
Reinventing the De Mayo reaction: synthesis of 1,5-diketones or 1,5-ketoesters via visible light [2+2] cycloaddition of β-diketones or β-ketoesters with styrenes†
Rebeca
Martinez-Haya
 a,
Leyre
Marzo
*b and
Burkhard
König
*b
a,
Leyre
Marzo
*b and
Burkhard
König
*b
aInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, E-46022, Valencia, Spain
bUniversität Regensburg, Fakultät für Chemie und Pharmazie, 93040 Regensburg, Germany. E-mail: burkhard.koenig@ur.de
First published on 20th September 2018
Abstract
A visible light mediated De Mayo reaction between 1,3-diketones and styrenes following a [2+2] cycloaddition pathway via a photosensitization mechanism gives access to 1,5-diketones. The reaction has been applied to substituted styrenes and aryl- and alkyl-substituted ketones. Moreover, the method converts β-ketoesters, β-amido esters, and β-cyano ketones. Seven membered rings, a frequent structural motif of natural products, are also accessible using this methodology.
The photochemical reaction between β-diketones and double bonds under UV light irradiation is known as the De Mayo reaction.1 In 1962 Paul Jose De Mayo reported that the enolic form of 1,3-diketones can undergo a [2+2] photocycloaddition with an olefin under UV irradiation affording a non-isolable cyclobutanol intermediate that evolves through a retro-aldol reaction yielding 1,5-diketones (Scheme 1). However, it was not until the late 1970s that its synthetic utility was fully realized when, via the intramolecular version, more complex macrocyclic structures became accessible.2 Examples are the total syntheses of (±)-ingenol3 or the alkaloid mesembrine4 that employ the De Mayo reaction as the key step, or the formal synthesis of vindorosine,5 among others.6 In these reactions β-diketone is directly excited by UV irradiation to its singlet excited state, which undergoes intersystem crossing to the excited triplet (Scheme 1). Further complexation of the triplet state with the double bond forms an exciplex that evolves to form the most stable 1,4-biradical. This 1,4-biradical generates the cyclobutanol intermediate that affords the desired products (Scheme 1).7a However, to the best of our knowledge a sensitized version of this reaction allowing the use of visible light has not been reported so far.7
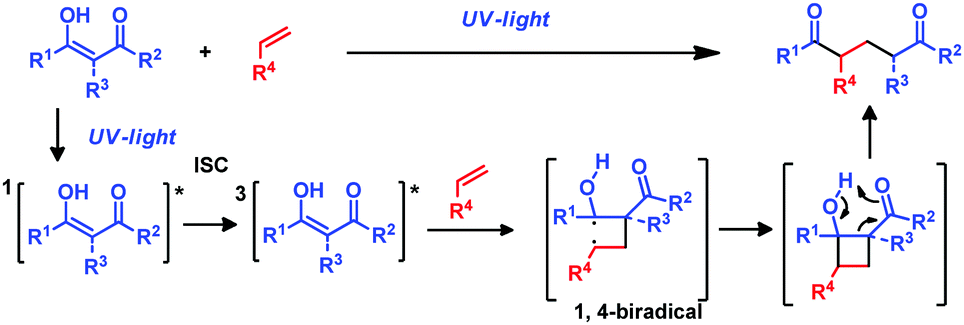 | ||
| Scheme 1 De Mayo photocycloaddition. | ||
Over the last few decades, visible light photocatalysis8 has developed into an important tool in synthesis. The lower cost and energy demand of the visible light sources, together with the selective excitation of the photocatalyst, thus avoiding undesired pathways, are some of the advantages. Photoredox catalysis has afforded several approaches for the [2+2] photocycloaddition between double bonds or Michael acceptors via photosensitization.9 In particular, Yoon reported the intramolecular [2+2] photocycloaddition of styrene derivatives by direct photosensitization using an iridium complex as a photosensitizer of the reaction under visible light irradiation.9a Based on this precedent, we envisaged that a visible light De Mayo reaction may be possible via the photosensitization of the styrene using a photocatalyst through energy transfer, triggering the [2+2] photocycloaddition with the enol of 1,3-diketones to obtain the desired 1,5-diketones.
