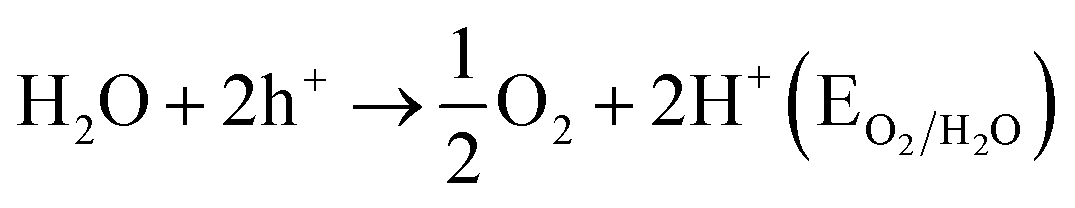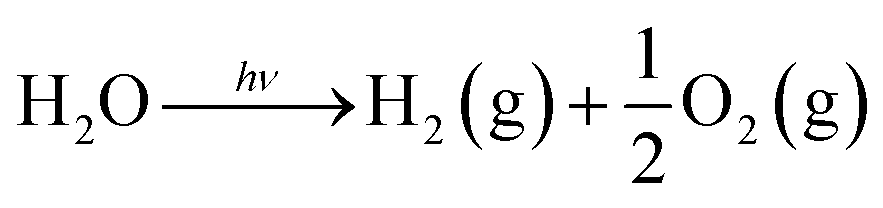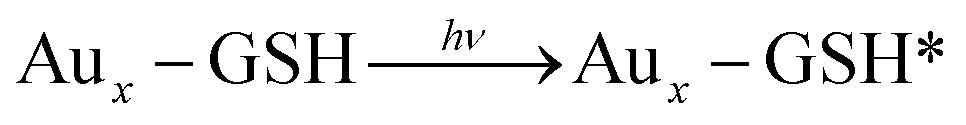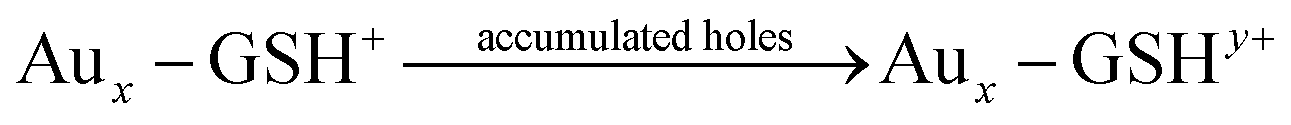DOI:
10.1039/C8RA06517A
(Review Article)
RSC Adv., 2018,
8, 30125-30147
Recent advances in syntheses, properties and applications of TiO2 nanostructures
Received
2nd August 2018
, Accepted 20th August 2018
First published on 24th August 2018
Abstract
TiO2 is a compound of great importance due to its remarkable catalytic and distinctive semiconducting properties. It is also a chemically stable, non-toxic and biocompatible material. Nano TiO2 is strong oxidizing agent with a large surface area and, hence, high photo-catalytic activities. With low production cost and a high dielectric constant, it is an inexpensive material. It can be prepared by diverse procedures such as solution and gas phase procedures. Nowadays, TiO2 is being used frequently for photo degradation of organic molecules and water splitting for hydrogen generation. Most important applications include purification, disinfection of waste water, self-cleaning coatings for buildings in urban areas and the production of the green currency of energy (hydrogen) by splitting water. The review describes the advances in the syntheses, properties and applications of TiO2 nano structures. Besides, efforts are also made to discuss the working mechanism and future challenges and perspectives.
1. Introduction
Nowadays, nano-structured materials are an important area of research owing to their several unique characteristic features. Among all the transition metal oxides, TiO2 nano-structures are the most attractive materials in modern science and technology. TiO2 has been widely used commercially in doughnuts, cosmetics, pigments,1 catalysts, sunscreens,2,3 solar cells,4 water splitting etc. TiO2 is being used in plastics, paints, varnishes, papers, medicines, inks, medications, toothpaste, food products, and industries.5–10 In 1972 first of all, Fujishima and Honda4 reported photo-assisted water splitting under UV light on a TiO2 photo anode as a semiconductor.4,11,12 The diverse claims can be separated into “environmental” and “energy” groups, several of which depend on the TiO2 properties itself as well as on the changes of the TiO2 material host (e.g. with organic and inorganic dyes). In previous years, the research activity expansion has been observed in nanotechnology and nanoscience.13–17 On the modification, preparation and properties of nano-materials, a significant amount of research, reports and reviews have been published recently4,11–40 to know and précis the progress in this field. TiO2 nanostructures in various forms, among the unique characteristics of nanomaterials, are gaining broader applications due to their size-related characteristics. For nanometer scale TiO2 the energy band structure becomes discrete due to its surface, photochemical and photo physical properties. Consequently, several works have focused on nano crystalline TiO2 syntheses with a high surface area. As a photocatalyst,15,16,27 TiO2 nanostructures have drawn much attention and are projected to show a significant role in serving to resolve several pollution and environmental problems. Thus, using TiO2 for H2 production and photo-assisted water splitting devices offers a way for hygienic and low price production of hydrogen by solar energy.13,41
This review aims to give an inclusive data on the advances in TiO2 based nanostructure, recent investigation and the development efforts, which tack energy and environmental challenges in consideration. Besides, the crystal structure, optical, electrical/electronic and adsorption, surface area, porosity properties of TiO2 nanostructure are discussed. The procedures of preparations, fabrications (nanoparticles, nanorods, nanowires, and nanotubes), the conditions of syntheses and accountability for regulation of titanate nanostructures morphology are also discussed. TiO2 nanostructures applications in electrocatalysis, environment and energy challenges are also highlighted. Finally, the mechanism of action and future challenges are also highlighted.
2. Properties of TiO2 nanostructures
2.1 Crystal structure of TiO2
Titanium dioxide [titanium(IV) oxide or titania] has a molecular formula TiO2 with 79.87 as molecular weight. TiO2, a non-toxic material, chemically stable, biocompatible and strong oxidizing agent (with large surface area) has very high photocatalytic activity. It is an inexpensive material with high dielectric constant and low production cost. TiO2 crystal exists in three common polymorphs in nature i.e. brookite, anatase, rutile,42,43 and some few common structures of TiO2 II i.e. columbite,44 TiO2 III: baddeleyite,45,46 TiO2 (R) (ramsdellite),47 TiO2 (B) (monoclinic)48 and TiO2 (H) (hollandite).49
Rutile is the most thermodynamically constant among the different polymorphs structures.50 From 400 to 1200 °C, it's critical temperature can vary which depends on the grain impurities and size.51 The optical and electrical properties and crystal structural of rutile, brookite and anatase are given in Table 1.51–63 These are discussed in the following paragraphs.
Table 1 Comparison of the crystal structural, optical and electrical properties for TiO2 nanostructures
| Properties |
Rutile |
Anatase |
Brookite |
| Crystal structure |
Tetragonal |
Tetragonal |
Orthorhombic |
| Lattice constant (Å) |
a = 4.5936 |
a = 3.784 |
a = 9.184 |
| c =2.9587 (ref. 312) |
c = 9.515 (ref. 312) |
b = 5.447 (ref. 312) |
| c = 5.154 |
| Space group |
P42/mnm52 |
I41/amd |
Pbca |
| Molecule (cell) |
2 |
2 |
4 |
| Volume/molecule (Å3) |
31.21 (ref. 54) |
34.061 |
32.172 |
| Density (g cm−3) |
4.13 |
3.79 |
3.99 |
| Ti–O bond length (Å) |
1.949 (4)53 |
1.937 (4) |
1.87–2.04 |
| 1.980 (2) |
1.965 (2) |
|
| O–Ti–O bond angle |
81.2° |
77.7° |
77.0–105° |
| 90.0° |
92.6° |
| Band gap at 10 K |
3.051 eV (ref. 54) |
3.46 eV (ref. 313) |
| Static dielectric constant (ε0, in MHz range) |
173 (ref. 314 and 315) |
48 (ref. 55) |
|
| High frequency dielectric constant, ε∞ (λ = 600 nm) |
8.35 (ref. 56) |
6.25 (ref. 57) |
| Nature of conductivity at room temperature (undoped) |
n-type semiconductor58–75 |
|
| Mott transition |
Not observed60 |
Observed313 |
| Electron effective mass |
9–13 m,61 10–30 m,62 12–32 m63 |
∼1 m (ref. 313) |
2.2 Rutile
In a tetragonal structure, with 6 atoms per unit cell (Fig. 1), rutile is the most stable having TiO6 octahedron showing a slight orthorhombic distortion.64–66 Rutile phase is stable while at these conditions TiO2 converts thermodynamically into auspicious phase.67 For unit sizes > 14 nm, rutile phase becomes more stable than anatase.68 Predominantly, the crystals of natural rutile exhibit (110) surface.42 This surface is the most stable stoichiometric rutile surface.69 In the unit cell of rutile, 4 oxygen atoms form a partial octahedron about Ti70 while 2 titanium atoms (at [0, 0, 0] and 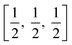 ) positions, separately are available. Distinctly, octahedron is linked to 10 near octahedra, out of which 2 parts an edge and eight share a bend with it. The octahedral edge shared are aligned along [001] direction as shows in Fig. 1.70
) positions, separately are available. Distinctly, octahedron is linked to 10 near octahedra, out of which 2 parts an edge and eight share a bend with it. The octahedral edge shared are aligned along [001] direction as shows in Fig. 1.70
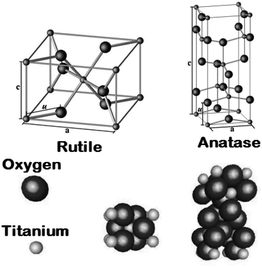 |
| | Fig. 1 Crystal structures of the rutile and anatase phases of TiO2. Small spheres represent Ti atoms, large spheres represent oxygen atoms. | |
2.3 Anatase
TiO6 octahedron distortion is significantly large for anatase phase while TiO2 has a tetragonal structure, hence, the symmetry of anatase is lower than orthorhombic (Fig. 1).70 The energy change between these 2 phases is minor closely (∼2 to 10 kJ mol−1).71 At 0 K, the rutile phase is not more thermodynamically constant than anatase. The unit cells of anatase contain 4 titanium atoms (at 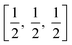 , [0, 0, 0],
, [0, 0, 0], 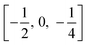 and
and 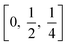 ) and 8 oxygen atoms, which form a partial TiO6 octahedron around Ti;70 with division of 4 edges of each octahedron.
) and 8 oxygen atoms, which form a partial TiO6 octahedron around Ti;70 with division of 4 edges of each octahedron.
2.4 Brookite
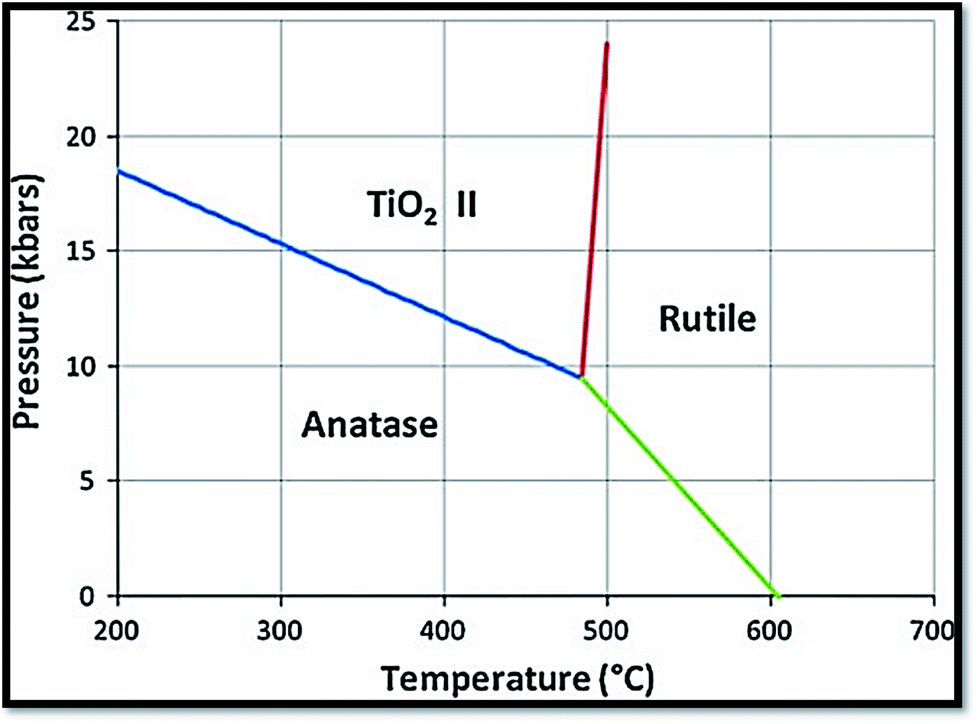
It is one of the 3 naturally occurring polymorphs of TiO2, which belongs to orthorhombic crystal system. It contains a larger cell volume with 8 TiO2 groups per unit cell (Fig. 2).53 Brookite is also the least dense form compared to anatase and rutile.65 Besides, it displays photocatalytic activity.72 Liu et al.73 investigated principled metal free photocatalytic water splitting. For this investigation the authors modified these surfaces by high pressure. Before and after modification, water splitting on usual anatase single crystal facets and on wafer slices of the [001] plane was investigated. Fig. 3 shows phase transition in TiO2 with the change in pressure and temperature.74 The temperature for transformation can be improved by accumulation impurities into TiO2. Anatase phase for powder samples containing V, Mo, & W respectively completely disappeared75 at temperature about 530, 680, and 830 °C.
