PA imaging depth.32–34
Stimulus-responsive nanomaterials that provide high performance in amplifying signals that are generated under peculiar pathophysiological conditions are key for various biomedical applications. Moreover, pH-responsive nanomaterials that utilize endo-or exogenous stimuli, e.g. acidic tumor microenvironments for inducing accurate and rapid responses to amplify pathophysiological signals without undesired side effects are important in many applications, such as biomedical imaging, drug delivery and biosensing.35 The acidic microenvironment is the most common feature of solid tumors, regardless of tumor types or development stages.36 This characteristic enables the development of ‘smart’ probes, which are switched on only in tumor tissues for achieving selective targeting, imaging, controlled drug release, etc.37,38 For example, Liu et al. developed pH-sensitive mixed-charge zwitterionic AuNPs for selective targeting of the acidic tumor microenvironment.39 The accumulation, retention, and cell uptake of their probe in tumors were significantly enhanced by the pH-induced aggregation effect when compared with nonsensitive PEGylated AuNPs. Further studies demonstrated that the mixed-charge AuNPs exhibited an encouraging aggregation-enhanced PTT efficacy in vitro when they formed aggregates.40 Sun et al. employed the salt-induced aggregation phenomenon of AuNPs in biological media for forming highly efficient and biocompatible NIR photothermal transducers for PTT and PA imaging of cancer.41 It was a simple and effective way to develop new classes of NIR photothermal transducers, but limited to intra-tumoral administration only which restricted the scope of applications. Without proper functionalisation on the particle surface, salt-induced aggregation often exhibits certain stability. Recently, Kim's group developed ‘smart’ AuNPs with a surface decorated with hydrolysis-susceptible citraconic amides.42,43 It is worth noting that Kim et al. only utilized their ‘smart’ AuNP platform in a tumor-xenografted mouse model using intratumoral injection which limits its ability to reflect real-life pharmacokinetic properties. Moreover, our preliminary data showed that LSC-modified AuNPs are unstable for in vivo intravenous injection studies; thus our study employs the tumor-xenografted mouse model with intravenous injection of c(RGDyK)-LSC and MHDA-functionalized AuNPs which shall offer and reflect a pharmacokinetic profile/property closer to real clinical situations. The citraconic amides were rapidly converted to positively charged amines by hydrolysis under mildly acidic conditions such that the particle surface acquired both positive and negative charges and thus aggregated rapidly via electrostatic attraction; the aggregates of nanoparticles therefore could be used for PTT and PA imaging. Highly sensitive and acid-labile conjugate molecules with AuNPs showed high potential for achieving precise tumor diagnosis and effective tumor detection in vivo.
Based on the above consideration, herein, we design and synthesize a gold-based nanoprobe with the aim of enhancing the detection accuracy of in vivo tumor-targeting PA imaging. AuNPs were surface modified with a self-assembled monolayer of both 4-(2-(5-(1,2-dithiolan-3-yl)pentanamido)ethylamino)-2-methyl-4-oxobut-2-enoic acid (LSC) and a cyclic RGDyK peptide conjugated with 16-mercaptohexadecanoic acid (c(RGDyK)-MHDA) at an optimal ratio. The “smart” LSC conjugate molecules bear a pH-responsive hydrolysis-susceptible citraconic amide moiety and a dithiol group for encouraging gold surface anchoring. The MHDA conjugate molecules bear a thiol at one end for anchoring to the gold surface and carboxylic group at the other end for conjugating with the αvβ3 integrin-targeting peptide, cyclic c(RGDyK) peptide. The conjugation of the c(RGDyK)-MHDA ligand endows the nanoprobe with an active tumor-targeting ability, and improved stability and biocompatibility of AuNPs in a mildly acidic environment. The obtained c(RGDyK)-MHDA/LSC@AuNP nanoprobe exhibited a strong photoacoustic signal at a wavelength of 680 nm owing to the pH-induced aggregation phenomenon. Previously, this novel nanoprobe was used as a PA imaging contrast agent to image the tumor microenvironment with a thorough evaluation of its tumor targeting efficacy and in vivo profile in a subcutaneous αvβ3-positive U87MG glioblastoma xenograft model using nude mice. This novel smart probe shall offer broad tumor specificity with a high signal-to-noise ratio, which enables a clear differentiation between blood and in vivo tumors.
Experimental section
Materials
All commercially available chemical reagents were used without further purification. Gold(III) chloride trihydrate (HAuCl4·3H2O, 99.9+%), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), N-(3-dimethylaminopropyl-N′-ethylcarbodiimide) hydrochloride (EDC), 16-mercaptohexadecanoic acid (MHDA) and N-hydroxysulfosuccinimide (sulfo-NHS) were obtained from Aldrich. Sodium citrate (HOC(COONa) (CH2COONa)2·2H2O) and (±)-α-lipoic acid were purchased from Sigma. Cyclic arginine-glycine-aspartic acid-D-tyrosine-cysteine (c(RGDyk)) was purchased from BankPeptide (China). All cell culture related media were purchased from ThermoFisher (Hong Kong, China). All cell lines were purchased from American Type Culture Collection (ATCC).Synthesis of 13 nm gold nanoparticles (AuNPs)
Gold nanoparticles (AuNPs) with a 13 nm diameter were synthesised by reducing a well-stirred solution of 100 mL of 1 mM chloroauric acid with 10 mL of 38.8 mM sodium citrate in deionized water under reflux, which was then cooled down to room temperature to obtain a red wine-coloured solution. The solution was kept at 4 °C until further use.Synthesis of 4-(2-(5-(1,2-dithiolan-3-yl) pentanamido)ethylamino)-2-methyl-4-oxobut-2-enoic acid (LSC)