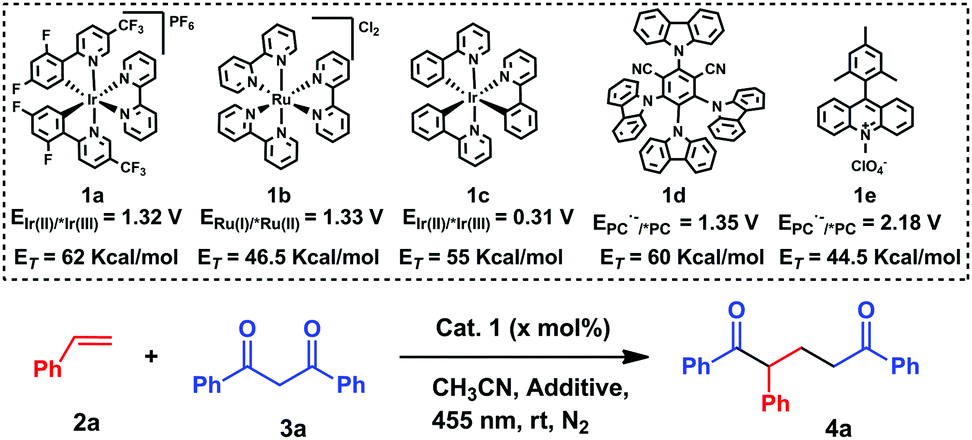
To prove this hypothesis, a solution of styrene 2a and 1,3-diphenylpropane-1,3-dione 3a in the presence of 2 mol% [Ir(dF(CF3)ppy)2(bpy)]PF61a, in CH3CN was subjected to blue LED irradiation over a period of 20 h, affording 1,5-diketone 4a in 69% yield (entry 1, Table 1). Then, different catalysts with different oxidation powers (see the scheme in Table 1)10 were tested in the reaction. Ru(bpy)3Cl21b, Ir(ppy)31c and Mes-Acr+1e did not react, while the carbazol derivative 1d afforded the desired product in 44% yield (entries 2–5, Table 1). Taking into account that the enol of 3a is the reactive species, tributylmethylammonium dibutyl phosphate was tested in the reaction, in order to shift the keto–enol equilibria to the enol form, obtaining 4a in 79% yield (entry 6, Table 1). Control reactions revealed that both the catalyst and light are necessary for the reaction (entries 7 and 8, Table 1). Finally, the use of the polar protic solvent EtOH improved the yield to 96% (entry 7, Table 1). Therefore, the optimized conditions are: 1a (2 mol%), 2a (5 equiv.), 3a (1 equiv.), (BuO)2P(O)ONBu3Me (25 mol%), EtOH (1 mL), N2, 25 °C, and 455 nm (for further details see Table S1, ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (mol%) | Additive (25 mol%) | Solvent | Yieldb (%) |
| a The reactions were performed using 0.1 mmol of 3a, 0.5 mmol of 2a, and 1 mL of CH3CN. b Isolated yields after 20 h. c Without light. | ||||
| 1 | 1a (2) | — | CH3CN | 69 |
| 2 | 1b (3) | — | CH3CN | 0 |
| 3 | 1c (2) | — | CH3CN | 0 |
| 4 | 1d (4) | — | CH3CN | 44 |
| 5 | 1e (4) | — | CH3CN | 0 |
| 6 | 1a (2) | (BuO)2P(O)ONBu3Me | CH3CN | 79 |
| 7 | 1a (2)c | (BuO)2P(O)ONBu3Me | CH3CN | 0 |
| 8 | — | (BuO)2P(O)ONBu3Me | CH3CN | 0 |
| 9 | 1a (2) | (BuO)2P(O)ONBu3Me | EtOH | 96 |
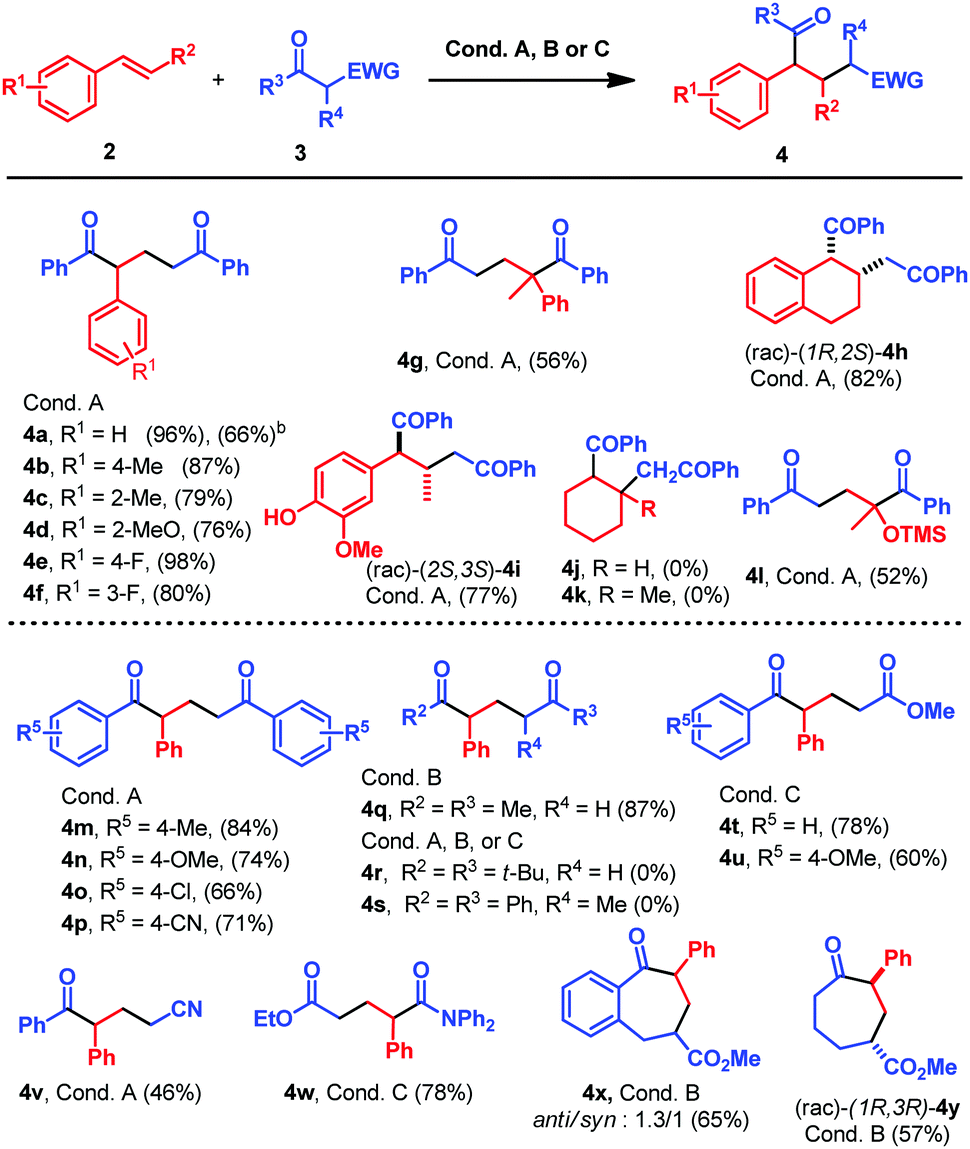
