
Control of tandem isomerizations: flow-assisted reactions of o-lithiated aryl benzyl ethers†
Hyune-Jea
Lee‡
 a,
Heejin
Kim‡
b,
Jun-ichi
Yoshida
*b and
Dong-Pyo
Kim
*a
a,
Heejin
Kim‡
b,
Jun-ichi
Yoshida
*b and
Dong-Pyo
Kim
*a
aCentre for Intelligent Microprocess of Pharmaceutical Synthesis, Department of Chemical Engineering, POSTECH (Pohang University of Science and Technology), Pohang, 790-784, South Korea. E-mail: dpkim@postech.ac.kr
bDepartment of Synthetic and Biological Chemistry Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: yoshida@sbchem.kyto-u.ac.jp
First published on 19th December 2017
Abstract
Tandem chemical changes are often difficult to control at will, because they proceed rapidly through multiple unstable reactive intermediates. It is desirable to develop a novel method for controlling such tandem changes to obtain desired products with high selectivity. Herein, we report a flow microreactor platform for controlling tandem isomerizations of o-lithiated aryl benzyl ethers based on precise residence time control.
The control of isomerization of reactive intermediates1 is an attractive research objective which will enhance product selectivity in chemical synthesis. In cases where an intermediate isomerizes rapidly to give another intermediate, the yield and selectivity of products depend on the controllability of the reaction time and temperature. In general, by using a batch-type reactor such as a flask, an isomerization is often hard to control, because it often occurs intramolecularly and is very fast even at very low temperature. However, by using flow chemistry based on microfluidics,2,3 the reaction time can be reduced to milliseconds or less.4 Therefore, intermediates can be used selectively at will by choosing the reaction time precisely. Based on this concept, there have been many reports on the control of a rapid isomerization5 and rearrangement6 of intermediates, as well as decomposition.7,8 To the best of our knowledge, however, there has been no attempt on the control of tandem isomerizations involving multiple reactive intermediates using flow microreactors. Moreover, many well-known anionic reactions such as the Favorskii rearrangement and benzilic acid rearrangement proceed through multiple unstable intermediates in organic synthesis. Thus, the study of the control of tandem isomerizations leads to deep understanding of such reactions.