(±)-α-Lipoic acid (500 mg, 2.42 mmol) was dissolved in 5 mL of anhydrous chloroform. 1,1-carbonyldiimidazole (700 mg, 4.31 mmol) was added to the lipoic acid solution and stirred for 15 min in an ice bath. Ethylenediamine (1.2 mL, 16.7 mmol) was added to the solution quickly. The resultant solution was stirred for 30 min in an ice bath and 40 min further at room temperature. The crude product was diluted to 30 mL with chloroform and washed with 15 mL of 10% NaCl solution three times and 10 mL of water two times. It was then dried with Na2SO4, and chloroform was removed by rotary evaporation. The product, a yellow viscous liquid, was obtained. The yield was 69% (420 mg). The product (420 mg, 1.7 mmol) was dissolved in 5 mL of anhydrous chloroform. 2 equivalents of citraconic anhydride (0.30 mL, 3.4 mmol) was added to the solution and stirred overnight at room temperature. A precipitate was obtained and washed with chloroform, and a yellow fine powder product was obtained. The yield was 63% (390 mg). 10 mg of NaOH was added to 0.5 mL of D2O to dissolve the sample for 1H-NMR characterization. 1H-NMR (400 MHz, D2O with 0.5 M NaOH): δ1.23–1.37 (m, 2H), 1.45–1.67 (m, 4H), 1.84 (s, 3H), 1.75–1.97 (m, 1H), 2.07–2.17 (t, 2H), 2.29–2.42 (m, 1H), 3.00–3.15 (m, 2H), 3.18 (s, 4H), 3.48–3.62 (m, 1H), 5.44 (s, 1H) (Fig. S1†).Synthesis of c(RGDyK)-MHDA
14.4 mg MHDA was dissolved in 5 mL DMF, and then 5 equivalents of NHS and EDC were added for activating the carboxylic group. Excess EDC and NHS were removed by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C and then dried overnight under nitrogen for further usage. 5 mg MHDA-NHS ester (12 μM) was added to the c(RGDyK) (7.5 mg, 12 μM) with a 1:1 molar ratio in 20 mM Na2HPO4 solution and stirred for 24 h at room temperature. The reaction mixture was centrifuged and washed three times at 14000 rpm for 10 min each at 4 °C to remove any unreacted peptide molecules. The reaction mixture was then analysed and purified by ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS): Agilent 1290 UHPLC-Agilent 6540 QTOF-MS (Agilent SB-C18 RRHD column, 1.8 μm, 2.1 × 100 mm), with the parameters setting of acquisition m/z range: 100–3200, fragmentor voltage: 175 V, Vcap voltage: 4500 V, and nozzle voltage: 1000 V. The flow rate was 0.3 mL min−1, with the mobile phase starting from solvent A (MilliQ water, 0.1% formic acid) to solvent B (ACN, 0.1% formic acid). Gradient: 0 min 10% B, 2 min 10% B, 30 min 90% B, 33 min 90% B, 34 min 10% B, and 40 min 10% B. UPLC-MS: m/z = 890.5 [M + H]+; calculated Mr for c(RGDyK)-MHDA = 890.2. The retention time for c(RGDyK)-MHDA and c(RGDyK) was 17 min and 0.6 min, respectively.
000 rpm for 10 min at 4 °C and then dried overnight under nitrogen for further usage. 5 mg MHDA-NHS ester (12 μM) was added to the c(RGDyK) (7.5 mg, 12 μM) with a 1:1 molar ratio in 20 mM Na2HPO4 solution and stirred for 24 h at room temperature. The reaction mixture was centrifuged and washed three times at 14000 rpm for 10 min each at 4 °C to remove any unreacted peptide molecules. The reaction mixture was then analysed and purified by ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS): Agilent 1290 UHPLC-Agilent 6540 QTOF-MS (Agilent SB-C18 RRHD column, 1.8 μm, 2.1 × 100 mm), with the parameters setting of acquisition m/z range: 100–3200, fragmentor voltage: 175 V, Vcap voltage: 4500 V, and nozzle voltage: 1000 V. The flow rate was 0.3 mL min−1, with the mobile phase starting from solvent A (MilliQ water, 0.1% formic acid) to solvent B (ACN, 0.1% formic acid). Gradient: 0 min 10% B, 2 min 10% B, 30 min 90% B, 33 min 90% B, 34 min 10% B, and 40 min 10% B. UPLC-MS: m/z = 890.5 [M + H]+; calculated Mr for c(RGDyK)-MHDA = 890.2. The retention time for c(RGDyK)-MHDA and c(RGDyK) was 17 min and 0.6 min, respectively.
Tailoring the pH-sensitive range of the MHDA/LSC@AuNPs by changing the feed ratio of conjugate molecules
A series of mixed self-assembled monolayer (SAM)-protected AuNPs with different feed ratios of the two thiol-containing conjugate molecules were prepared while keeping the total thiol concentration unchanged. Here, these AuNPs are referred to as MHDA/LSC@AuNPs X:Y (X:Y means the feed ratio of LSC to MHDA). Taking MHDA/LSC@AuNPs 96:1 as an example, briefly, a mixed thiol aqueous solution of MHDA (0.087 mg, 0.30 μM) dissolved in methanol and LSC (10 mg, 28 μM) dissolved in 0.5 M NaOH were added to the citrate-coated AuNPs (10 mL) at pH 9. The pH of the solution was adjusted to pH 9 with 1 M NaOH, as LSC has better solubility under alkaline conditions. This also helped to keep the AuNPs stable through the ligand exchange reaction. After stirring at room temperature for 24 h, the modified AuNPs were purified with a centrifugal filter (Amicon, ultra 30 kDa MW cutoff).
To determine the sensitivity and stability of modified AuNPs (7 nM) with respect to different pH values (10 mM phosphate buffer; pH 5.8, 6.5 and 7.4), the zeta potential and hydrodynamic size (DLS) were measured in buffer solutions at different pH. The stability of gold nanoparticles in different pH buffer solutions was monitored by UV-vis absorption.
Synthesis of the targeted nanoprobe c(RGDyk)-MHDA/LSC@AuNPs and non-targeted nanoprobe MHDA/LSC@AuNPs
The targeted nanoprobe was obtained by a one-step modification of AuNPs with LSC and c(RGDyk)-MHDA molecules with an optimal surface conjugate molecule ratio of 96:1. The resultant product, c(RGDyk)-MHDA/LSC@AuNPs, was stored at 4 °C for further experiments. The final c(RGDyk)-MHDA/LSC@AuNPs were reconstituted in PBS and filtered through a 0.22 μm filter for cell and animal experiments. The non-targeted nanoprobe was synthesized under the same conditions except for using MHDA instead of c(RGDyk)-MHDA. The molar concentration of gold in the targeted nanoprobe solution was calculated to be 144 nM by ICP-OES analysis. The conjugated RGD was determined by measuring the differences in the concentration of RGD using UPLC-MS before and after addition of gold nanoparticles during the coupling reaction which indicated a complete coupling reaction (details shown in Fig. S2†). UHPLC-MS was used to measure the quantity of c(RGDyk)-MHDA molecules in the filtrate after ultra-filtration. The amount of unconjugated molecules in the filtrate was too low to be quantified as indicated in Fig. S3,† so the conjugation yield is approximated to unity. The yield of the targeted nanoprobe c(RGDyk)-MHDA/LSC@AuNPs and non-targeted nanoprobe MHD/LSC@AuNPs was estimated to be 92% and 86% according to the difference in the gold concentration before and after conjugation measured by ICP-OES.