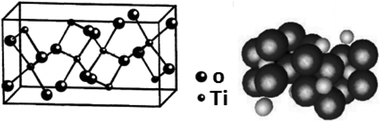 |
| | Fig. 2 Lattice structure of brookite TiO2. | |
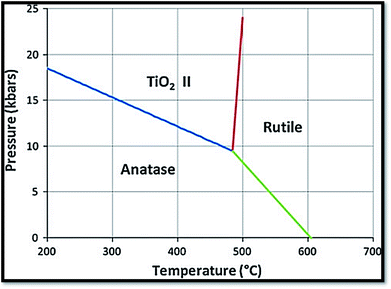 |
| | Fig. 3 Reaction boundaries of phase transitions in TiO2.74 | |
2.5 Optical properties of TiO2
In the range from IR to visible spectra, the rutile is more anisotropic than anatase. Contrarily, anatase shows important anisotropy in the band gap region.76 At about 0.75 to 1.18 eV (ref. 77–80) for rutile, a blue color appearance dependent on the amount of reduction of TiO2, arises from the visible part infrared absorption band peaking. The blue color appearance in IR has also been observed in anatase too.81,82 Besides, at 3.0 eV an oxygen vacancy causing a yellow color is a color center.81
2.6 Electrical/electronic properties of TiO2
The electrical/electronic properties of TiO2 nanostructures are dependent on the crystallographic directions. Titanium dioxide nanostructure (titania) is an important photocatalytic material, which exists in to 2 chief polymorphs i.e. rutile and anatase with tetragonal coordinations. Table 1 shows the several TiO2 electrical properties. A semiconductor, TiO2 has high resistivity (∼1015 Ω cm),83 titanium interstitials and bulk oxygen vacancies. These are considered to generate low electron donor levels that donate to the electric conductivity of TiO2.84
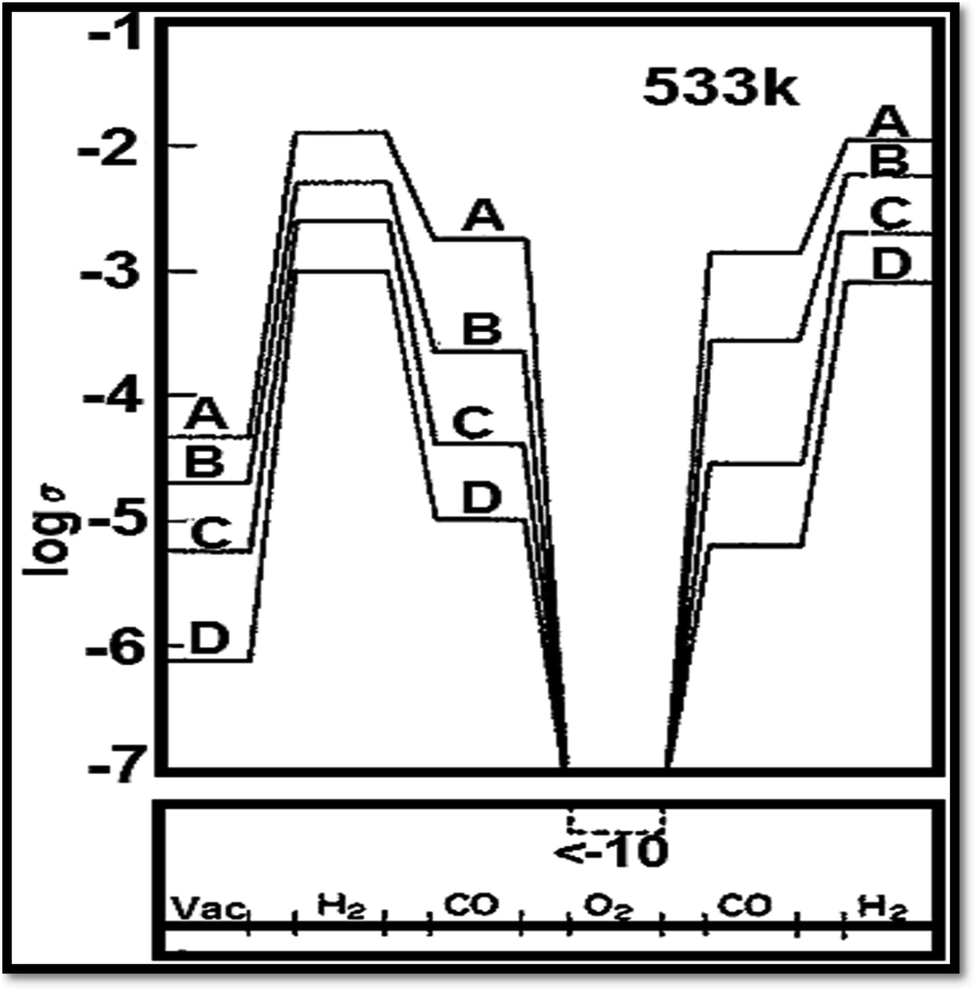
Vacancies for oxygen are the main fault in TiO2.70 The absence of oxygen introduces electrons excess in the solid ensuing in a rise of conductance of electricity.70 Fig. 485 shows the influence of gas cycle of sequence on electrical conductivity of 0.5% Pt/TiO2 doped with (A) W6+ (B) Ta5+(C) undoped and (D) Mg2+. Another thing to be added is that Nb (niobium) or tantalum atoms act as electron donors86 when incorporated into TiO2. Besides, Nb (niobium) or tantalum atoms increase the electrical conductivity. On the other hand, chromium, manganese, and iron act as electron acceptors,87 decreasing or increasing the TiO2 electrical conductivity. On the ratio between the oxygen vacancy concentration and their concentration, the augment or reduction in electrical conductivity depends.88 To the electrical conductivity, the point defects typed imperfection plays a crucial role. It is reported that the ionization energies for titanium interstitials89 is 0.007–0.08 eV. To the electronic conduction, the contribution of oxygen vacancies is also important.90 Several reports recommended the presence of both phenomena, with an activation energy of carrier generation around 4 meV.91
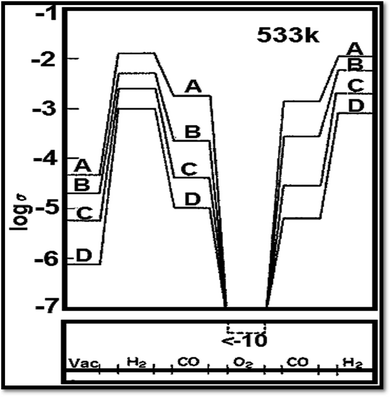 |
| | Fig. 4 Influence of gas cycle of sequence on electrical conductivity of 0.5% Pt/TiO2 doped with (A) W6+ (B) Ta5+ (C) undoped and (D) Mg2+.85 | |
After studying the formation energies and the electronic states of 4 types of lattice point defects in rutile, it was found that in the forbidden band, the existence of vacancy for oxygen leads to a deep donor defect level. Moreover, to establish that the band gap is ∼2.86 eV, the band building of the pure rutile TiO2 was studied.92 The valence band (VB) of wide gap rutile and anatase are formed by O2p states, while titanium 3d states forms the conduction band (CB).70 On these states, the detailed calculations can be found.45 Recently, several researches have been reported that the electronic structure of rutile bulk can also be described by means of additional techniques used in the experiment such as electrical resistivity, electroabsorption,93,94 photoconductivity and photoluminescence,95,96 X-ray absorption spectroscopy (XAS),97–102 resonant Raman spectra96,103 and photoelectrochemical analysis.58,104
3. Synthesis of TiO2 nanostructures
There are some methods of making TiO2 nanostructures. Gas phase and solution routes methods are the main ones. The solution route is able to coat complex shapes, to govern on stoichiometry, and preparation of composite materials. The presence of carbon as an impurity is an expensive precursor, and long processing times are the difficulties of solution route. There are two methods of synthesis (i) physical and (ii) chemical methods. Both methods have disadvantages and advantages. Physical procedures produce large quantities of material, but their resolution is inadequate to nanometers. The chemical procedures can approach the atomic layer limit105 through the preparation of minor (e.g., <100 nm and often <10 nm) structures. Hence, at a smaller scale, chemical procedures are typically completed.
3.1 Solution procedures
3.1.1 Sol gel route. In materials science and ceramic engineering, a wet-chemical technique widely used is sol–gel process. The advantage of sol–gel process is inexpensiveness. With sol–gel method, nanostructured TiO2 has been synthesized from titanium precursor hydrolysis. Usually, this method involve an acid catalyzed hydrolysis of titanium(IV) alkoxide tracked by condensation.106–119 This process includes 4 steps i.e. hydrolysis, polycondensation, drying and thermal decomposition. The sol–gel procedure includes the metal alkoxides usage, which suffer hydrolysis and condensation polymerization reactions to give gels as shown in these equations.120Hydrolysis:
| | |
Ti(OR)4 + 4H2O → Ti(OH)4 + 4ROH
| (1) |
Condensation:
| | |
Ti(OH)4 → TiO2 + 2H2O
| (2) |
where, R = ethyl, iso-propyl,
etc. For the change of liquid sol into a solid gel phase, the whole polymerization leads to loss of solvent. The sol particles size depends on the composition of solution, temperature and pH.
20 Ti–O–Ti chains development is preferred with little amount of water, additional titanium alkoxide in the reaction combination and stumpy hydrolysis rates.
The advantages of sol–gel method are as follows:
• Product homogeneity.
• Good rheostat above powder particle shape and size and size distribution.
• Easiness of manufacturing multi-component materials.
• No particular apparatus with low price.
• Low processing temperature.
• Synthesis of films with high photocatalytic activity.
Utilizing sol–gel template method, the syntheses of TiO2 nanotubes, nanorod and nanowires has been reported. This synthesis was carried out from the porous anodic alumina membranes (AAMs), supramolecular template and organogel. The first step for synthesis of nanorod is tumbling porous AAMs into a warmed TiO2 sol following by drying and heating processes.121 Ghamsari and colleagues122 synthesized TiO2 nanorod by sol–gel Template Process. Briefly, the porous anodic alumina of 60 μm in thickness and pores of 100 nm in diameter were used as templates.
Fig. 5 and 6 show SEM images of AAM template and TiO2 nanorods grown in the AAM template. By changing the dipping time, nanorods length can be changed. An individual TiO2 nanorods picture is shown in Fig. 6(b). Through the full length of the rods and continuous yet the rode is uniform. To obtain the nanowire arrays of TiO2, electrophoretic deposition of TiO2 colloidal suspensions can be used into the pores of an AAM.123 At room temperature; in another procedure; titanium tetraisopropoxide (TTIP) is dissolved in ethanol followed by the addition of glacial CH3COOH (mixed with demineralised water and EtOH) using HNO3 as pH 2–3 controller. AAM is used as the cathode and platinum as an anode. TiO2 nanowires are obtained after dissolving AAM template in 5% NaOH solution. Using titanium butoxide, titanium isopropoxide [TIPO] and titanium tetrachloride; as Ti precursors in HCl solution; have been synthesized.124 Nanotubes were also synthesized by Kasuga and colleagues125–127 using sol–gel method with innermost and outside diameters of approximately 5 and 8 nm, respectively. The spine designed products were nanotubes.
 |
| | Fig. 5 SEM of AAM template, (a) top and (b) side views. | |
 |
| | Fig. 6 SEM of TiO2 nanorods growth in AAM template, (a) low magnification image, (b) high magnification image.123 | |
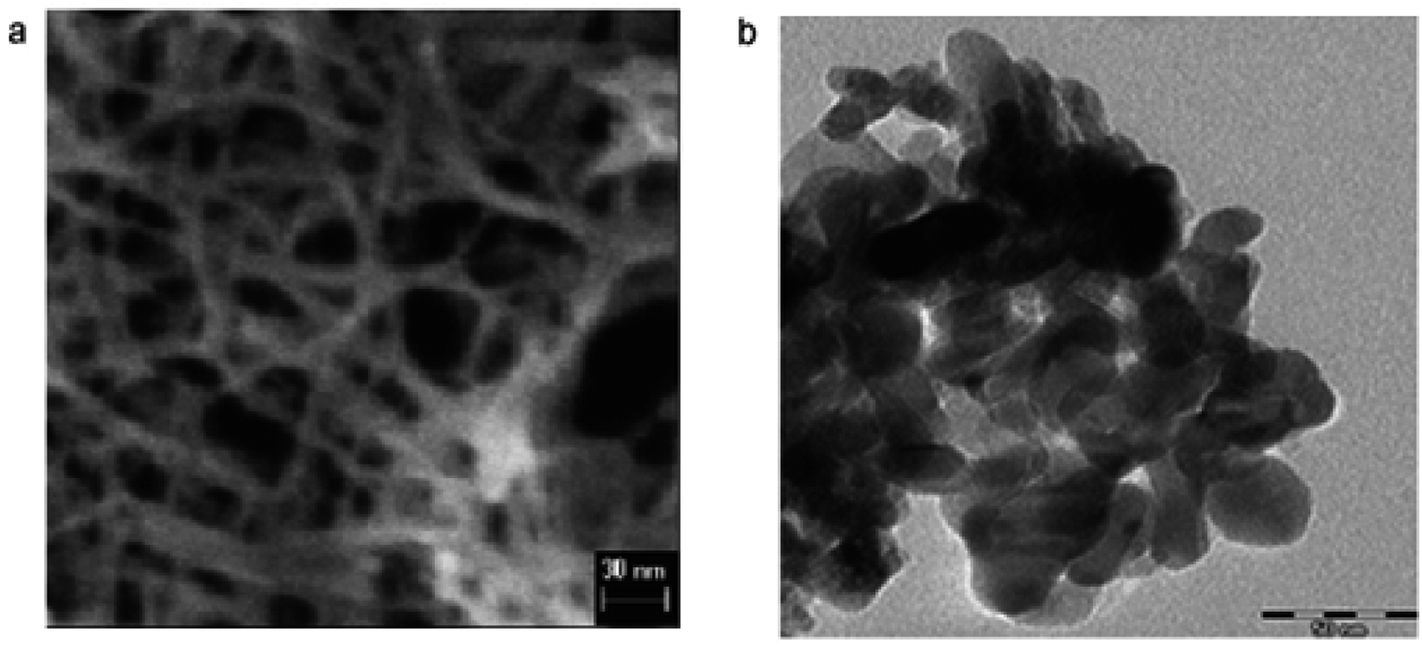
3.1.2 Hydrothermal procedures. Hydrothermal syntheses are promising procedures to gain titania nanocrystalline particles. The hydrothermal process is effective to attain the crystalline phase at lower temperatures. Hydrothermal is broadly used for the manufacture of minor units in the ceramics industry. Generally, hydrothermal synthesis is known as crystal synthesis or crystal growth. Below 300 °C, the hydrothermal synthesis is usually carried out. 374 °C is the critical temperature and 22.1 MPa is the critical pressure of water under supercritical conditions.Some studied indicated application of hydrothermal method to prepare TiO2 nanoparticles.128–132 To synthesize TiO2 nanoparticles, hydrothermal treatment of titanium precursor (peptized precipitates) was also used.129 Besides, reaction of hydrothermal of titanium alkoxide under acidic ethanol–water solution was also used to prepare TiO2 nanoparticles. Under EtOH–H2O environment, TiO2 nanoparticles synthesized were mainly primary structures of anatase. The sizes of the units were in range of 7–25 nm. Besides, TiO2 nanoparticles133 were synthesized using hydrothermal method. With increasing stirring for 45 minutes into distilled water at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volume ratio, 0.5 M solution of titanium butoxide Ti(OBu)4 in 2-propanol was gradually added. To prepare nanoparticles for further examination, the gels prepared were treated hydrothermally. TEM images of TiO2 nanoparticles prepared using hydrothermal method are shown in Fig. 7.
:4 volume ratio, 0.5 M solution of titanium butoxide Ti(OBu)4 in 2-propanol was gradually added. To prepare nanoparticles for further examination, the gels prepared were treated hydrothermally. TEM images of TiO2 nanoparticles prepared using hydrothermal method are shown in Fig. 7.
 |
| | Fig. 7 TEM images of TiO2 nanoparticles after hydrothermal treatment of TBA peptized gel at (a) 210 °C and (b) 270 °C.133 | |
Vijayalakshmi and Rajendran134 synthesized TiO2 nanoparticles using hydrothermal method by making a water solution of titanium. This solution was made by mingling one molar stoichiometric ratio of TTIP in 50 mL deionized water. TiO2 nanowire in the solutions of 10 to 15 M sodium hydroxide were synthesized; using TiO2 white powders; without stirring within an autoclave at 150–200 °C for 24–72 h by Zhang and colleagues.135 Fig. 8 shows SEM and TEM images of nanowires of TiO2 prepared by Zhang and colleagues.135 Titania nanotubes136 were synthesized via hydrothermal process. TiO2 powders were heated at 20–110 °C for 20 h. TiO2 nanotubes achieved were washed using a dil. HCl aqueous solution and deionized water. With NaOH aqueous solution, raw TiO2 material was treated. Besides, titania nanotubes137 were synthesized via hydrothermal. 5.0 g anatase TiO2 nanopowders were mixed with 180 mL 10 M NaOH in a perfluoroalkoxy (PFA) bottle and stirred for 1 h. The resultant precipitate was separated by centrifugation after hydrothermal reaction. Until pH value reached approximately 7, precipitate was rinsed with diluted HCl and demineralised water several times. Fig. 9 shows electron microscopy pictures of titania nanotubes and titania powders. Fig. 10 indicates the TEM pictures of TNTs; revealing that the TNTs have tubular, hollow and open structures. 3–6 nm is the inner diameter size of the TNTs and 1.9 nm is the average wall thickness. Besides, some other studies for syntheses of TiO2 nanowires by hydrothermal are also available.110–115
 |
| | Fig. 8 SEM images of TiO2 nanowires and a TEM image of a single nanowire prepared by Zhang and colleagues.135 | |
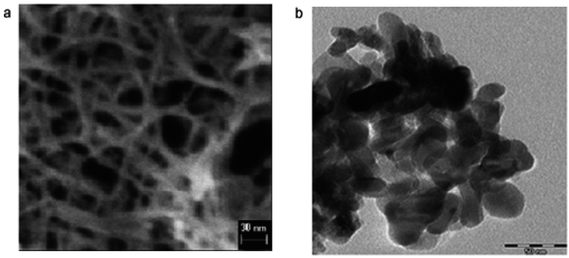 |
| | Fig. 9 Electron microscopy images of (a) titania powders and (b) titania nanotubes.312 | |
 |
| | Fig. 10 TEM images of titania nanotubes at (a) low magnification and (b) high magnification.338 | |
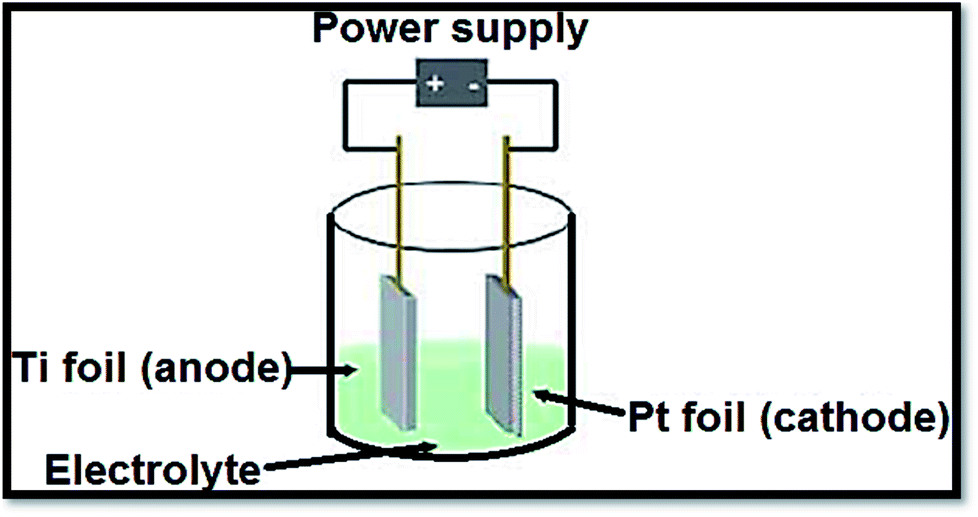
3.1.3 Electrochemical procedures. To produce advanced thin films such as superlattice, epitaxial, nanoporous and quantum dot ones by anodization, electrochemical procedures provided a diversity and low temperature approach. The distinctive states of the films are measured by electrolysis parameters such as temperature, current density, potential and pH.138 In a typical process, the conducting piece (undergoing anodization) is positioned in an electrolytic bath serving as anode. The cathode was a rod of platinum. From electrolyte to positive anode, at power supply the electrons are forced. Drifting through the power source, the electrons return to cathode. With hydrogen ions, these electrons react due to which bubbles off hydrogen gas are occurred. If oxide film is porous, the primary determinant is the electrolyte composition. Fig. 11139 shows a representation of an electrochemical anodization cell.