Herein we report a conceptual study aimed at the complete control of tandem isomerizations involving three chemical intermediates using a flow microreactor system (Scheme 1). The sequential reactions of o-lithiated aryl benzyl ethers in the flask were studied by Barluenga and his co-workers.9 However, the complete control of these isomerizations was not achieved in conventional flasks, because tandem reactions including an intramolecular H–Li exchange10 and [1,2]-Wittig rearrangement11 were so fast. In this study, we prove that tandem isomerizations of o-lithiated aryl benzyl ethers can be effectively controlled by varying the residence time in a flow-assisted manner. Furthermore, the dual functionalization of aryl benzyl ether was successfully accomplished via sequential halogen–lithium exchange and deprotonation reactions.
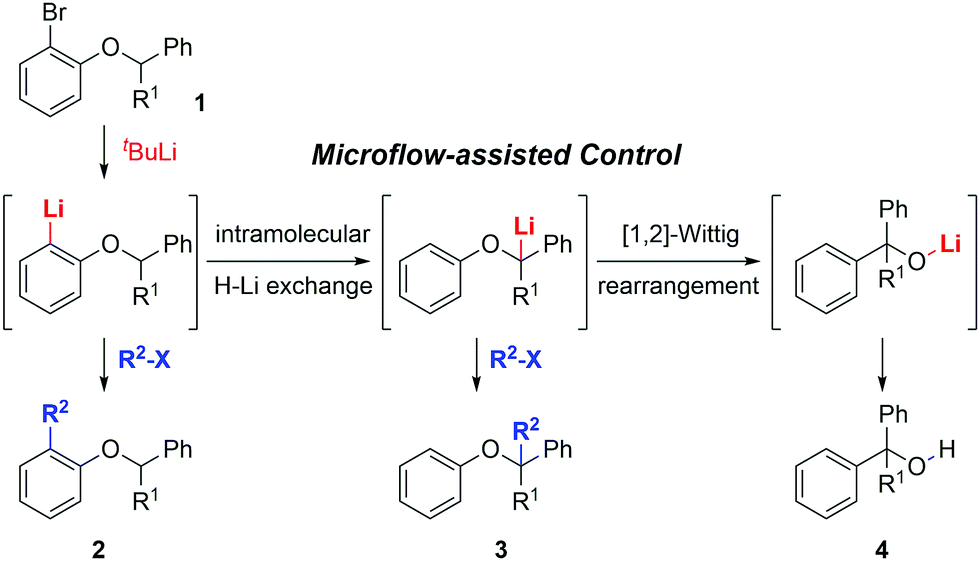 | ||
| Scheme 1 Control of tandem isomerization of o-lithiated aryl benzyl ethers involving three intermediates. | ||
In preliminary studies using a flask, we carried out the sequential reaction of compound 1a as a starting material. Compound 1a was lithiated using 2 equiv. of t-BuLi at −78 °C for 10 min, and methyl iodide was added. A mixture of compounds 2a and 3a was obtained in 48% and 42% respectively (Table S1, ESI†). These results indicate that the control of intramolecular H–Li exchange was not efficient. When the reaction time was increased to 1 h, 3a was obtained as the sole product, while compound 2a was not observed at all (Table S1, ESI†). The result indicated that the Br–Li exchange reaction and intramolecular H–Li exchange reaction were complete within 1 h even at −78 °C.
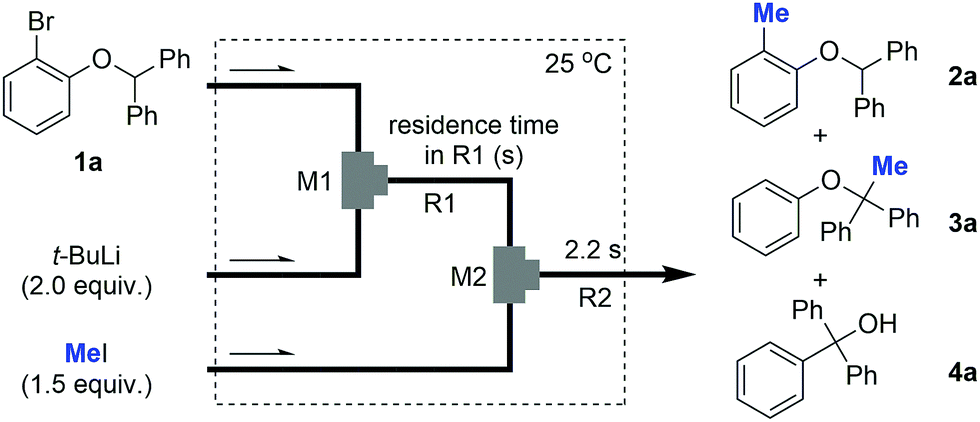
Next, we conducted the same reactions using flow microreactors including two micromixers (inner diameter of M1: 250 μm, M2: 500 μm) and two microtube reactors (R1 and R2) as illustrated in Table 1. A solution of 1a and t-BuLi was reacted in M1 and R1 and the resulting solution was reacted with methyl iodide in M2 and R2. The reaction temperature was kept at 25 °C and the residence time in R1 was changed from 3 ms to 6.3 s by changing the diameter and length of the microtube reactor (entries 1–6, Table 1). The results clearly show the progress of the H–Li exchange reaction with time. With the residence time of 3 ms, 2a was obtained in excellent yield (96%) (entry 1, Table 1). An increase in the residence time causes a decrease in the yield of 2a and an increase of the yield of 3a. With a residence time of 6.3 s, 3a was obtained in 96% yield as a sole product (entry 6, Table 1). In addition, the solution obtained by the flow reaction for 6.3 s was further reacted in a flask for 1 h at 50 °C to obtain compound 4a in 74%, which was derived from [1,2]-Wittig rearrangement (entry 7, Table 1). These results indicate that the optimization of the residence time and temperature using a flow-assisted microreactor guarantees the high controllability of fast tandem isomerizations.