General characterization
TEM measurements were performed on a JEOL JEM-2011 TEM with an accelerating voltage of 200KV. The sample was prepared by pipetting a drop of an aqueous solution of nanoparticles onto a 400-mesh copper grid coated with carbon, and the sample was allowed to dry in air before measurement. UV/VIS absorption spectra were obtained using an Agilent Cary 8454 UV-visible spectrophotometer. 1H NMR was performed using a 400 Hz Bruker Plus with D2O as the solvent, and mass spectral data were obtained from a Bruker amazon speed ESI-ion trap-ETD MS high resolution mass spectrometer. The zeta potential and hydrodynamic sizes of AuNPs and c(RGDyk)-MHDA/LSC@AuNPs were measured by DLS using a Nano ZetaSizer (Malvern Instruments, Worcestershire, UK) with a 90° scattering angle at 25 °C. The data were analyzed with Zetasizer software 6.32.Cell culture
U-87MG (human malignant glioma) cancer cells were cultured in MEM cell medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C in a humidified atmosphere of 5% CO2. MCF-7 (breast cancer cells) was cultured in DMEM medium at 37 °C in a humidified atmosphere of 5% CO2.PA instrumentation
PA spectra and PA/US coregistered images were acquired with a Vevo LAZR Photoacoustic Imaging System (FUJIFILM VisualSonics) using a 21 MHz transducer. It is equipped with a tunable Nd:YAG laser system (OPOTEK Inc., 680–980 nm, 20 Hz repetition rate, 5 ns pulse width, 50 mJ pulse peak energy) to trigger the system acquisition and excite the tissue with optical pulses for generating the PA effect.PA signal measurement of the nanoprobe in tube phantoms
The PA signals of nanoprobe (1.44 nM) in 10 mM PBS buffer of pH 5.8, 6.5, 7.4 and 8.0 were amplified. Tube phantoms were constructed from polyethylene tubes (PE50, Intramedic, Becton Dickinson, Sparks, MD) of 2 cm length and an inner diameter of 0.7 mm. The tubes were heat-sealed at one end, filled with either solution of nanoparticles, and then heat-sealed on the opposite side. To each testing solution, 50 μL of c(RGDyk)-MHDA/LSC@AuNPs was added to a phosphate buffer solution with an equal volume (10 mM, 40 °C), and the resulting mixture was added to PE tubes which was placed in the water tank of the PA imaging system. Typical PA spectra were acquired from 680 to 970 nm with a 5 nm step size.PA detection of tissue-mimicking phantoms
The U87MG and MCF-7 cells were plated with a density of 1 × 104 cells per well in a 6-well plate. After 24 h, the cells were exposed to a final concentration of the 14.4 nM gold nanoprobe and incubated for 3 h, 6 h and 12 h separately. The MHDA/LSC@AuNPs were used as the control group. For PA detection of tissue-mimicking phantoms under pH 5.8 and pH 7.4, the culture medium was replaced with 400 μL of fresh medium containing the c(RGDyk)-MHDA/LSC@AuNPs with 600 μL pH 7.4 or 5.8 buffer solution. Cells were washed three times with 1× PBS buffer prior to trypsinization. Cells that contained AuNPs were gathered by centrifugation for 3 min at 1000 rpm to remove the liquid. Prior to tissue-mimicking phantom construction, the cells were first pre-treated with c(RGDyk)-MHDA/LSC@AuNPs.The cells (400 μL) were blended with 1% agarose gel (400 μL) and transferred into a cylindrical mold (8 mm diameter × 10 mm height). The gel was allowed to solidify followed by extraction from the mold and placed into a customized plastic box (2 cm width × 3 cm length). The gel was positioned at the centre of the plastic box such that the distance between the sample and the phantom's edges were 0.5 cm in width and 1.0 cm in length.
To prevent the dissolution of the sample, 5% agarose gel was employed to seal the cylindrical sample within the plastic box. Finally, the tissue-mimicking phantoms were moved into a customized water tank with the bottom sealed with an Aquaflex ultrasound gel pad (2 cm × 9 cm). The US gel was applied between the sample and gel pad while a 3D motor and US transducer were aligned and mounted on a rail moved together by controlling a scanning stage. Thick slab processing was employed to visualize the accumulated signal over 5 mm. The PA amplitude of the phantom was measured at 680–970 nm.
PA signal measurement in subcutaneous xenograft tumors in mice
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “The Special Health Service, Department of Health, Hong Kong Special Administrative Region” and approved by the Animal Ethics Committee of “The Hong Kong Polytechnic University”. Female nude mice were obtained from the Chinese University of Hong Kong at ages of 4–6 weeks. The mice were first anesthetized with 3% isoflurane followed by subcutaneous injection of 107 U87MG glioblastoma cells suspended in 200 μL MEM on the right hind limb. When the tumor reached 5 mm in diameter, as measured with a digital calliper (within 2 weeks of subcutaneous injection of U87MG), c(RGDyk)-MHDA/LSC@AuNPs (144 nM; 200 μL) were injected into the subject through intravenous injection (tail-vein) and imaged for 48 h (2 h per imaging episode). Thus, the animals were imaged 24 times; each image recorded under anaesthesia with 3% isoflurane. The first time-point (pre-IV) tumors were imaged at 680, 710, 730, 750, 830, 870, and 950 nm wavelength to obtain the 2D and 3D micro-ultrasound and multi-spectral PA images at 24 and 48 h post-injection.Spectral unmixing was demonstrated using Vevo LAZR software based on the photoacoustic spectra of c(RGDyk)-MHDA/LSC@AuNPs. Oxygenated and deoxygenated haemoglobin were obtained, multiplexed and overlaid on the B-mode ultrasound image according to a previous method.44 The resulting PA signal intensity for all unmixed components was quantified using Vevo software by drawing a region of interest (ROI) around the tumor on image slices. The analytical PA intensity was the average PA intensity signal between all image slices covering the whole tumor volume. After that, the mice were sacrificed immediately after the imaging process by an overdose of anaesthetic (a mixture of 90 mg kg−1 ketamine and 9 mg kg−1 xylazine). During nanoparticle administration, the body weight and tumor size were measured. After 2 days of such treatment, the mice were then sacrificed and the tumors were isolated for weighing and immunohistochemistry analysis.
ICP measurements
c(RGDyk)-MHDA/LSC@AuNPs were digested with aqua regia and then diluted with 1% HNO3. For the biodistribution study, nude mice with U87MG xenografts (n = 3/group) were i.v. injected first with 200 μL of 144 nM c(RGDyk)-MHDA/LSC@AuNPs and sacrificed at 4, 24, 48 h post-injection. The liver, spleen, kidneys, heart and tumor were collected, weighed and digested with aqua regia under heating. 1, 2, 5, 10, and 20 ppm Au solutions were prepared for calibration. Every data point was expressed as a mean (±SD) from triplicate measurement. The gold concentration in solution was determined using an Agilent 7500 series inductively coupled plasma mass spectrometer (ICP-MS).Results and discussion
Design and synthesis
The design and fabrication of an activatable nanoprobe for tumor-targeting PA imaging is illustrated in Scheme 1. The smart nanoprobe, c(RGDyK)-MHDA/LSC@AuNPs, was synthesized by conjugating both the pH-sensitive LSC conjugate molecules and c(RGDyK)-MHDA ligand onto 13 nm citrate-capped AuNPs (TEM images are shown in Fig. S4†) at pH 9. The pH-sensitive linker LSC was synthesized based on a previous method,37 while the c(RGDyK)-MHDA ligand was synthesized by a coupling reaction between the –NH2 group of the peptide and the –COOH group of MHDA through the common NHS/EDC conjugation (Fig. S2†). The role of the self-assembled monolayer of the c(RGDyK)-MHDA ligand is to improve the stability of nanoparticles because longer carbon chain n-alkanethiolates have a stronger reactive binding probability to gold by decreasing the activation energy barrier for the S–H bond dissociation.45,46 Concurrently, the nanoprobe also possesses an active tumor-targeting ability through the function of the arginine-glycine-aspartic acid (RGD) tripeptide, as it and its derivatives are widely reported for targeting the αvβ3 integrin in angiogenic vessels.26 Once the probe was placed under mildly acidic conditions, a mimic of the tumor environment, the susceptible citraconic amide moieties hydrolysed to a positive surface potential from negative as its terminal functional group changes from a carboxylate anion to a protonated amine. | ||
| Scheme 1 (A) Schematics of the pH-responsive c(RGDyk)-MHDA/LSC@AuNP nanoprobe modified with two thiol ligands c(RGDyk)-MHDA and LSC. (B) Schematic illustration of the working mechanism of the intravenous administration of the nanoprobe to form AuNP depots with ultra-strong plasmonic coupling for PA imaging; nanoparticles produce an enhanced PA signal (red arrow) upon irradiation at 680 nm (blue arrow) in a tumor microenvironment. | ||
Tailoring the pH-induced aggregation behaviour of the MHDA/LSC@AuNPs by changing the feed ratio of conjugated molecules
To optimize and regulate the response time of AuNP aggregation at pH 5.8, different ratios of LSC to MHDA were first used to modify 13 nm AuNPs and obtain various MHDA/LSC@AuNPs. The degree of nanoprobe aggregation and pH sensitivity were studied using UV-vis spectroscopy under pH 5.8 and 7.4 conditions. As shown in Fig. 1, when the feed molar ratio of LSC to MHDA ligands ranges from 8:1 to 128:1, the modified AuNPs showed different aggregation performances (Fig. S5 and S6†). It was noted the AuNPs modified with a molar ratio of 64:1 could disperse well at the normal tissue pH 7.4 (Fig. 1B) while they slightly aggregated in response to the tumor acidic pH 5.8 (Fig. 1A) after 2 h incubation. When the molar ratio was increased to 96:1, the modified AuNPs presented slight aggregation in pH 7.4 PB solution (Fig. 1D) while they quickly aggregated at pH 5.8 (Fig. 1C) after 2 h of incubation. When the molar ratio was further increased to 128:1, the modified AuNPs aggregated in both pH 7.4 and pH 5.8 solutions (Fig. 1E and F). It should be emphasized that the kinetics of hydrolysis is the key factor influencing the behaviour of the nanoprobe. If the aggregation is too fast, e.g. within a few minutes, it would not be compatible with the various prevailing interferences caused by electrostatic attraction to the biomolecules. Previous data revealed that the hydrolysis lifetime of the citraconic amide is 120 min in pH 5.5 buffer solution at 35 °C, which is adequate for the in vivo application of PA imaging.47 It is thought that the acidic tumour microenvironment leads a charge inversion of MHDA/LSC@AuNPs by hydrolysing the leaving group of LSC which alters the charge from negative to positive. Thus, the surface of the nanoparticles possesses both positive and negative charge after the conversion. The opposite electrostatic attraction, therefore, promotes the aggregation within the tumour cells. The change of surface charge can be monitored by the zeta potential change of MHDA/LSC@AuNPs before and after hydrolysis (Fig. S9A†). The zeta potential became less negative under mildly acidic conditions (pH 5.8 and 6.5), while the zeta potential presents little change under neutral conditions (pH 7.4) (Fig. S9A†), which is consistent with the gradual change of the terminal carboxylic group to the amine group of MHDA/LSC@AuNPs. DLS analysis indicated that the nanoprobe (MHDA/LSC = 96:1) was monodisperse in deionized water with a hydrodynamic radius of 16.4 nm, while it forms aggregates with a significantly increased hydrodynamic radius of over 500 nm at pH 6.5 and 5.8 (Fig. S9B†). The absolute UV intensity was employed to assess modified AuNPs' UV sensitivity with LSC molarity ranging from 64 to 128 while MHDA molarity was kept at 1. The molar ratio of 96:1 was capable of inducing a distinct red shift at a steady rate while the other feed ratio failed to provide such constant performance (Fig. 1E). Therefore, the molar ratio of 96:1 was selected for in vivo experiments.
 | ||
| Fig. 1 pH-induced aggregation behaviours of MHDA/LSC@AuNPs can be tailored by changing the feed ratio of LSC to MHDA ligands attached to the AuNPs' surface: (A, C and E) pH 5.8 and (B, D and F) pH 7.4 PB solution. | ||
Characterization of the c(RGDyk)-MHDA/LSC@AuNP targeted nanoprobe
The c(RGDyk)-MHDA/LSC@AuNP targeted nanoprobe was modified with a mixture of LSC and c(RGDyk)-MHDA ligands with the molar ratio of 96:1. The pH sensitivity and the degree of aggregation were studied by monitoring the UV-vis spectra of the nanoprobe solution. At pH 5.8, the nanoprobe was expected to exhibit both positive and negative charges. Aggregation therefore took place, and the localized surface plasmon resonance (LSPR) peaks of the nanoprobe started to red-shift and broaden in the NIR region, which is a characteristic property of surface plasmon coupling effect of gold nanoparticles (Fig. 2A). The absorption maximum red-shifted to reach 650 nm with its red tail reaching over 800 nm upon 120 min incubation at 37 °C. In contrast, the nanoprobe presented only a slight red-shift in pH 7.4 solution (Fig. 2B). With the absorption maxima shifting to a longer wavelength over time, the colour of the sample gradually changes from red to violet and grey. A control experiment using MHDA@AuNPs showed no obvious red-shift even after 4 h incubation at pH 5.8 (Fig. S6†), since MHDA itself was not susceptible to pH-dependent hydrolysis. This indicated that the nanoprobe would not aggregate by merely the colloidal instability of the carboxylic acid surface upon pH change, as demonstrated by its distinct zeta potential and hydrodynamic size when compared to those of c(RGDyk)-MHDA/LSC@AuNPs. The stability of the nanoprobe in PBS (pH 7.4), DMEM, plasma, 35 mg mL−1 HAS protein (usual serum HAS level) and DI water was investigated, and the results are shown in Fig. 2C and S10.† The results demonstrated that HSA protein and DMEM medium will not lead to aggregation of the nanoprobe. TEM analysis further confirmed the aggregation of the nanoprobe in mildly acidic solutions (Fig. 2D–F).