With the optimized conditions in hand, we proceed to study the scope of the reaction between β-diketone 3a and different styrene derivatives 2 (Table 2). Styrenes bearing electron donating or electron withdrawing groups in the ortho, meta or para position yielded the final products in good to very good isolated yields (4a–f, Table 2). Therefore, the electronic nature of the substituents does not seem to have a big impact in the reaction. The reaction could also be performed on a 1 mmol scale of 3a using a different set up (see the ESI,† Fig. S11), obtaining 4a in 66% yield, thus proving the robustness of the methodology (Table 2). More hindered styrene derivatives also underwent this reaction satisfactorily. Thus, α-methyl styrene afforded 4g in a moderate 56% yield, while dialin yielded 4h as a single stereoisomer in a very good yield (Table 2). The reaction was also performed with the E/Z-mixture of isoeugenol that afforded 4h as a single diastereoisomer. This stereoselectivity can be explained attending to the fast (E)/(Z) geometric isomerization of the triplet alkenes, and assuming that only (E)-isoeugenol would react in a stereoconvergent manner.9a Alkyl substituted double bonds such as cyclohexene or 1-methyl-1-cyclohexene did not react (4j and 4k, Table 2). This lack of reactivity is due to the higher triplet energy of these compounds (see the mechanistic proposal).11 (Isopropenyloxy)trimethylsilane afforded the desired product in good yield (4l, 52% yield, Table 2). Next, we studied the scope of the reaction between β-diketones 3 and styrene 2a (Table 2). First, differently substituted 1,3-diketones were studied. Under the optimized reaction conditions, aromatic diketones bearing electron withdrawing or electron donating substituents in the aromatic ring reacted smoothly, affording the desired products 4m–p in good to excellent yields (Table 2). Alkyl substituents are also tolerated in the reaction, but in this case slightly different reaction conditions are required: compound 4q is obtained in an excellent yield when 4 mol% of 1d is used as a catalyst in CH3CN as a solvent (conditions B, Table 2). The steric hindrance afforded by the t-Bu or the methyl group in the reactive methylene completely suppresses the reactivity (4r and 4s, Table 2).
| a All reactions were carried out using 0.1 mmol 3, 0.5 mmol 2a and the reaction conditions indicated in each case. Reaction conditions A: 0.002 mmol Ir[dFCF3ppy]2(bpy)PF61a, 0.025 mmol n-Bu2PO4NMe(n-Bu)3, EtOH, N2, 25 °C, 455 nm; reaction conditions B: 0.004 mmol 4CzIPN 1d, 0.025 mmol n-Bu2PO4NMe(n-Bu)3, CH3CN, N2, 25 °C, 455 nm; reaction conditions C: 0.004 mmol 4CzIPN 1d, 0.1 mmol K2CO3, CH3CN, N2, 25 °C, 455 nm. b Reaction carried out with 1 mmol 3a, 5 mmol 2a, 0.02 mmol Ir[dFCF3ppy]2(bpy)PF61a, 0.25 mmol n-Bu2PO4NMe(n-Bu)3, EtOH (10 mL), N2, 25 °C and 455 nm LED. |
|---|
|
We extended the reaction scope to other ketones bearing different electron withdrawing substituents in the β-position. β-Keto esters also underwent this reaction, but a combination of 1 equivalent of a stronger base (K2CO3) and 4 mol% 1d in CH3CN was necessary to obtain 4t and 4u in good yields (conditions C, Table 2). β-Cyano ketones reacted under the standard conditions giving 4v in a moderate 46% yield, while from a β-amido ester, 4w was isolated in 78% yield using reaction conditions C (Table 2). To prove that the reaction could be applied for the synthesis of larger ring systems, such as compounds bearing 7 membered rings,12 the reaction was performed with cyclic β-keto esters. Thus, under reaction conditions B, 4x was obtained as a diasteromeric mixture 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of the (rac)-(6S,8R) and (rac)-(6R,8R) δ-keto esters in good yield, while 4y was isolated as a single regioisomer in good yield (Table 2).
:1 of the (rac)-(6S,8R) and (rac)-(6R,8R) δ-keto esters in good yield, while 4y was isolated as a single regioisomer in good yield (Table 2).
Then, the elucidation of the reaction mechanism was attempted, considering either an electron transfer or an energy transfer as the initial step of the reaction. Tobita et al. reported that, in solution, 3a exists mainly as the enol form,13 therefore, this will be the initial reactive species considered for the reaction mechanism. Regarding the radical mechanism an initial oxidation step was considered. The oxidation potentials of the three initial species are 1.43 V for 3a (see Fig. S5, ESI†), 1.97 V for 2a,14 and 0.6 V vs. SCE for the enolate of 3a (H-NMR experiments revealed the presence of the enolate in solution in the presence of the base, see the ESI,† Section 5 and Fig. S7 for CV). Reported redox potentials for the photocatalysts, which afford the final products, are 1.32 V and 1.35 V vs. SCE for 1a and 1d, respectively (see Table 1).10 Thus, in principle, an electron transfer reaction (photoredox) between any of the photocatalysts and the enolate of the β-diketone could be thermodynamically favoured, but it would not be favoured in the case of an electron transfer with the styrene (see the ESI,† Table S2). With this assumption, photocatalyst 1e, with a redox potential of 2.18 V vs. SCE,10f,g should even work better than the active ones. However no product formation was observed when 1e was used (Table 1, entry 5). Besides, previous studies by Sharp15 revealed that the radical reaction between β-diketones and double bonds under oxidative conditions affords linear α-substituted β-diketones,16 while the De Mayo reaction (photochemical conditions) yields the 1,5-diketone derivative.17 Under these precedents, a photocycloaddition pathway promoted by photosensitization is more likely than a radical pathway promoted by oxidation of the β-diketone.
Regarding the photosensitization mechanism, the excited photocatalyst can transfer the energy either to 2a or to 3a (Scheme 2). According to the triplet energy values (ET(enol-3a) = 59 kcal mol−1;18ET(2a) = 60 kcal mol−1;9aET(1a) = 62 kcal mol−1;10aET(1d) = 60 kcal mol−110e), sensitization19 of enol-3a or styrene 2, with triplet energies in the range of 1a, is feasible. This statement was corroborated by the efficient quenching observed in the time resolved luminescence quenching experiments of 1a* with 2a and 3a (see the ESI,† Fig. S2 and S3). In addition, the lack of reactivity in the reactions with photocatalysts that are well known photosensitizers20 such as Ru(ppy)3Cl21b (ET = 46.5 kcal mol−110b,c), Ir(ppy)31c (ET = 55 kcal mol−110d) or 1e (ET = 55 kcal mol−110f), but whose triplet energies are below the triplet energies of 2a and 3a support an energy transfer as the most likely mechanism (see the ESI,† Section 4). Therefore, our proposed mechanism starts with the excitation of the photocatalyst by irradiation with blue light, followed by energy transfer to 2a or 3a, reaching the triplet excited state 3(2a)* or 3(3a)*. Then, 3(2a)* reacts with 3a, while 3(3a)* would react with 2a, leading to a common 1,4-biradical intermediate I, with the radicals in the most stable positions (α to the OH and in the benzylic position), explaining the observed regioselectivity of the final products. From this biradical intermediate, cyclobutanol II is generated, followed by retro aldol condensation affording the final product 4a (Scheme 2).
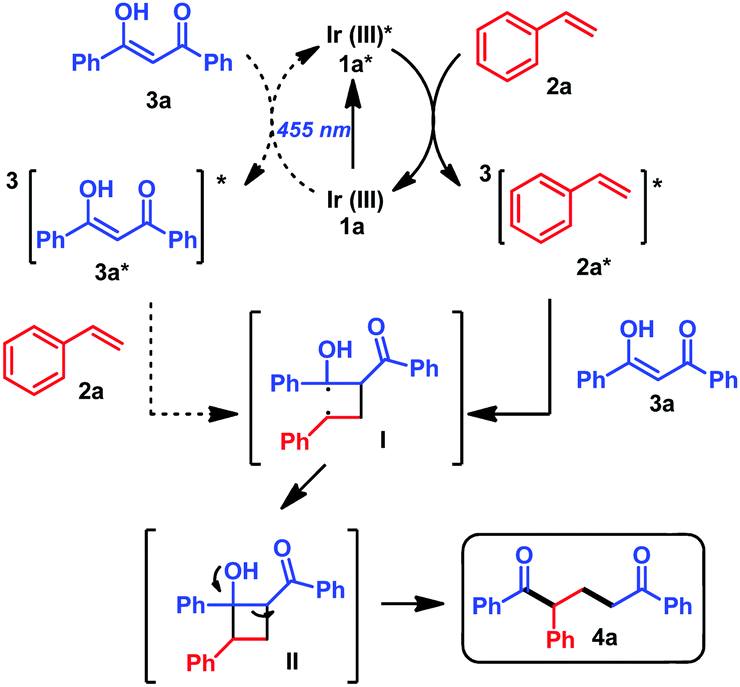 | ||
| Scheme 2 Visible light mediated photosensitized approach of the De Mayo photocycloaddition. | ||
According to the time resolved luminescence quenching experiments it is difficult to elucidate whether the energy transfer to styrene 2a or 3a would be predominant. However, 2a is 5 times more concentrated in the reaction mixture than 3a, thus energy transfer to 2a seems more likely. In addition, GC-MS analysis (see the ESI,† Fig. S10) of the reaction between 3a and 2a under the optimized conditions (entry 9, Table 1) revealed the formation of the dimerization product of styrene9c as side product of the reaction. On the other hand, the lack of reactivity with cyclohexene derivatives (4j, 4k, Table 2) that present triplet energies around 80 kcal mol−1 suggested that photosensitization of 3a is not enough to trigger the reaction (a time resolved luminescence quenching experiment of 1a with cyclohexene shows no interaction between 1a* and the olefin, see the ESI,† Fig. S4). Therefore, although photosensitization of 3a cannot be discarded as a possible mechanistic pathway, all this indirectly proves the indicated photosensitization of styrene 2a as the predominant pathway under these reaction conditions. Finally, the quantum yield of the reaction was determined to be Φ = 1.1%, suggesting that the mechanism does not contain significant radical chains.21
In conclusion, the first visible light mediated De Mayo reaction through a photosensitization mechanism has been developed. α or β-substituted styrenes undergo this reaction smoothly, but the higher triplet state energy of alkyl substituted olefins made them unreactive under this condition. β-Diketone, β-ketoester, β-cyano ketone and β-amido ester derivatives could be successfully functionalized and 7 membered rings are readily accessible using this method. Mechanistic studies support photosensitization as the key step of the reaction, and experimental observations suggested photosensitization of styrenes as the most likely mechanistic pathway.
This work was supported by the Deutsche Forschungsgemein-schaft DFG (GRK 1626, Chemical Photocatalysis). L. M. thanks the Alexander von Humboldt foundation for a postdoctoral fellowship. R. M.-H. thanks the DAAD for a short-term research grant. We thank Ms Regina Hoheisel (University of Regensburg) for her assistance in cyclic voltammetry measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. De Mayo, H. Takashita and A. B. M. A. Satter, Proc. Chem. Soc., 1962, 119 Search PubMed; P. De Mayo and H. Takeshita, Can. J. Chem., 1963, 41, 440 CrossRef CAS.
- (a) M. J. Begley, V. Mellor and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1983, 1, 1905 RSC; (b) W. Oppolzer, Acc. Chem. Res., 1982, 15, 135 CrossRef CAS; (c) M. T. Crimmins, Chem. Rev., 1988, 88, 1453 CrossRef CAS; (d) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed.
- J. D. Winkler, M. B. Rouse, M. F. Greaney, S. J. Harrison and Y. T. Jeon, J. Am. Chem. Soc., 2002, 124, 9726 CrossRef CAS PubMed.
- J. D. Winkler, C. L. Muller and R. D. Scott, J. Am. Chem. Soc., 1988, 110, 4832 CrossRef.
- J. D. Winkler, R. D. Scott and P. G. Williard, J. Am. Chem. Soc., 1990, 112, 8971 CrossRef CAS.
- J. D. Winkler, C. M. Bowen and F. Liotta, Chem. Rev., 1995, 95, 2003 CrossRef CAS.
- (a) Y.-J. Wu, in Name Reactions for Carbocyclic Ring Formations, ed. J. J. Li, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, ch. 5, pp. 451–488 Search PubMed; (b) A. C. Weedon, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and P.-S. Song, CRC Press, Boca Raton, 1995, pp. 670–684 Search PubMed.
- For reviews on visible light photocatalysis, see: (a) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (c) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 49, 1924 Search PubMed; (d) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed; (e) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307 CrossRef CAS PubMed; (f) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997 CrossRef CAS PubMed; (g) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS PubMed; (h) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed; (i) E. Meggers, Chem. Commun., 2015, 51, 3290 RSC; (j) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380 RSC; (k) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed.
- (a) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329 CrossRef CAS PubMed; (b) V. Mojr, E. Svobodoá, K. Straková, T. Neveselý, J. Chudoba, H. Dvořáková and R. Cibulka, Chem. Commun., 2015, 51, 12036 RSC; (c) S. K. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072 RSC; (d) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807 CrossRef CAS PubMed; (e) F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jacob and T. Bach, Angew. Chem., Int. Ed., 2017, 57, 827 CrossRef PubMed ; for examples of enantioselective [2+2] photocycloaddition via triplet sensitization see: ; (f) R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368 CrossRef CAS PubMed; (g) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed; (h) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891 CrossRef CAS PubMed.
- For values of Eox and ET of 1a see: (a) D. Hanss, J. C. Freys, G. Bernardinelli and O. S. Wenger, Eur. J. Inorg. Chem., 2009, 4850 CrossRef CAS ; for 1b see: ; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156 CrossRef CAS PubMed ; for 1c see; (d) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigel-leti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS ; for 1d see: ; (e) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS ; for 1e see: ; (f) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC; (g) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- E T (cyclohexene) = 81.2 kcal mol−1, ET (1-methyl-1-cyclohexene) = 75.2 kcal mol−1. For values of the ET of different olefines see: (a) T. Ni, R. A. Caldwell and L. A. Melton, J. Am. Chem. Soc., 1989, 111, 457 CrossRef CAS; (b) G. V. Luokova, S. E. Starodubova and V. A. Smirnov, Khim. Vys. Energ., 2007, 41, 434 Search PubMed.
- (a) Z.-F. Xie, H. Suemune and K. Sakai, Synth. Commun., 1989, 19, 987 CrossRef CAS; (b) C.-J. Li, D.-L. Chen, Y.-Q. Lu, J. X. Haberman and J. T. Mague, Tetrahedron, 1998, 54, 2347 CrossRef CAS; (c) B. C. Hong, S. H. Chen, E. S. Kumar, G.-H. Lee and K.-J. Lin, J. Chin. Chem. Soc., 2003, 50, 917 CrossRef CAS; (d) C. Roscini, D. M. E. Davies, M. Berry, A. J. Orr-Ewing and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2008, 47, 2283 CrossRef CAS PubMed.
- (a) S. Tobita, J. Ohba, K. Nakagawa and H. J. Shizuka, J. Photochem. Photobiol., A, 1995, 92, 61 CrossRef CAS; (b) M. Moriyasu, A. Kato and Y. Hashimoto, J. J. Chem. Soc., Perkin Trans. 2, 1986, 515 RSC.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714 CAS.
- J. I. G. Cadogan, D. H. Hey and J. T. Sharp, Proc. Chem. Soc., 1964, 142 CAS.
- For an example of the oxidative coupling of dibenzoyl methane and styrene see: (a) B. M. Casey, C. A. Eakin, J. Jiao, D. V. Sadasivam and R. A. Flowers, Tetrahedron, 2009, 65, 10762 CrossRef CAS PubMed ; for an additional example of the oxidative coupling of β-diketones or β-ketoesters and styrene derivatives see: ; (b) T. Y. Ko and S. W. Youn, Adv. Synth. Catal., 2016, 358, 1934 CrossRef CAS.
- For an example of the photochemical reaction between dibenzoyl methane and styrene see: P.-F. Casals, J. Ferard and R. Ropert, Tetrahedron Lett., 1976, 35, 3077 CrossRef.
- A. Kikuchi, N. Oguchi and M. Yagi, J. Phys. Chem. A, 2009, 113, 13492 CrossRef CAS PubMed.
- (a) N. J. Turro, J. Chem. Educ., 1966, 43, 13 CrossRef CAS; (b) W. L. Dilling, Chem. Rev., 1969, 69, 845 CrossRef CAS; (c) N. J. Turro, in Modern Molecular Photochemistry, Benjamin/Cummings, California, 1978, ch. 9, pp. 296–359 Search PubMed; (d) A. Albini, Synthesis, 1981, 249 CrossRef CAS.
- (a) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS; (b) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07044j |
| This journal is © The Royal Society of Chemistry 2018 |