|
|
||||
|---|---|---|---|---|
| Entry | Residence time in R1 [s] | Yield of 2aa [%] | Yield of 3aa [%] | Yield of 4aa [%] |
| a Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. b The resulting solution from R2 was stirred in a flask for 1 h at 50 °C. For details, see the ESI. | ||||
| 1 | 0.003 | 96 | 0 | 0 |
| 2 | 0.016 | 93 | 5 | 0 |
| 3 | 0.25 | 78 | 20 | 0 |
| 4 | 0.63 | 60 | 38 | 0 |
| 5 | 3.1 | 6 | 91 | 0 |
| 6 | 6.3 | 0 | 96 | 0 |
| 7b | 1b h | 0 | 0 | 74 |
Moreover, in addition to 1a, we conducted flow reactions of various aryl benzyl ethers (Fig. 1a), including 1b (Ar = naphthyl, R = phenyl), 1c (Ar = phenyl, R = hydrogen) and 1d (Ar = naphthyl, R = hydrogen) at 25 °C (Fig. 1b–d). In the case of the reaction of 1b, both H–Li exchange and [1,2]-Wittig rearrangement seemed to be faster than those of 1a (Fig. 1b), presumably because of the enhanced reactivity of naphthyllithium by peri-interaction.12 Even with a residence time of 3 ms, a mixture of 2b (77%) and 3b (22%) was obtained.
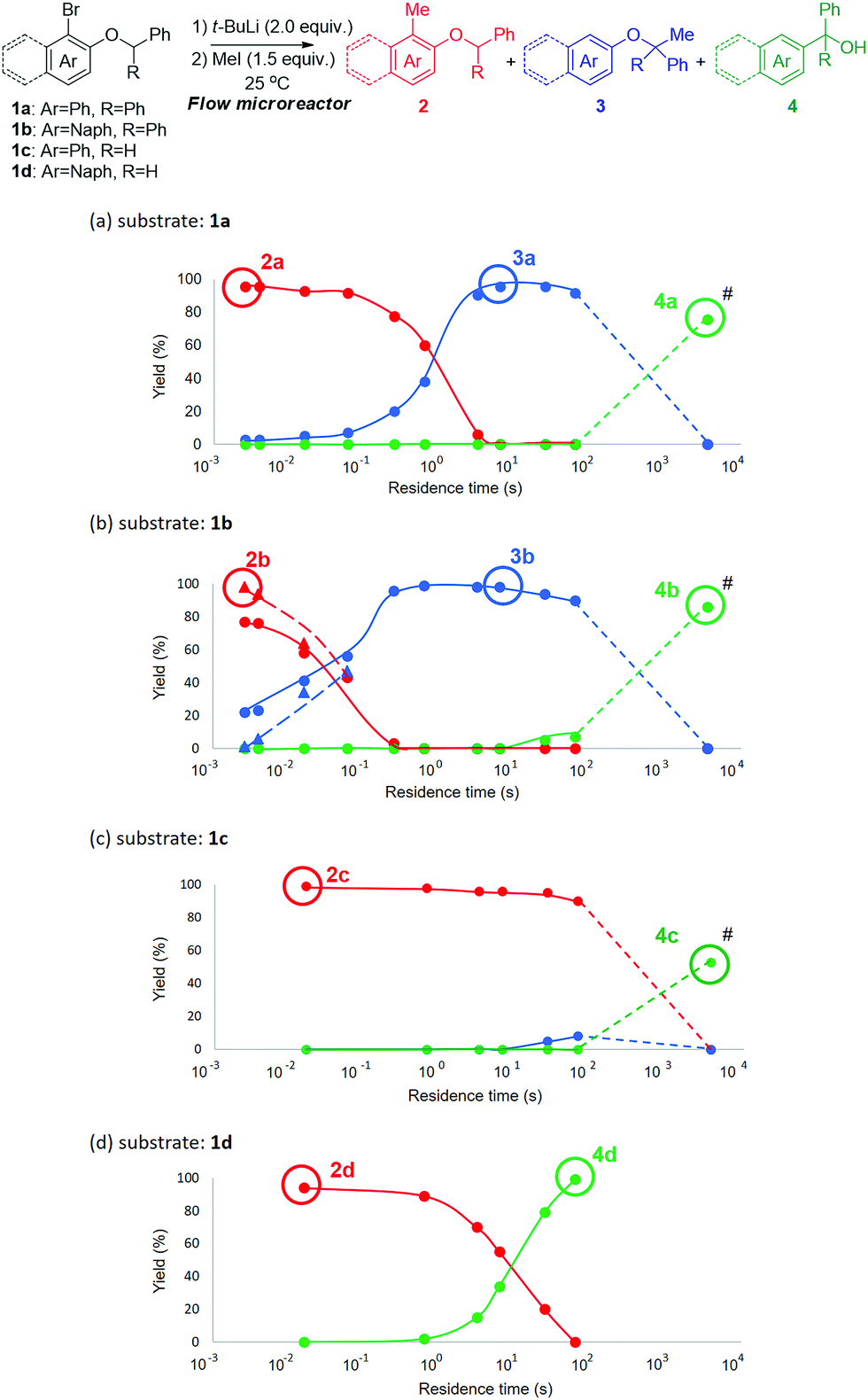 | ||
| Fig. 1 Tandem isomerizations of various aryl benzyl ethers by changing the residence time in R1 in a flow microreactor system for products 2, 3 and 4d, followed by further stirring in a flask at 50 °C for 1 h to obtain products 4a–c (#). Methyl triflate was used as an electrophile (symbol ▲ with a dashed line), instead of methyl iodide (symbol ● with a solid line). | ||
