
Automated reaction progress monitoring of heterogeneous reactions: crystallization-induced stereoselectivity in amine-catalyzed aldol reactions†
Céline
Rougeot
a,
Henry
Situ
a,
Blessing Huynh
Cao
b,
Vaso
Vlachos
c and
Jason E.
Hein
 *a
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, V6T1Z1, Canada. E-mail: jhein@chem.ubc.ca
bChemistry and Chemical Biology, University of California, Merced, 95343, USA
cMettler-Toledo AutoChem, Inc., Columbia, MD 21016, USA
First published on 26th January 2017
Abstract
A prototype automated system has been developed, which is capable of acquiring accurate kinetic reaction profiles from heterogeneous reactions. The device is capable of monitoring the composition and concentration of either the dissolved components (solution phase) or slurry (solid and solution phases) in parallel. This prototype was used to study the diastereo- and enantioselectivity of the aldol reaction between 4-tert-butyl cyclohexanone and p-nitrobenzaldehyde, catalyzed by either pyrrolidine or L-proline. With both catalysts, the observed high diastereoselectivity involves an interplay between solution phase reaction/epimerization and crystallization. These observations were possible due to the facile method of tandem solution and slurry analysis.
Introduction
Detailed kinetic analysis represents the gold standard for delineating a chemical mechanism. In general, the most comprehensive understanding of a new chemical process can be obtained through reaction progress measurements, where changes in starting materials, intermediates, products and by-products are charted over the entire course of the reaction.1 Crucial information brought forth from reaction progress analysis provides the key to unlocking the full potential of the reaction and lays the groundwork for the discovery of new reactivity paradigms.Despite the value offered by reaction progress measurements, they are not generally considered routine practice when initiating a mechanistic investigation. In part, this barrier to widespread adoption stems from the lack of easy-to-deploy analytical technology capable of interrogating the chemical transformation under standard synthetic conditions. This is particularly true for reactions involving heterogeneous reagents or products, especially with solid/liquid slurry systems. Moreover, liquid manipulation of a heterogeneous system is complicated. Advances in pump technology have improved this situation, but problems with changes in viscosity over the reaction course, and particle size led to serious variation in sample composition.
A number of existing process analytical technique can be utilized to obtain accurate concentration vs. time data.2 In recent years, application of flow NMR, or FT-IR spectroscopy has become much more routine, allowing the conversion of a chemical process to be monitored in real time.3 Other useful operando techniques include EPR spectroscopy, UV-vis spectroscopy.4 In addition to these in situ approaches, a range of off-line analyses have been deployed, which utilize rapid sampling and analysis of aliquots by direct ESI-MS/MS, HPLC-MS or MALDI-TOF.5 However, these techniques only monitor soluble components from the solution phase. Insoluble reagents are, at best, not visible to the analytical method and can complicate or entirely prevent reaction analysis by these techniques.