 | ||
| Fig. 2 pH-sensitivity of AuNPs: (A) time evolution of the UV-vis spectra of nanoprobes as soon as they are added to the PB solution (10 mM, pH 5.8). (B) UV-vis spectra of the nanoprobe at pH 7.4, 6.5 and 5.8 after 1 h incubation. (C) UV-vis spectra of the nanoprobe incubated in the PB solution (10 mM, pH 7,4), DMEM culture medium with 10% FBS (pH 7.4) and serum (plasma). (D–F) TEM images of the nanoprobes at pH 7.4, 6.5 and 5.8 PB buffer solutions. | ||
Photoacoustic characterization of the c(RGDyk)-MHDA/LSC@AuNP targeted nanoprobe
Before using PA to detect the tumor microenvironment in vivo, the PA signals of the nanoprobe in different pH buffer solutions were first measured for investigating the signal change upon pH-induced aggregation (Fig. S11†). The full PA spectra from 680 to 970 nm of the nanoprobe scanned with a 5 nm step size are shown in Fig. S10.† Similar to organic optical dyes or other exogenous contrast agents, our nanoprobe was shown to be a promising PA contrast agent in the “ON” stage when they aggregated in an acidic environment.Moreover, the nanoparticle aggregation induced strong absorption peaks at 680 nm. This is especially advantageous in multiplexed PA imaging, as demonstrated here, by eliciting a PA signal that is well separated from the blood oxygenation signal at 750 nm (Fig. 5). 680 and 800 nm were chosen for PA imaging in our study. It could be observed that the PA signals of the nanoprobe at 680 nm were stronger than those at 800 nm under mildly acidic conditions (Fig. 3A). However, the PA signals were barely detectable at pH 8.0. The change of PA signal enhancement over time was monitored and is shown in Fig. 3B. The nanoprobe at pH 5.8 exhibited a sharp increase in the PA signal during the first 30 min followed by a slow increase until 2 h. In contrast, the nanoprobe at pH 7.4 did not show any obvious changes in signal enhancement over time. Overall, the PA signals of the nanoprobe at pH 5.8 were 3.6 times larger than those of the nanoprobe tested at pH 7.4 after 2 h incubation.
 | ||
| Fig. 3 (A) Solution PA images of the nanoprobe (14.4 nM) after mixing with 10 mM PB solution for 1 h in PE tubes. Dashed lines indicate the position of PE tubes. (B) Time evolution of the PA signals of the nanoprobe after mixing with various buffer solutions at 680 nm. | ||
In vitro photoacoustic imaging of the c(RGDyk)-MHDA/LSC@AuNP nanoprobe
The PA images of the targeted nanoprobes interacted with two different cell lines were also taken in tissue mimic agarose gel (Fig. 4A and B). To test whether the nanoprobe could be used as a cancer-cell specific PA-imaging contrast agent, in vitro PA monitoring was carried out using αvβ3 integrin-overexpressed U87MG cancer cells. This cell line demonstrated an overexpressed αvβ3 integrin level, while MCF-7 which only showed a low expression of the αvβ3 integrin was also examined as a control. The cells were incubated either with the 14.4 nM targeted nanoprobe or non-targeted nanoprobe for different time periods (0–12 h). MTT assay was performed to confirm the cellular toxicity of the nanoprobe (Fig. S13†). The cells were then washed to remove free nanoprobes, followed by mixing with an agar solution to form a tissue-mimicking phantom. The quantitative increase of PA signal over time is illustrated in Fig. 4A and B. At the beginning of the measurement, the PA signal was very weak in both cell types. Shortly after the incubation, a sharp increase in the PA signal was observed in the time period 0 to 12 h. Moreover, the signals obtained from cancerous U87MG cells increased more rapidly than those of MCF-7 cells under the same conditions. For example, the PA signal was 3.5 times stronger for the U87MG cells compared to that for the MCF-7 cells after 6 h incubation. These results suggested that the cellular labelling of the nanoprobe was greatly enhanced due to the selective targeting of the integrin αvβ3 receptor on the U87MG cell surface. | ||
| Fig. 4 PA response of quantitative measurement of each mimic tissue consisting of U87MG (A) and MCF-7 cells (B) co-incubated with 14.4 nM c(RGDyk)-MHDA/LSC@AuNPs (targeted nanoprobe) and MHDA/LSC@AuNPs (non-targeted nanoprobe) at different time points. (C and D) PA images and quantitative measurement of the targeted nanoprobe phantom containing same U87MG cells with and without pre-injection of 5 μL of 0.1 mM c(RGDyK) inhibitor at 680 nm. (E and F) PA images and quantitative measurement of the targeted nanoprobe phantom containing same U87MG cells in pH 5.8 and pH 7.4 buffer solution. | ||
The results reveal that the nanoprobe in the nM concentration can achieve aggregation in a tumor microenvironment. To further confirm the targeting ability of the nanoprobe, competitive inhibition was carried out using free c(RGDyK) peptide for co-incubation to inhibit the nanoprobe's binding to U87MG cells. The PA images and signal enhancement of the phantom containing the same cell number after incubation with 0.9 mg mL−1 targeted nanoprobe with and without the free peptide are shown in Fig. 4C and D.
The targeted nanoprobe led to an exponential increase of the PA signals of U87 cells (Fig. 4C, bottom). In contrast, the competing group showed nearly no PA signal (Fig. 4C, upper), indicating that the free peptide pre-treatment appeared to have blocked most of the surface receptors for binding with the nanoprobe, and it even precluded any significant passive accumulation of nanoparticles. The results confirmed that the targeting ability and specificity of the nanoprobe originated from the recognition between c(RGDyK) and the αvβ3 receptor.
The PA images and signal of each mimic tissue consisting of U87MG co-incubated with the targeted nanoprobe and non-targeted nanoprobe under pH 5.8 and 7.4 are shown in Fig. 4E and F. They revealed that each mimic tissue consisting of U87MG co-incubated with the targeted probe at pH 5.8 exhibited a higher PA signal than that at pH 7.4 at different time points, which further demonstrated that the targeted nanoprobe is a good pH-responsive PA contrast agent.
In vivo photoacoustic imaging of the c(RGDyk)-MHDA/LSC@AuNP nanoprobe
The PA imaging ability of the nanoprobe to the αvβ3 integrin-positive tumor was then evaluated in the U87MG tumor-bearing nude mice. Expressions of αvβ3 in U87MG tumor and MCF7 cells (control) were determined by RT-PCR (Fig. S14†). Before the PA signal measurement in subcutaneous xenograft tumors in mice, we utilized croconic acid dyes (Croc) to analyse the acidity of xenograft tumors in mice as they are known to exhibit unique pH responsive absorption. As illustrated in Fig. S15C,† the Croc generated strong absorbance under acidic pH (pH 5.8 and 6.5), while it produced weak absorbance under pH 7.4 and 8. Furthermore, the in vivo experiment indicated that the PA signal from the tumor was far more intense than that from intramuscular injection (Fig. S15D†). Therefore, this evidence confirms the acidic nature of the tumor.48PA signals could be determined via multispectral scanning and wavelength deconvolution because of the strong absorbance maximum at 680 nm from aggregated AuNPs, which facilitated the deconvolution from other tissue signals (Fig. 5B). PA images of oxygenated haemoglobin (OxH) and deoxygenated haemoglobin (deOxH) signals could also be simultaneously captured and overlaid with the nanoprobe signal in PA images, since OxH and deOxH have unique absorption maxima at 750 and 850 nm that could be deconvoluted from that of the nanoprobe (680 nm) (Fig. 5C).49,50 When being overlaid with the ultrasound images of the tumor (Fig. 5D), the PA signals of the nanoprobe could be seen to be focused predominately around the periphery of the tumor. The contrast signal of the OxH was located on the surface of the tumor and that of deOxH was predominately on the tumor interior as expected of the tumor with a hypoxic core. Thus, the nanoprobes were suitable for in vivo PA imaging and studying the tumor microenvironment.
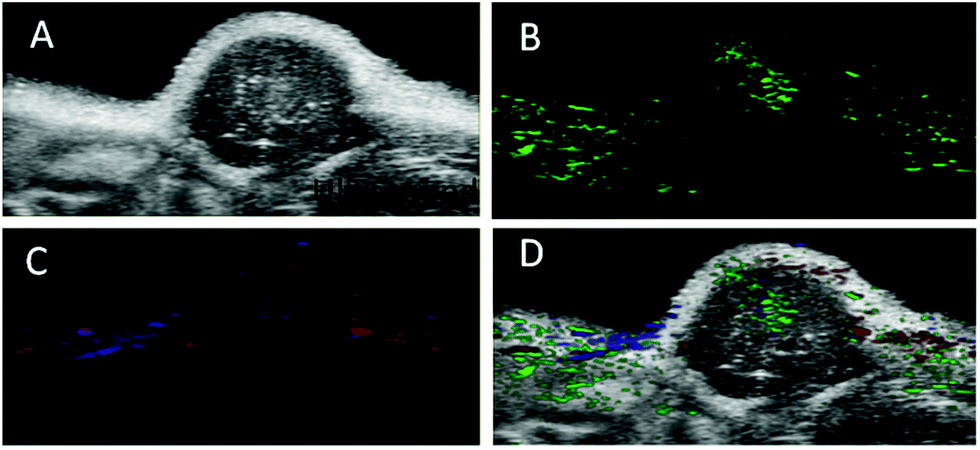 | ||
| Fig. 5 In vivo multiplexed imaging of targeted nanoprobe accumulation in the U87MG tumor: (A) an in vivo ultrasound image of a subcutaneous implanted U87MG tumor. (B) Heat map of PA signals from the targeted nanoprobe injected in mice implanted with a U87MG tumor (green). (C) Heat map of oxygenated haemoglobin (red) and deoxygenated haemoglobin (blue) of the same U87MG tumor. (D) Overlaid with the ultrasound image and targeted nanoprobe, oxygenated haemoglobin, deoxygenated haemoglobin PA signals. | ||
The accumulation level of the targeted nanoprobe in the tumor was tracked over time with PA imaging to study the pharmacokinetic properties of the nanoparticles. Before the injection, the photoacoustic and ultrasound images of the mice were recorded. Pre-injection scans indicated that only a few major blood vessels in the tumor were recorded using the Vevo system with a low contrast (indicated as ‘0 h’). 0.1 mmol c(RGDyk)/kg (50 μL) per mice body weight before targeted nanoprobe administration was injected via i.v. injection. The U87MG tumor-bearing nude mice were then injected with 200 μL of the targeted nanoprobe (144 nM per kg mouse body weight) in PBS through the tail vein. After injection, photoacoustic images were acquired at 680 nm at 1, 2, 4, 6, 24 and 48 h post-injection. For the receptor-blocking experiment, mice were pre-injected with free c(RGDyK) 1 h earlier followed by post-injection of the nanoprobe.
A non-targeting control group, MHDA/LSC@AuNPs, was also studied. Quantitative analysis of the PA signal from the tumor was performed by drawing and analysing more than twenty slides of regions-of-interest (ROIs) around the tumors on the basis of the ultrasound images. The perspective views of 3D volume rendering of PA images pre-injection and 24 h post-injection were recorded using the Vevo system to confirm the accurate size of the U87 tumor (Fig. S16†). Since tumors showed weak intrinsic PA signals because of the absorption of haemoglobins in the NIR region, the PA signal increment at 680 nm (defined as the PA signals of the tumor site after probe injection deducted by the background signal of the tumor before injection) was utilized in data analysis to minimize the interference of the tissue background. The PA signal acquired at 680 nm for free c(RGDyK)-treated and untreated mice gradually increased after injection of the targeted nanoprobe, both of which reached the maximum at 4 h post-injection followed by a small reduction and finally levelling-off in the next 4 h (Fig. 6A and B). This indicated that the targeted nanoprobe efficiently accumulated at the tumor site. However, at each time point, the mice injected with the targeted nanoprobe showed a significantly higher PA signal in the tumor compared to the blocking group pre-injected with c(RGDyk). The mice injected with targeted nanoparticles after 4 h showed a 2.8-fold higher PA signal in the tumor than the blocking group. For the control group of MHDA@AuNPs, no significant increase of signal was observed in the tumour region under the same concentration, which demonstrates the responsiveness of LSC conjugate molecules and c(RGDyk) for increasing tumor accumulation (Fig. S17†). To further understand the biodistribution and pharmacokinetics of the nanoprobe in vivo, the gold concentration of various organs including tumours was determined by ICP-MS at 4, 24 and 48 h separately after one dose injection. Ex vivo biodistribution revealed a high accumulation in the reticuloendothelial system (RES) including the liver and spleen (Fig. 7). Over 9.7 μg g−1 targeted nanoprobe, the second highest accumulation, was successfully retained in the tumor at 4 h post-injection.