On the other hand, 2b was obtained in 98% yield when methyl triflate was used as an electrophile (dashed lines, Fig. 1b) instead of methyl iodide. This means that the trapping with methyl iodide competes with the H–Li exchange. However, the trapping with methyl triflate is faster than the H–Li exchange. In the reaction of 1c (Fig. 1c), the H–Li exchange was slow, probably because the acidity of the benzylic proton is much lower. Even with a residence time of 63 s, only a small amount of 3c was obtained. When 1d was used for the reaction (Fig. 1d), the H–Li exchange is somewhat slower than that of 1a; however, the [1,2]-Wittig rearrangement is much faster than that of 1a. In this case, the [1,2]-Wittig rearrangement is faster than the H–Li exchange, and therefore 3d was not obtained with any residence time. With longer residence times, 4d was obtained in a good yield but at the expense of 2d.
The rate of the intramolecular H–Li exchange increases in the order 1c < 1d < 1a < 1b. This tendency is consistent with the acidity of protons (R = Ph or H) and reactivity by peri-interaction. The rate of the [1,2]-Wittig rearrangement increases in the order 1a and 1c < 1b < 1d. The stabilization effect of radicals (Ar = Ph or Naph) seems to be responsible for this tendency. These tendencies are consistent with a reported reaction mechanism.13
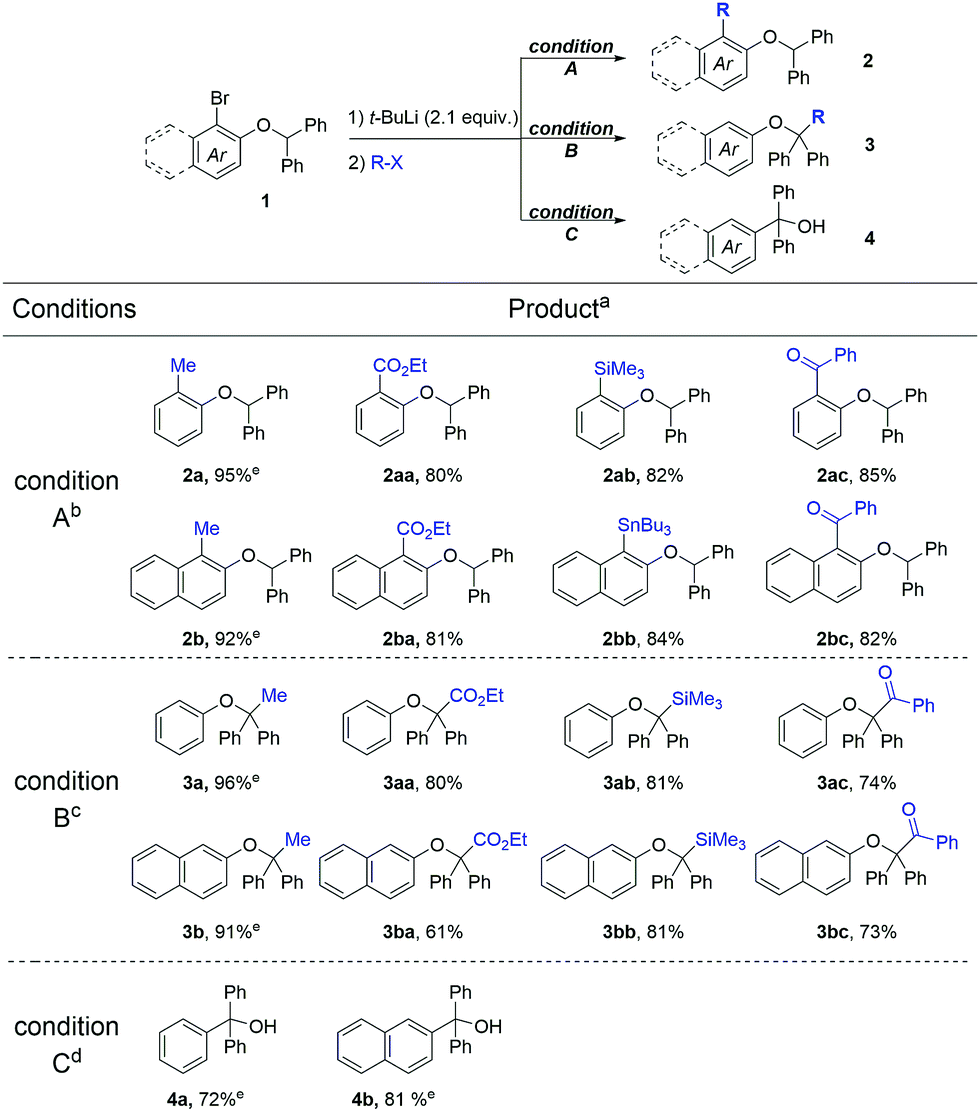
Under three optimized conditions (conditions A for aryl-functionalization, conditions B for benzyl-functionalization, and conditions C for triaryl alcohols; for details, see the ESI†), we conducted selective functionalization with various electrophiles such as methyl iodide (or triflate), ethyl chloroformate, tributyltin chloride, trimethylsilyl chloride, and benzoyl chloride (Table 2). Compounds 2 derived from the non-isomerized intermediate, compounds 3 derived from the deprotonated intermediate and compounds 4 derived from the rearranged intermediate were selectively obtained in good isolated yields.
| a Yields of isolated products. b Residence time for lithiation was 3 ms at 25 °C. c Residence time for lithiation was 6.3 s at 25 °C. d After the lithiation for 6.3 s at 25 °C in flow, the resulting solution was stirred in a flask at 50 °C for 1 h. For details, see the ESI. e Determined by 1H NMR spectroscopy using 1,3,5-trimethoxy benzene as an internal standard. |
|---|
|
Based on the present method for the control of tandem isomerizations, dual functionalization of aryl benzyl ethers was achieved using a flow microreactor system (Fig. 2). By controlling the residence times precisely for two lithiation-trapping sequences, doubly functionalized products 5a and 5b were successfully obtained from substrates 1c and 1d, respectively, within 15 s of the total reaction time.
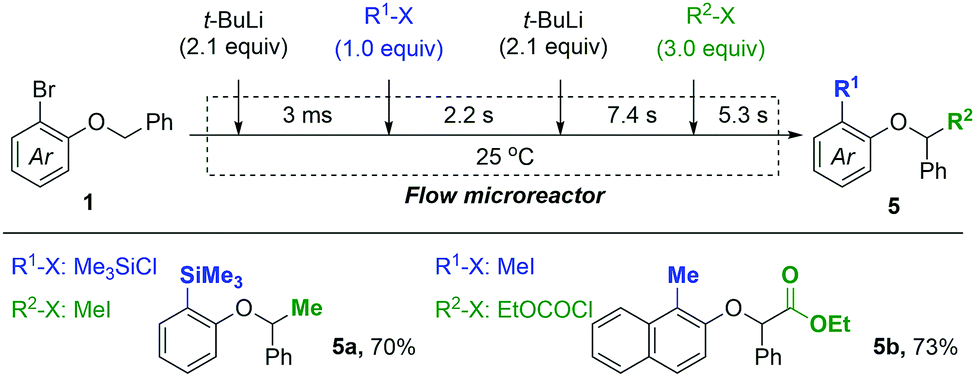 | ||
| Fig. 2 Dual functionalization of aryl benzyl ethers in a one-flow microreactor via controlled sequential isomerization reactions. | ||
In summary, we have demonstrated the flow-assisted control of tandem isomerizations of H–Li exchange and [1,2]-Wittig rearrangement. Under the optimized conditions based on the kinetic studies, three intermediates can be selectively trapped at will to obtain the desired products. The present study opens a new possibility of chemical synthesis using tandem isomerizations. We hope that the method will be widely applied for the sequential control of various reaction intermediates.
We acknowledge the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2017R1A3B1023598). This work was also supported by a Grant in-Aid for Scientific Research (S) 26220804 and a Grant-in-Aid for Young Scientists (B) 16K17898 funded by the Japan Society for the Promotion of Science (JSPS).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For example, R. A. Moss, M. S. Platz and M. Jones Jr., Reactive Intermediate Chemistry, Wiley, Hoboken, 2004 Search PubMed.
- Selected reviews on flow microreactor synthesis: (a) P. Watts and S. J. Haswell, Chem. Soc. Rev., 2005, 34, 235–246 RSC; (b) A. J. deMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed; (c) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed; (d) T. Fukuyama, M. T. Rahman, M. Sato and I. Ryu, Synlett, 2008, 151–163 CAS; (e) Z. Qian, I. R. Baxendale and S. V. Ley, Chem. – Eur. J., 2010, 16, 12342–12348 CrossRef CAS PubMed; (f) K. S. Elvira, X. C. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed; (g) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (h) S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed; (i) P. L. Heider, S. C. Born, S. Basak, B. Benyahia, R. Lakerveld, H. Zhang, R. Hogan, L. Buchbinder, A. Wolfe, S. Mascia, J. M. B. Evans, T. F. Jamison and K. F. Jensen, Org. Process Res. Dev., 2014, 18, 402–409 CrossRef CAS; (j) P. Zhang, M. G. Russell and T. F. Jamison, Org. Process Res. Dev., 2014, 18, 1567–1570 CrossRef CAS; (k) K. Gilmore, D. Kopetzki, J. W. Lee, Z. Horvath, D. T. McQuade, A. Seidel-Morgenstern and P. H. Seeberger, Chem. Commun., 2014, 50, 12652–12655 RSC; (l) S. Newton, C. F. Carter, C. M. Pearson, L. de C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRef CAS PubMed; (m) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (n) D. R. Snead and T. F. Jamison, Angew. Chem., Int. Ed., 2015, 54, 983–987 CrossRef CAS PubMed; (o) T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed; (p) D. Ghislieri, K. Gilmore and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 678–682 CAS; (q) M. Brzozowski, M. O’Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed; (r) A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed; (s) R. Singh, H.-J. Lee, A. K. Singh and D.-P. Kim, Korean J. Chem. Eng., 2016, 33, 2253–2267 CAS; (t) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- (a) I. R. Baxendale, S. V. Ley, A. C. Mansfield and C. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 4017–4021 CrossRef CAS PubMed; (b) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (c) R. A. Maurya, C. P. Park, J. H. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952–5955 CrossRef CAS PubMed; (d) S. Sharma, R. Maurya, K.-I. Min, G. Jeong and D.-P. Kim, Angew. Chem., Int. Ed., 2013, 52, 7564–7568 CrossRef CAS PubMed; (e) C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794–2797 CrossRef CAS PubMed; (f) I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211–230 CAS; (g) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed; (h) R. A. Maurya, K.-I. Min and D.-P. Kim, Green Chem., 2014, 16, 116–120 RSC; (i) A. K. Singh, D.-H. Ko, N. K. Vishwakarma, S. Jang, K.-I. Min and D.-P. Kim, Nat. Commun., 2016, 7, 10714 CrossRef PubMed.
- (a) J. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed; (b) J. Yoshida, Flash Chemistry. Fast Organic Synthesis in Microsystems, Wiley-Blackwell, 2008 Search PubMed; (c) J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- (a) A. Nagaki, H. Kim and J. Yoshida, Angew. Chem., Int. Ed., 2009, 48, 8063–8065 CrossRef CAS PubMed; (b) A. Nagaki, E. Takizawa and J. Yoshida, J. Am. Chem. Soc., 2009, 131, 1654–1655 CrossRef CAS PubMed and 3787; (c) A. Nagaki, E. Takizawa and J. Yoshida, Chem. – Eur. J., 2010, 16, 14149–14158 CrossRef CAS PubMed; (d) Y. Tomida, A. Nagaki and J. Yoshida, J. Am. Chem. Soc., 2011, 133, 3744–3747 CrossRef CAS PubMed; (e) A. Nagaki, S. Kim, N. Miuchi, H. Yamashita, K. Hirose and J. Yoshida, Org. Chem. Front., 2016, 3, 1250–1253 RSC.
- H. Kim, K.-I. Min, K. Inoue, D. J. Im, D.-P. Kim and J. Yoshida, Science, 2016, 352, 691–694 CrossRef CAS PubMed.
- (a) A. Nagaki, H. Kim and J. Yoshida, Angew. Chem., Int. Ed., 2008, 47, 7833–7836 CrossRef CAS PubMed; (b) A. Nagaki, H. Kim and J. Yoshida, Angew. Chem., Int. Ed., 2009, 48, 8063–8065 CrossRef CAS PubMed; (c) A. Nagaki, H. Kim, H. Usutani, C. Matsuo and J. Yoshida, Org. Biomol. Chem., 2010, 8, 1212–1217 RSC; (d) H. Kim, A. Nagaki and J. Yoshida, Nat. Commun., 2011, 2, 264 CrossRef PubMed; (e) W. Ren, H. Kim, H.-J. Lee, J. Wang, H. Wang. and D.-P. Kim, Lab Chip, 2014, 14, 4263–4269 RSC; (f) H. Kim, H.-J. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2015, 54, 1877–1880 CrossRef CAS PubMed; (g) H. Kim, H.-J. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2016, 55, 1422–1426 CrossRef CAS PubMed.
- (a) L. J. Martin, A. L. Marzinzik, S. V. Ley and I. R. Baxendale, Org. Lett., 2011, 13, 320–323 CrossRef CAS PubMed; (b) D. N. Tran, C. Battilocchio, S. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC; (c) D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 RSC; (d) J. S. Poh, D. N. Tran, C. Battilocchio, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 7920–7923 CrossRef CAS PubMed; (e) J.-S. Poh, S. Makai, T. V. Keutz, D. N. Tran, C. Battilocchio, P. Pasau and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 1864–1868 CrossRef CAS PubMed.
- J. Barluenga, F. J. Fañanás, R. Sanz, C. Marcos and M. Trabada, Org. Lett., 2002, 4, 1587–1590 CrossRef CAS PubMed.
- (a) A. Ahmed, J. Clayden and M. Rowley, Tetrahedron Lett., 1998, 39, 6103–6106 CrossRef CAS; (b) I. Fleming, S. R. Mack and B. P. Clark, Chem. Commun., 1998, 715–716 RSC.
- G. Wittig and H. Doser, Justus Liebigs Ann. Chem., 1942, 550, 260–268 CrossRef CAS.
- (a) V. Balasubramaniyan, Chem. Rev., 1966, 66, 567–641 CrossRef CAS; (b) J.-B. Robert, J. S. SHerfinski, R. E. Marsh and J. D. Roberts, J. Org. Chem., 1974, 39, 1152–1156 CrossRef CAS; (c) D. Pla, O. Sadek, S. Cadet, B. Mestre-Voegtlé and E. Gras, Dalton Trans., 2015, 44, 18340–18346 RSC.
- (a) P. T. Lansbury, V. A. Pattision, J. D. Sidler and J. B. Bieber, J. Am. Chem. Soc., 1966, 88, 78–84 CrossRef CAS; (b) J. F. Garst and C. D. Smith, J. Am. Chem. Soc., 1976, 98, 1526–1537 CrossRef CAS; (c) U. Schöllkopf, Angew. Chem., Int. Ed., 1979, 18, 563–572 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc08460a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |