
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis and engineering of substrate shuttling by the acyl carrier protein (ACP) in fatty acid synthases (FASs)†
Emanuele
Rossini‡
 a,
Jan
Gajewski‡
b,
Maja
Klaus
b,
Gerhard
Hummer
*ac and
Martin
Grininger
*b
a,
Jan
Gajewski‡
b,
Maja
Klaus
b,
Gerhard
Hummer
*ac and
Martin
Grininger
*b
aDepartment of Theoretical Biophysics, Max Planck Institute of Biophysics, Max-von-Laue-Straße 3, 60438 Frankfurt am Main, Germany. E-mail: gerhard.hummer@biophys.mpg.de
bInstitute of Organic Chemistry and Chemical Biology, Buchmann Institute for Molecular Life Sciences, Goethe University Frankfurt, Max-von-Laue-Str. 15, 60438 Frankfurt am Main, Germany. E-mail: grininger@chemie.uni-frankfurt.de
cInstitute of Biophysics, Goethe University Frankfurt, Max-von-Laue-Straße 9, 60438 Frankfurt am Main, Germany
First published on 25th September 2018
Abstract
Perturbations of domain–domain interactions impact the function of type I fatty acid synthases. We identify interface point mutations that modulate fatty acid chain lengths, and explain their effect in changes of domain–domain binding energetics. Engineering of similar interfaces in related megasynthases may be exploited for custom synthesis of natural products.
Living cells produce fatty acids (FA) in an iterative yet tightly controlled process (Fig. 1A).1–4 As the decisive step in chain length control by fatty acid synthase (FAS), the fully reduced acyl chain, bound to the acyl carrier protein (ACP) (Fig. 1B),5 is either transferred to the condensing ketoacyl synthase (KS) for another elongation, or released by a transferase (MPT) or a thio-esterase (TE) (Fig. S1, ESI†).6–8 In bacterial and fungal multi-enzyme (type I) FASs, chain length is controlled by the competitive substrate acceptance of the KS domain and the MPT domain, and as such is influenced by the interfaces ACP:KS and ACP:MPT.9,10 We recently engineered the G2559S-M2600W-mutated Corynebacterium ammoniagenes FAS, hereafter termed FASGSMW, to produce a bimodal spectrum of C8- and C14/C16-CoA in vitro.11,12 The two mutations of FASGSMW are located in the KS-binding channel, where they attenuate the loading of octanoyl in the KS binding channel and promote the off-loading of the acyl chain by the substrate-promiscuous MPT to produce C8-CoA. With its bimodal product spectrum, the FASGSMW mutant is a well-suited reporter system for elaborating the impact of domain–domain interactions (DDIs) in substrate shuttling (Fig. 1C). We expected shifts in the proportion of C8- and C14/C16-CoA by perturbing the interfaces between the carrier protein ACP with domains KS (ACP:KS) and MPT (ACP:MPT). Mutations decreasing the affinity between the ACP and KS domains should steer synthesis towards short-chain products by promoting the release from the MPT domain. Conversely, increased ACP:KS affinity should decrease the share of short-chain products. In the following, we report data collected on the ACP:KS interface. For ACP:MPT interface engineering, see Note S1, and Fig. S2, S3 (ESI†).