 | ||
| Fig. 6 Targeting of integrin αvβ3-positive U87MG tumors in mice by the targeted nanoprobe: (A) photoacoustic images and ultrasound images of nude mice bearing U87MG tumors were obtained before injection or at 2, 4, 8, and 24 h after intravenous injection of the targeted nanoprobe (144 nM) (B) average PA intensity increment at 680 nm as a function of the post-injection time of the targeted nanoprobe in free c(RGDyK) pre-treated and untreated mice (n = 3). Error bars represent standard deviation. *p < 0.05. | ||
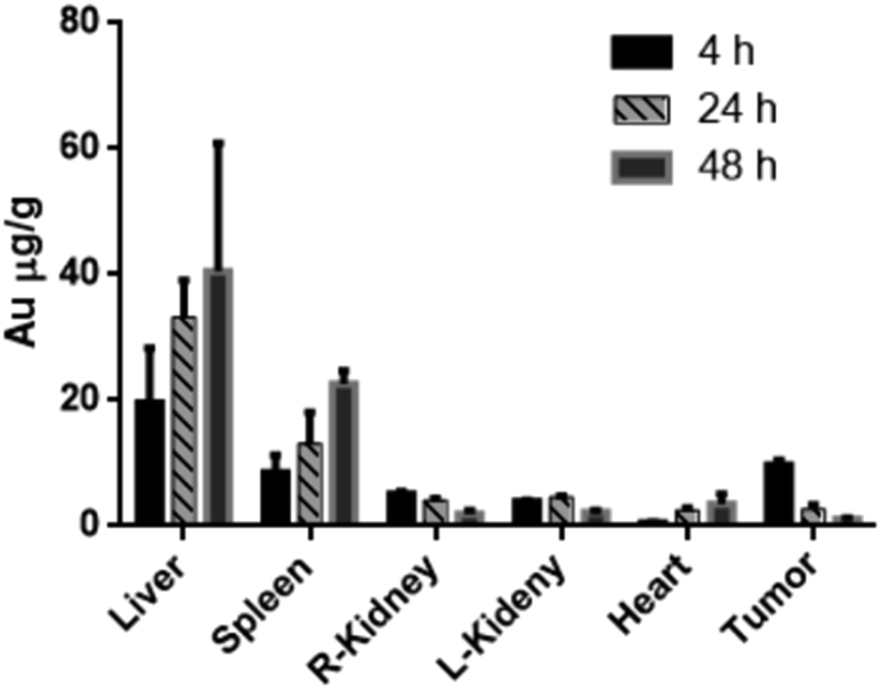 | ||
| Fig. 7 Biodistribution of the targeted nanoprobes in U87MG tumor-bearing mice 4, 24 and 48 h after i.v. injection (200 μL). The gold concentration in dry organs was determined by ICP-MS (n = 4). The error bars represent standard derivation (SD). | ||
Conclusions
In summary, pH-responsive gold nanoparticles possess specific targeting properties towards cancer cells and provide distinct signals in acidic tumour microenvironments when compared with another conventional PA contrast agent. The in vivo experiment using a tumor-xenograft mouse model with intravenous injection demonstrated its utility in PA imaging which suggests its potential in future biomedical applications.We have taken full advantage of the intrinsically mildly acidic tumor microenvironment and developed a pH-stimuli-responsive PA nanoprobe based on careful control of the ratio of two thiol ligands for AuNP surface decoration to achieve tumor-targeting PA imaging in live animals. The nanoprobe, once internalized into the tumor tissue due to the active tumor-targeting c(RGDyK) towards the overexpressed integrin αvβ3 receptors, could smartly respond to the tumor acidic microenvironment which converts the citraconic amides to positively charged primary amines. As a result, the nanoparticles aggregate rapidly via electrostatic attraction, lighting up the NIR signal for specific PA imaging of tumors. The stability test performed by incubation of c(RGDyk)-MHDA/LSC@AuNPs with DI water, DMEM, PBS (pH 7.4) and HSA solution demonstrates that the nanoprobe is stable enough under normal biological conditions. The addition of MHDA ligands also improves the stability of the nanoprobe, prolonging its circulation time.
Moreover, its activatable nature effectively overcomes the shortcomings of poor signal-to-noise ratios and ‘false positive’ results in ‘always on’ imaging strategies. Its high pH-sensitivity makes it superior to commercial molecular probes in providing PA signals at low concentrations down to the nM level. Our strategy for the design of activatable probes for PA imaging provides a valuable approach to construct smart PA imaging-guided resection of tumours and a target strategy for post-therapy monitoring of drug efficacy for clinical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Hong Kong Research Grants Council (PolyU 153012/15P), Area of Excellence Grants (1-ZVGG) of Hong Kong Polytechnic University, Basic Research Program of Shenzhen Grants (No. JCYJ20160531184120814), Hong Kong Polytechnic University (University Research Facility in Materials Characterization and Device Fabrication (UMF)), and Hong Kong Polytechnic University [University Research Facility for Chemical and Environmental Analysis (UCEA) and University Research Facility in Life Science (ULS)].References
- L. V. Wang and S. Hu, Photoacoustic tomography, Science, 2012, 335, 1458–1462 CrossRef CAS PubMed.
- S. Zackrisson, S. M. W. Y. vande Ven and S. S. Gambhir, Cancer Res., 2014, 74, 979–1004 CrossRef CAS PubMed.
- V. Ntziachristos and D. Razansky, Chem. Rev., 2010, 110, 2783–2794 CrossRef CAS PubMed.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- C. Kim, C. Favazza and L. V. Wang, Chem. Rev., 2010, 110, 2756 CrossRef CAS PubMed.
- Y. Jiang and K. Pu, Small, 2017, 13, 1700710 CrossRef PubMed.
- Y. Liu, Y. Yang and M. Sun, et al. , Chem. Sci., 2017, 8, 2710–2716 RSC.
- Q. Che, C. Liang, X. Sun, J. Chen, Z. Yang, H. Zhao, L. Feng and Z. Liu, PNAS, 2017, 114, 5343–5348 CrossRef PubMed.
- X. Hu, C.-W. Wei, J. Xia, I. Pelivanov, M. O'Donnell and X. Gao, Small, 2013, 9, 2046–2052 CrossRef CAS PubMed.
- K. Yang, L. Zhu, L. Nie, X. Sun, L. Cheng, C. Wu, G. Niu, X. Chen and Z. Liu, Theranostics, 2014, 4, 134–141 CrossRef CAS PubMed.
- R. Zhang, D. Pan, X. Cai, X. Yang, A. Senpan, J. S. Allen, G. M. Lanza and L. V. Wang, Theranostics, 2015, 5, 124–133 CrossRef PubMed.
- A. de la Zerda, S. Bodapati, R. Teed, S. Y. May, S. M. Tabakman, Z. Liu, B. T. Khuri-Yakub, X. Chen, H. Dai and S. S. Gambhir, ACS Nano, 2012, 6, 4694–4701 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- C. Xie, X. Zhen, Y. Lyu and K. Pu, Adv. Mater., 2017, 29, 1703693 CrossRef PubMed.
- W. Li and X. Chen, Nanomedicine, 2015, 10, 299–320 CrossRef CAS PubMed.
- Y.-S. Chen, W. Frey, S. Kim, P. Kruizinga, K. Homan and S. Emelianov, Nano Lett., 2011, 11, 348–354 CrossRef CAS PubMed.
- P. P. Joshi, S. J. Yoon, W. G. Hardin, S. Emelianov and K. V. Sokolov, Bioconjugate Chem., 2013, 24, 878–888 CrossRef CAS PubMed.
- X. Cai, W. Li, C.-H. Kim, Y. Yuan, L. V. Wang and Y. Xia, ACS Nano, 2011, 5, 9658–9667 CrossRef CAS PubMed.
- W. Li, P. K. Brown, L. V. Wang and Y. Xia, Contrast Media Mol. Imaging, 2011, 6, 370–377 CrossRef CAS PubMed.
- G. D. Moon, S.-W. Choi, X. Cai, W. Li, E. C. Cho, U. Jeong, L. V. Wang and Y. Xia, J. Am. Chem. Soc., 2011, 133, 4762–4765 CrossRef CAS PubMed.
- C. Liu, S. Li, Y.-J. Gu, H. Xiong, W.-T. Wong and L. Sun, Mol. Imaging Biol., 2018, 20, 919–929 CrossRef CAS PubMed.
- L. Zu, L. Liu, Y. Qin, H. Liu and H. Yang, Nanomedicine, 2016, 12, 1805–1813 CrossRef CAS PubMed.
- L. Nie, S. Wang, X. Wang, P. Rong, A. Bhirde, Y. Ma, G. Liu, P. Huang, G. Lu and X. Chen, Small, 2014, 10, 1585–1593 CrossRef CAS PubMed.
- M. Sun, F. Liu, Y. Zhu, W. Wang, J. Hu, J. Liu, Z. Dai, K. Wang, Y. Wei, J. Bai and W. Gao, Nanoscale, 2016, 8, 4452–4457 RSC.
- J. Song, J. Kim, S. Hwang, M. Jeon, S. Jeong, C. Kim and S. Kim, Chem. Commun., 2016, 52, 8287–8290 RSC.
- E. Petryayeva and J. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- L. Nie, S. Wang and X. Wang, Small, 2014, 10, 1585–1593 CrossRef CAS PubMed.
- R. Lin, K. Dnyananda and T. Chan, Contrast Media Mol. Imaging, 2012, 7, 7–18 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- D. Pan, M. Pramanik, A. Senpan, J. S. Allen, H. Zhang, S. A. Wickline, L. V. Wang and G. M. Lanza, FASEB J., 2011, 25, 875–882 CrossRef CAS PubMed.
- X. Cheng, R. Sun, L. Yin, Z. Chai, H. Shi and M. Gao, Adv. Mater., 2017, 29, 1604894 CrossRef PubMed.
- D. Pan, M. Pramanik, S. A. Wickline, L. V. Wang and G. M. Lanza, Recent advances in colloidal gold nanobeacons for molecular photoacoustic imaging, Contrast Media Mol. Imaging, 2011, 6, 378–388 CrossRef CAS PubMed.
- J. F. Hainfeld, M. J. O'Connor, P. Lin, L. Qian, D. N. Slatkin and H. M. Smilowitz, PLoS One, 2014, 9, e88414 CrossRef PubMed.
- S.-E. Kim, B.-R. Lee, H. Lee, S. D. Jo, H. Kim, Y.-Y. Won and J. Lee, Sci. Rep., 2017, 7, 17327 CrossRef PubMed.
- T.-H. Tsoi, Y.-J. Gu, W.-S. Lo, W.-T. Wong, C.-F. Ng, C.-S. Lee and K.-L. Wong, Chempluschem, 2017, 82, 802–809 CrossRef CAS.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- T. Ji, Y. Zhao, Y. Ding and G. Nie, Adv. Mater., 2013, 25, 3508–3525 CrossRef CAS PubMed.
- W. Wu, L. Luo, Y. Wang, Q. Wu, H. Dai, J. Li, C. Durkan, N. Wang and G. Wang, Theranostics, 2018, 8, 3038–3058 CrossRef CAS PubMed.
- X. Liu, Y. Chen, H. Li, N. Huang, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS PubMed.
- H. Li, X. Liu, N. Huang, K. Ren, Q. Jin and J. Ji, ACS Appl. Mater. Interfaces, 2014, 6, 18930–18937 CrossRef CAS PubMed.
- M. Sun, F. Liu, Y. Zhu, W. Wang, J. Hu, J. Liu, Z. Dai, K. Wang, Y. Wei, J. Bai and W. Gao, Nanoscale, 2016, 8, 4452–4457 RSC.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639–13645 CrossRef CAS.
- J. Song, J. Kim, S. Hwang, M. Jeon, S. Jeong, C. Kim and S. Kim, Chem. Commun., 2016, 52, 8287–8290 RSC.
- H. D. Lu, B. K. Wilson, A. Heinmiller, B. Faenza, S. Hejazi and R. K. Prud'homme, ACS Appl. Mater. Interfaces, 2016, 8, 14379–14388 CrossRef PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- S.-F. Lai, H.-R. Tan, E. S. Tok, Y.-H. Chen, E. B. L. Ong, M.-T. Li, Y.-Y. Chen, F.-C. Chien, P. Chen, G. Margaritondo and Y. Hwu, Chem. Commun., 2015, 51, 7954–7959 RSC.
- A. C. Brinegar and J. E. Kinsella, J. Agric. Food Chem., 1980, 28, 818–824 CrossRef CAS PubMed.
- Q. Chen, X. Liu and J. Zeng, Biomaterials, 2016, 98, 23–30 CrossRef CAS PubMed.
- M. L. Li, J.-T. Oh, X. Xie, G. Ku, W. Wang, C. Li, G. Lungu, G. Stoica and L. V. Wang, Proc. IEEE, 2008, 96, 481–489 CAS.
- R. Fainchtein, B. Stoyanov and J. Murphy, Biomedical Optoacoustics, 2000, vol. 3916, pp. 19–33 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00190a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2019 |