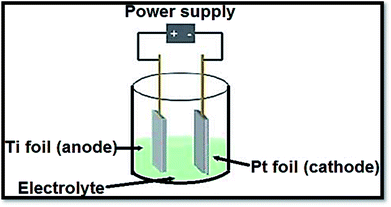 |
| | Fig. 11 Depiction of an electrochemical cell in which the Ti samples are anodized. Fabrication variables include temperature, voltage, pH and electrolyte composition. | |
In 2001, TiO2 nanotube arrays fabrication through anodic Ti oxidation was reported first time.140 Advanced studies engrossed on extension of morphology,141 length, pore size142 and wall thickness143 of the nanotube were carried out. Fig. 12144 shows SEM of TA6V anodised in CA electrolyte under 5 V for 20 min. The usage of different electrolytic diluents permitted the architecture control from well detached, standalone nanotubes to tightly packed arrays. The wall thickness of typical nanotubes ranged from 5 to 30 nm, with 20 to 350 nm pore size and 0.2 to 1000 mm length. Zwilling and co-workers144 used fluorinated electrolyte to produce a porous surface of TiO2 films via electrochemically anodization of titanium. Shimizu et al.145 reported that dil. H2SO4 could be used to bond porous TiO2 thin films. Yamamoto et al.146 observed that the act of dye-sensitized solar cell device was better by cathodically electro-synthesized TiO2 films; having sealed commercial Degussa P25 nanoparticles. Anodization or anodic oxidation of titanium foils was conducted by Patermarakis et al.147 Platinum and titanium were used as cathode and anode. Using formamide–water mixtures (having fluoride ions), Shankar and co-workers148 studied the effect of five dissimilar cationic species on development of TiO2 nanotube array.
 |
| | Fig. 12 Scanning electron micrograph of TA6V anodised in CA electrolyte.144 | |
In 2006, Ruan and co-workers149 synthesized titania nanotube by titanium anodization in an electrolyte. In 2006, nanotube arrays of protracted length from DMSO electrolytes was described by Paulose and co-workers.150 The importance of diethylene glycol (DEG) for nanotube array synthesis was reported by Yoriya and co-workers.151 These authors investigated tube formation. The necessity of tube separation and crystallization in the DEG-based electrolytes were required. The benefit of TiO2 nanotubes produced by anodization is readily attachment capability in a perpendicular orientation onto the substrate; improving electron transfer pathways as compared to the non-oriented structure.152 In one study, glycerol pH and water (75–25, v/v) electrolyte having 0.5% NH4F were modified using H2SO4.153 The length of nanotube was 950 nm on adjusting pH 5.6 by H2SO4. Nanotubes of 16 mm were found by anodizing at 20 V for 8 h after adding of 0.1 M CH3COONa by maintaining pH at 5.6. TiO2 nanostructured as photoanodes were prepared by nanotubes of TiO2 by anodic oxidation of foil of titanium140,154–156. In a model experimentation, a plate of clean titanium was anodized in a 0.5% hydrogen flouride solution under 10–20 V for 10–30 min. The counter electrode used in the experiment was platinum. Crystallized TiO2 nanotubes were obtained by annellation of plate of anodized Ti at 500 °C for 6 hours in oxygen.157 The diameter and length of TiO2 nanotubes may be optimized with the utilized potential between 1.0 and 25.0 V in optimized phosphate/HF electrolytes.158 SEM pictures of TiO2 nanotubes produced with this approach are shown in Fig. 13.159 For the first time, H generation efficiency, phase evolution and electrochemical synthesis of anatase, anatase–rutile and anatase–rutile–brookite (ARB) TiO2 nanotubes was reported Preethi et al.160 The samples tubular morphology was confirmed by TEM and SEM.
 |
| | Fig. 13 SEM images of TiO2 nanotubes prepared with anodic oxidation.159 | |
3.2 Gas phase procedures
3.2.1 Chemical vapor deposition. The vapor deposition processes to form coatings to alter the mechanical, electrical are normally used. Also, these methods can be castoff to produce free-stand bodies, fibers and films, or to form fused materials. Recently, chemical vapor depositions have received great attention in the manufacture of nanomaterials. These processes occur in a vacuum space and if chemical reaction doesn't occur, these processes are called physical vapor deposition (PVD). In this process, gases must be heated by suitable thermal. By using liquid precursor and titanium tetraisopropoxide (TTIP), pyrolysis in a mixed gases (consisting of helium/oxygen atmosphere) leads to prepare bushy crystalline of TiO2 films with sizes below 10 nm.161 Ayllon et al.162 described the synthesis of amorphous nanoparticles of TiO2. Pradhan, et al.163 prepared nanorod arrays of TiO2 with a length of 0.5–2 μm and a diameter of about 50–100 nm (Fig. 14). Metal organic CVD was used by the authors on a WC-Co substrate using (TTIP) as the precursor. Similarly, on the particle size distribution of aerosol particles prepared by gas-phase chemical reactions, Okuyama and colleagues164–166 described the effects of the primary concentration of TTIP vapor and temperature profile of the furnace. They found that in the controlled cylindrical furnace, the thermal decomposition of TTIP vapor led to form 10–60 nm sized ultrafine TiO2 nanoparticles. Likewise; using TTIP as precursor; the formation of TiO2 nanoparticles reinforced on porous silica gel (60–100 mesh) via metalorganic chemical vapor deposition MOCVD process was reported by Ding and colleagues.167 A tubular furnace controlled the reactor temperature. Pretreatment of the support materials, CVD reaction and calcinations process were involved in the synthesis of TiO2 nanoparticle/silica gel photocatalyst. Contrarily, the fabrication of TiO2 nanorods grown on fused silica substrates was reported by Wu et al.168 Directly, the nanostructures of TiO2 were grown on the substrates. The only one crystalline anatase and rutile TiO2 nanorods were formed, respectively, when the temperature ranged between 630 and 560 °C at a pressure of 5 torr. Furthermore, for preparing of TiO2 nanomaterial, the foregoing chemical vapor deposition approaches were also used. These included electrostatic spray hydrolysis,169 diffusion flame pyrolysis,170 ultrasonic spray pyrolysis, thermal plasma pyrolysis171–173 laser-induced pyrolysis174,175 and ultra-sonicated hydrolysis etc.
 |
| | Fig. 14 SEM micrograph of TiO2 nanowires deposited without NH3 at 500 °C; (a) with 800 sccm argon flow, and (b) with 500 sccm argon flow.163 | |
3.2.2 Physical vapor deposition. Among several techniques for the syntheses of TiO2 nanowires, primary PVD procedures include ion implantation.176–178 Before growth of TiO2 nanowires, a layer of Ti nanopowders deposited on the substrate was also reported by this group.176,177 These authors used gold as catalyst.176 Fig. 15 displays a model SEM image of nanowires of TiO2 made by physical vapor deposition method.176 Xiang et al.179 reported deposit of TiO2 nanowire on a Si substrate via thermal evaporation of Ti powder in a precise furnace. These authors molded SiO2/TiO2 shell–core nanostructures by monitoring the preparation surroundings such as the time of reaction and the substrate position.179 On a Si substrate, a two-steps thermal evaporation technique was used to grow TiO2 nanowires. By a radio frequency coil, Ti powder was heated inside a quartz reactor under mixed gases composed of Ar-diluted O2 atmosphere.177–180
 |
| | Fig. 15 SEM images of the TiO2 nanowire arrays prepared by the PVD method.176 | |
4. Applications of TiO2 nanostructures
TiO2 nanostructures have several advantages making them as the most important existing resources with promising usage in the deferent areas. These unique features of TiO2 include electronic and short diffusion path. Generally, TiO2 nanomaterials are wide band gap semiconductors containing Eg-values ranging from 3.0 to 3.2 eV with high absorption in the UV region. Titanium dioxide (nanomaterial) is used an important material in high-tech applications. It is the most bio-compatible material. Its optical and biologically properties are responsible for UV protection applications.181–184 As implant materials, TiO2 nanostructures are successfully used for dental, orthopedic and osteosynthesis applications.185 TiO2 suitable for optical applications.70 Besides, photochromism has also been observed in single crystal iron doped rutile.186 Today, the largest practical research on TiO2 is its use for photo assisted organic molecules degradation.187 The applications utilizing this process are purification of waste water, disinfection and self-cleaning coatings in urban areas buildings. The following sub-sections describe different applications of nano TiO2 structures.
4.1 Photocatalytic applications
Various semiconductor materials such as WO3, ZrO2, BiVO4, Fe2O3 and NaTaO3 have been developed and showed photocatalytic activities over the past few periods. Among of these, TiO2 is the most photo-catalytically active material used for water splitting as well as decomposition of organic materials.65,188–190 Hydrogen is environmental friendly and clean and, hence, it is considered as the future fuel. Numerous studies have been developed to produce hydrogen by splitting water. First of all in 1972, Fujishima and Honda4 reported the photocatalytic splitting of water in the occurrence of a TiO2 catalyst. These materials have also been used to kill bacteria in water treatment191 and tumor cells in cancer treatment.14,192 Some reviews are available on the photocatalytic mechanisms.11,16,24,33 By the absorption of photon hv1, the photocatalytic mechanism is initiated manufacturing an electron–hole pair on the surface of TiO2 nanoparticle. An electron is encouraged to the conduction band (CB) with formation of a positive hole with the valence band (VB). One or more radicals or intermediate species involved in this photodecomposition process play a crucial role in the photocatalytic mechanisms of reaction. The different factors play significant roles in controlled photocatalytic activities of semiconductors. With constant adsorbents density on the surface, the semiconductors having large surface area lead to faster reactions on the surface.193
The size of titanium dioxide particles is much correlated to the fraction of surface located atoms. Due to the decrease in particles dimensions, the atomic fraction enhancing the catalytic activities on the surface increases. The band gap energy depends on the size of nanoparticle. It increases with decreasing nanoparticle sizes, which can be exploited in the optimization of redox potential. The electrons in transmission band and the holes in valence band allow photoredox reactions easily. For high photocatalytic activity, the band gap permits only UV light to be proficiently used. Kormann and colleagues112 investigated the decomposition of 2-propanol using different sizes of TiO2 nanoparticles. The authors observed that photocatalytic activities of 7.0 nm particles were 1.6 times better than 15–30 nm size.
Wang et al.194 studied the decomposition of chloroform by TiO2 nanoparticles. They prepared TiO2 nanoparticles with different size particles and observed the best size of TiO2 nanoparticles for chloroform photocatalytic decomposition. By decreasing particle size from 21 to 11 nm, an enhancement in activity was observed. Contrarily, by reducing to 6 nm, the activity decreased completely. The authors concluded that the optimal particle size was about 10 nm for this reaction. In different types of TiO2 nanomaterials, TiO2 nanorods, mesoporous TiO2 and nanotubes have revealed high photocatalytic performances under the appropriate circumstances.195–197 Rhodamine B was oxidized by mesoporous TiO2 by Peng et al.196 On the oxidation reaction of desired materials, TiO2 mesoporous showed significant activity. The nanotubes of TiO2 treated with H2SO4 solutions were prepared by Yang et al.197 The authors observed that the TiO2 nanotubes treated with different concentration of H2SO4 solutions showed photocatalytic activities for degradation of acid orange II. It was due to small particles having high specific surface areas of TiO2 nanotubes after treated with H2SO4 solution.
To improve the photocatalytic activities under UV irradiation, metal doping TiO2 nanomaterials have been primarily studied. In current years, wide research work has attentive on visible light tempted photocatalysis by metal doped semiconductor for improved photocatalytic act on the various organic pollutants degradation.196–202 As potential dopants, several metal ions including iron203–205 nickel206,207 vanadium208 chromium209 platinum210 ruthenium and cobalt ions211 have been investigated. There are contradictory consequences on the effects of doping on the visible-light photoactivities of titanium dioxide. Non-metals such as N, C and S (as an impurity) have also been studied for their visible light photocatalytic activities.212–221 These studies informed that the non-metallic insertion enhanced photocatalytic actions as related to those for pure TiO2 nanomaterials; particularly in the visible light region.213 To obtain a charge balance, there are several procedures if an anion is substituted with a high valence anion. In TiO2 lattice, nitrogen ions substitute oxygen atoms. Titanium dioxide nanotubes photocatalysts have been successfully prepared by Tokudome et al.219 The author investigated breakdown of gaseous isopropanol into carbon dioxide and acetone by N-doped TiO2 nanotubes. They observed that N-doped TiO2 showed high photocatalytic oxidation activity when illuminated with visible light. Other points related to investigated photocatalytic activities of TiO2 doped sulfur(s) have also been reported.222 The results exhibited that in region of visible light S doped TiO2 showed a high photocatalytic activity but lower in UV region. Recently, carbon has received significant interest as a nonmetallic dopant in TiO2 materials. In visible light region, the decompositions of methylene blue and isopropanal have been demonstrated by C-doped TiO2.216 Using C-doped TiO2 nanoparticles, Shen and colleagues223 reported the degradation of trichloroacetic acid under visible light. They observed that C-doped titanium dioxide exhibited a remarkable photocatalytic activity for the reaction involved in degradation. Flouride doped TiO2 was prepared by Yu et al.224 The obtained F-TiO2 displayed higher photocatalytic activity than undoped TiO2 under proper preparation conditions.224 For acetaldehyde and trichloroethylene decomposition, other comprehensive studies provided the investigated of N/F-doped TiO2 nanomaterials. It was reported that TiO2 nanomaterials (N/F-doped) had high visible light photocatalytic activities for decomposition reactions.225,226 The different dopants moieties metal and nonmetal doping titanium dioxide and preparation procedures are given in Table 2. On the other hand, this table comprises splitting of water by photochemical cell reaction over numerous photo catalysts (Table 3).
Table 2 The different dopants moieties metal and nonmetal-doping titanium dioxide and preparation methods
| Kind of dopants |
Doped elements |
Preparation methods |
Potential applications |
Ref. |
| Metal dopants |
Ag |
Silver nitrate was mixed with reduction agent (sodium citrate tribasic dihydrate) and the reaction temperature was raised to 80 °C with continuous stirring. Then TIP and HNO3 were added and the reaction was maintained at 50 °C for 24 h. The prepared sol was dried at 105 °C for 24 h and calcined at 300 °C |
Degradation of nitrophenol in aqueous phase |
316 |
| Fe |
The reactive magnetron sputtering method: 99.99% titanium target and 99.9% iron pieces were placed in the reaction chamber and mixture of argon and oxygen was introduced into the chamber during discharging |
Wastewater decoloring |
317 |
| V |
Sol–gel method: solution 1 (vanadyl acetylacetonate dissolved in n-butanol) was mixed with solution 2 (acetic acid in titanium butoxide) and hydrolyzed (24 h) by the water generated via the esterification of acetic and butanol. The suspension as dried at 150 °C, pulverized and calcined at 400 °C for 0, 5 h |
Wastewater decoloring |
318 |
| Au |
Titanium(IV) butoxide dissolved in absolute ethanol was added to solution containing tetrachloroauric acid (HAuCl4·4H2O), acetic acid and ethanol. The resulting suspension was aged (2 days), dried under vacuum, grinding and calcinated at 650 °C |
Wastewater decoloring |
319 |
| Pt |
Photoreduction process: TiO2 was suspended in a mixture of hexachloroplatinic acid in methanol. The suspension was irradiated with a 125 W mercury lamp (60 min). Pt–TiO2 was separated by filtration, washed with distilled water and dried at 100 °C for 24 h |
Wastewater decoloring |
320 |
| Nonmetal dopants |
N |
Titanium nitride (TiN) oxidation: Heating of TiN at 450–550 °C for 2 h in air (heating and cooling temperature rate: 2 °C min−1) |
Photooxidation of aromatic compounds (e.g. toluene) |
321 |
| Treating anatase TiO2 powder ST01 in the NH3 (67%)/Ar atmosphere at 600 °C for 3 h |
Photooxidation of acetaldehyde in gas phase |
322 |
| S |
Oxidation annealing of titanium disulfide (TiS2) at 300–600 °C |
Wastewater decoloring |
323 |
| N,S |
Hydrolysis of Ti(SO4)2 in NH3 aqueous solution. Precipitate was centrifuged, washed with distilled water and alcohol. Obtained gels were dried under vacuum at 80 for 10 h and were ground to obtain xerogel. The xerogel was calcinated at 400–800 °C in air for 3 h |
Photooxidation of volatile compounds in gas phase (e.g. acetone and formaldehyde) |
324 |
| C |
Sol–gel method: TBOT was hydrolyzed in the presence of ethanol, water and nitric acid; precipitated titanium hydroxide was dried at 110 °C and calcinated in air at 150–200 °C |
Photooxidation of phenol compounds in aqueous phase |
325 |
| Acid-catalyzed sol–gel process. Alkoxidide precursor was dissolved in corresponding alcohol, mixed with hydrochloric acid aqueous solution. Obtained gel was aged for several days and calcinated in air (3 h at 65 °C and 3 h at 250 °C) and grounded |
Photooxidation of phenol compounds in aqueous phase |
326 |
| B |
Anatase TiO2 powder (ST01) was grinding with boric acid triethyl ester and calcinated in air at 450 °C |
Improved photocatalytical activity |
327 |
| P |
Sol–gel method: TIP was hydrolyzed in the presence of isopropanol and water, after hydrolysis phosphoric acid was added. Dispersion was stirred for 2 h, centrifuged at 3500 rpm and dried at 100 °C. Obtained powder was calcinated at 300 °C |
Photooxidation of phenol compounds in aqueous phase |
328 |
Table 3 Water splitting by photochemical–cell reaction over various photocatalysts
| Photocatalysts |
Weights |
Reaction solutions |
Light sources |
Rate of evolutions (μmol h−1) |
Ref. |
| O2 |
H2 |
| Pt/TiO2 |
0.3 g |
2.17 M Na2CO3 |
400 W Hg lamp |
568 |
287 |
329 |
| ZrO2 |
1 g |
Distilled water |
400 W Hg lamp |
72 |
36 |
330 |
| ZrO2 |
1 g |
1.09 M Na2CO3 |
400 W Hg lamp |
142 |
75 |
330 |
| Pt/ZrO2 |
1 g |
0.94 M NaHCO3 |
400 W Hg lamp |
120 |
61 |
330 |
| Ru2O/ZrO2 |
1 g |
Distilled water |
400 W Hg lamp |
11 |
5 |
330 |
| Cu/ZrO2 |
1 g |
Distilled water |
400 W Hg lamp |
14 |
6 |
330 |
| NiO/Sr2Nb2O7 |
1 g |
Distilled water |
400 W Hg lamp |
110 |
36 |
331 |
| NiO/Sr2Ta2O7 |
1 g |
Distilled water |
400 W Hg lamp |
1000 |
480 |
331 |
| (Tetra)BaTa2O6 |
1 g |
Distilled water |
400 W Hg lamp |
21 |
10 |
332 |
| (Ortho)BaTa2O6 |
1 g |
Distilled water |
400 W Hg lamp |
33 |
15 |
332 |
| (Ortho)BaTa2O6 |
1 g |
0.0005 M Ba(OH)2 |
400 W Hg lamp |
126 |
59 |
332 |
| (Ortho)BaTa2O6 |
1 g |
0.001 M KOH |
400 W Hg lamp |
24 |
11 |
332 |
| (Ortho)BaTa2O6 |
1 g |
0.0005 M BaCl2 |
400 W Hg lamp |
15 |
6 |
332 |
| NiO/BaTa2O6 |
1 g |
Distilled water |
400 W Hg lamp |
629 |
303 |
332 |
| Ni/Rb4Nb6O17 |
1 g |
Distilled water |
400 W Hg lamp |
936 |
451 |
333 |
| Ni/K4Nb6O17 |
1 g |
Distilled water |
400 W Hg lamp |
403 |
197 |
333 |
| Pt/TiO2, TiO2 |
12 mg |
2 M KBr, 6.5 mM FeCl2 |
500 W Hg |
2.8 |
1.3 |
334 |
| Pt–TaON, Pt–WO3 |
0.2 g |
5 mM NaI |
300 W Xe lamp with filters: λ > 420 nm |
24 |
12 |
335 |
| Pt/BaTaO2N, Pt/WO3 |
0.1 g |
5 mM NaI |
300 W Xe lamp with filters: λ > 420 nm |
6.6 |
3.1 |
336 |
| Pt/SrTiO3:Rh, BiVO4 |
0.1 g |
2 mM FeCl3 |
300 W Xe with filter: λ > 420 nm |
15 |
7.2 |
337 |
| Pt/SrTiO3:Rh, Bi2MoO6 |
0.1 g |
2 mM FeCl3 |
300 W Xe with filter: λ > 420 nm |
19 |
8.9 |
235 |
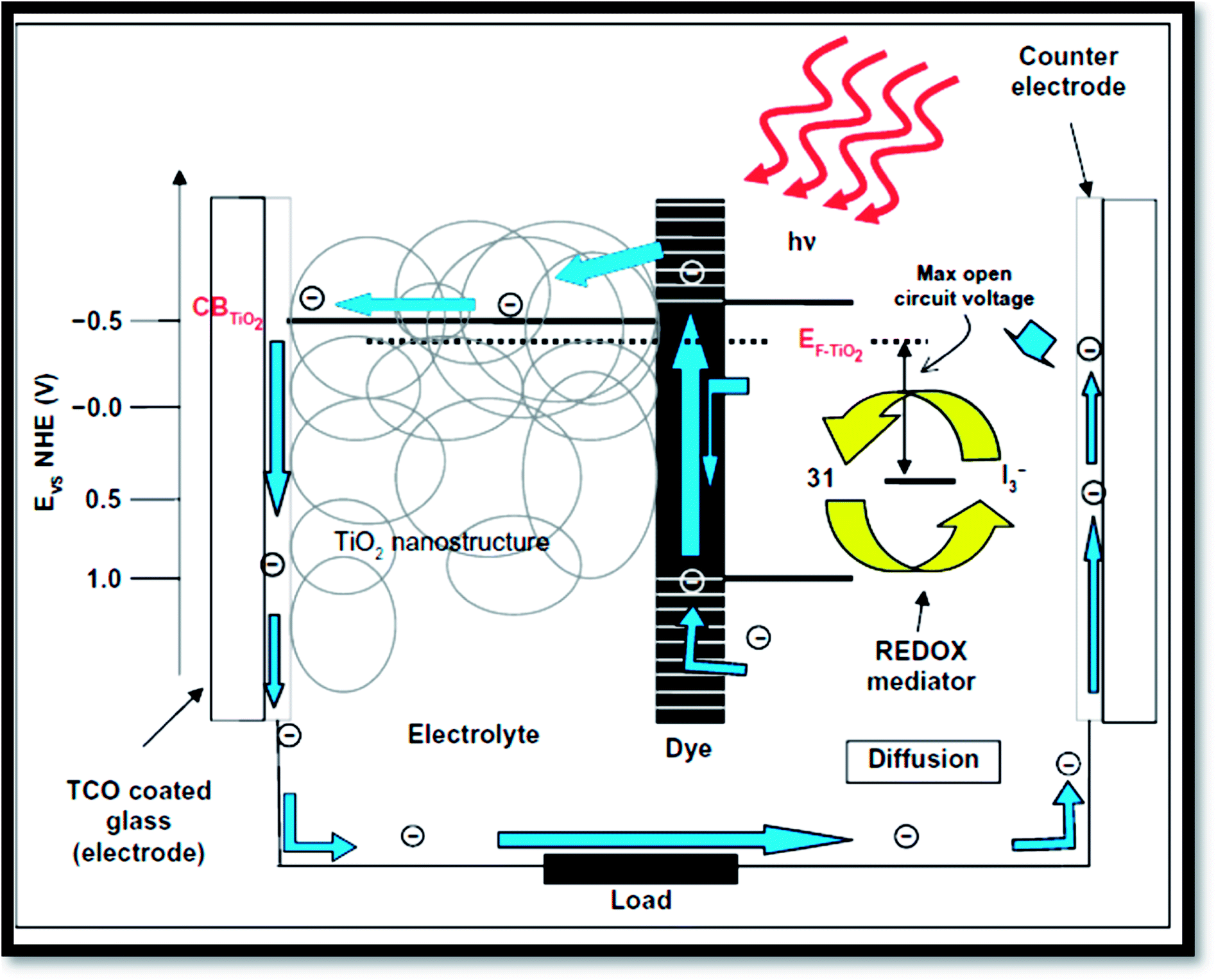
4.2 Photovoltaic applications
Fig. 16 exhibits the principle of a dye sensitized solar cells (DSSC).41 Dye sensitized solar cells have involved much consideration as reformative low priced substitutes to usual solid state devices.227 A DSSC composed a nanoporous film prepared from mesoporous oxide layer of anatase TiO2. Firstly, through a transparent conducting oxide (TCO) electrode, cell is illuminated where TiO2 is deposited. Into the metal oxide conduction band, photo excited dye molecules insert electrons. I−/IO3− couple acts as redox species in the electrolyte reducing the oxidized dye molecules back to their inventive state. Overall, the device produces electric power from light.4,32 The surface of nanoporous TiO2 film is bounded by more cations than the electrons. Henceforth, electron transport mechanism is measured to be purely diffusive.228,229 From solid-state p–n junction, the process of absorption of light and the transport in the case of DSSCs are different due to distinct phenomenon. Through eqn (3) to (6), the process is described:| | |
Sensitizer (S) + hv → S*(excited)
| (3) |
| | |
S* + TiO2 → e−(TiO2) + oxidized sensitizer+
| (4) |
| | |
Oxidized sensitizer+ + 3/2 I− → S + 1/2 I3−
| (5) |
| | |
1/2 I3− + e−(counter electrode) → 3/2 I−
| (6) |
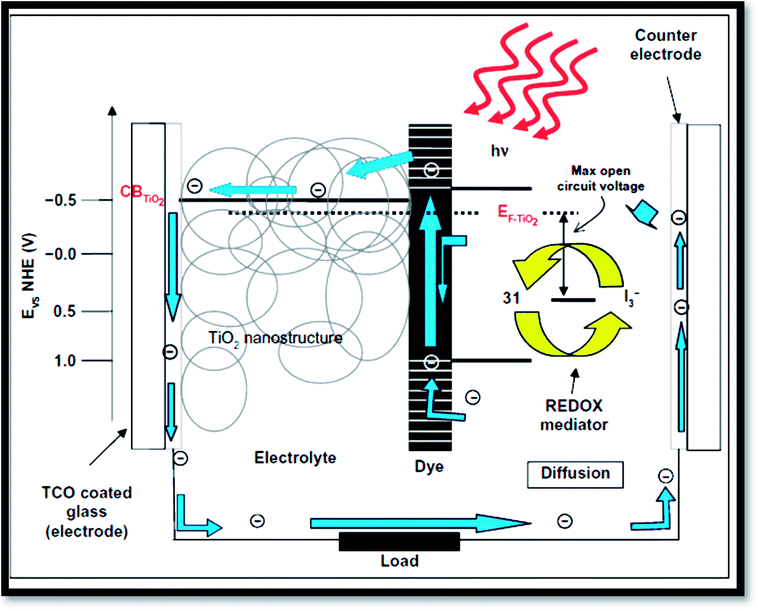 |
| | Fig. 16 Operation principles and energy levels of nanocrystalline dye-sensitized solar cell.41 | |
From the equations, it is clear that photosensitizer (S) adsorbs light on the exterior of the semiconductor TiO2 (eqn (3)). Then the photo-excited dye (S*) hand over electrons into titanium dioxide (eqn (4)). After that the reduced species found in the electrolyte (I−) recovers electrons losing in the sensitizer (eqn (5)). Finally, by the mediator (I3−) that gains electrons (eqn (6)), the circuit is closed.
4.3 Photocatalytic water splitting
Photo-electrochemical and photo-catalytic water splitting to produce hydrogen are considered as potential procedures to achieve hydrogen. These are discussed below.
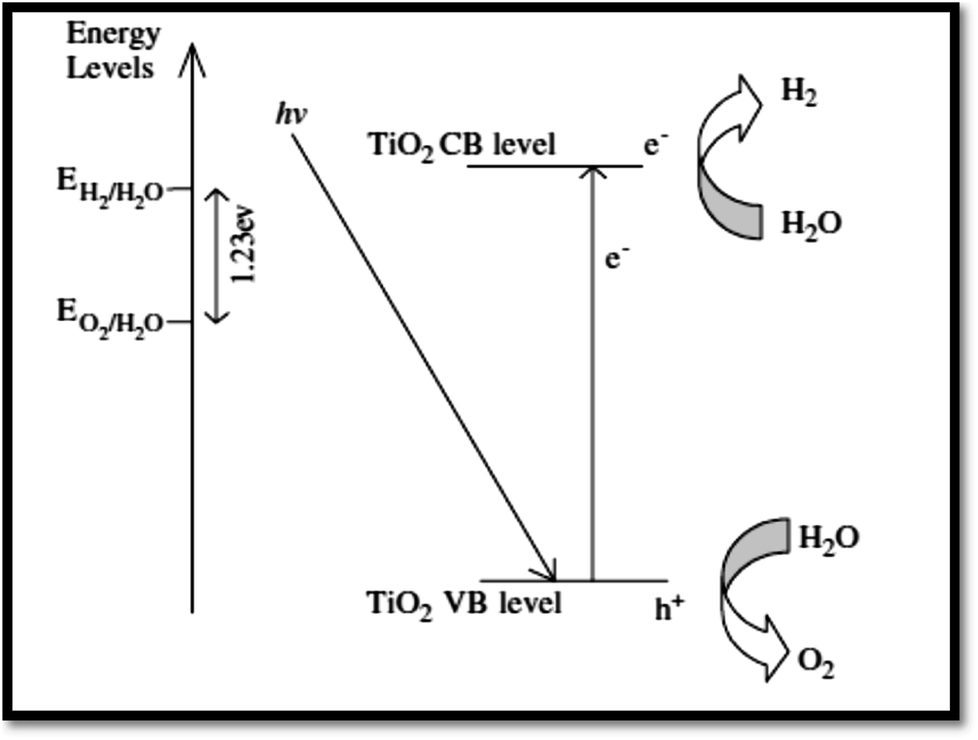
4.3.1 Fundamentals of photocatalytic water splitting. At semiconductor electrodes, breakdown of water is the main aim of lively research in current years. First, on a TiO2 electrode, photocatalytic splitting of water was reported by Fujishima and Honda4 in 1972. For green and sustainable energy sources,230 photocatalytic water splitting into H2 and O2 using TiO2 nanomaterials remains to be challenge. To drive chemical reactions, the generated electron and hole can be used. Water molecules for overall water splitting231–233 are oxidized as well as reduced. The oxidation of water takes place by the holes to form O2. While the reduction of water takes place by the electrons to form H2. Of course, there are several features of TiO2 but under solar energy, its photocatalytic water splitting efficacy is still quite low. Consequently, to activate the photocatalyst, only UV light can be utilized. It enhances its visible light response including upgraded bulk properties of the material. Water splitting mechanism on TiO2 for hydrogen production is shown in Fig. 17.
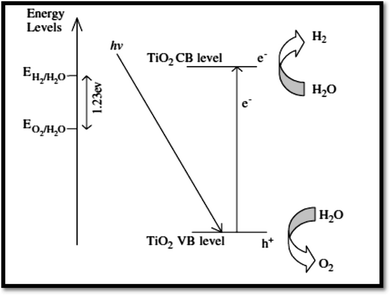 |
| | Fig. 17 Mechanism of TiO2 photocatalytic water-splitting for hydrogen production.230 | |
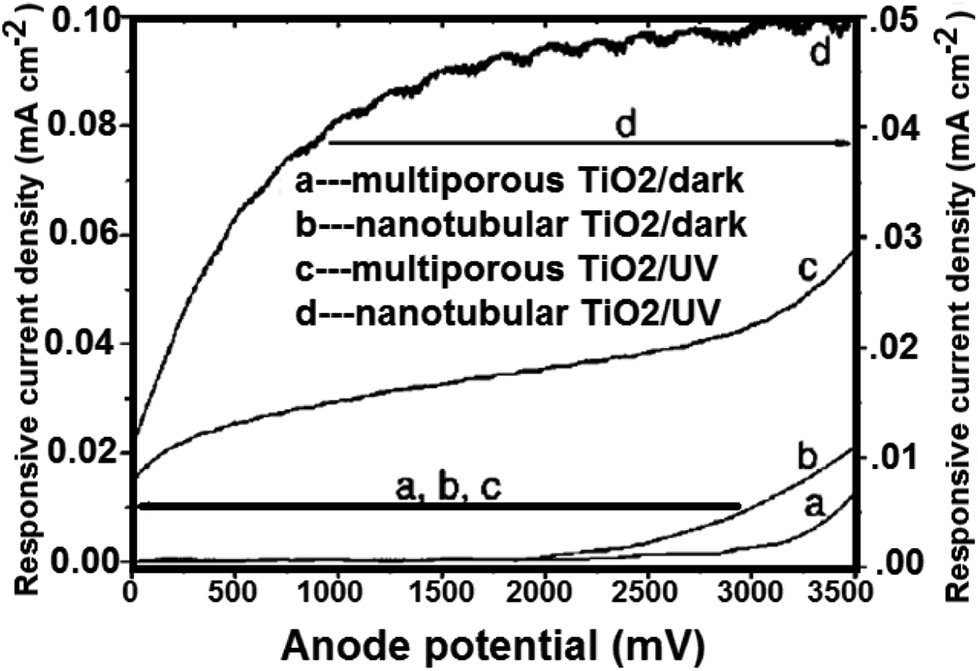
4.3.2 Water splitting on TiO2 nanotube arrays. TiO2 nanotubes photocatalysts was investigated for water splitting under UV illumination by Shankar et al.143 By potentiostatic anodization of titanium foil at 10 V in an electrolyte consist of (0.5% hydrofluoric acid, CH3COOH mixed in a 7:1 ratio),143 the TiO2 nanotubes were prepared. Besides, four different electrolyte bath temperatures i.e. 5 °C, 25 °C, 35 °C, and 50 °C were tried to obtain the variation in the nanotubes length and thickness. TiO2 nanotubes having pore diameter of 22 nm and wall thickness of 34 nm (ref. 156) were used to generate hydrogen gas. The total adaptation efficiency was 6.8%.156 At 320–400 nm (98 mW cm−2), the authors also reported that for illumination. The nanotube array of TiO2 photoanodes may generate hydrogen from water with 12.25% photoconversion efficiency. On titanium foil; nanotube arrays with 570 nm in length and 70 nm in pore size; were prepared in 0.15 M hydrofluoric acid–0.5 M nitric acid by Xie et al.217 Fig. 18 shows the comparison between the photoelectrochemical current of annealed TiO2 nanotube array films and that of porous TiO2. To higher surface area, the nanotube array samples with higher electrochemical current density were also ascribed.
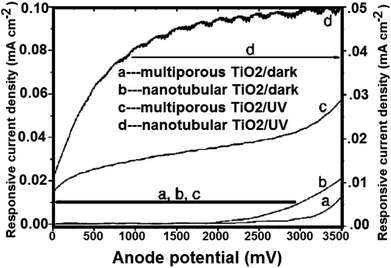 |
| | Fig. 18 Measured current of crystallized porous and nanotubular TiO2/Ti electrodes under linearly swept potential from 0 to 3500 mV vs. SEC.217 | |
4.3.3 Water splitting on doped TiO2. Splitting of water has also been studied on TiO2 obtained after doping with metal–non metals. Exactly doping is the incorporation of foreign elements into TiO2 crystal lattice. The aim of doping is to enhance photocatalytic activities.234 With the potential for H2 evolution, certainly the band gaps of titanium dioxide semiconductor photocatalysts become extensive.In a photoelectrochemical cell to oxidation and reduction of water to H2 and O2, Wrighton and colleagues235 investigated the use of an n-type semiconductor SrTiO3 electrode. Without any external bias, decomposition of H2O can be driven photochemically. Jin and co-workers236 reported that TiO2 doped with platinum/boron was a good system for splitting of water. Besides, Khan and colleagues237 described the response for splitting of water on TiO2 doped with carbon. The total conversion efficiency was 11%, which had a high water splitting performance. The authors noted that at 40 mW cm−2 illumination, an extreme photo-conversion efficiency of 8.35% could be obtained. The synthesis of TiO2 nanocrystalline thin films; with Pt-loaded through use of radio frequency magnetron sputtering method (RF); was described by Matsuoka et al.238 They investigated that visible light was responsive for water decomposed in the occurrence of TiO2 nanocrystalline.239 Bocharov et al.240 estimated theoretically the photocatalytic fitness of tinniest single wall fluorite structured titania (4,4) nanotube (NT) possessing three layers each (O–Ti–O) and doped by V, Ni, Mn, Sc, Cr, Fe, Co, Zn and Cu atoms substituted for host Ti atoms. They had associated electronic structure of ground state. For splitting of water, the results were in favor of Sc-doped titania nanotubes only for a potential photocatalytic usage. Using the sol–gel technique, Guayaquil et al.241 synthesized a series of mesoporous TiO2 (meso-TiO2). At 500° and 550°, they were calcined following synthesis. For H generation, the mesoporous 2.50 wt% Pt–TiO2 had the maximum photoactivity that was displayed by the prepared semiconductors. Alitabar et al.242 increased the photocatalytic activity of TiO2 nanotube arrays used as a photoanode in water splitting by doping it with sodium and carbon.
4.3.4 Water splitting on dye sensitized TiO2. To increase the photocatalytic reaction efficiency, one of the ways is to couple TiO2 semiconductor nanomaterials to an organic dye to employ visible light for energy change. Some dyes with visible light sensitivities can be utilized in photocatalytic systems and solar cells.243–245 Duonghong and co-workers246 studied the effect of using Ru(bipy)32+ as a dye sensitizer under visible light radiation for hydrogen gas production. They reported water decomposition by using [Ru(bipy)3]2+ and rhodamine B under visible light. Besides, the effect of utilizing [Ru(dcpy)2(dpq)]2+ as a dye sensitizer, Alitabar and Yoozbashizadeh242 carried out a parametric investigation on photocatalytic splitting of water for hydrogen gas manufacture under visible light irradiation. Hydrogen production was increased when dye molecules were adsorbed on to TiO2. With the help of structure directing surfactant, Sreethawong et al.247 prepared mesoporous-assembled TiO2/Pt. Furthermore, the nitrogen doped TiO2/Pt (N–TiO2) was prepared via calcination of the hydrolysis product of Ti(SO4)2 with aqueous ammonia followed by platinum loading by Li et al.248 N–TiO2/Pt was sensitized and used for hydrogen production in occurrence of triethanolamine using anhydrous EtOH solution of eosin Y. Under comparable experimental conditions, it was observed that eosin Y–N–TiO2/Pt exhibited advanced photocatalytic activity than eosin Y–TiO2/Pt.
4.3.5 Water splitting on modified TiO2. Many scientists have modified TiO2 by different compounds to augment the process of water splitting. Over the surface of TiO2, when silver oxide nano clusters were impregnated, Mandari et al.249 synthesized plasmonic Ag2O/TiO2 photocatalysts. Zhang et al.250 synthesized (g-C3N4) graphitic carbon nitride nanosheets/titanium dioxide (TiO2) nanoparticles hetero-structure. The prepared g-C3N4/TiO2 hetero-structured composites showed excellent photocatalytic hydrogen generation. Besides, on the g-C3N4 nanosheets the TiO2 nanoparticles were well dispersed. The as obtained g-C3N4 coupled with TiO2 not only increased the surface area of g-C3N4, but also promoted the separation of photo-generated charge carriers. Hou et al.251 used two typed gold films (i) coated with TiO2 (ii) and uncoated with TiO2. After that, they reported measurements of photocatalytic water splitting. For hydrogen gas production with Pt and CoOx as dual cocatalysts, Zhang et al.252 reported a new CoOx/TiO2/Pt photocatalyst. This type of catalyst was prepared by template assisted atomic layer deposition (ALD). Under UV light irradiation, the generated electrons and holes transfer to the different surfaces of TiO2 nanotubes. Photocatalytic efficiency of CoOx/TiO2/Pt photocatalysts was 275.9 μmol h−1, while that pristine TiO2 nanotubes was 56.5 μmol h−1. Xu et al.253 synthesized novel plate like Co(OH)2 decorated TiO2 nanosheets for photocatalytic hydrogen generation illustrating Co(OH)2 decorated TiO2 samples of high rate of hydrogen generation than native TiO2 sample. Sui et al.254 prepared a Pt/TiO2 catalyst, where Pt was atomically and selectively dispersed on the (1 0 1) facets of nanosized TiO2 single crystals. For H2 production, a remarkably higher photocatalytic activity was compared to frequently used Pt/TiO2. Si et al.255 synthesized anatase TiO2, thin TiO2 (B) nano-sheets and their composites. The properly aligned structure of band, with the conduction band level of ultra-thin TiO2 (B) was about 0.6 eV higher than that of the anatase. Wang et al.256 reported that very small amount of fluorine inhibited hydrogen and oxygen recombination. The detailed study showed that inhibition was by occupation of hydrogen and oxygen sorption and activation sites by fluorine ion over platinum. Liu et al.257 fabricated a composite photoanode having TiO2 nanotube arrays altered with polyoxometalate (POM) and Co9S8.
5. Mechanism of water splitting
On surface modification of TiO2, a number of efforts have been made to expand the photocatalytic activities as simple change can eagerly adjust the mechanisms and accelerates the kinetics of photocatalysis.258,259 In the UV regime, the band gap for TiO2 lies. Besides, the latter has lower photocatalytic activities due to the lower ECB of ritual by ∼0.2 eV. For modification of TiO2 nanomaterials, the goals are to improve their optical activities.213,214 There are several methods to recover the performance of TiO2 nanomaterials. In visible light, other colorful compounds sensitizing TiO2 might be improved optical activity. Second, doping with other elements can also change the optical properties of TiO2 nanomaterials. At the atomic level, Hussain et al.260 explored the interfacial structure between a rutile TiO2(110) surface pre-characterized and liquid water. The mechanism of splitting of water occurring in the occurrence of semiconductor photocatalyst was explained by Salvador in 1–4 equations as follows.261
| Semiconductor + 2hν → 2h+ + 2e− |
EtOH serves as a sacrificial reagent which also helps to increase the entire process efficiency.64 Water splitting mechanism on Ag/TiO2 was explained by Liu et al.262
According to Salvador et al.261 during the process of water splitting, formation of H+ and OH− can take place through the reaction of H2O molecules with holes in TiO2 valence band. The consumption of ·OH can occur through chemical reaction with CH3CH2OH. With enhanced energy, the electrons react with H+ at the surface of Ag NPs where H+ can take an electron through SPR (surface plasmon resonance) to generate an H atom, and then 2H atoms associate with each other for the formation of an H2 molecule.
| eAg− + hν(visible) → eSPR− |
| CH3CH2OH + ·OH → CH3COOH + H+ |
With an I−/IO3− (iodide/iodate) redox-mediator system, water splitting mechanism on TiO2 was explained by Nishijima et al.263 Besides, they studied the photoassisted oxidation of water continued with impartially high efficiency when Fe3+ ions were used as electron acceptors. This whole process of splitting of water on doped titanium dioxide; containing a rutile phase; was compared to that on doped titanium dioxide with an anatase phase. After all, they proposed the mechanism of water splitting in following equations:
| C2H5OH + 3H2O + 6h+ → 2CO2 + 6H+ |
| IO3− + 3H2O + 6e− → I− + 6OH− |
| I− + 6OH− + 6h+ → IO3− + 3H2O |
Mechanism for water splitting on TiO2 photosensitized by glutathione capped metal nanoclusters was explained by Chen and Kamat.264 In aqueous buffer solution (pH = 7), a mesoscopic TiO2 film photosensitized by glutathione capped metal nanoclusters (Aux-GSH NCs) was utilized as the photoanode with platinum counter electrode. After all, they also proposed the water splitting mechanism as follows:
| Aux − GSH*⋯TiO2 → Aux − GSH+⋯TiO2(e) |
| TiO2(e) + Pt → TiO2 + Pt(e) |
| Aux − GSH+ + 1/2H2O → Aux − GSH + 1/4O2 + H+ |
| Aux − GSH+ + EDTA → Aux − GSH + oxidized EDTA |
6. Lithium batteries
As the most promising energy storage technologies, lithium ion batteries (LIBs) are measured as the best for renewable energy, electric vehicles and mobile electronics. In recent years, as a possible anode, TiO2 (B) has received growing interest for Li ion batteries. As compared to commercialized Li4Ti5O12, it offers higher energy storage. From the micron to the nanoscale, it is better than rutile and anatase. The important factors playing an important role and greatly affect the lithium ion batteries (LIB) performance are particle size and crystallographic alignment of the nanostructured.265–267 The crystallographic orientations [such as TiO2(B)] and the theoretical studies268,269 confirmed that lithium ion mobility favors the direction in the order of b > c ≫ a-axis channel.270
7. Gas sensors
The other application of TiO2 nanocrystalline is as gas sensor because gas adsorption causes a change in electrical conductivity like ZnO semiconductors.155,270–272 Thus, TiO2 is usually used as an oxygen gas instrument, e.g. to gauge the burning process of fuel in car engines to control fuel environmental pollution and consumption. Grimes et al.155 considered the use of titanium dioxide as sensor.
8. White pigments
The pigment being the most abundant component in the coating affects the properties of the coating materials. Because of its highest refractive index of stability, colorless, and relatively low and uniform absorption of visible light, titanium dioxide has newly become a core white pigment source of scattering of light. By two procedures i.e. hydrothermal and hydrolysis, Samya El-Sherbiny et al.273 created anatase and rutile. In paper coating, the usage of the prepared nano-pigments disclosed that a slight amount of titanium dioxide was satisfactory to attain important improvements in brightness and opacity. It is due to ability of scattering light.
9. TiO2-based nanocomposites as catalysts
TiO2 photo catalysts have been widely studied for the uptake of organic contaminants in water and air.274 The present article also concentrated on hybrid nanocrystals based on TiO2, describing different examples of synthetic methods and deliberating their applications in water treatment. Some papers are reported in the area of hybrid nanocrystals preparation. The papers on the photocatalytic features of nanocatalysts indicated their huge measure applications as challenges. However, these nanomaterials hold good assurance for the dilapidation of organic and inorganic contaminants in water or gas phase. The main features of nano sized TiO2 materials are enormously high surface to volume ratio and turning into a great density of catalytically surface sites.275 The another fact is the size reliant band gap of nano sized semiconductors. It is likely to finely adjust the redox potentials of photogenerated electron hole couples to selectively control photochemical reactions. Besides, the catalyst surface is occupied easily by charges photogenerated in nano-catalysts.276 The hybrid nanocrystals formed by two or more components hold great promise for the development of multifunctional nanocatalysts among the embarrassment of methods proposed in the literature so far.277–281 Indeed, the opportunity is offered by hybrid nanocrystals to merge in one material resulting in countless possible combinations.282–285 Under visible light irradiation, spatial separation of e−/h+ could also be provide by the photoactivity hybrid nanocrystals. Thus, improving is an occasion to magnetically recuperate the photocatalysts or stimulate biocidal utility even in the dark. The surface properties, the particle size of the catalysts, their morphology, the composition, organization of the metal and TiO2 are the factors, which also affect the photocatalytic efficiency.286 Therefore, for the preparation of metal nanoparticle hybrid hetero-structures, numerous synthetic methods have been described, which include impregnation,287 UV irradiation,288 electrodeposition,289 sonochemistry,290 hydrothermal,291 sol–gel,292 and flame-spray synthesis.293 Wenqing et al.294 prepared nanocomposites of titanium dioxide (P25) and reduced graphene oxide (RGO) utilizing numerous methods such as hydrazine reduction, UV-assisted photocatalytic reduction and hydrothermal. Those nanocomposites were studied as photocatalysts for the progress of hydrogen from alcohol solution under UV-vis irradiation. It was found that the assimilation of RGO into P25 considerably increased the photocatalytic activity for H2 evolution, and the P25–RGO composite synthesized by the hydrothermal method showed the excellent presentation. Deepa et al.295 prepared Au/TiO2 nanocomposites for photocatalytic hydrogen generation in the company of a sacrificial electron donor like ethanol or methanol under UV-visible and visible light irradiation. These nanocomposites exhibited excellent photocatalytic activity for hydrogen generation under UV-visible conditions. Amount of hydrogen evolved using Au/TiO2 nanocomposites under dark condition was zero. Anna and Jerzy296 studied the hydrogen generation by water splitting on Pt–TiO2 catalyst. Besides, they also examine the influence of numerous sacrificial chemicals like MeOH, Na2S, and ethylene diamine tetraacetic acid (EDTA), I− and IO3− ions on the photocatalytic efficacy in water splitting reactions in ultraviolet (UV) illumination. Photocatalytic water splitting was achieved at EDTA and Na2S utilization as the sacrificial reagents. Yatskiv et al.297 reported that the photocatalytic systems having mesoporous TiO2 and Pd/SiO2, produced extra quantities of molecular hydrogen at keeping in dark after the ending of irradiation. Václav and Daniela298 prepared TiO2/ZnS/CdS composites by homogeneous hydrolysis of aqueous solutions mixture of TiO2SO4, ZnSO4, and CdSO4 with thioacetamide for photocatalytic hydrogen production from water. Hydrogen generation was seen in the company of Pd and Pt nanoparticles deposited on TiO2/ZnS/CdS composites. The excellent photocatalytic activity for H2 evolution indicated with TiZnCd7 on surface deposited with palladium, which had 78.5% ZnS, 20.21% TiO2, and 1.29% CdS·TiO2/ZnS/CdS. Quanjun et al.299 reported new composite material having of TiO2 nanocrystals via two steps hydrothermal process utilizing thiourea, sodium molybdate and graphene oxide as precursors with tetrabutylorthotitanate and MoS2/graphene hybrid as titanium precursors. TiO2/MoS2/graphene composite attained high H2 generation rate of 165.3 μmol h−1 without a noble metal co-catalyst. Anna et al.300 interpreted the development of the quantum yield of H2 generation using TiO2/Ag0 to TiO2/Ni0 to TiO2/Cu0 in terms of alterations in the electronic interface between the semiconductor surface and metal nanoparticles. They observed a controlled metal amount range for maximum quantum yield of hydrogen generation. Ke et al.301 examined Pt-loaded nanocomposites, pristine MWNTs and TiO2 for their photocatalytic activities for water splitting with triethanolamine as an electron donor. Hydrogen was effectively generated on Pt/MWNT–TiO2 in visible light illumination (λ > 420 nm). Hydrogen production rate up to 8 mmol g−1 h−1 under full spectral irradiation of a Xe-lamp, or more was attained. Takuya et al.302 investigated the photocatalytic hydrogen generation in MeOHn with CuO/TiO2, SnO/TiO2, ZnO/TiO2, CuO/Al2O3/TiO2 and Al2O3/TiO2 nanocomposites. The maximum hydrogen generation was achieved with CuO/Al2O3/TiO2 nanocomposites having 0.3 wt% Al2O3/TiO2/0.2 wt% CuO. Nitish et al.303 demonstrated the higher hydrogen evolution reaction (HER) activity of Pt nanoparticles (Pt NPs) reinforced on titanium dioxide (TiO2) nanocrystals (Pt–TiO2) and nitrogen doped reduced graphene oxide (N-rGO) constructed TiO2 nanocomposite (Pt–TiO2–N-rGO). Lu et al.304 prepared TiO2 photocatalysts with nickel sulfide co-catalyst by stocking nickel sulfide on TiO2 with solvothermal synthesis method. It was reported that NiS was used as a co-catalyst with TiO2 for the photocatalytic generation of H2. High hydrogen generation was achieved with NiS as hexagonal structure with content in the composite of 7 at% in relation to TiO2. The rate of H2 generation was augmented by 30 times than that of TiO2 alone. Fuyun et al.305 prepared nanocomposite of N-doped TiO2 with graphene oxide (NTG) to improve the photocatalytic efficiency. NTG exhibited high photocatalytic efficiency hydrogen evolution. It was of 716.0 or 112.0 μmol h−1 g−1 at high pressure Hg or Xe lamp, which was around 9.2 or 13.6 times higher than P25 photocatalyst.
Recently, gold nanoparticles were reinforced on TiO2–C3N4 for CO oxidation in visible light illumination exploiting hydrothermal method; beginning with titanium glycolate and graphitic C as precursors. TiO2–C3N4 microspheres were prepared and then decorated with gold nanoparticles by letting HAuCl reaction at alkaline pH (pH = 10) in the attendance of sodium carbonate. After 2 h, the dried and washed powder was calcinated at 350 °C.306 Besides, visible light dynamic silver modified titania catalyst was described for the utilization in the decomposition of MeOH, CH3COOH, 2-PrOH and Escherichia coli.307 A plasmonic gold silver alloy on TiO2 photocatalyst was also described that indicated elevated degradation of stearic acid at 490 nm than reported using pristine TiO2.308. Qiu et al.309 have described that CuxO/TiO2 photocatalyst resulted into effective VOCs uptake. Furthermore, Wang et al.310 prepared and used TiO2 NRs/FexOy/Ag core shell nanostructures for photodegradation of rhodamine B in solar light illumination. Recently, a reviews described successful removal of benzene, methylene blue and carbamazepine by photodegradation using CNT/TiO2 nanostructured composites.311
10. Future challenges and perspectives
Recently, the applications of TiO2 nanostructures have been exploited to clean environment and produce hydrogen. But there are certain limitations, which we have to overcome to make the structures applicable in real life problems. The efficiencies of these structures are not excellent; especially for decomposing of persistent organic volatile pollutants and production of hydrogen at large scale. Generally, doped TiO2 nanostructures result into poor photoactivity. Other challenges are to augment spectral sensitivity of these structures to visible and NIR regions and the bio-compatibility of TiO2 nanostructures. Therefore, there is a great need of future research focusing constant photoactivity in the long run. These can be achieved by modifying the synthetic routs. Nonmetal doped TiO2 nanostructures have low photocatalytic activity under visible light UV radiation. Therefore, some materials such as polymers, glass, ceramics, or metals may serve as magical identities in this area for economic and eco-friendly applications.
Future research needs the development of new synthetic procedures and nanostructures with higher surface states. It may be served by non-lithographic complementary metal oxide semiconductor compatible techniques. This technique may be applicable for new doping materials, dopant incorporation into TiO2 nanostructures and applications for environmental and alternate energy areas. Besides, visible to near infra-red activated titanium nanostructures should be designed. TiO2 nanostructures may serve as the ideal materials in biological and medicinal science. Therefore, there is a great need to study the bio-compatibility of these structures at supra molecular level. In a nut shell, the researchers have several challenges to tackle with them in near future. Therefore, there is a great need to improve the structures and properties of these materials. The basic knowledge of chemistry, physics and computer modeling may help to achieve the task.
11. Conclusion
An inclusive review of the syntheses, properties and applications of TiO2 nanostructures is offered. These nanostructures can be prepared by different synthesis procedures as per the requirements. It was observed that the physico-chemical properties of titanium dioxide nanostructures are responsible for wide applications in several fields such as gas sensors, white pigments, lithium batteries, photocatalytic applications (photodegradation of organic compounds), photovoltaic applications and water splitting. TiO2 nanostructures play a great role in water purification by degrading biological and organic pollutants. The use in generating hydrogen as a green currency is the biggest asset of nano TiO2 structures. It was observed that the decomposing and water splitting properties of TiO2 nanostructures are not good enough, which can be exploited at a commercial level economically. It may be predicted that these materials may be the choice in water purification. Most interestingly, we believe that TiO2 nanostructures will achieve a great reputation in generation of hydrogen fuel – a need of the next century.
Conflicts of interest
There is no conflict of interest.
References
- G. Pfaff and P. Reynders, Chem. Rev., 1999, 99, 1963–1982 CrossRef PubMed.
- A. Salvador, M. C. Pascual-Martí, J. R. Adell, A. Requeni and J. G. March, J. Pharm. Biomed. Anal., 2000, 22, 301–306 CrossRef PubMed.
- M. D. Newman, M. Stotland and J. I. Ellis, J. Am. Acad. Dermatol., 2009, 61, 685–692 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- T. Kaida, K. Kobayashi, M. Adachi and F. Suzuki, J. Cosmet. Sci., 2004, 55, 219–220 Search PubMed.
- J. J. Wang, B. J. S. Sanderson and H. Wang, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2007, 628, 99–106 CrossRef PubMed.
- R. A. Schwartz, Acta Dermatovenerol. Croat., 2004, 12(4), 249 Search PubMed.
- J. J. Jacobs, A. K. Skipor, J. Black, R. M. Urban and J. O. Galante, J. Bone Jt. Surg., Am. Vol., 1991, 73, 1475–1486 CrossRef.
- Y. Sul, Int. J. Nanomed., 2010, 5, 87–100 CrossRef.
- A. Patri, T. Umbreit, J. Zheng, K. Nagashima, P. Goering, S. Francke-Carroll, E. Gordon, J. Weaver, T. Miller, N. Sadrieh, S. McNeil and M. Stratmeyer, J. Appl. Toxicol., 2009, 29, 662–672 CrossRef PubMed.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef.
- D. Tryk, A. Fujishima and K. Honda, Electrochim. Acta, 2000, 45, 2363–2376 CrossRef.
- M. Gratzel, J. Sol-Gel Sci. Technol., 2001, 22, 7–13 CrossRef.
- S. Ivanković, M. Gotić, M. Jurin and S. Musić, J. Sol-Gel Sci. Technol., 2003, 27, 225–233 CrossRef.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef.
- A. P. Alivisatos, J. Phys. Chem., 1996, 100, 13226–13239 CrossRef.
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49–68 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef PubMed.
- D. M. Adams, L. Brus, C. E. D. Chidsey, S. Creager, C. Creutz, C. R. Kagan, P. V. Kamat, M. Lieberman, S. Lindsay, R. A. Marcus, R. M. Metzger, M. E. Michel-Beyerle, J. R. Miller, M. D. Newton, D. R. Rolison, O. Sankey, K. S. Schanze, J. Yardley and X. Zhu, J. Phys. Chem. B, 2003, 107, 6668–6697 CrossRef.
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Nanopart. Res., 1999, 1, 439–458 CrossRef.
- X. Chen, Y. Lou, S. Dayal, X. Qiu, R. Krolicki, C. Burda, C. Zhao and J. Becker, J. Nanosci. Nanotechnol., 2005, 5, 1408–1420 CrossRef PubMed.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef PubMed.
- M. S. Dresselhaus and G. Dresslhaus, Annu. Rev. Mater. Sci., 1995, 25, 487–523 CrossRef.
- M. S. Dresselhaus, G. Dresselhaus, A. Jorio, A. G. Souza Filho, M. A. Pimenta and R. Saito, Acc. Chem. Res., 2002, 35, 1070–1078 CrossRef PubMed.
- M. S. Dresselhaus, G. Dresselhaus and A. Jorio, Annu. Rev. Mater. Res., 2004, 34, 247–278 CrossRef.
- A. L. Efros and M. Rosen, Annu. Rev. Mater. Sci., 2000, 30, 475–521 CrossRef.
- M. A. El-Sayed, Acc. Chem. Res., 2004, 37, 326–333 CrossRef PubMed.
- M. Gratzel, Prog. Photovoltaics, 2000, 8, 171–185 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef PubMed.
- S. S. Mao, Int. J. Nanotechnol., 2004, 1, 42 CrossRef.
- M. Nirmal and L. Brus, Acc. Chem. Res., 1999, 32, 407–414 CrossRef.
- A. J. Nozik, Inorg. Chem., 2005, 44, 6893–6899 CrossRef PubMed.
- A. J. Nozik, Annu. Rev. Phys. Chem., 2001, 52, 193–231 CrossRef PubMed.
- M. Ouyang, J.-L. Huang and C. M. Lieber, Acc. Chem. Res., 2002, 35, 1018–1025 CrossRef PubMed.
- L. Vayssieres, Int. J. Nanotechnol., 2004, 1(1–2), 1–41 Search PubMed.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- V. E. Henrich, Rep. Prog. Phys., 1985, 48, 1481–1541 CrossRef.
- J. F. Banfield, B. L. Bischoff and M. A. Anderson, Chem. Geol., 1993, 110, 211–231 CrossRef.
- P. Y. Simons and F. Dachille, Acta Crystallogr., 1967, 23, 334–336 CrossRef.
- J. Tang and S. Endo, J. Am. Ceram. Soc., 1993, 76, 796–798 CrossRef.
- J. S. Olsen, L. Gerward and J. Jiang, J. Phys. Chem. Solids, 1999, 60, 229–233 CrossRef.
- M. Latroche, L. Brohan, R. Marchand and M. Tournoux, J. Solid State Chem., 1989, 81, 78–82 CrossRef.
- J. Akimoto, Y. Gotoh, Y. Oosawa, N. Nonose, T. Kumagai, K. Aoki and H. Takei, J. Solid State Chem., 1994, 113, 27–36 CrossRef.
- R. Marchand, L. Brohan and M. Tournoux, Mater. Res. Bull., 1980, 15, 1129–1133 CrossRef.
- J. Haines and J. M. Léger, Phys. B, 1993, 192, 233–237 CrossRef.
- M. Horn, C. F. Schwebdtfeger and E. P. Meagher, Z. Kristallogr. Cryst. Mater., 1972, 136, 273–281 CrossRef.
- W. H. Baur, Acta Crystallogr., 1961, 14, 214–216 CrossRef.
- S.-D. Mo and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef.
- J. Pascual, J. Camassel and H. Mathieu, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 5606–5614 CrossRef.
- A. Eucken and A. Büchner, Z. Phys. Chem., 1934, 27B, 321–349 Search PubMed.
- G. E. Jellison, F. A. Modine and L. A. Boatner, Opt. Lett., 1997, 22, 1808 CrossRef PubMed.
- N. Hosaka, T. Sekiya, C. Satoko and S. Kurita, J. Phys. Soc. Jpn, 1997, 66, 877–880 CrossRef.
- D. C. Cronemeyer, Phys. Rev., 1952, 87, 876–886 CrossRef.
- L. Forro, O. Chauvet, D. Emin, L. Zuppiroli, H. Berger and F. Lévy, J. Appl. Phys., 1994, 75, 633–635 CrossRef.
- R. R. Hasiguti and E. Yagi, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 7251–7256 CrossRef.
- G. A. Acket and J. Volger, Physica, 1966, 32, 1680–1692 CrossRef.
- R. G. Breckenridge and W. R. Hosler, Phys. Rev., 1953, 91, 793–802 CrossRef.
- H. P. R. Frederikse, J. Appl. Phys., 1961, 32, 2211–2215 CrossRef.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef PubMed.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef.
- A. Navrotsky, J. C. Jamieson and O. J. Kleppa, Science, 1967, 158, 388–389 CrossRef PubMed.
- Q. Zhang, Appl. Catal., B, 2000, 26, 207–215 CrossRef.
- L. Thiên-Nga and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13233–13241 CrossRef.
- G. Cangiani, Ab-initio Study of the Properties of TiO2 Rutile and Anatase Polytypes, PhD thesis, EPFL – Environmental Chemistry Modeling Laboratory, SISSA, Trieste, Italy, 2003.
- J. Muscat, V. Swamy and N. M. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 224112 CrossRef.
- A. Di Paola, M. Addamo, M. Bellardita, E. Cazzanelli and L. Palmisano, Thin Solid Films, 2007, 515, 3527–3529 CrossRef.
- N. Liu, H. G. Steinrück, A. Osvet, Y. Yang and P. Schmuki, Appl. Phys. Lett., 2017, 110, 072102-1–072102-4 Search PubMed.
- J. Jamieson and B. Olinger, Miner Notes, 1969, 54, 1477 Search PubMed.
- F. Bregani, C. Casale, L. E. Depero, I. Natali-Sora, D. Robba, L. Sangaletti and G. P. Toledo, Sens. Actuators, B, 1996, 31, 25–28 CrossRef.
- M. E. Straumanis, T. Ejima and W. J. James, Acta Crystallogr., 1961, 14, 493–497 CrossRef.
- O. R. S. da Rocha, R. F. Dantas, M. M. M. B. Duarte, M. M. L. Duarte and V. L. da Silva, Chem. Eng. J., 2010, 157, 80–85 CrossRef.
- G. K. Mor, O. K. Varghese, M. Paulose, K. Shankar and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 2011–2075 CrossRef.
- Y. Ao, J. Xu, S. Zhang and D. Fu, Appl. Surf. Sci., 2010, 256, 2754–2758 CrossRef.
- L. Dong, G. Cao, Y. Ma, X. Jia, G. Ye and S. Guan, Trans. Nonferrous Met. Soc. China, 2009, 19, 1583–1587 CrossRef.
- S. Banerjee, J. G. Pal, P. Muraleedharan, A. K. Tyagi and B. Raj, Res. Commun., 2006, 90, 1378–1383 Search PubMed.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef PubMed.
- H. K. Ardakani, Thin Solid Films, 1994, 248, 234–239 CrossRef.
- A. T. Paxton and L. Thiên-Nga, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1579–1584 CrossRef.
- E. C. Akubuiro and X. E. Verykios, J. Phys. Chem. Solids, 1989, 50, 17–26 CrossRef.
- S. Fujitsu and T. Hamada, J. Am. Ceram. Soc., 1994, 77, 3281–3283 CrossRef.
- N. G. Eror, J. Solid State Chem., 1981, 38, 281–287 CrossRef.
- J.-L. Carpentier, A. Lebrun and F. Perdu, J. Phys. Chem. Solids, 1989, 50, 145–151 CrossRef.
- P. Knauth and H. L. Tuller, J. Appl. Phys., 1999, 85, 897–902 CrossRef.
- K. Mizushima, M. Tanaka, A. Asai, S. Iida and J. B. Goodenough, J. Phys. Chem. Solids, 1979, 40, 1129–1140 CrossRef.
- N. Yu and J. W. Halley, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4768–4776 CrossRef.
- H. X. Zhu, P. X. Zhou, X. Li and J.-M. Liu, Phys. Lett. A, 2014, 378, 2719–2724 CrossRef.
- F. Arntz and Y. Yacoby, Phys. Rev. Lett., 1966, 17, 857–860 CrossRef.
- A. Frova, P. J. Boddy and Y. S. Chen, Phys. Rev., 1967, 157, 700–708 CrossRef.
- A. K. Ghosh, F. G. Wakim and R. R. Addiss, Phys. Rev., 1969, 184, 979–988 CrossRef.
- A. Amtout and R. Leonelli, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 6842–6851 CrossRef.
- S. Hüfner and G. K. Wertheim, Phys. Rev. B: Solid State, 1973, 7, 2333–2336 CrossRef.
- D. W. Fischer, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 5, 4219–4226 CrossRef.
- F. M. F. de Groot, J. Faber, J. J. M. Michiels, M. T. Czyżyk, M. Abbate and J. C. Fuggle, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 2074–2080 CrossRef.
- L. A. Grunes, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 2111–2131 CrossRef.
- G. van der Laan, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 12366–12368 CrossRef.
- F. M. F. de Groot, M. Grioni, J. C. Fuggle, J. Ghijsen, G. A. Sawatzky and H. Petersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 5715–5723 CrossRef.
- K. Watanabe, K. Inoue and F. Minami, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 2024–2033 CrossRef.
- L. Kavan, M. Grätzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef PubMed.
- Y. Bessekhouad, D. Robert and J. V. Weber, J. Photochem. Photobiol., A, 2003, 157, 47–53 CrossRef.
- G. Oskam, A. Nellore, R. L. Penn and P. C. Searson, J. Phys. Chem. B, 2003, 107, 1734–1738 CrossRef.
- T. Sugimoto, Adv. Colloid Interface Sci., 1987, 28, 65–108 CrossRef.
- M. A. Anderson, M. J. Gieselmann and Q. Xu, J. Membr. Sci., 1988, 39, 243–258 CrossRef.
- E. A. Barringer and H. K. Bowen, Langmuir, 1985, 1, 414–420 CrossRef.
- J. H. Jean and T. A. Ring, Langmuir, 1986, 2, 251–255 CrossRef.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, J. Phys. Chem., 1988, 92, 5196–5201 CrossRef.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef.
- J.-L. Look and C. F. Zukoski, J. Am. Ceram. Soc., 1995, 78, 21–32 CrossRef.
- J. L. Look and C. F. Zukoski, J. Am. Ceram. Soc., 1992, 75, 1587–1595 CrossRef.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef.
- D. Vorkapic and T. Matsoukas, J. Am. Ceram. Soc., 2005, 81, 2815–2820 CrossRef.
- D. Vorkapic and T. Matsoukas, J. Colloid Interface Sci., 1999, 214, 283–291 CrossRef PubMed.
- C.-S. Kim, B. K. Moon, J.-H. Park, S. Tae Chung and S.-M. Son, J. Cryst. Growth, 2003, 254, 405–410 CrossRef.
- L. Miao, S. Tanemura, S. Toh, K. Kaneko and M. Tanemura, J. Cryst. Growth, 2004, 264, 246–252 CrossRef.
- M. S. Ghamsari, F. Hajiesmaeilbaigi and S. Mirdamadi, Semicond. Phys., Quantum Electron. Optoelectron., 2007, 10, 36–39 Search PubMed.
- Y. Lin, G. S. Wu, X. Y. Yuan, T. Xie and L. D. Zhang, J. Phys.: Condens. Matter, 2003, 15, 2917–2922 CrossRef.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef PubMed.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef.
- F. Sayilkan, Turk. J. Chem., 2007, 31, 211–221 Search PubMed.
- S. Y. Chae, M. K. Park, S. K. Lee, T. Y. Kim, S. K. Kim and W. I. Lee, Chem. Mater., 2003, 15, 3326–3331 CrossRef.
- F. Cot, A. Larbot, G. Nabias and L. Cot, J. Eur. Ceram. Soc., 1998, 18, 2175–2181 CrossRef.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Colloid Interface Sci., 2003, 260, 82–88 CrossRef PubMed.
- J. Yang, S. Mei and J. M. F. Ferreira, Mater. Sci. Eng., C, 2001, 15, 183–185 CrossRef.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Mater. Res., 2002, 17, 2197–2200 CrossRef.
- M. Rehan, X. Lai and G. M. Kale, CrystEngComm, 2011, 13, 3725 RSC.
- R. Vijayalakshmi and V. Rajendran, Scholars Research Library, 2012, 4, 1183–1190 Search PubMed.
- Y. X. Zhang, G. H. Li, Y. X. Jin, Y. Zhang, J. Zhang and L. D. Zhang, Chem. Phys. Lett., 2002, 365, 300–304 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef.
- Z. Liu, Y. G. Andreev, A. Robert Armstrong, S. Brutti, Y. Ren and P. G. Bruce, Prog. Nat. Sci.: Mater. Int., 2013, 23, 235–244 CrossRef.
- M. Cargnello, T. R. Gordon and C. B. Murray, Chem. Rev., 2014, 114, 9319–9345 CrossRef PubMed.
- A. Craig and K. Gopal, TiO2 Nanotube Arrays, Synthesis, properties and Application, Springer, Dordrecht Heidelberg, London, New York, 2009 Search PubMed.
- D. Gong, C. A. Grimes, O. K. Varghese, W. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef.
- G. K. Mor, O. K. Varghese, M. Paulose and C. A. Grimes, Sens. Lett., 2003, 1, 42–46 CrossRef.
- Q. Cai, M. Paulose, O. K. Varghese and C. A. Grimes, J. Mater. Res., 2005, 20, 230–236 CrossRef.
- G. K. Mor, K. Shankar, O. K. Varghese and C. A. Grimes, J. Mater. Res., 2004, 19, 2989–2996 CrossRef.
- V. Zwilling, M. Aucouturier and E. Darque-Ceretti, Electrochim. Acta, 1999, 45, 921–929 CrossRef.
- Y. Shimizu, N. Kuwano, T. Hyodo and M. Egashira, Sens. Actuators, B, 2002, 83, 195–201 CrossRef.
- J. Yamamoto, A. Tan, R. Shiratsuchi, S. Hayase, C. R. Chenthamarakshan and K. Rajeshwar, Adv. Mater., 2003, 15, 1823–1825 CrossRef.
- G. Patermarakis, J. Electrochem. Soc., 1995, 142, 737 CrossRef.
- K. Shankar, G. K. Mor, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 21–26 CrossRef.
- C. Ruan, M. Paulose, O. K. Varghese and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 1283–1295 CrossRef.
- M. Paulose, K. Shankar, S. Yoriya, H. E. Prakasam, O. K. Varghese, G. K. Mor, T. A. Latempa, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. B, 2006, 110, 16179–16184 CrossRef PubMed.
- S. Yoriya, G. K. Mor, S. Sharma and C. A. Grimes, J. Mater. Chem., 2008, 18, 3332 RSC.
- R. Subasri, M. Tripathi, K. Murugan, J. Revathi, G. V. N. Rao and T. N. Rao, Mater. Chem. Phys., 2010, 124, 63–68 CrossRef.
- Y. Yin, Z. Jin, F. Hou and X. Wang, J. Am. Ceram. Soc., 2007, 90, 2384–2389 CrossRef.
- G. K. Mor, O. K. Varghese, M. Paulose, N. Mukherjee and C. A. Grimes, J. Mater. Res., 2003, 18, 2588–2593 CrossRef.
- G. K. Mor, M. A. Carvalho, O. K. Varghese, M. V. Pishko and C. A. Grimes, J. Mater. Res., 2004, 19, 628–634 CrossRef.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191–195 CrossRef PubMed.
- O. K. Varghese, D. Gong, M. Paulose, C. A. Grimes and E. C. Dickey, J. Mater. Res., 2003, 18, 156–165 CrossRef.
- S. Bauer, S. Kleber and P. Schmuki, Electrochem. Commun., 2006, 8, 1321–1325 CrossRef.
- O. K. Varghese, D. Gong, M. Paulose, K. G. Ong, E. C. Dickey and C. A. Grimes, Adv. Mater., 2003, 15, 624–627 CrossRef.
- L. K. Preethi, T. Mathews, M. Nand, S. N. Jha, C. S. Gopinath and S. Dash, Appl. Catal., B, 2017, 218, 9–19 CrossRef.
- S. Seifried, M. Winterer and H. Hahn, Chem. Vap. Deposition, 2000, 6, 239–244 CrossRef.
- J. A. Ayllón, A. Figueras, S. Garelik, L. Spirkova, J. Durand and L. Cot, J. Mater. Sci. Lett., 1999, 18, 1319–1321 CrossRef.
- S. K. Pradhan, P. J. Reucroft, F. Yang and A. Dozier, J. Cryst. Growth, 2003, 256, 83–88 CrossRef.
- K. Okuyama, Y. Kousaka, N. Tohge, S. Yamamoto, J. J. Wu, R. C. Flagan and J. H. Seinfeld, AIChE J., 1986, 32, 2010–2019 CrossRef.
- K. Okuyama, R. Ushio, Y. Kousaka, R. C. Flagan and J. H. Seinfeld, AIChE J., 1990, 36, 409–419 CrossRef.
- K. Okuyama, J.-T. Jeung, Y. Kousaka, H. V. Nguyen, J. J. Wu and R. C. Flagan, Chem. Eng. Sci., 1989, 44, 1369–1375 CrossRef.
- Z. Ding, X. Hu, G. Q. Lu, P.-L. Yue and P. F. Greenfield, Langmuir, 2000, 16, 6216–6222 CrossRef.
- J.-J. Wu and C.-C. Yu, J. Phys. Chem. B, 2004, 108, 3377–3379 CrossRef.
- D. G. Park and J. M. Burlitch, Chem. Mater., 1992, 4, 500–502 CrossRef.
- H. D. Jang and S.-K. Kim, Mater. Res. Bull., 2001, 36, 627–637 CrossRef.
- Y.-L. Li and T. Ishigaki, Thin Solid Films, 2002, 407, 79–85 CrossRef.
- S.-M. Oh and T. Ishigaki, Thin Solid Films, 2004, 457, 186–191 CrossRef.
- X. H. Wang, J.-G. Li, H. Kamiyama, M. Katada, N. Ohashi, Y. Moriyoshi and T. Ishigaki, J. Am. Chem. Soc., 2005, 127, 10982–10990 CrossRef PubMed.
- M. Grujić-Brojčin, M. J. Šćepanović, Z. D. Dohčević-Mitrovi, I. Hinić, B. Matović, G. Stanišić and Z. V Popović, J. Phys. D: Appl. Phys., 2005, 38, 1415–1420 CrossRef.
- M. Šćepanović, Z. Dohčević-Mitrović, I. Hinić, M. Grujić-Brojčin, G. Stanišić and Z. V. Popović, Mater. Sci. Forum, 2005, 494, 265–270 Search PubMed.
- J.-M. Wu, H. C. Shih and W.-T. Wu, Chem. Phys. Lett., 2005, 413, 490–494 CrossRef.
- J.-M. Wu, H. C. Shih, W.-T. Wu, Y.-K. Tseng and I.-C. Chen, J. Cryst. Growth, 2005, 281, 384–390 CrossRef.
- B. Xiang, Y. Zhang, Z. Wang, X. H. Luo, Y. W. Zhu, H. Z. Zhang and D. P. Yu, J. Phys. D: Appl. Phys., 2005, 38, 1152–1155 CrossRef.
- H. Zhang, X. Luo, J. Xu, B. Xiang and D. Yu, J. Phys. Chem. B, 2004, 108, 14866–14869 CrossRef.
- J.-M. Wu, H. C. Shih and W.-T. Wu, Nanotechnology, 2006, 17, 105–109 CrossRef.
- D. K. Hwang, J. H. Moon, Y. G. Shul, K. T. Jung, D. H. Kim and D. W. Lee, J. Sol-Gel Sci. Technol., 2003, 26, 783–787 CrossRef.
- J.-W. Kim, J.-W. Shim, J.-H. Bae, S.-H. Han, H.-K. Kim, I.-S. Chang, H.-H. Kang and K.-D. Suh, Colloid Polym. Sci., 2002, 280, 584–588 CrossRef.
- B. Mahltig, H. Böttcher, K. Rauch, U. Dieckmann, R. Nitsche and T. Fritz, Thin Solid Films, 2005, 485, 108–114 CrossRef.
- A. P. Popov, A. V Priezzhev, J. Lademann and R. Myllylä, J. Phys. D: Appl. Phys., 2005, 38, 2564–2570 CrossRef.
- C. E. Sittig, Charakterisierung der Oxidschichten auf Titan und Titanlegierungen sowie deren Reaktionen in Kontact mit biologisch relevanten Modellösungen, PhD thesis, ETH Nr, 12657, Zürich, 1998.
- W. Clark and P. Broadhead, J. Phys. C: Solid State Phys., 1970, 3, 1047 CrossRef.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431–432 CrossRef.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef.
- Y. Xie, Electrochim. Acta, 2006, 51, 3399–3406 CrossRef.
- Y. Lai, L. Sun, Y. Chen, H. Zhuang, C. Lin and J. W. Chin, J. Electrochem. Soc., 2006, 153, D123 CrossRef.
- N. M. Mahmoodi, M. Arami, N. Y. Limaee and N. S. Tabrizi, Chem. Eng. J., 2005, 112, 191–196 CrossRef.
- A.-P. Zhang, World J. Gastroenterol., 2004, 10, 3191–3193 CrossRef PubMed.
- A. O. Ibhadon, Multifunctional TiO2 Catalysis and Applications, in Proceedings of Green Chemistry and Engineering International Conference, Washington, DC, USA, 24–26 June 2008 Search PubMed.
- C.-C. Wang, Z. Zhang and J. Y. Ying, Nanostruct. Mater., 1997, 9, 583–586 CrossRef.
- J.-M. Wu, T.-W. Zhang, Y.-W. Zeng, S. Hayakawa, K. Tsuru and A. Osaka, Langmuir, 2005, 21, 6995–7002 CrossRef PubMed.
- T. Peng, D. Zhao, K. Dai, W. Shi and K. Hirao, J. Phys. Chem. B, 2005, 109, 4947–4952 CrossRef PubMed.
- S. Yang, X. Quan, X. Li, N. Fang, N. Zhang and H. Zhao, J. Environ. Sci., 2005, 17, 290–293 Search PubMed.
- Y. Bessekhouad, D. Robert, J.-V. Weber and N. Chaoui, J. Photochem. Photobiol., A, 2004, 167, 49–57 CrossRef.
- Y. Cao, W. Yang, W. Zhang, G. Liu and P. Yue, New J. Chem., 2004, 28, 218 RSC.
- W. Li, Y. Wang, H. Lin, S. Ismat Shah, C. P. Huang, D. J. Doren, S. A. Rykov, J. G. Chen and M. A. Barteau, Appl. Phys. Lett., 2003, 83, 4143–4145 CrossRef.
- S. Peng, Y. Li, F. Jiang, G. Lu and S. Li, Chem. Phys. Lett., 2004, 398, 235–239 CrossRef.
- Y. Sakatani, H. Ando, K. Okusako, H. Koike, J. Nunoshige, T. Takata, J. N. Kondo, M. Hara and K. Domen, J. Mater. Res., 2004, 19, 2100–2108 CrossRef.
- X. Zhang and L. Lei, Mater. Lett., 2008, 62, 895–897 CrossRef.
- M. S. Nahar, K. Hasegawa and S. Kagaya, Chemosphere, 2006, 65, 1976–1982 CrossRef PubMed.
- K. Iketani, R.-D. Sun, M. Toki, K. Hirota and O. Yamaguchi, Mater. Sci. Eng., B, 2004, 108, 187–193 CrossRef.
- J. Zhu, F. Chen, J. Zhang, H. Chen and M. Anpo, J. Photochem. Photobiol., A, 2006, 180, 196–204 CrossRef.
- D. Hyun Kim, K. Sub Lee, Y.-S. Kim, Y.-C. Chung and S.-J. Kim, J. Am. Ceram. Soc., 2006, 89, 515–518 CrossRef.
- J. C.-S. Wu and C.-H. Chen, J. Photochem. Photobiol., A, 2004, 163, 509–515 CrossRef.
- C.-C. Pan and J. C. S. Wu, Mater. Chem. Phys., 2006, 100, 102–107 CrossRef.
- S. Kim, S.-J. Hwang and W. Choi, J. Phys. Chem. B, 2005, 109, 24260–24267 CrossRef PubMed.
- R. Amadelli, L. Samiolo, A. Maldotti, A. Molinari, M. Valigi and D. Gazzoli, Int. J. Photoenergy, 2008, 2008, 1–9 CrossRef.
- T. Lindgren, J. M. Mwabora, E. Avendaño, J. Jonsson, A. Hoel, C.-G. Granqvist and S.-E. Lindquist, J. Phys. Chem. B, 2003, 107, 5709–5716 CrossRef.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049–1051 CrossRef.
- X. Chen, Y.-B. Lou, A. C. S. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef.
- Y. Choi, T. Umebayashi and M. Yoshikawa, J. Mater. Sci., 2004, 39, 1837–1839 CrossRef.
- H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003, 32, 772–773 CrossRef.
- Y. Xie, L. M. Zhou and H. Huang, Mater. Lett., 2006, 60, 3558–3560 CrossRef.
- S. Sakthivel, M. Janczarek and H. Kisch, J. Phys. Chem. B, 2004, 108, 19384–19387 CrossRef.
- H. Tokudome and M. Miyauchi, Chem. Lett., 2004, 33, 1108–1109 CrossRef.
- C. Shifu, C. Lei, G. Shen and C. Gengyu, Chem. Phys. Lett., 2005, 413, 404–409 CrossRef.
- O. Diwald, T. L. Thompson, T. Zubkov, S. D. Walck and J. T. Yates, J. Phys. Chem. B, 2004, 108, 6004–6008 CrossRef.
- T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115–121 CrossRef.
- M. Shen, Z. Wu, H. Huang, Y. Du, Z. Zou and P. Yang, Mater. Lett., 2006, 60, 693–697 CrossRef.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef.
- D. Li, H. Haneda, N. K. Labhsetwar, S. Hishita and N. Ohashi, Chem. Phys. Lett., 2005, 401, 579–584 CrossRef.
- D. Li, H. Haneda, S. Hishita, N. Ohashi and N. K. Labhsetwar, J. Fluorine Chem., 2005, 126, 69–77 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- F. Cao, G. Oskam, G. J. Meyer and P. C. Searson, J. Phys. Chem., 1996, 100, 17021–17027 CrossRef.
- A. Solbrand, H. Lindström, H. Rensmo, A. Hagfeldt, S.-E. Lindquist and S. Södergren, J. Phys. Chem. B, 1997, 101, 2514–2518 CrossRef.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- A. Hameed and M. A. Gondal, J. Mol. Catal. A: Chem., 2004, 219, 109–119 CrossRef.
- A. Yamakata, T. Ishibashi and H. Onishi, J. Mol. Catal. A: Chem., 2003, 199, 85–94 CrossRef.
- W. Y. Teoh, J. A. Scott and R. Amal, J. Phys. Chem. Lett., 2012, 3, 629–639 CrossRef PubMed.
- M. S. Wrighton, A. B. Ellis, P. T. Wolczanski, D. L. Morse, H. B. Abrahamson and D. S. Ginley, J. Am. Chem. Soc., 1976, 98, 2774–2779 CrossRef.
- Z.-L. Jin and G.-X. Lu, Energy Fuels, 2005, 19, 1126–1132 CrossRef.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef PubMed.
- M. Matsuoka, M. Kitano, M. Takeuchi, M. Anpo and J. M. Thomas, Top. Catal., 2005, 35, 305–310 CrossRef.
- M. Matsuoka, M. Kitano, M. Takeuchi, M. Anpo and J. M. Thomas, Mater. Sci. Forum, 2005, 486–487, 81–84 Search PubMed.
- D. Bocharov, S. Piskunov, Y. F. Zhukovskii, E. Spohr and P. N. D’yachkov, Vacuum, 2017, 146, 562–569 CrossRef.
- J. F. Guayaquil-Sosa, B. Serrano-Rosales, P. J. Valadés-Pelayo and H. de Lasa, Appl. Catal., B, 2017, 211, 337–348 CrossRef.
- M. Alitabar and H. Yoozbashizadeh, Mater. Res. Bull., 2017, 95, 169–176 CrossRef.
- K. Dhanalakshmi, Int. J. Hydrogen Energy, 2001, 26, 669–674 CrossRef.
- R. Argazzi, N. Y. Murakami Iha, H. Zabri, F. Odobel and C. A. Bignozzi, Coord. Chem. Rev., 2004, 248, 1299–1316 CrossRef.
- A. S. Polo, M. K. Itokazu and N. Y. Murakami Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef.
- D. Duonghong, E. Borgarello and M. Graetzel, J. Am. Chem. Soc., 1981, 103, 4685–4690 CrossRef.
- T. Sreethawong, C. Junbua and S. Chavadej, J. Power Sources, 2009, 190, 513–524 CrossRef.
- Y. Li, C. Xie, S. Peng, G. Lu and S. Li, J. Mol. Catal. A: Chem., 2008, 282, 117–123 CrossRef.
- K. K. Mandari, B. S. Kwak, A. K. R. Police and M. Kang, Mater. Res. Bull., 2017, 95, 515–524 CrossRef.
- H. Zhang, F. Liu, H. Wu, X. Cao, J. Sun and W. Lei, RSC Adv., 2017, 7, 40327–40333 RSC.
- B. Hou, L. Shen, H. Shi, R. Kapadia and S. B. Cronin, Phys. Chem. Chem. Phys., 2017, 19, 2877–2881 RSC.
- J. Zhang, Z. Yu, Z. Gao, H. Ge, S. Zhao, C. Chen, S. Chen, X. Tong, M. Wang, Z. Zheng and Y. Qin, Angew. Chem., Int. Ed., 2017, 56, 816–820 CrossRef PubMed.
- H. Xu, S. Li, L. Ge, C. Han, Y. Gao and D. Dai, Int. J. Hydrogen Energy, 2017, 42, 22877–22886 CrossRef.
- Y. Sui, S. Liu, T. Li, Q. Liu, T. Jiang, Y. Guo and J.-L. Luo, J. Catal., 2017, 353, 250–255 CrossRef.
- J. Si, Y. Wang, X. Xia, S. Peng, Y. Wang, S. Xiao, L. Zhu, Y. Bao, Z. Huang and Y. Gao, J. Power Sources, 2017, 360, 353–359 CrossRef.
- M. Wang, Z. Li, Y. Wu, J. Ma and G. Lu, J. Catal., 2017, 353, 162–170 CrossRef.
- R. Liu, Z. Sun, X. Song, Y. Zhang, L. Xu and L. Xi, Appl. Catal., A, 2017, 544, 137–144 CrossRef.
- P. V Kamat and D. Meisel, Curr. Opin. Colloid Interface Sci., 2002, 7, 282–287 CrossRef.
- W. Choi, J. Lee, S. Kim, S. Hwang, M. C. Lee and T. K. Lee, J. Ind. Eng. Chem., 2003, 9, 96–101 Search PubMed.
- H. Hussain, G. Tocci, T. Woolcot, X. Torrelles, C. L. Pang, D. S. Humphrey, C. M. Yim, D. C. Grinter, G. Cabailh, O. Bikondoa, R. Lindsay, J. Zegenhagen, A. Michaelides and G. Thornton, Nat. Mater., 2017, 16(4), 461–466 CrossRef PubMed.
- P. Salvador, New J. Chem., 1988, 12, 35–43 Search PubMed.
- E. Liu, L. Kang, Y. Yang, T. Sun, X. Hu, C. Zhu, H. Liu, Q. Wang, X. Li and J. Fan, Nanotechnology, 2014, 25, 165401 CrossRef PubMed.
- K. Nishijima, T. Kamai, N. Murakami, T. Tsubota and T. Ohno, Int. J. Photoenergy, 2008, 2008, 1–7 CrossRef.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef PubMed.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588–598 CrossRef.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- D. Panduwinata and J. D. Gale, J. Mater. Chem., 2009, 19, 3931–3940 RSC.
- C. Arrouvel, S. C. Parker and M. S. Islam, Chem. Mater., 2009, 21, 4778–4783 CrossRef.
- C.-C. Tsai and H. Teng, Chem. Mater., 2006, 18, 367–373 CrossRef.
- H. Miyazaki, T. Hyodo, Y. Shimizu and M. Egashira, Sens. Actuators, B, 2005, 108, 467–472 CrossRef.
- A. M. Ruiz, A. Cornet and J. R. Morante, Sens. Actuators, B, 2004, 100, 256–260 CrossRef.
- S. El-Sherbiny, F. Morsy, M. Samir and O. A. Fouad, Appl. Nanosci., 2014, 4, 305–313 CrossRef.
- R. I Bickley, T. Gonzalezcarreno, J. S. Lees, L. Palmisano and R. J. D. Tilley, J. Solid State Chem., 1991, 92, 178–190 CrossRef.
- R. Comparelli, E. Fanizza, M. L. Curri, P. D. Cozzoli, G. Mascolo, R. Passino and A. Agostiano, Appl. Catal., B, 2005, 55, 81–91 CrossRef.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne and K. O'Shea, et al., Appl. Catal., B, 2012, 125, 331–349 CrossRef.
- M. Long, L. Zheng, B. Tan and H. Shu, Appl. Surf. Sci., 2016, 386, 434–441 CrossRef.
- Y. Ben-Shahar and U. Banin, Top. Curr. Chem., 2016, 374, 54 CrossRef PubMed.
- S. Murgolo, F. Petronella, R. Ciannarella, R. Comparelli, A. Agostiano, M. L. Curri and G. Mascolo, Catal. Today, 2015, 240, 114–124 CrossRef.
- Y. Tang, G. Zhang, C. Liu, S. Luo, X. Xu, L. Chen and B. Wang, J. Hazard. Mater., 2013, 252–253, 115–122 CrossRef PubMed.
- F. Petronella, A. Truppi, C. Ingrosso, T. Placido, M. Striccoli, M. L. Curri, A. Agostiano and R. Comparelli, Catal. Today, 2017, 281, 85–100 CrossRef.
- L. Karimi, M. E. Yazdanshenas, R. Khajavi, A. Rashidi and M. Mirjalili, Cellulose, 2014, 21, 3813–3827 CrossRef.
- F. Petronella, A. Truppi, T. Sibillano, C. Giannini, M. Striccoli, R. Comparelli and M. L. Curri, Catalysts, 2017, 7, 100 CrossRef.
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449–493 CrossRef.
- Á. Kmetykó, Á. Szániel, C. Tsakiroglou, A. Dombi and K. Hernádi, React. Kinet., Mech. Catal., 2016, 117, 379–390 CrossRef.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely and G. J. Hutchings, et al., ACS Nano, 2012, 6, 6284–6292 CrossRef PubMed.
- A. Takai and P. V. Kamat, ACS Nano, 2011, 5, 7369–7376 CrossRef PubMed.
- L. Francioso, D. S. Presicce, P. Siciliano and A. Ficarella, Sens. Actuators, B, 2007, 123, 516–521 CrossRef.
- Y. Mizukoshi, Y. Makise, T. Shuto, J. Hu, A. Tominaga, S. Shironita and S. Tanabe, Ultrason. Sonochem., 2007, 14, 387–392 CrossRef PubMed.
- H. Wang, T. You, W. Shi, J. Li and L. Guo, J. Phys. Chem. C, 2012, 116, 6490–6494 CrossRef.
- R. S. Sonawane and M. K. Dongare, J. Mol. Catal. A: Chem., 2006, 243, 68–76 CrossRef.
- G. L. Chiarello, E. Selli and L. Forni, Appl. Catal., B, 2008, 84, 332–339 CrossRef.
- F. Wenqing, L. Qinghua, Z. Qinghong and W. Ye, J. Phys. Chem. C, 2011, 115, 10694–10701 CrossRef.
- J. Deepa, M. S. Christopher, S. R. Sadhana, M. S. Khadga and J. K. Kenneth, Int. J. Photoenergy, 2013, 2013, 10 Search PubMed.
- G. Anna and W. Jerzy, Energy Fuels, 2005, 3, 1143–1147 Search PubMed.
- V. I. Yatskiv, A. V. Korzhak, V. M. Granchak, A. S. Kovalenko and S. Ya. Kuchmiy, Theor. Exp. Chem., 2003, 39, 167–171 Search PubMed.
- Š. Václav and K. Daniela, Int. J. Photoenergy, 2011, 2011, 14 Search PubMed.
- X. Quanjun, Y. Jiaguo and J. Mietek, J. Am. Chem. Soc., 2012, 15, 6575–6578 Search PubMed.
- V. K. Anna, I. E. Natalia, L. S. Alexander, K. B. Viktor, E. R. Alexandra, I. L. Valentina, Y. Stepan, V. G. I. Kuchmiy and A. M. Piotr, J. Photochem. Photobiol., A, 2008, 198, 126–134 CrossRef.
- D. Ke, P. Tianyou, Ke. Dingning and W. Bingqing, Nanotechnology, 2009, 20, 12 CrossRef PubMed.
- M. Takuya, K. Satoshi, K. Hideyuki, S. Tohru, O. Kiyohisa, C. V. Suresh and S. Kunihiro, Int. J. Hydrogen Energy, 2010, 35, 6554–6560 CrossRef.
- R. Nitish, T. L. Kam and P. Debabrata, J. Phys. Chem. C, 2015, 33, 19117–19125 Search PubMed.
- Z. Lu, T. Baozhu, C. Feng and Z. Jinlong, Int. J. Hydrogen Energy, 2012, 37, 17060–17067 CrossRef.
- P. Fuyun, L. Yingliang, X. Shengang, L. Jing, W. Chenxu and C. Shaokui, Int. J. Hydrogen Energy, 2013, 38, 2670–2677 CrossRef.
- K. Yang, C. Meng, L. Lin, X. Peng, X. Chen, X. Wang, W. Dai and X. Fu, Catal. Sci. Technol., 2016, 6, 829–839 RSC.
- E. Kowalska, Z. Wei, B. Karabiyik, A. Herissan, M. Janczarek, M. Endo, A. Markowska-Szczupak, H. Remita and B. Ohtani, Catal. Today, 2015, 252, 136–142 CrossRef.
- S. W. Verbruggen, M. Keulemans, M. Filippousi, D. Flahaut, G. van Tendeloo, S. Lacombe, J. A. Martens and S. Lenaerts, Appl. Catal., B, 2014, 156–157, 116–121 CrossRef.
- X. Qiu, M. Miyauchi, K. Sunada, M. Minoshima, M. Liu, Y. Lu, D. Li, Y. Shimodaira, Y. Hosogi and Y. Kuroda, ACS Nano, 2012, 6, 1609–1618 CrossRef PubMed.
- Y. Wang, F. Pan, W. Dong, L. Xu, K. Wu, G. Xu and W. Chen, Appl. Catal., B, 2016, 189, 192–198 CrossRef.
- S. Mallakpour and E. Khadem, Chem. Eng. J., 2016, 302, 344–367 CrossRef.
- M. Horn, C. F. Schwerdtfeger and E. P. Meagher, Z. Kristallogr., 1972, 136, 273–281 CrossRef.
- H. Tang, Electronic Properties of Anatase TiO2 Investigated by Electrical and Optical Measurements on Single Crystals and Thin Films, Département de physique, Ecole polytechnique fédérale de Lausanne EPFL, Lausanne, France, 1994 Search PubMed.
- N. Tsuda, K. Nasu, A. Yanase and K. Siratori, Electronic Conduction in Oxides, Springer-Verlag, Berlin, Gerseveral, 1991 Search PubMed.
- A. Eucken and W. Dannöhl, Die thermisce Ausdehnung einiger Alkalihalogenide and Metalle bei hohen Temperaturen, Z. Elektrochem., 1934, 40, 814–821 Search PubMed.
- M. S. Lee, S.-S. Hong and M. Mohseni, J. Mol. Catal. A: Chem., 2005, 242, 135–140 CrossRef.
- J. O. Carneiro, V. Teixeira, A. Portinha, L. Dupák, A. Magalhães and P. Coutinho, Vacuum, 2005, 78, 37–46 CrossRef.
- J. C.-S. Wu and C.-H. Chen, J. Photochem. Photobiol., A, 2004, 163, 509–515 CrossRef.
- X. Z. Li and F. B. Li, Environ. Sci. Technol., 2001, 35, 2381–2387 CrossRef PubMed.
- F. B. Li and X. Z. Li, Chemosphere, 2002, 48, 1103–1111 CrossRef PubMed.
- Z. Wu, F. Dong, W. Zhao and S. Guo, J. Hazard. Mater., 2008, 157, 57–63 CrossRef PubMed.
- R. Asahi, Science, 2001, 293, 269–271 CrossRef PubMed.
- K. Takeshita, A. Yamakata, T. Ishibashi, H. Onishi, K. Nishijima and T. Ohno, J. Photochem. Photobiol., A, 2006, 177, 269–275 CrossRef.
- J. Yu, M. Zhou, B. Cheng and X. Zhao, J. Mol. Catal. A: Chem., 2006, 246, 176–184 CrossRef.
- S. Y. Treschev, P.-W. Chou, Y.-H. Tseng, J.-B. Wang, E. V. Perevedentseva and C.-L. Cheng, Appl. Catal., B, 2008, 79, 8–16 CrossRef.
- C. Lettmann, K. Hildenbrand, H. Kisch, W. Macyk and W. F. Maier, Appl. Catal., B, 2001, 32, 215–227 CrossRef.
- A. Zaleska, J. W. Sobczak, E. Grabowska and J. Hupka, Appl. Catal., B, 2008, 78, 92–100 CrossRef.
- L. Kőrösi and I. Dékány, Colloids Surf., A, 2006, 280, 146–154 CrossRef.
- K. Sayama and H. Arakawa, J. Chem. Soc., Faraday Trans., 1997, 93, 1647–1654 RSC.
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533 CrossRef.
- A. Kudo, H. Kato and S. Nakagawa, J. Phys. Chem. B, 2000, 104, 571–575 CrossRef.
- H. Kato and A. Kudo, Chem. Phys. Lett., 1998, 295, 487–492 CrossRef.
- K. Sayama, H. Arakawa and K. Domen, Catal. Today, 1996, 28, 175–182 CrossRef.
- K. Fujihara, T. Ohno and M. Matsumura, J. Chem. Soc., Faraday Trans., 1998, 94, 3705–3709 RSC.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829–3931 RSC.
- M. Higashi, R. Abe, T. Takata and K. D omen, Chem. Mater., 2009, 21, 1543–1549 CrossRef.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef.
- L. B. Fen, T. K. Han, M. Nee, B. C. Ang and M. R. Johan, Appl. Surf. Sci., 2011, 258, 431–435 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Mohd Suhailb,
Zied A. Alothmanc and
Abdulrahman Alwarthanc
*ab,
Mohd Suhailb,
Zied A. Alothmanc and
Abdulrahman Alwarthanc
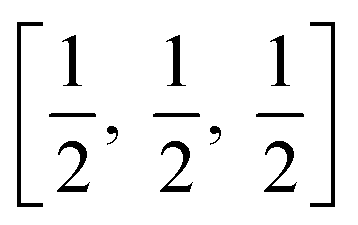 ) positions, separately are available. Distinctly, octahedron is linked to 10 near octahedra, out of which 2 parts an edge and eight share a bend with it. The octahedral edge shared are aligned along [001] direction as shows in Fig. 1.70
) positions, separately are available. Distinctly, octahedron is linked to 10 near octahedra, out of which 2 parts an edge and eight share a bend with it. The octahedral edge shared are aligned along [001] direction as shows in Fig. 1.70
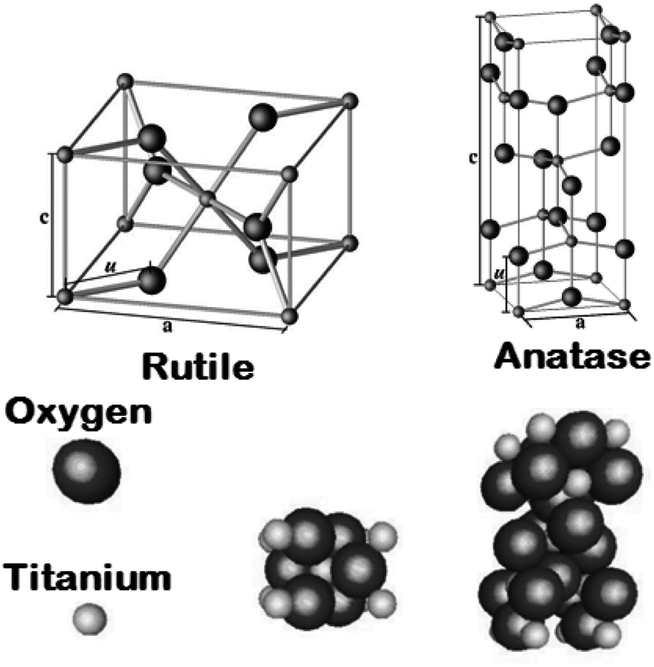
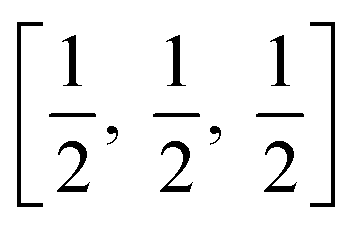 , [0, 0, 0],
, [0, 0, 0],  and
and  ) and 8 oxygen atoms, which form a partial TiO6 octahedron around Ti;70 with division of 4 edges of each octahedron.
) and 8 oxygen atoms, which form a partial TiO6 octahedron around Ti;70 with division of 4 edges of each octahedron.