Herein, we describe a prototype apparatus capable of automated sampling of both the soluble (solution only) and slurry (solid and solution) components of heterogeneous reactions in parallel. Integrating the sampling technology with an automated liquid handling robot provides highly reproducible and accurate measurements of the reaction. The combination of solution and slurry compositional data allows us to visualise and monitor both chemical and physical processes during the course of the reaction. This system allows robust kinetic analysis on reactions that would normally be intractable using conventional tools. The prototype has been applied to study the enantio- and diastereoselectivity of the aldol reaction between 4-substituted cyclohexanones and p-nitrobenzaldehyde, catalyzed by secondary amine organocatalysts.
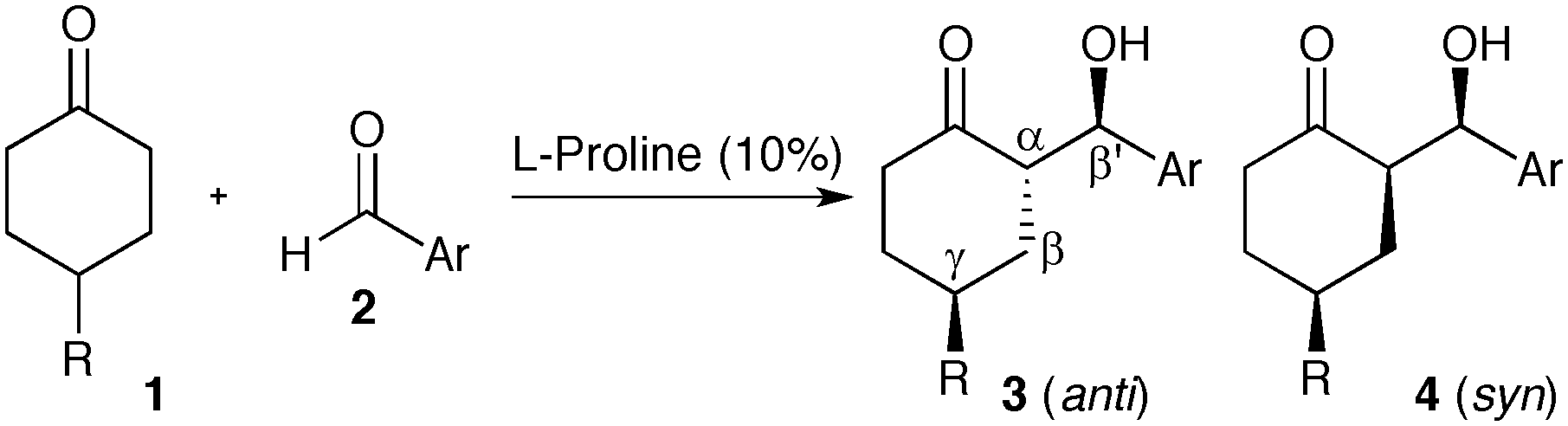
Achiral 4-substituted cyclohexanones can readily be desymmetrized via aldol reactions catalyzed by L-proline or prolinamide organocatalysts.6 This strategy results in an impressive increase in complexity, providing access to advanced intermediates bearing three new stereocenters (Scheme 1). Although four diastereotopic pairs of enantiomers can be generated, numerous reports have demonstrated that L-proline catalyzes reactions with aromatic aldehydes resulting in excellent diastereo- and enantioselectivity. In most cases, ketoalcohol 3 is isolated as a single stereoisomer, with 4 being occasionally reported as a minor product. This exceptional stereocontrol has been exploited to access numerous chiral building blocks, including a key fragment required in the production of novel Alzheimer's therapeutics.7
 | ||
| Scheme 1 L-Proline-catalysed aldol reaction between 4-substiuted cyclohexanones and aryl aldehydes. | ||
The stereochemical outcome of the proline-catalyzed reaction has been explained by the Houk–List model,8 where a combination of steric and non-covalent interactions produces a favourable diastereomeric transition state (Scheme 2, 6).9 In particular, a strong hydrogen bonding interaction between the carboxylic acid in the enamine intermediate and the aldehyde electrophile is key to realizing high enantioselectivity and diastereoselectivity.6c Thus, the Houk–List model predicts that the product geometry is kinetically controlled, involving fast irreversible formation of the key α-stereocentre. This explains the selective formation of the thermodynamically less favourable product 3, with its axial/equatorial arrangement of the two cyclohexyl-substituents due to the 1,3-anti geometry at the α,γ carbons.
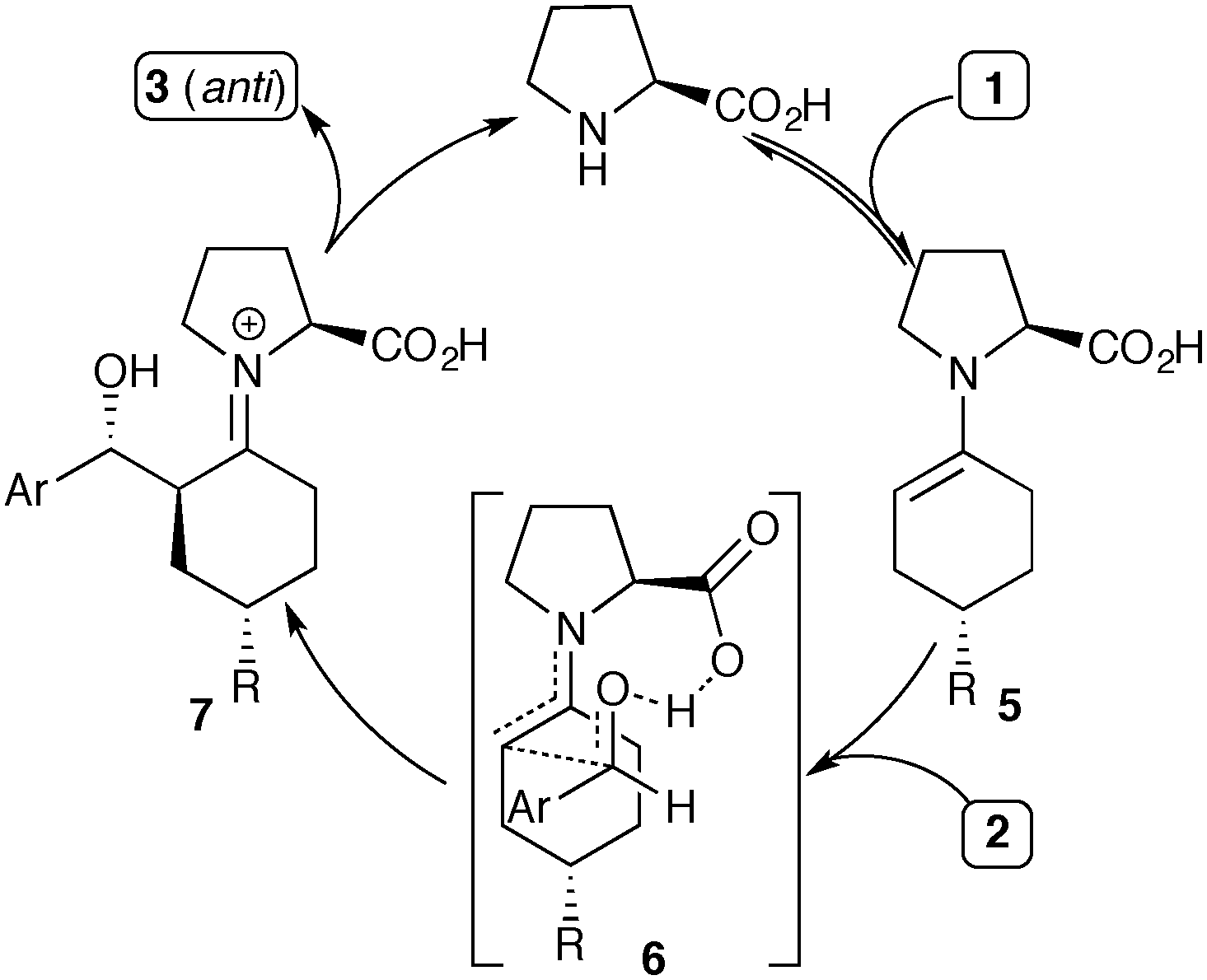 | ||
| Scheme 2 Catalytic cycle for L-proline-catalysed aldol reaction. | ||
However, aldol reactions catalysed by achiral secondary amines, such as pyrrolidine have been reported to still offer very high diastereoselectivity. Furthermore, many synthetic procedures utilize heterogeneous conditions where reaction components (ketone, aldehyde, catalyst or products) are not fully soluble over the course of the reaction.10 These features suggest that a dynamic interplay involving the solution phase reaction and solid–solution solubility equilibria may play an important role in the observed stereochemical outcome. In addition, HPLC analysis of reaction aliquots reveals a complex mixture, including minor diastereomers, as well as products from aldol condensation and Mannich addition.11 While these by-products only become a complicating factor after extended reactions times, high conversion typically requires 2–3 days for these systems. These observations belie the excellent stereo- and chemoselective outcome normally expected of this aldol reaction.
Results and discussion
Schematic of the automated sampling device
A definitive understanding of the multitude of interconnected chemical and physical processes at work in this deceptively simple reaction can be gained through reaction progress measurements. However, accurately analyzing concentration changes for a slurry presents a significant challenge. To address this issue a prototype automated sampling apparatus based on a Gilson GX-281 liquid handling robot has been developed (Fig. 1). The apparatus incorporates two parallel sampling channels; one utilizing a Mettler-Toledo EasySampler reaction sampling probe and the second comprised of a 6-port 2-position Rheodyne valve (Gilson 218 injector unit) attached to a programmable syringe pump (New Era Pump Systems Inc.).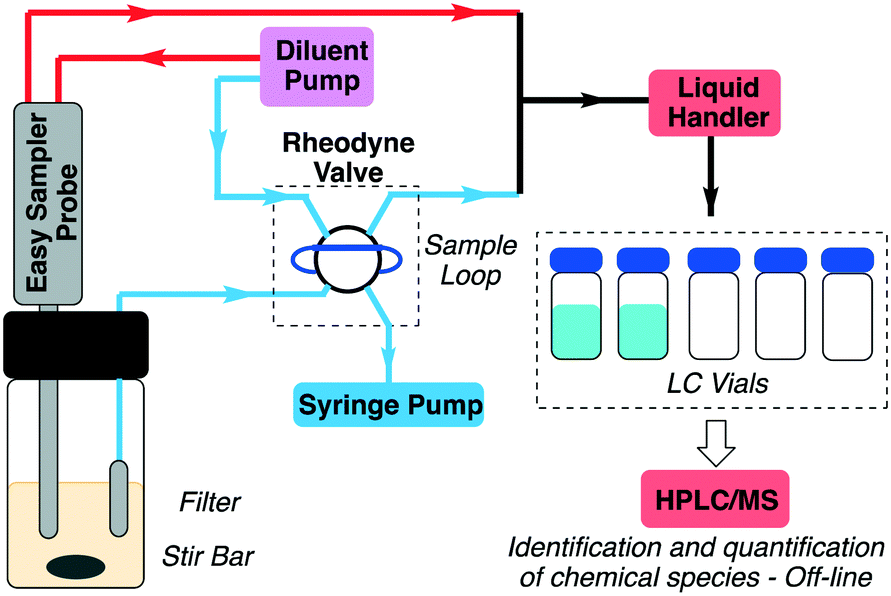 | ||
| Fig. 1 Schematic of the prototype automated sampling device. | ||
The EasySampler probe consists of a PTFE sleeve, with a hastelloy sampling head bearing a fixed 20 μL sampling pocket. When triggered, a linear actuator extends the sample head into the reaction media, allowing the solid and liquid component of the slurry to enter the sample pocket. The fluid lines are primed with MeOH, allowing the slurry to be dissolved and diluted once the sampling head is withdrawn back into the probe head. A schematic showing details of the operation and setup of the probe sampling head can be found in the ESI† (Fig. S2).
By contrast, the solution sampling channel relies on a PEEK capillary fitted with a sintered metal filter (Upchurch Scientific), permitting only the crystal-free supernatant to be drawn into the sample loop by the syringe pump. It should be noted that preventing cavitation in the sample lines can be a significant challenge, however, we have found that by minimizing the dead volume in the sampling lines and adjusting the syringe pull speed reproducible sampling can be achieved.
The action of all components is controlled via the GX-281, including valve actuation as well as triggering the syringe pump and EasySampler probe. In this configuration, Gilson's Trilution software package can be used to script all actions and events, resulting in excellent reproducibility in sample timing. Also, both sampling channels utilize a fixed volume device to create the reaction aliquot; a 20 μL sampling pocket for the slurry and a 15 μL sample loop for the solution channel. This removes errors due to metering and, when coupled with the GX-281 liquid handling pump, the system is capable of very reproducible aliquot preparation. Moreover, the system can reliably operate under a variety of conditions, performing rapid sampling (∼1 per minute), high sample counts (up to 300 aliquots) and over extended reaction times (up to 90 hours in some cases).
Application to the aldol reaction
The reaction between 4-tert-butylcyclohexanone (8) and 4-nitrobenzaldehyde (9) catalyzed by pyrrolidine was performed in MTBE (Fig. 2). The ability to automatically monitor the concentration and compositing of both the solid and solution components provides insight into both the reaction rate and stereoselectivity. The slurry sampling channel (Fig. 2, filled circles), which reports on the solid and solution components of the reaction, shows that initially three of the four possible diastereomers are generated, all with the same rate. After approximately 5 hours major diastereomer rac-11 continues to form, diastereomer rac-12 reaches steady state while rac-13 begins to decline. The solution phase channel (Fig. 2, hollow circles), shows a very different profile for the major product rac-11. While the reaction trends for diastereomers rac-12 and rac-13 are nearly identical in both the solution and slurry sampling profiles, product rac-11 is nearly absent from the solution phase. This definitively shows that while diastereomers rac-12 and rac-13 remain dissolved over the course of the reaction, rac-11 is isolated in the solid phase.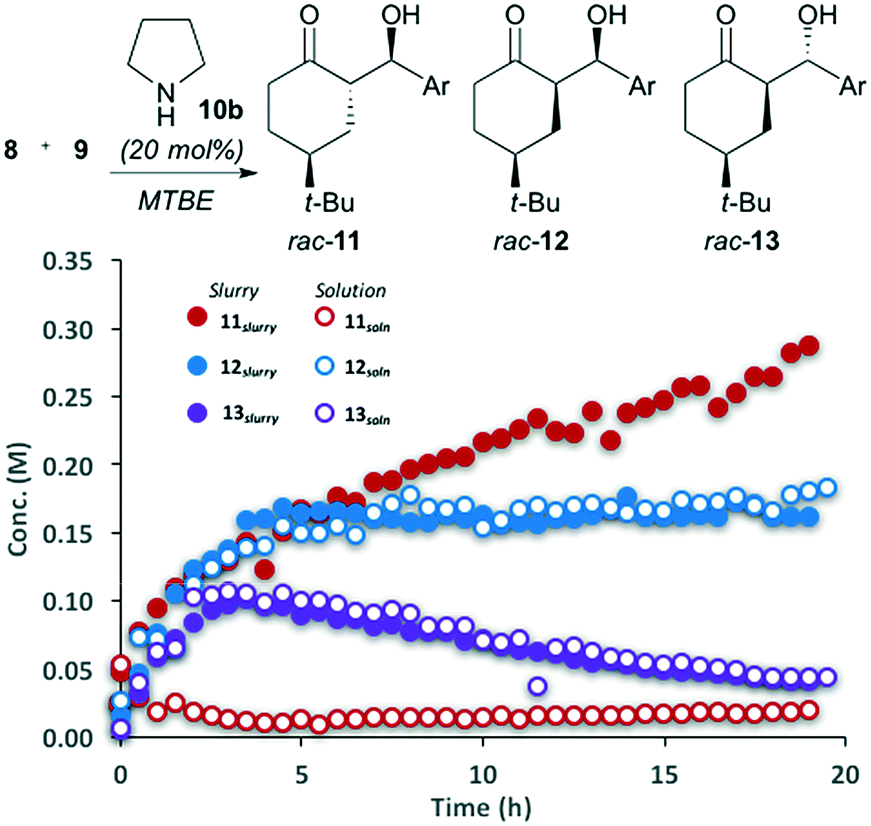 | ||
| Fig. 2 Slurry and solution reaction progress curves for the pyrrolidine-catalyzed aldol reaction; Ar = p-nitrobenzaldehyde. Curves generated by HPLC-MS; products 11, 12 and 13 are isolated a racemic compounds. | ||
The reaction progress measurements indicates that the overall catalytic network can be considered as a series of equilibria, with very low diastereoselectivity in the addition of the aldehyde to the key enamine (14) (Scheme 3). As ketone 8 becomes limiting, diastereomer rac-13 undergoes decomposition via either retro aldol reaction or condensation to yield α,β-unsaturated ketones (not shown). In parallel, rapid interconversion between diastereomers rac-11 and rac-12 occurs via base-catalyzed epimerization, without C–C bond scission. While diastereomer rac-12 is thermodynamically more stable, the extremely low solubility of rac-11 drives the equilibrating network, ultimately causing the crystallization of rac-11 to control the observed diastereoselectivity.
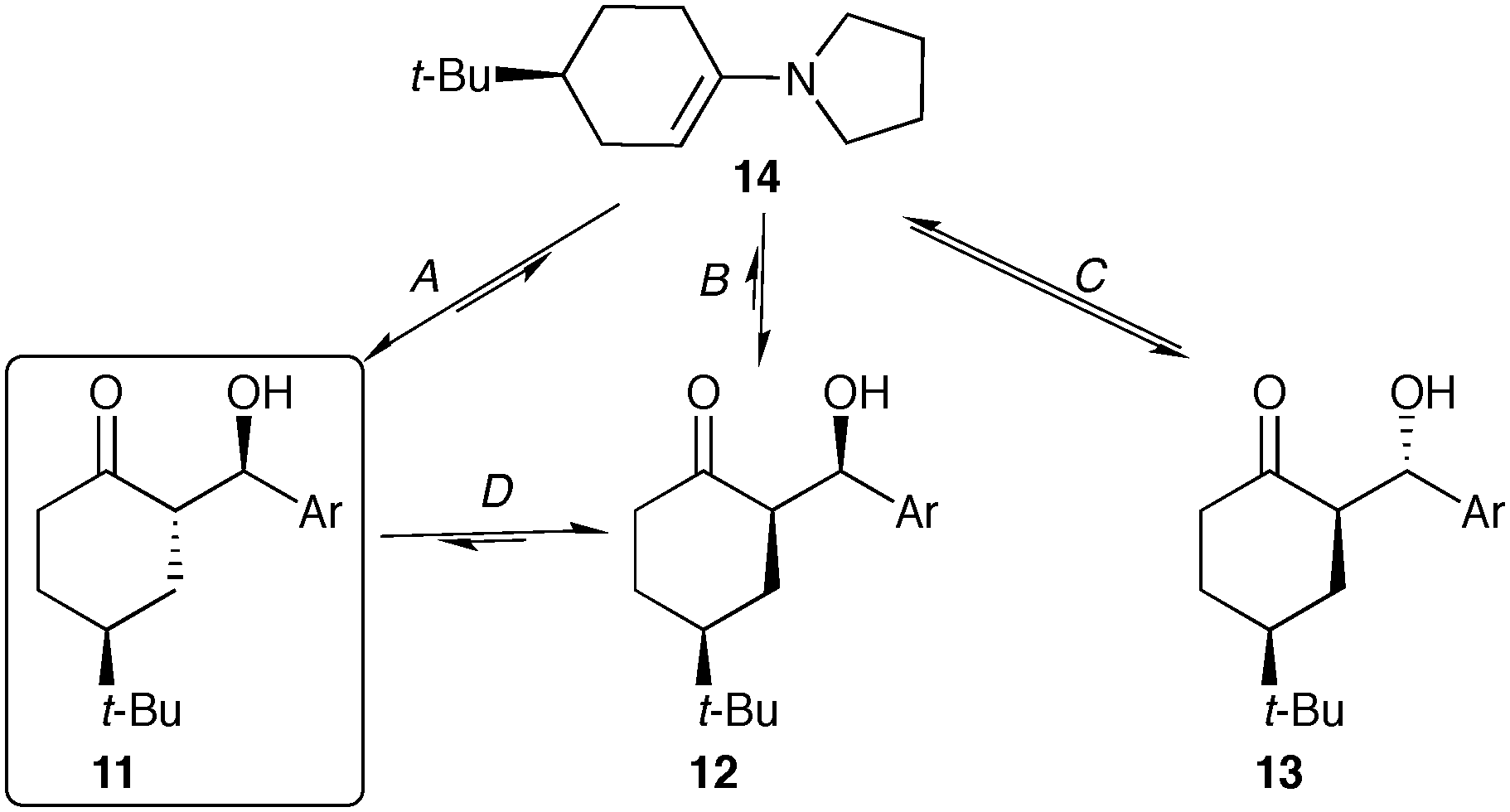 | ||
| Scheme 3 Interconversion between aldol diastereomers; Ar = p-nitrobenzaldehyde. | ||
Reports have stated that isomerization of 11 proceeds via an aldol/retro-aldol sequence,12 however, our observations suggested that rapid interconversion between 11 and 12 primarily proceeds via deprotonation at the α-stereocenter. To validate this hypothesis, enantiomerically pure aldol product S,S,R-11 was dissolved in d6-DMSO and treated with catalytic pyrrolidine. The solution was monitored by 1H NMR, showing rapid conversion to the thermodynamically more stable diastereomer R,S,R-12 over the course of one hour, giving an equilibrium mixture of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 3). More importantly, both remaining 11 and diastereomer 12 where present as single enantiomers. Treating 11 with tertiary amine bases (TEA or DBU) gives a similar reaction profile. These results eliminate the involvement of an enamine intermediate, and instead confirm that the major pathway involves epimerization via enolate 15. While 1H-NMR provides a tentative assignment of the diastereomer geometry, the stereochemistry for both 11 and 12 was unambiguously confirmed by single crystal X-ray analysis. In each cases, the enantiopure samples of 11 and 12 crystallized in a non-centrosymmetric P212121 space group (Fig. 5).
:1 (Fig. 3). More importantly, both remaining 11 and diastereomer 12 where present as single enantiomers. Treating 11 with tertiary amine bases (TEA or DBU) gives a similar reaction profile. These results eliminate the involvement of an enamine intermediate, and instead confirm that the major pathway involves epimerization via enolate 15. While 1H-NMR provides a tentative assignment of the diastereomer geometry, the stereochemistry for both 11 and 12 was unambiguously confirmed by single crystal X-ray analysis. In each cases, the enantiopure samples of 11 and 12 crystallized in a non-centrosymmetric P212121 space group (Fig. 5).
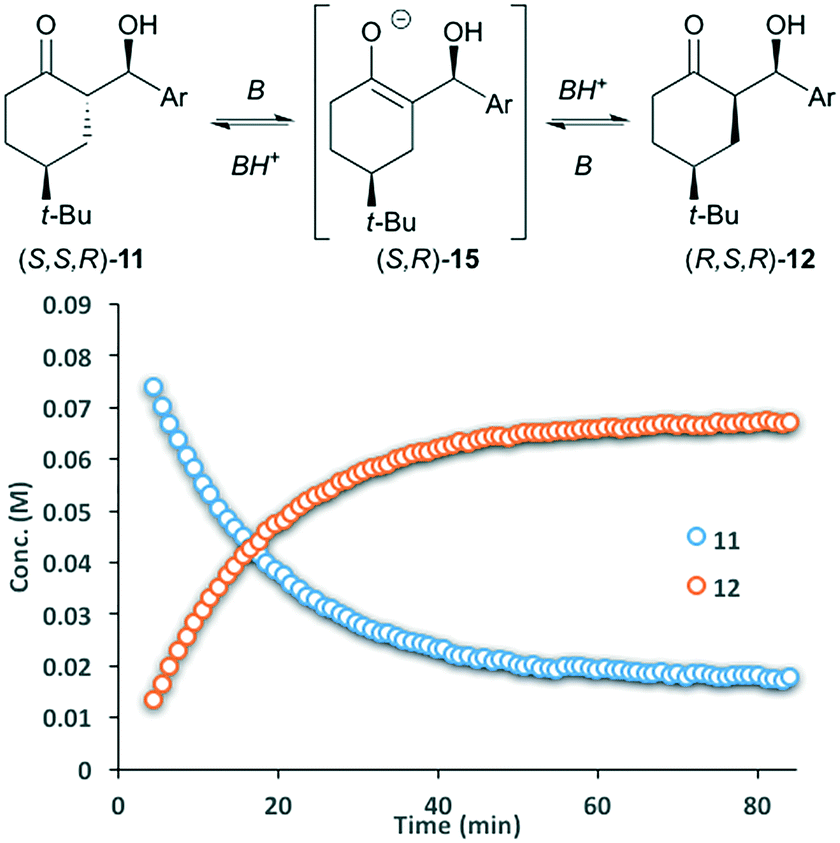 | ||
| Fig. 3 Base-catalyzed isomerization between diastereomers (S,S,R)-11 and (R,S,R)-12 under homogenous conditions; Ar = p-nitrobenzaldehyde. Curves generated by 1H-NMR. | ||
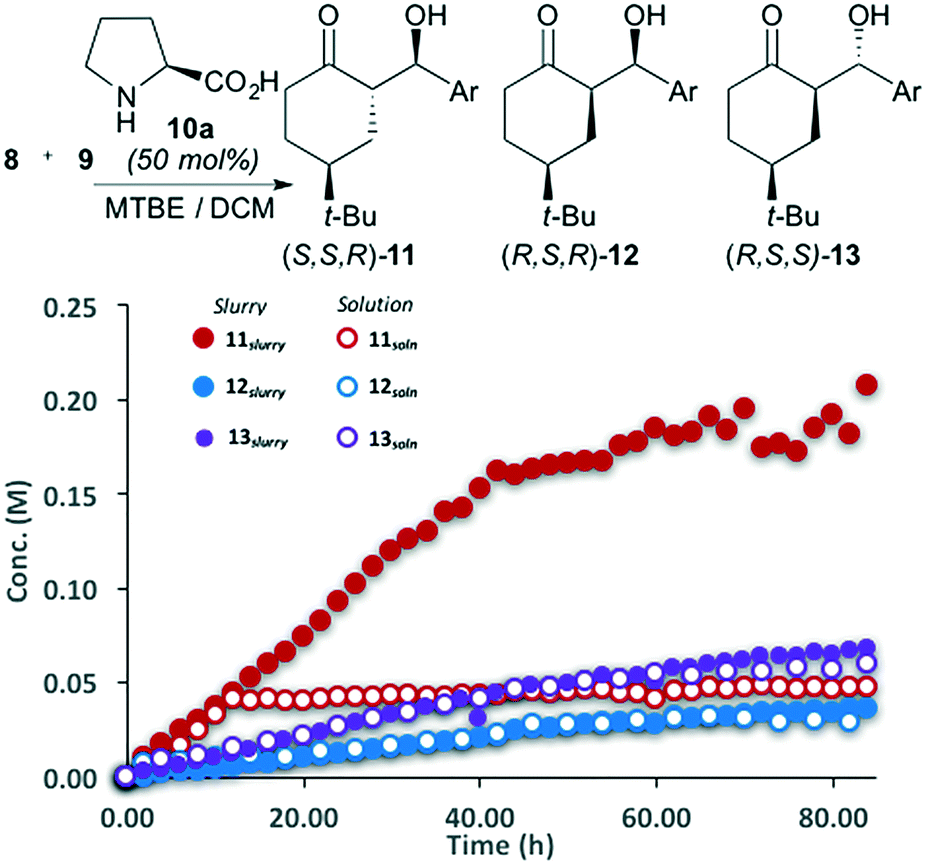 | ||
| Fig. 4 Slurry and solution reaction progress curves for the L-proline-catalyzed aldol reaction; Ar = p-nitrobenzaldehyde. Curves generated by HPLC-MS; products 11, 12 and 13 are isolated a single enantiomers. | ||
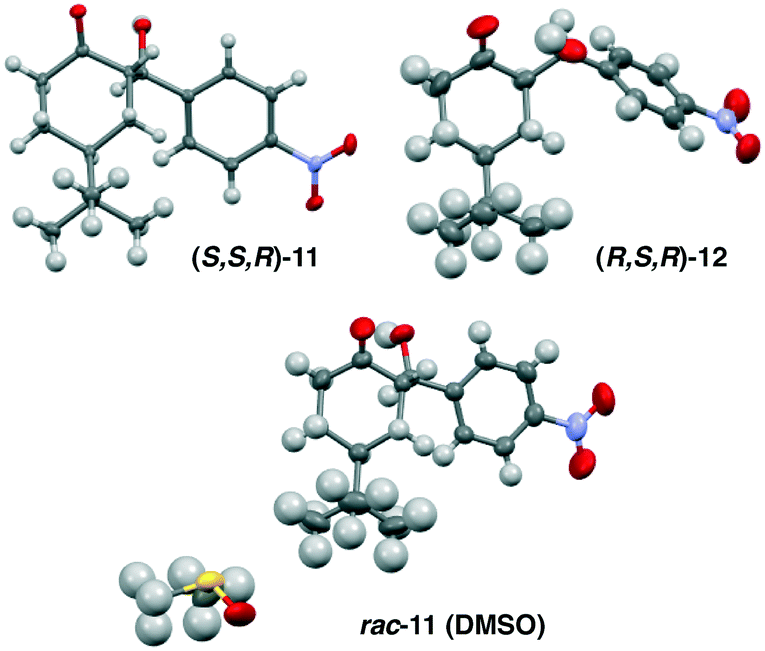 | ||
| Fig. 5 X-Ray structures for both homochiral and heterochiral crystals for diastereomers 11 and 12, only the asymmetric unit is displayed for clarity. | ||
The rapid isomerization and decomposition observed in the homogenous solution phase chemistry begins to highlight the synthetic importance of the heterogeneous reaction environment. Crystallization aids in sequestering the thermodynamically less-stable diastereomer 11, both protecting it from subsequent decomposition but also guiding the equilibrating mixture of diastereomers to a single stereochemical fate. The low solubility of rac-11 (∼0.25% by mass in MTBE at 25 °C) is critically important to this process, allowing efficient mass transfer to this diastereomer. The interplay between chemical (aldol reaction and isomerization) and physical (solubility and crystallization) equilibria is well known in the context of Dimroth's principle,13 and has been applied in numerous crystallization-induced stereoselective transformations (CIST).14 Based on our observations, the reported chemo- and stereoselectivity for the aldol reaction between 4-tert-butyl cyclohexanone and p-nitrobenzaldehyde can not be solely due to kinetic control. Instead, CIST plays a dominant role when operating under heterogenous conditions.
The aldol reaction between ketone 8 and aldehyde 9 shows a similar reaction profile when catalyzed by L-proline (Fig. 4). Attempting to perform the reaction using MTBE failed due to proline's exceptionally low solubility in this solvent. Instead, replacing the solvent with 1:1 MTBE:DCM alleviated this issue. This new solvent mixture also increased the solubility of diastereomer 11, which reached a concentration of ∼0.05 M before crystallization occurred. Diastereomers 12 and 13 remained minor components in this reaction. As with the pyrrolidine catalysed process, the slurry and solution aliquots for 12 and 13 are identical, confirming that these diastereomers remain in the solution phase throughout the reaction.
Unlike with pyrrolidine, L-proline plays a role with respect to the aldol diastereoselectivity. Throughout the reaction, S,S,R-11 is formed more rapidly than R,S,R-12 or R,S,S-13. However, the product ratio, prior to the crystallization of 11 (t = 0–10 h), was only 3:1.5:1 for diastereomers 11, 12 and 13 respectively. On stirring for extended time the ratio improved to 8:2:1. This suggests that again, kinetic control alone is not responsible for the observed product distribution and solid–liquid phase behavior plays a significant role.
Catalyzing the reaction with L-proline allows S,S,R-11 to be generated as a single enantiomer (e.e. > 99%). Analyzing the slurry phase aliquots by chiral HPLC revealed that unlike the diastereomeric ratio, the e.e. of the major product 11 does not change throughout the reaction. This observation indicates that while dynamic equilibration between the solid and solution components is an important factor in the diastereoselectivity, the enantioselectivity is primarily under catalyst control. In a similar fashion as the pyrrolidine-catalyzed reaction, the solid phase sequesters 11, preventing epimerization and decomposition, which primarily degrade or interconvert minor isomers 12 and 13.
The possibility of solution phase racemization coupled to preferential crystallization was intriguing, as it presents the opportunity to exploit attrition-enhanced deracemization to guide or improve the e.e. of the crystalline product.15 Indeed, Bolm and coworkers have reported that Veidma-type ripening could be used to resolve racemic slurries of rac-11 by exploiting an aldol/retro-aldol process.12b However, this pathway does not play a significant role in our system. Performing the aldol reaction in the presence of zirconium beads (to aid in attrition) failed to produce enantiomerically enriched 11 even after extended time. In addition, studies on the decomposition of 11 (Fig. 3) indicates that retro-aldol/aldol reaction is minor compared to base-catalyzed epimerization to diastereomer 12, thus preventing interconversion between enantiomers of 11.
Furthermore, in order for product rac-11 to participate in attrition-enhanced deracemization a racemic solution must spontaneously crystallize into separate R- or S-homochiral solid phases.16 While, as expected, enantiopure samples did give rise to non-centrosymmetric homochiral crystals, all attempts to crystallize racemic solution produced a heterochiral crystal phase. In particular, crystallizing rac-11 from DMSO generated a stable DMSO-heterochiral solvate crystal structure. Similar crystal behaviour was observed with diastereomer rac-12, which also produces heterochiral crystals from racemic solutions. Thus, product rac-11 is not a stable conglomerate, but rather a racemate-forming compound. The existence of the stable DMSO-heterochiral solvate crystal and the overall preference to form racemate crystal phases would generally preclude this compound from participating in attrition-enhanced deracemization, as the equilibrating network of enantiomers would tend to precipitate as the more stable, and less soluble racemate crystal.
Conclusions
This study highlights the benefit and practical value of parallel solution/slurry sampling for studying heterogenous reaction systems. The automated platform is capable of accurate and robust reaction sampling, allowing the reaction kinetics and selectivity to be readily acquired. Furthermore, the ability to capture data from the isolated solution phase and solid/solution slurry allows us to delineate both the reaction and crystallization kinetics.Applying this tool to the pyrrolidine or L-proline-catalyzed aldol reaction has illuminated the critical role that solubility plays in determining the reaction diastereoselectivity. When utilizing pyrrolidine, the stereochemical outcome is primarily controlled by solid–solution effects. This includes both mass transfer to the least soluble diastereomer via solution phase equilibration and non-specific decomposition of the minor isomers remaining in the solution phase. This observation explains how the product bearing the thermodynamically-less favourable axial/equatorial arrangement of the two cyclohexyl-substituents can be formed selectively. While solubility effects are important for the diastereoselectivity of the L-proline-catalyzed reaction, the enantioselectivity, at least with regard to the major diastereomer, is controlled by the catalyst. The solid phase still plays an important role in sequestering the product and preventing decomposition.
Acknowledgements
The authors gratefully acknowledge Mettler-Toledo Autochem for the donation of the EasySampler probe. Financial support for this work was provided by the University of British Columbia and the Natural Sciences and Engineering Resource Council of Canada (Engage, 2016-RGPIN-04613). Additionally, we thank Pfizer Worldwide Research and Development for student research fellowships (to C. R. and H. S.).Notes and references
- (a) D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed; (b) E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 78–88 CrossRef CAS PubMed; (c) D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- (a) U. Bentrup, Chem. Soc. Rev., 2010, 39, 4718–4730 RSC; (b) R. Chung, D. Yu, V. T. Thai, A. F. Jones, G. K. Veits, J. R. de Alaniz and J. E. Hein, ACS Catal., 2015, 4579–4585 CrossRef CAS; (c) A. Chanda, A. M. Daly, D. A. Foley, M. A. LaPack, S. Mukherjee, J. D. Orr, G. L. Reid III, D. R. Thompson and H. W. Ward II, Org. Process Res. Dev., 2015, 19, 63–83 CrossRef CAS; (d) R. Theron, Y. Wu, L. P. E. Yunker, A. V. Hesketh, I. Pernik, A. S. Weller and J. S. McIndoe, ACS Catal., 2016, 6, 6911–6917 CrossRef CAS.
- (a) D. A. Foley, C. W. Doecke, J. Y. Buser, J. M. Merritt, L. Murphy, M. Kissane, S. G. Collins, A. R. Maguire and A. Kaerner, J. Org. Chem., 2011, 76, 9630–9640 CrossRef CAS PubMed; (b) F. Susanne, D. S. Smith and A. Codina, Org. Process Res. Dev., 2012, 16, 61–64 CrossRef CAS; (c) M. T. Drexler, D. A. Foley, H. W. Ward II and H. J. Clarke, Org. Process Res. Dev., 2015, 19, 1119–1127 CrossRef CAS.
- (a) K. Grabow and U. Bentrup, ACS Catal., 2014, 4, 2153–2164 CrossRef CAS; (b) J. Rabeah, U. Bentrup, R. Stößer and A. Brückner, Angew. Chem., Int. Ed., 2015, 54, 11791–11794 CrossRef CAS PubMed.
- (a) K. L. Vikse, Z. Ahmadi, C. C. Manning, D. A. Harrington and J. S. McIndoe, Angew. Chem., Int. Ed., 2011, 50, 8304–8306 CrossRef CAS PubMed; (b) D. A. Foley, J. Wang, B. Maranzano, M. T. Zell, B. L. Marquez, Y. Xiang and G. L. Reid, Anal. Chem., 2013, 85, 8928–8932 CrossRef CAS PubMed; (c) G. A. Bailey and D. E. Fogg, ACS Catal., 2016, 6, 4962–4971 CrossRef CAS.
- (a) J. Jiang, L. He, S.-W. Luo, L.-F. Cun and L.-Z. Gong, Chem. Commun., 2007, 736–738 RSC; (b) X. Companyó, G. Valero, L. Crovetto, A. Moyano and R. Rios, Chem. – Eur. J., 2009, 15, 6564–6568 CrossRef PubMed; (c) H. Yang, S. Mahapatra, P. H.-Y. Cheong and R. G. Carter, J. Org. Chem., 2010, 7279–7290 CrossRef CAS PubMed; (d) J. Agarwal and R. K. Peddinti, Eur. J. Org. Chem., 2012, 6390–6406 CrossRef CAS; (e) B. H. Lipshutz and S. Ghorai, Org. Lett., 2012, 14, 422–425 CrossRef CAS PubMed.
- T. C. Nugent, F. T. Najafian, H. A. E. D. Hussein and I. Hussain, Chem. – Eur. J., 2016, 22, 14342–14348 CrossRef CAS PubMed.
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS PubMed.
- (a) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS PubMed; (b) A. Armstrong, R. A. Boto, P. Dingwall, J. Contreras-García, M. J. Harvey, N. J. Mason and H. S. Rzepa, Chem. Sci., 2014, 5, 2057–2071 RSC.
- (a) B. Rodríguez, A. Bruckmann and C. Bolm, Chem. – Eur. J., 2007, 13, 4710–4722 CrossRef PubMed; (b) Á. Martínez-Castañeda, B. Poladura, H. Rodríguez-Solla, C. Concellón and V. del Amo, Org. Lett., 2011, 13, 3032–3035 CrossRef PubMed; (c) G. D. Yadav and S. Singh, Tetrahedron: Asymmetry, 2015, 26, 1156–1166 CrossRef CAS.
- N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100–15101 CrossRef CAS PubMed.
- (a) S. Luo, P. Zhou, J. Li and J.-P. Cheng, Chem. – Eur. J., 2010, 16, 4457–4461 CrossRef CAS PubMed; (b) A. M. Flock, C. M. M. Reucher and C. Bolm, Chem. – Eur. J., 2010, 16, 3918–3921 CrossRef CAS PubMed.
- (a) O. Dimroth, Justus Liebigs Ann. Chem., 1910, 377, 127–163 CrossRef; (b) E. Hein, D. Gherase and D. G. Blackmond, Top. Curr. Chem., 2013, 333, 83–108 CrossRef PubMed.
- K. M. J. Brands and A. J. Davies, Chem. Rev., 2006, 106, 2711–2733 CrossRef CAS PubMed.
- (a) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed; (b) C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed.
- (a) J. E. Hein, B. Huynh Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 12629–12636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, crystallographic data and specifics for protoype equipment. See DOI: 10.1039/c6re00211k |
| This journal is © The Royal Society of Chemistry 2017 |