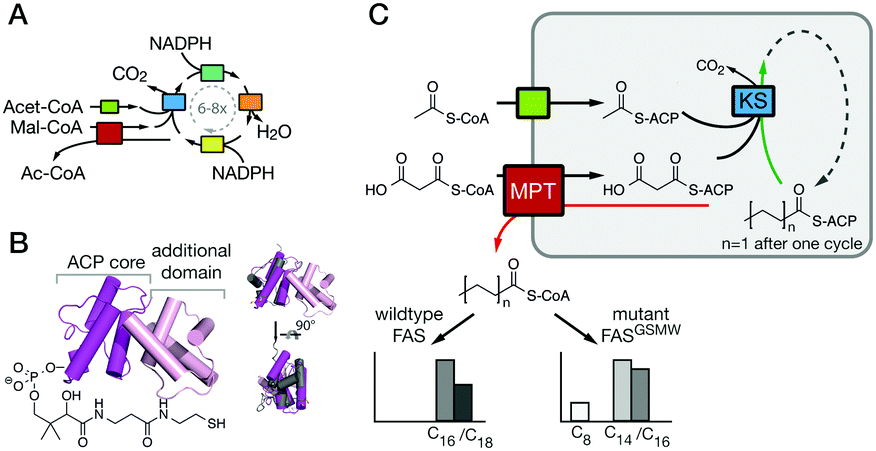 | ||
| Fig. 1 FA synthesis. (A) Iterative type I bacterial and fungal FA synthesis produces acyl-CoA esters. (B) Phosphopantetheinylated ACPs covalently bind substrates and intermediates. ACPs of type I bacterial and fungal FASs are comprised of two domains (yeast FAS ACP magenta; pdb 2uv817), in contrast to other ACPs (rat FAS ACP in grey; 2png25). (C) Modulated chain length regulation in FASGSMW induces a bimodal product spectrum. | ||
In light of the need for microbial short-chain FA production,12–14 we aimed at mutating the ACP:KS interface to shift the FASGSMW product spectrum towards short-chain products. To modulate the ACP:KS interface, mutations were introduced on the surface of the KS domain only, since an intact ACP leaves other DDIs during synthesis unaffected (Fig. 2). We selected mutation sites D2553, D2556 and N2557 on the KS domain surface to target interactions previously identified for FabB, a KS homologous protein in E. coli,15 and its ACP. Mutation site A2696 was identified from a crosslinking study on FabF, a KS homologue in E. coli.16 On the basis of the crystal structure of the S. cerevisiae FAS with ACP docked to the KS domain,17,18 we selected two novel interaction sites, N2621 and D2622 (Fig. 2 and Table S1, ESI†). The selected residues were mutated to alanine and/or to residues with opposite charges. The mutant proteins were analyzed in activity and product spectra (ESI,† Material and methods).
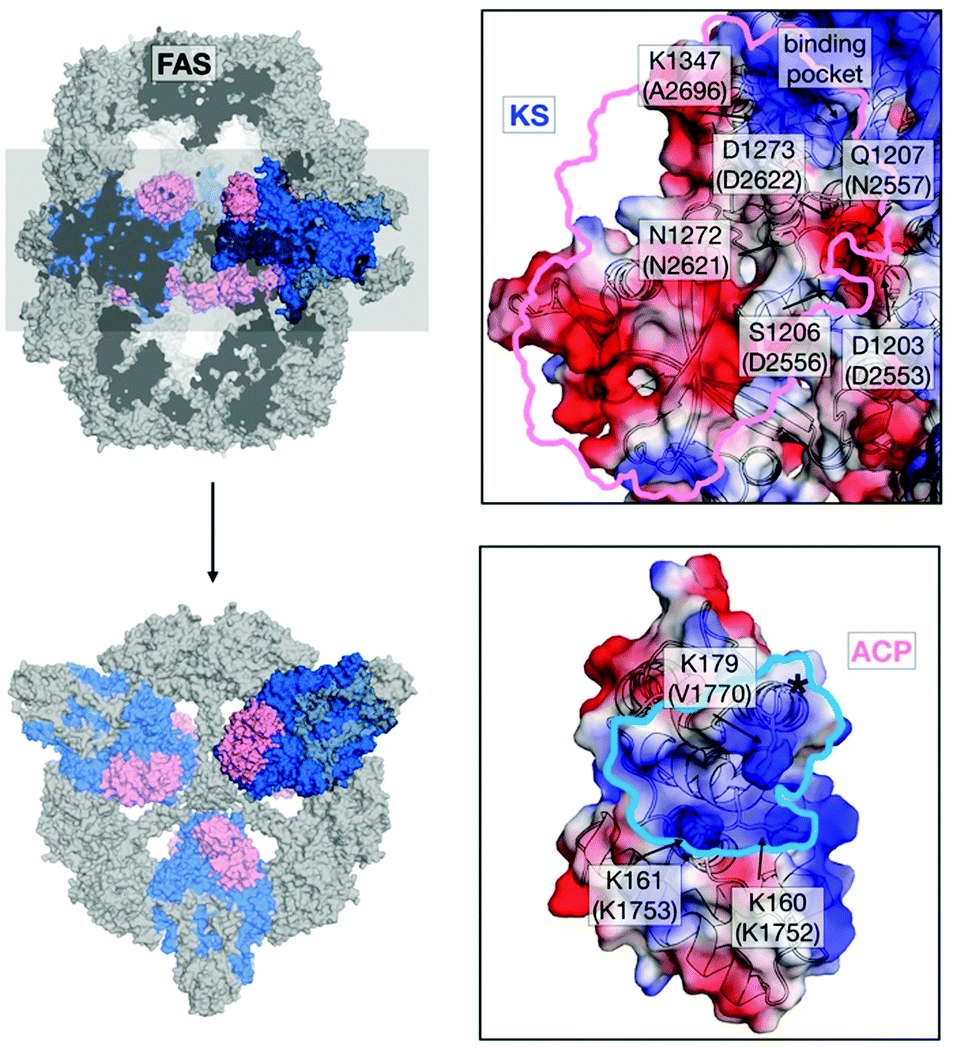 | ||
| Fig. 2 Interface engineering. Structural overview of fungal FAS (pdb: 3hmj26) depicted as cross-section along the threefold axis and in substructures as indicated. ACP and KS are colored according to its vacuum electrostatics (generated with PyMOL; red, negatively charged; blue, positively charged). Active-site serine of ACP is highlighted (*). KS mutations are indicated in S. cerevisiae FAS numbering and in C. ammoniagenes FAS equivalent numbering in brackets. | ||
Our data clearly demonstrate the sensitivity of substrate shuttling in FAS to mutations in the ACP:KS interface. For several constructs, a significant increase in the fraction of short acyl-CoA esters was observed (Fig. 3A). Mutations N2557E, N2621D and D2622A were most efficient in C8-CoA production, yielding up to 77% C8-CoA of all detected CoA esters. The constructs with the highest yields in short-chain acyl-CoA were most severely impaired in their activity; i.e., N2557E, N2621D and D2622A showed only a fraction of the specific activity of the template construct (Fig. 3B). The correlation of selective short acyl-CoA production and poor activity implies that changes in the product spectrum result from the suppression of specific DDIs. D2553A, D2553N and D2556A showed effects opposite to what was anticipated (Fig. 3A), and increased the fraction of long chain acyl-CoA.
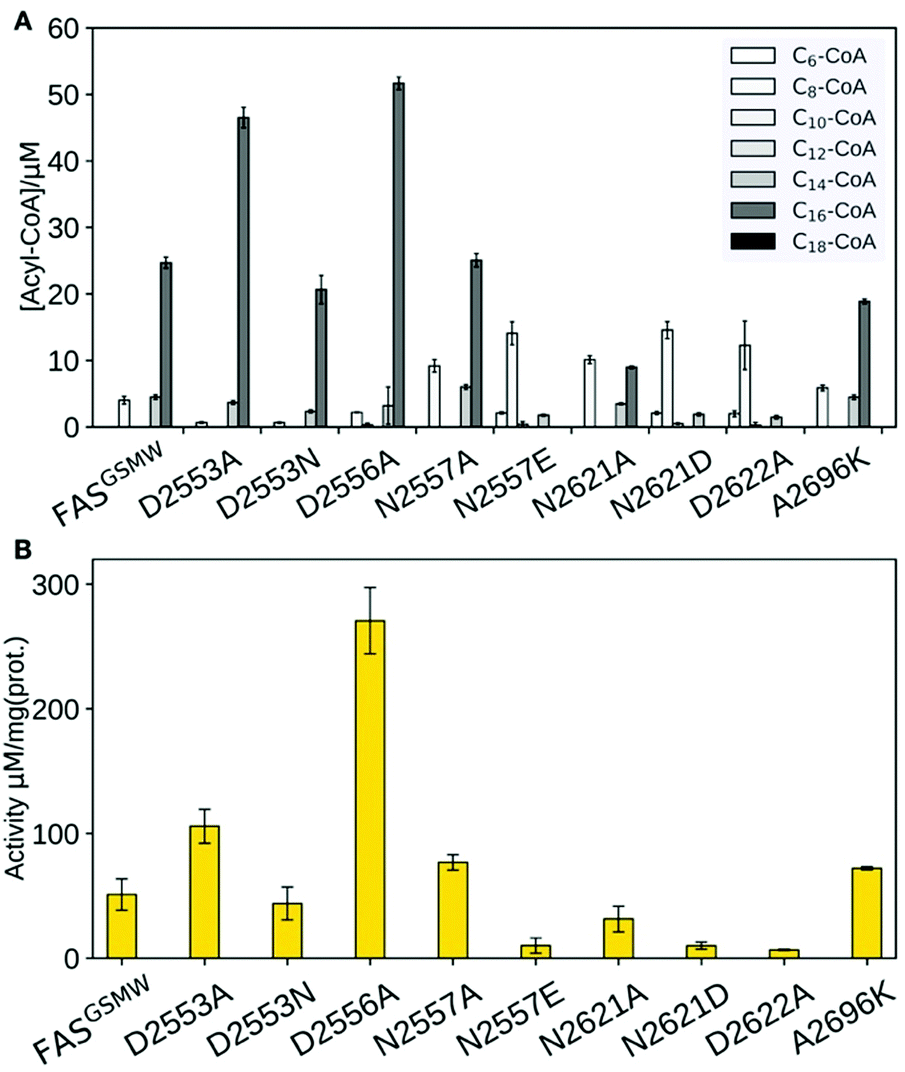 | ||
| Fig. 3 Product spectra and overall enzymatic activity of FAS. (A) Product spectra of KS surface mutations introduced in FASGSMW. Error bars show the standard deviation of three assays for each construct. (B) Overall fatty acid synthetic activities of FAS constructs. Error bars show the standard deviation of three assays for each construct. | ||
We used structure-based homology modeling to gain a deeper understanding of the relation between ACP:KS domain interactions. We constructed three-dimensional models for ACP:KS using the S. cerevisiae FAS crystallographic structure (pdb: 2uv8) as a template (Note S2 and Fig. S4–S8, ESI†).17 High crystallographic B-factors and low sequence conservation in a loop between K160 and P167 of ACP suggested local structural disorder (Fig. 4A). To account for possible flexibility in this region, we constructed two different models (A and B). In model A, we fully retained the secondary structure of the template. In model B, we locally relaxed the secondary structure restriction to account for a variation in bacterial and fungal systems (Fig. 4B). The multiple sequence alignment indicates that target loop residues V1753 and R1754 correspond to template K161 and S162, respectively (S. cerevisiae FAS numbering (2uv8); Fig. S5, ESI†). In S. cerevisiae FAS, K161 forms a salt bridge with KS D1203 that is thus not maintained in C. ammoniagenes, with V1753 and D2553 at the positions of K161 and D1203. R1754 in the ACP loop emerges as a possible salt-bridge partner of D2553 on KS. Model B enables this missing non-covalent interaction at the interface. After molecular dynamics (MD) relaxation, both models A and B remained close to the yeast crystal structure, with Cα backbone Root Mean Square Deviations (RMSD) of 0.74 Å and 0.93 Å, respectively (Fig. 4B). For models A and B, we performed binding-energy calculations using structures sampled in MD simulations of FASGSMW and of surface mutants, as obtained by modifying the wild type model.
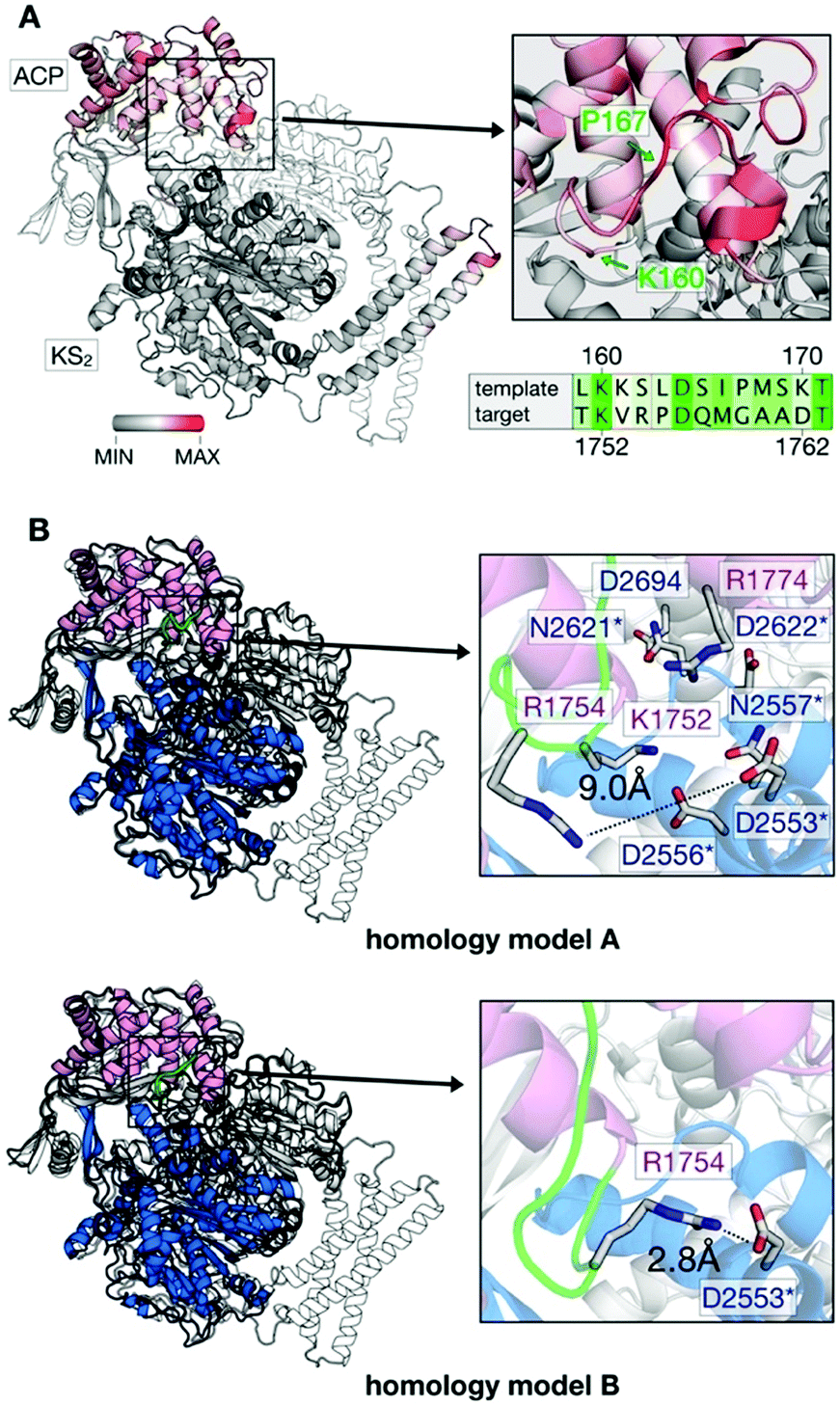 | ||
| Fig. 4 Generation of in silico models. (A) B factor representation of experimental template structure (B-factor range between 0 and 202). ACP binds to dimeric KS (KS2) as indicated (protomer colored in white). Close-up of the ACP loop between K160 and P167, and sequence alignment (green coloring reflects similarity (Fig. S5, ESI†)). (B) Structural comparison of template (S. cerevisiae) and target (C. ammoniagenes) ACP:KS complexes. S. cerevisiae FAS template shown in gray and C. ammoniagenes FAS in magenta (ACP) and blue/white (KS2). The ACP loop between K1752 and G1759 of ACP is highlighted in green. Models A and B differ in interactions between the interface residues R1774 and D2553 (see Note S2 for more details on the modeling approach, ESI†). | ||
The calculated binding affinities correlate both with the measured FAS activities and the observed product spectra (Fig. 5A). Strong ACP:KS binding is associated with the production of long acyl chains and high FAS activity. The good correlation implies that the stability of the ACP:KS interaction indeed modulates C. ammoniagenes FAS function, which allows us to establish detailed relations between structure, energetics and FAS product spectrum.
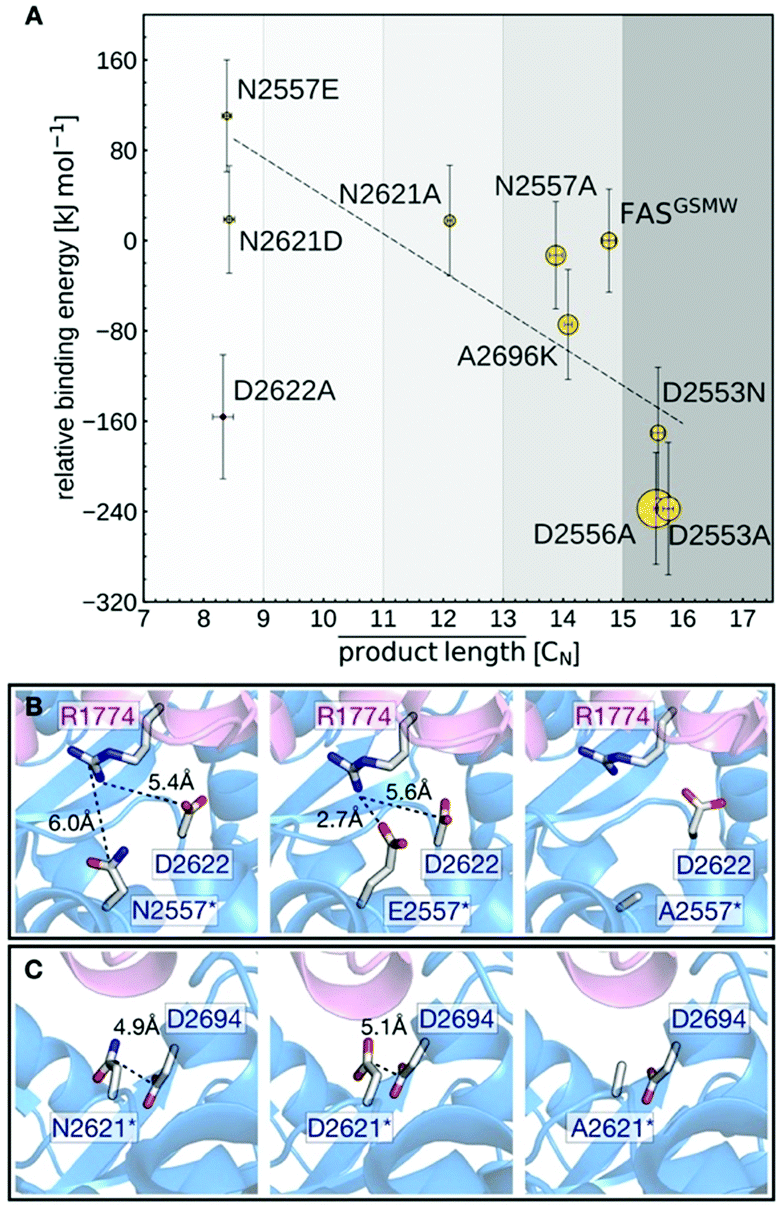 | ||
| Fig. 5 Analysis of mutated ACP:KS interfaces. (A) Calculated binding free energies (vertical axis) and measured FAS enzymatic activity (symbol radius) versus mean acyl chain length of the FAS product spectra. Error bars reflect the standard deviations from the experimental replicates (horizontal) and binding energy computations (vertical). (B) Interaction network of interface residue N2557 in the native (left) and N2557E-mutated (middle) and N2557A-mutated (right) interfaces. (C) Interface residue N2621 in the native (left) and N2621D-mutated (middle) and N2621A-mutated (right) interfaces. | ||
Mutants N2557E and N2621D are most effective in producing C8-CoA and display the highest (i.e., least favorable) relative binding energies. This can be explained by mutations introducing negative charges at the DDI that affect a network of electrostatic interactions across residues 2557, 2621, 2622 and 2694 (KS) and 1774 (ACP) (Fig. 5B and C). In the unmodified interface, the polar residue R1774 is located at distances of 6.0 Å and 5.4 Å from N2557 and D2622, respectively. By contrast, the mutant E2557 sequesters R1774 in a salt-bridge interaction. The resulting charge imbalance around D2622 weakens the electrostatic interaction network and the interface stability (Fig. 5B).
Similarly, the change in charge of N2621D disrupts the electrostatic interaction between N2621 and D2694 (Fig. 5C). Both mutations lower FAS enzymatic activity. By contrast, mutations N2557A and N2621A do not alter the charge balance of the interface and have no effect on FAS function (Fig. 5B and C). A2696 is located at the interface periphery and its mutations do not trigger the modulation of FAS function (Note S3 and Fig. S9, ESI†). Mutants D2553A, D2553N and D2556A revert the FASGSMW spectrum to long chain production and display the highest (i.e., most favorable) relative binding energies. Interestingly, the energy shift of D2553A is only captured when combining the two alternative configurations of model A (the mutant) and model B (the wild-type). In such a scheme, the polarity of position 2553 (KS) influences the orientation of R1754 (ACP) that forms a salt-bridge with D2553 and is repelled by A2553 (Note S4 and Fig. S10, ESI†). Mutant D2553N, despite a possible change in charge, has a minor impact on R1754 orientation (Note S4 and Fig. S10, ESI†). In D2556A, the negative charge deletion leads to a minor but effective structural reorganization of the electrostatic network at the interface (Note S5 and Fig. S11, ESI†). In case of the charged-to-neutral mutation D2622A at the center of a network of electrostatic interactions, the limited sampling of motions in the computation of binding affinities is likely insufficient to capture a major reorganization of the interface, providing an explanation for this single outlier (Note S6 and Fig. S12, ESI†).
As a further challenge to modeling substrate shuttling, binding energies are not the only determinant of DDI kinetics. Effects of accessibility and confinement will impact the frequency and duration of productive interaction between ACP and KS. As a first step towards capturing substrate shuttling kinetics, we set up coarse-grained simulations to count frequencies of competitive binding events of ACP to wild type and surface mutated KS (Note S7 and Fig. S13, ESI†).19 The kinetic approach did, however, not lead to conclusive data, reflecting both the challenges in simulating binding kinetics in a confined environment and the paucity of experimental information required for model generation.
As its main result, this study shows that single mutations at domain–domain interfaces can severely perturb substrate shuttling in FASs. In silico modeling demonstrates that mutations are particularly invasive when inducing charge imbalances at the interfaces, which agrees with earlier finding on the importance of electrostatic complementarity of ACP:catalytic domains interfaces.5,17,20 Engineering of DDIs proved to be powerful in modulating the chain length spectrum of C. ammoniagenes FAS, and some mutations, i.e., N2557E, N2621D and N2622A, produced C8-CoA with high selectivity. However, the strong decrease in activity of the mutated FASs questions a true biotechnological relevance of DDI engineering in FASs, and rather illustrates an inherent “low-resolution” problem of the approach. While engineering of substrate binding channels12 or the thioesterase-mediated hydrolyzation of acyl-ACP of certain chain length13,21,22 hijacks FA synthesis at a specific acyl-ACP chain length, ACP surface mutations interfere already in initial FA cycles, causing the overall drop in activity. Extensive and refined27 mutation strategies may alleviate or overcome these problems. The observed sensitivity of substrate shuttling is also interesting for the related PKSs. Similar to FASs, engineering of substrate shuttling may enable modulation of product synthesis in iterative polyketide synthases (PKSs).23 In modular PKSs, ACPs are involved in the substrate shuttling within one multienzyme complex (module), but also in the translocation of the substrate to a downstream module. The strong influence by even single mutations implies that a successful assembly of such modules for the design of new chimeric biosynthetic pathways will strongly rely on the effective adaptation of interfaces across the borders of the chimeric assembly lines.24
This work was supported by a Lichtenberg grant of the Volkswagen Foundation to M. G. (grant number 85701). Further support was received from the LOEWE program (Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz) of the state of Hesse conducted within the framework of the MegaSyn Research Cluster and by the Max Planck Society. E. R. acknowledges Dr Ramachandra Bhaskara for many useful inputs and valuable comments. Open Access funding provided by the Max Planck Society.
Conflicts of interest
Concerning competing financial interest, J. G. and M. G. declare that they are inventors of two patents concerning the production of short fatty acids (EP patent application 15 162 192.7 filed on April 1st, 2015 and EP patent application 15 174 342.4 filed on June 29th, 2015).Notes and references
- E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68(3), 501–517 CrossRef CAS PubMed.
- M. Grininger, Curr. Opin. Struct. Biol., 2014, 25, 49–56 CrossRef CAS PubMed.
- K. Finzel, D. J. Lee and M. D. Burkart, ChemBioChem, 2015, 16(4), 528–547 CrossRef CAS PubMed.
- T. Maier, M. Leibundgut, D. Boehringer and N. Ban, Q. Rev. Biophys., 2010, 43, 373–422 CrossRef CAS PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29(10), 1111–1137 RSC.
- E. Schweizer and H. Bolling, Proc. Natl. Acad. Sci. U. S. A., 1970, 67(2), 660–666 CrossRef CAS.
- H. Engeser, K. Hubner, J. Straub and F. Lynen, Eur. J. Biochem., 1979, 101(2), 413–522 CrossRef CAS PubMed.
- W. Pirson, L. Schuhmann and F. Lynen, Eur. J. Biochem., 1973, 36(1), 16–24 CrossRef CAS PubMed.
- M. Sumper, D. Oesterhelt, C. Riepertinger and F. Lynen, Eur. J. Biochem., 1969, 10(2), 377–387 CrossRef CAS PubMed.
- A. Kawaguchi, K. Arai, Y. Seyama, T. Yamakawa and S. Okuda, J. Biochem., 1980, 88(2), 303–306 CrossRef CAS PubMed.
- J. Gajewski, F. Buelens, S. Serdjukow, M. Janssen, N. Cortina, H. Grubmuller and M. Grininger, Nat. Chem. Biol., 2017, 13, 363–365 CrossRef CAS PubMed.
- J. Gajewski, R. Pavlovic, M. Fischer, E. Boles and M. Grininger, Nat. Commun., 2017, 8, 14650 CrossRef PubMed.
- Y. J. Choi and S. Y. Lee, Nature, 2013, 502(7472), 571–574 CrossRef CAS PubMed.
- L. d'Espaux, D. Mendez-Perez, R. Li and J. D. Keasling, Curr. Opin. Chem. Biol., 2015, 29, 58–65 CrossRef PubMed.
- J. G. Borgaro, A. Chang, C. A. Machutta, X. Zhang and P. Tonge, J. Biochem., 2011, 50(49), 10678–10686 CrossRef CAS PubMed.
- Z. Ye and G. Williams, J. Biochem., 2014, 53(48), 7494–7502 CrossRef CAS PubMed.
- M. Leibundgut, S. Jenni, C. Frick and N. Ban, Science, 2007, 316(5822), 288–290 CrossRef CAS PubMed.
- P. Johansson, B. Wiltschi, P. Kumari, B. Kessler, C. Vonrhein, J. Vonck, D. Oesterhelt and M. Grininger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12803–12808 CrossRef CAS PubMed.
- Y. C. Kim and G. Hummer, J. Mol. Biol., 2008, 375(5), 1416–1433 CrossRef CAS PubMed.
- C. Anselmi, M. Grininger, P. Gipson and J. Faraldo-Gomez, J. Am. Chem. Soc., 2010, 132, 12357–12364 CrossRef CAS PubMed.
- C. Leber and N. A. Da Silva, Biotechnol. Bioeng., 2014, 111(2), 347–358 CrossRef CAS PubMed.
- Z. Zhu, Y. J. Zhou, A. Krivoruchko, M. Grininger, Z. K. Zhao and J. Nielsen, Nat. Chem. Biol., 2017, 13, 360–362 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- M. Klaus and M. Grininger, Nat. Prod. Rep., 2018 10.1039/c8np00030a.
- E. Ploskon, C. Arthur, S. Evans, C. Williams, J. Crosby, T. Simpson and M. Crump, J. Biol. Chem., 2008, 283, 518–528 CrossRef CAS PubMed.
- P. Johansson, B. Mulinacci, C. Koestler, R. Vollrath, D. Oesterhelt and M. Grininger, Structure, 2009, 17(8), 1063–1074 CrossRef CAS PubMed.
- S. Schneider, D. Provasi and M. Filizola, Biochemistry, 2016, 55, 6456–6466 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational methods are given. See DOI: 10.1039/c8cc06838k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |