
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular self-assembly of a polyelectrolyte chain based on step-growth polymerization of hydrophobic and hydrophilic monomers†
Li
Sun
ab,
Shang
Ma
a,
Chen
Wang
a,
Yongmei
Chi
ab and
Jian
Dong
 *ab
*ab
aCollege of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing, Zhejiang Province 312000, China. E-mail: jiandong@usx.edu.cn; Fax: +86-575-88341521; Tel: +86-575-88342511
bSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang Province 315211, China
First published on 15th November 2017
Abstract
A novel amphiphilic polyelectrolyte (APE) prepared by step growth polymerization of hydrophobic and hydrophilic monomers is shown to have self-assembly structures. The polymerization reaction between citric acid and hexamethylene diisocyanate led to poly(hexamethylene citramide imide) (PHMCI), which was characterized by two-dimensional NMR, vibrational spectroscopies and gel permeation chromatography. Hydrolysis of PHMCI through a ring opening reaction resulted in a polyelectrolyte, poly(hexamethylene citramide) (PHMC) with three key elements of self-organization, i.e., ionizable side chains, hydrophobic groups and hydrogen bonding. PHMC is able to self-assemble to nanoparticles in water with variable hydrodynamic diameters and surface structures. The presence of noncovalent cross-linking of cooperative H-bonding units facilitates the formation of the polymer nanoparticles. The cooperative H-bonding is maintained from pH 5.5 to 10.0, which is verified by quantum chemical calculations. The nanoparticles are highly efficient metal chelators, offering potential as new remedies for metal sequestration in water or soil.
Introduction
Polymeric nanoparticles (NPs) with controlled morphologies are playing vital roles in materials separation, catalysis, targeted delivery of pharmaceuticals and biomedical imaging technology, where particles with a precise arrangement of chemical compositions and tailored morphology and size are required.1–4 Amphiphilic block copolymers,1,5–10 alternating, random, graft copolymers,2,11–23 and homopolymers composed of both hydrophilic and hydrophobic components in repeating units,24 are able to self-associate into various morphologies through an intra- or intermolecular fashion.Amphiphilic polyelectrolytes (APEs) consist of hydrophobic and ionizable monomer units.11,12,15,16,25,26 While the strong hydrophobic attraction force may help the hydrophobic units to associate with each other in aqueous media, the electrostatic repulsion force among the ionizable units causes the latter to extend through the space as far as possible. Thus, APEs may demonstrate distinct association behavior depending on the delicate geometric and energetic parameters of the hydrophobic and electrostatic forces.25 Studies of the self-organization behavior of APEs can shed light on the formation of higher order structures of biopolymers and culminate in a broad range of smart nano-materials for applications in personal care products, coatings, drug delivery systems, and environmental remedies.2
Past examples of APEs, including a neutral-cationic block copolymer–polyelectrolyte complex, a zwitterionic triblock copolymer, and a copolymer grafted with both hydrophobic and ionizable side chains, prepared by chain growth polymerization, have been shown to form interconvertible micelles/aggregates in aqueous solutions.7b,8a,b There is a dearth of studies on APEs based on step growth polymerization, however, such investigations may help to provide a better understanding of supramolecular structures of certain types of natural or artificial polypeptides where multiple cooperative hydrogen H-bonding, metal-coordination, ionic bridging, as well as van der Waals forces, can trigger chain misfolding or even neurodegenerative diseases.27–31 If the self-assembly is assisted by the presence of H-bonding and interpolyelectrolyte complexation, stable nano-scale structures can be formed by polymers with only weak or little hydrophobic interactions.
Here, we report a novel type of single APE chain based on step growth polymerization and post-polymerization hydrolysis (Scheme 1). The obtained polycitramide has a hydrophilic citric acid monomer unit with a hydroxyl group and an ionizable COOH pendant group, a short hydrophobic hexamethylene unit, and H-bonding forces between the main chains, but without complications of sequence heterogeneity or subtle imbalance of the hydrophobic and hydrophilic forces which might be caused by slight changes in block lengths prepared via a chain growth polymerization. Such a polyelectrolyte with three key driving forces for formation of nanoparticles has not been reported in the literature.
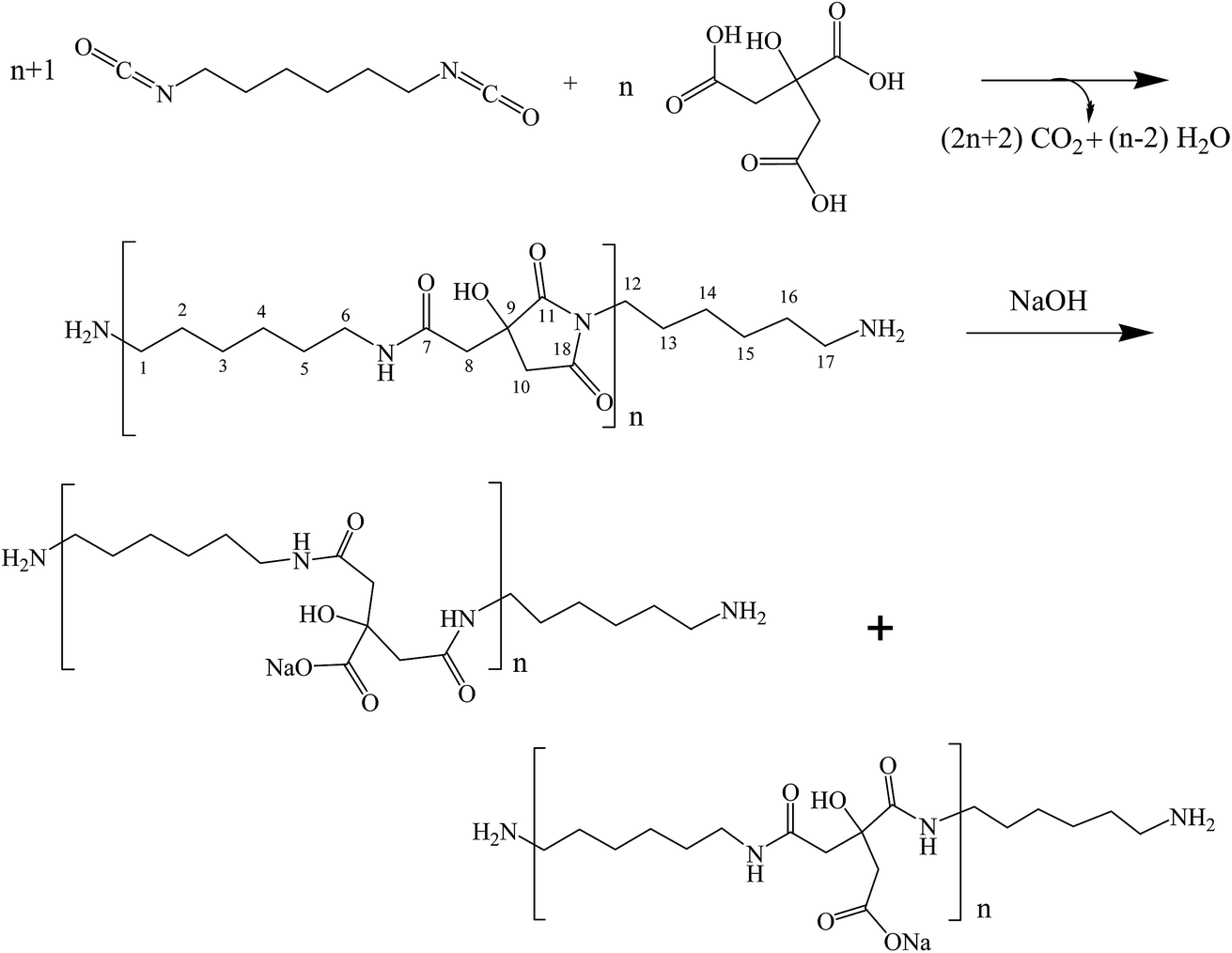 | ||
| Scheme 1 Synthesis of poly(hexamethylene citramide imide) (step 1) and its hydrolysis to poly(hexamethylene citramide) (step 2). | ||
Experimental section
Materials
Citric acid (CA), 4-dimethylaminopyridine (DMAP), N,N-dimethylacetamide (DMAc), hexamethylene diisocyanate (HMDI), ethanol, LiCl, NaOH, HCl and acetone, all in analytical grade, were purchased from Aladdin Reagent Co., Shanghai.Preparation of poly(hexamethylene citramide imide) and hydrolysis to polycitramide
CA (1.9214 g), HMDI (2.40 mL), freshly distilled DMAc (30.0 mL), and DMAP (0.0122 g) are added to a 100 mL round-bottom flask, which was placed in a silicone oil bath at 70 °C. The reactants were refluxed for 6 h and a viscous brown solution was formed. The reactants were poured into 30 mL of ethanol in a 250 mL beaker and agitated for half an hour on a magnetic stirrer. 180 mL of distilled water was slowly added to the mixture solution while stirring, resulting in the formation of white precipitates. The solid was isolated by filtration, transferred to a beaker and washed by a mixture of ethanol and water repeatedly. The product was dried in a vacuum oven at 60 °C for 6 h, giving poly(hexamethylene citramide imide) at a yield of 75% (see Scheme 1).0.050 g of the above product was added to a 100 mL round bottom flask and 50 mL of 0.010 mol L−1 NaOH solution was added. The mixture was heated in a silicone oil bath with a temperature of 100 °C for 1 h, and the solid was gradually dissolved in the aqueous solution, giving an opaque solution. The water in the solution was removed by using a rotary evaporator at 80 °C and the concentrated solution was precipitated with acetone. The resulting solid was washed with acetone by filtration and dried in the vacuum oven at 80 °C for 10 h to yield the hydrolyzed product poly(hexamethylene citramide) (see Scheme 1).
A series of buffer solutions (at 100 mM concentration) with different pH values were prepared. The hydrolyzed product poly(hexamethylene citramide) was mixed with a buffer solution thoroughly in a Millipore ultracentrifuge concentrator and centrifuged for 15 min at a speed of 4000 rpm. The sample solution was refilled with the buffer solution and centrifuged repeatedly at least for 4 times in order to fully exchange the aqueous medium with the buffer solution. The particle distributions of the polycitramides thus obtained were analyzed by dynamic light scattering (DLS) technique.
Instrumental analysis
FT-IR analysis was undertaken on Thermo Fisher Scientific Nicolet 6700 infrared spectrometer. FT-Raman spectra were collected on Bruker FT-Raman spectrometer after scanning for 200 times with a liquid nitrogen cooled Ge detector, at a laser excitation wavelength of 1064 nm. 1H NMR, 13C NMR, 1H–1H COSY, 13C–1H HSQC NMR spectra were recorded on Bruker AVANCE III 400 MHz NMR spectrometer, by using a mixed solvent of D2O and hexafluoroisopropyl alcohol. Molecular weight analysis was performed on an Agilent 1100 chromatograph system by using PLgel 5 μm MIXED-C column and DMAc eluent with 0.5 wt% LiCl, at a flow rate of 1.0 mL min−1. Transmission electron microscopic analysis was performed by JEM-1011 electron microscope at an acceleration voltage of 100 kV. The aqueous samples were spread on copper grids, slowly evaporating the solvent in the air at room temperature. Analysis of the particles in the TEM images was assisted by the ImageJ software (National Institutes of Health, USA). Particle size distribution analysis was also measured by a dynamic light scattering (DLS) analyzer based on photon cross-correlation spectroscopy (PCCP) (Nanophox, Sympatec GmbH, Germany) equipped with a He–Ne laser. Measurement of surface charges of the nanoparticles was carried out by using the Malvern Zetasizer Nano ZS. The sample solvents were replaced by buffer solutions with different pH values by using Millipore ultracentrifuge concentrators with a nominal molecular weight cut-off value of 30 kD. The metal ion chelating capabilities of the polycitramides were measured by using a Ca2+- and a Pb2+-selective electrode (Shanghai Precision & Scientific Instrument Co., Ltd.), respectively, and an ion meter (Shanghai Precision & Scientific Instrument Co., Ltd., Model PXB 720A) according to the procedures described in the previous study.32 A sample (10.0 mg) was dissolved in 50 mL of a pH = 7.0 aqueous solution, which had been adjusted to give a CaCl2 concentration of 1.0 mmol L−1 (or a PbNO3 concentration of 1.0 mmol L−1) and a potassium chloride concentration of 0.08 mol L−1. The resulting mixture was stirred at 30 °C for 10 min and the Ca2+ ions (or Pb2+ ions) in the solution were determined using the Ca2+ ion electrode (or Pb2+ ion electrode). The chelated metal amount was calculated by subtracting the free metal amount from the initial total amount of the metal. The metal concentrations were found by comparison with standard log[M2+] versus millivolt plots.Quantum chemical calculations
The calculations were performed using Gaussian 09 packages.33 The gas-phase geometries were optimized using M06-2X functional with 6-31G(d) basis set.34Results and discussion
Preparation of poly(hexamethylene citramide imide) and its hydrolysis to poly(hexamethylene citramide)
The step growth polymerization between citric acid and hexamethylene diisocyanate yields amide–imide linkages as shown in step 1 of Scheme 1. FTIR and FT-Raman spectroscopic analysis of the polymer obtained from step 1 confirmed the formation of poly(hexamethylene citramide imide) (PHMCI). The FTIR spectrum of the polymer shown in Fig. 1a displays an N–H stretching band at 3323 cm−1 and C–H stretching bands at 2933 and 2856 cm−1. Amide I, amide II and amide III bands appear at 1620, 1574 and 1252 cm−1, respectively. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of the citric acid reactant expected at 1750–1720 cm−1 is absent in the product spectrum of Fig. 1a. The bands at 1702 and 1781 cm−1 are attributed to asymmetric and symmetric CO stretching vibrations of the cyclic imide group (Scheme 1), respectively.32,35 This is confirmed by Raman spectroscopic analysis of the polymer. As shown in Fig. 1b, the peak at 1781 cm−1 due to the symmetric stretching of the imide ring CO groups become an intense Raman peak, whereas the asymmetric stretching around 1702 cm−1 has a very low Raman activity and is nearly invisible. The CO intensity pattern in the Raman spectrum of the imide ring is complementary to the infrared intensity pattern in Fig. 1.32,35
O stretching band of the citric acid reactant expected at 1750–1720 cm−1 is absent in the product spectrum of Fig. 1a. The bands at 1702 and 1781 cm−1 are attributed to asymmetric and symmetric CO stretching vibrations of the cyclic imide group (Scheme 1), respectively.32,35 This is confirmed by Raman spectroscopic analysis of the polymer. As shown in Fig. 1b, the peak at 1781 cm−1 due to the symmetric stretching of the imide ring CO groups become an intense Raman peak, whereas the asymmetric stretching around 1702 cm−1 has a very low Raman activity and is nearly invisible. The CO intensity pattern in the Raman spectrum of the imide ring is complementary to the infrared intensity pattern in Fig. 1.32,35
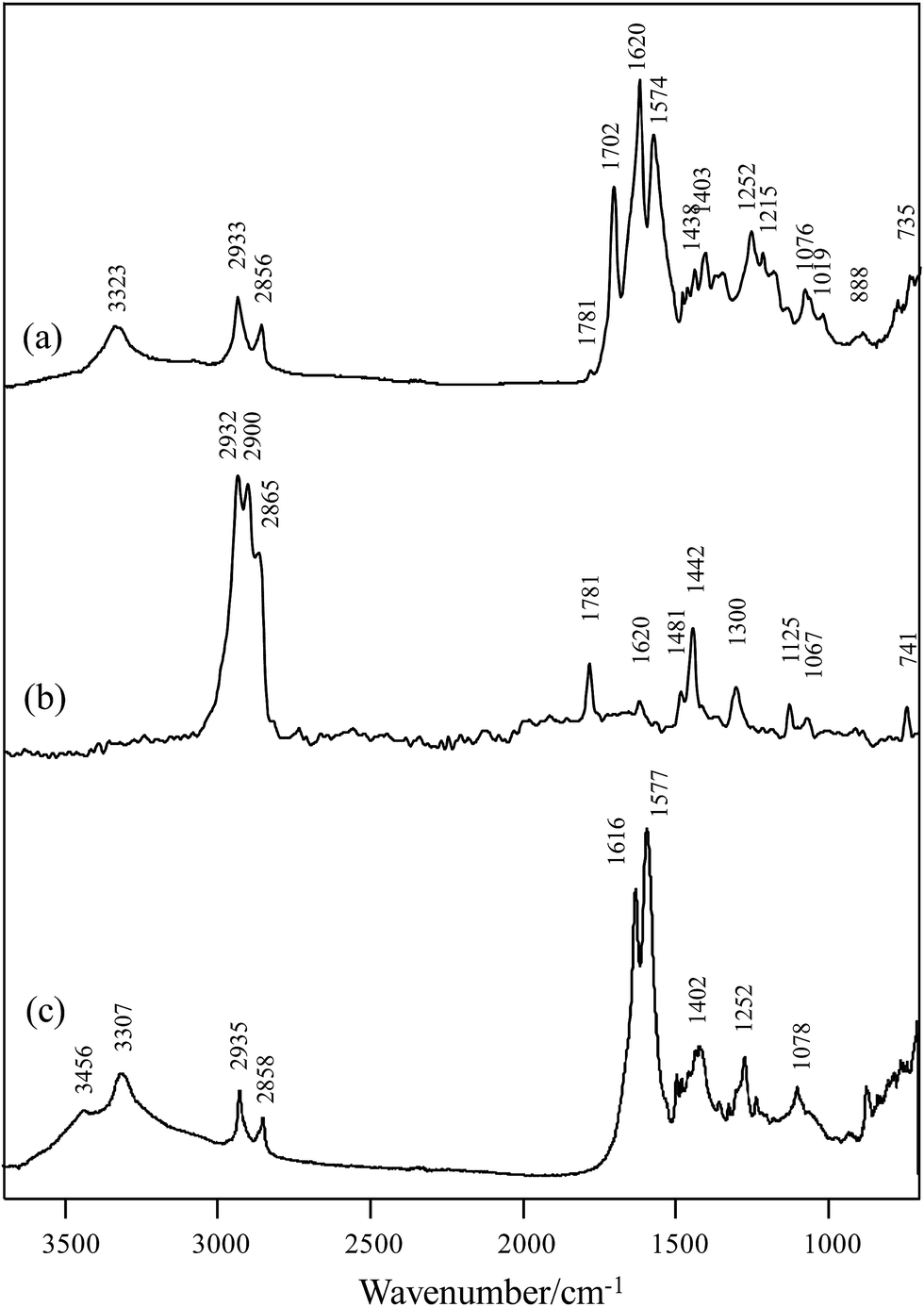 | ||
| Fig. 1 (a) FTIR and (b) FT-Raman spectra of poly(hexamethylene citramide imide); (c) FTIR spectrum of the hydrolyzed polymer. | ||
NMR spectroscopic analysis verified the structure of PHMCI. Fig. 2 (top panel) shows 1H NMR spectrum of the polymer. The peaks at 3.14 and 3.23 ppm are due to the protons on C8 and C10 of the citric moiety (see the chemical structure in Fig. 2 top for atomic numbering). In the hexamethylene moiety, protons on C3 and C4 appear at 1.57 ppm while protons on C2 and C5 show at 1.74 ppm. The resonance at 3.33 ppm is due to the protons on C1 and C6 atoms next to the N atoms. The resonance at 3.77 ppm is ascribed to the protons in the terminal –NH3+. Fig. 2 (bottom panel) shows 13C NMR spectrum of PHMCI. In the hexamethylene group, C1 and C6 carbons next to the N atoms (see Scheme 1) appear at 40.2 ppm, C2 and C5 carbons appear at 29.0 ppm, while C3 and C4 carbons emerge at 25.8 ppm. The peaks at 39.0 ppm and 39.6 ppm are attributed to the C10 in the citric acid moiety and C8 in the exocyclic carbon. The peak at 72.5 ppm is due to the C9 in the citric acid moiety. The peaks at 174.0, 176.9 and 179.7 ppm correspond to the C7, C11 and C18 carbonyls, respectively.
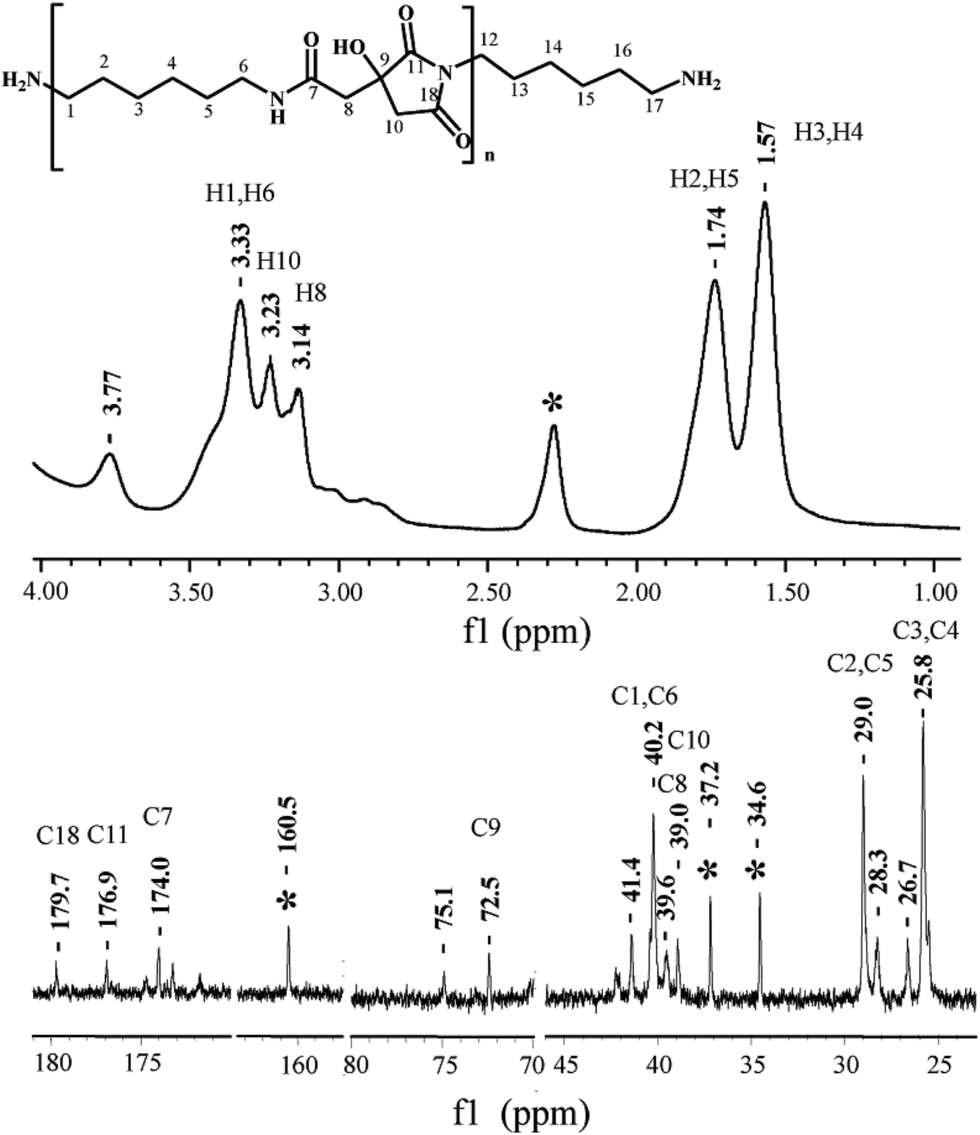 | ||
| Fig. 2 1H (top) and 13C (bottom) NMR spectra of poly(hexamethylene citramide imide) (note the peak at 2.3 ppm in the 1H spectrum and the peaks at 34.6, 37.2 and 160.5 ppm in the 13C spectrum are due to the residual solvent DMAc). | ||
The 1H–1H COSY NMR spectrum of the polymer provides further support to its chemical structure. In Fig. 3 (top panel), the cross peaks between the protons on the C4 and C5 at 1.53 and 1.69 ppm indicate their coupling to each other. Similar cross peaks between the protons on C5 and C6 at 1.69 and 3.38 ppm also show their coupling. The cross-peaks in the 1H–13C HSQC NMR spectrum in Fig. 3 (bottom panel) show that the proton at 1.57 ppm is connected to the carbon at 25.8 ppm, the proton at 1.74 ppm is connected to the carbon at 29.0 ppm, while the proton at 3.33 ppm is connected to the carbon at 40.2 ppm, corroborating their assignments to the hexamethylene structure. The proton at 3.14 ppm in the citric acid moiety is indeed connected to the carbon at 39.6 ppm, while the proton at 3.20 ppm is connected to the carbon at 38.9 ppm. These data illustrate the reaction between the OCN-group in the HMDI and the –COOH in the citric acid leads to the PHMCI structure shown in Scheme 1.
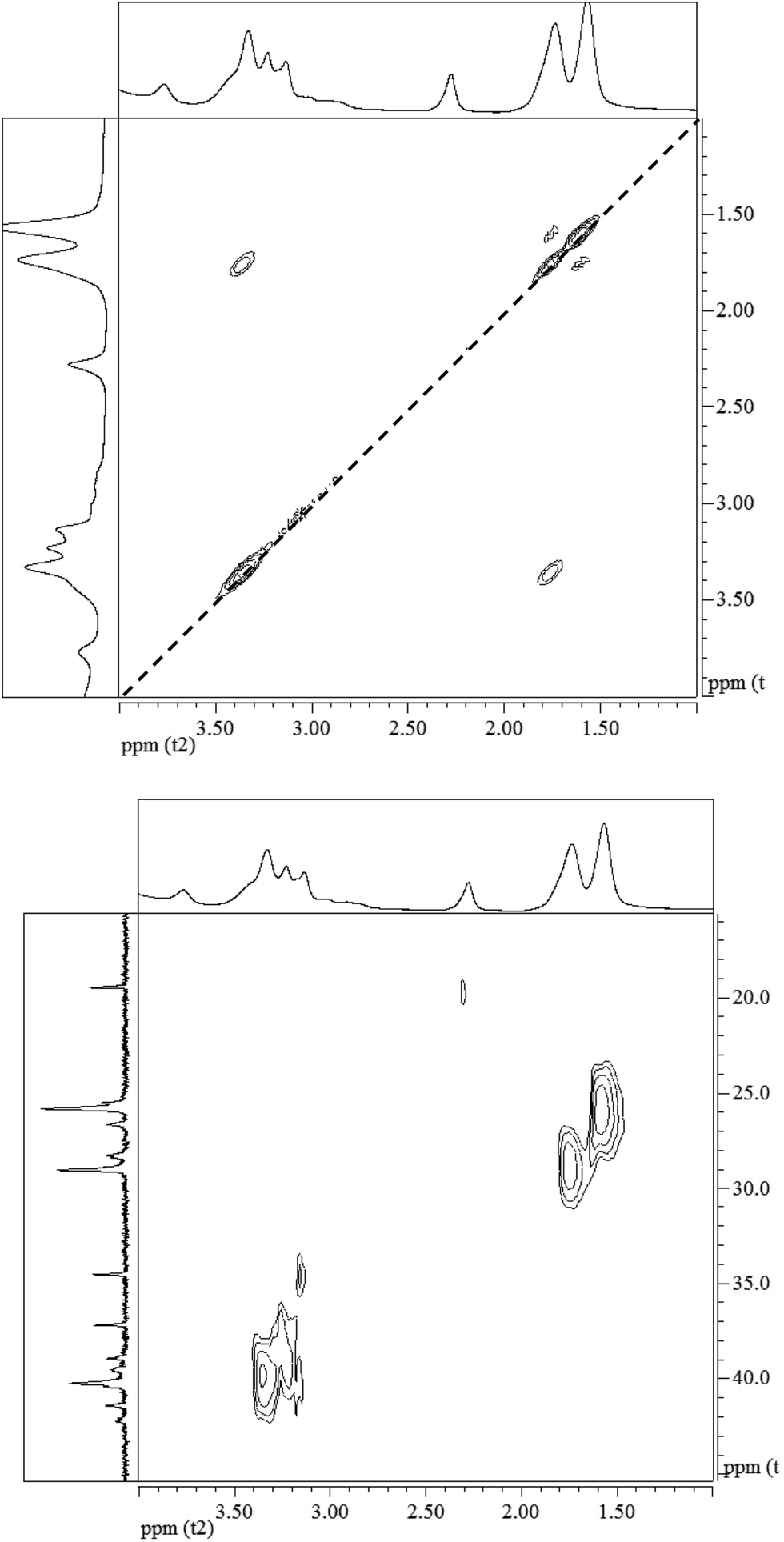 | ||
| Fig. 3 (Top) 1H–1H COSY NMR spectrum and (bottom) 1H–13C HSQC NMR spectrum of poly(hexamethylene citramide imide). | ||
The molecular weights of the PHMCI prepared under different conditions were determined by GPC analysis. The samples all have unimodal distributions of their MW (see Fig. 1S in ESI†). The Mn of PHMCI are in the range of 4 × 103–5 × 103 Da, while the dispersity of the MW is in the range of 1.31–1.67 (see Table 1S in ESI†).
Hydrolysis of PHMCI in alkaline solutions gives rise to poly(hexamethylene citramide) (PHMC) with a carboxylate side chain (see step 2 of Scheme 1). This reaction occurs by ring opening of the imide group through scission of either C–N bond.32 FTIR spectrum of the hydrolyzed polymer (Fig. 1c) shows disappearance of the imide CO band at 1702 cm−1 and appearance of bands due to the COO− group, when compared with Fig. 1a. The strong band at 1577 cm−1 and a medium band at 1402 cm−1 in Fig. 1c confirm the formation of the –COO− group, while the new band at 1616 cm−1 in Fig. 1c is attributed to the amide I band of the citramide formed. Hence, inter- or intrachain H-bonds between the amide groups in backbone and those between the COOH groups can be formed during self-assembly, besides the hydrophobic interactions between the –(CH2)6– groups and electrostatic forces involving the carboxylate side chain and terminal –NH3+. It is of note that the position of the amide I band at 1616 cm−1 in Fig. 1c indicates that the amide groups form β-pleated sheet structures.36
Self-assembly behavior of PHMC
The self-assembly behavior of PHMC in buffer solutions with different pH ranging from 5.5 to 10.0 was investigated by dynamic light scattering (DLS) method. The samples have unimodal distributions of their particle sizes at all of the pH points measured, as illustrated in Fig. 4. In Fig. 5, the hydrodynamic diameters (Dh) of the particles are plotted versus solution pH values. Interestingly, PHMC forms self-assembled structures with varying sizes depending on the pH. With the increase of the pH from 5.5 to 6.8, the Dh increases from ca. 60 nm to a maximum value of 2370 nm. Further increasing the pH lowers the Dh of the particles to 380 nm. The polydispersity index (PDI) values of the Dh change from 0.31 to 0.44 within the pH range from 6.0 to 10.0 (see Fig. 2S in ESI†). The origin of the unique pH dependent profile of Dh mainly comes from the ionization states of the carboxylate side group and terminal –NH3+ group, and consequently, the significant aggregation of the particles due to changes in the surface charges. Electrostatic interactions and H-bonding between the surface acidic and basic groups form the inter-particle forces that tether the particles into aggregated states in various pH buffer solutions. Different ionization states in PHMC particles in acidic, intermediate and basic pH values are supported by zeta-potential measurement.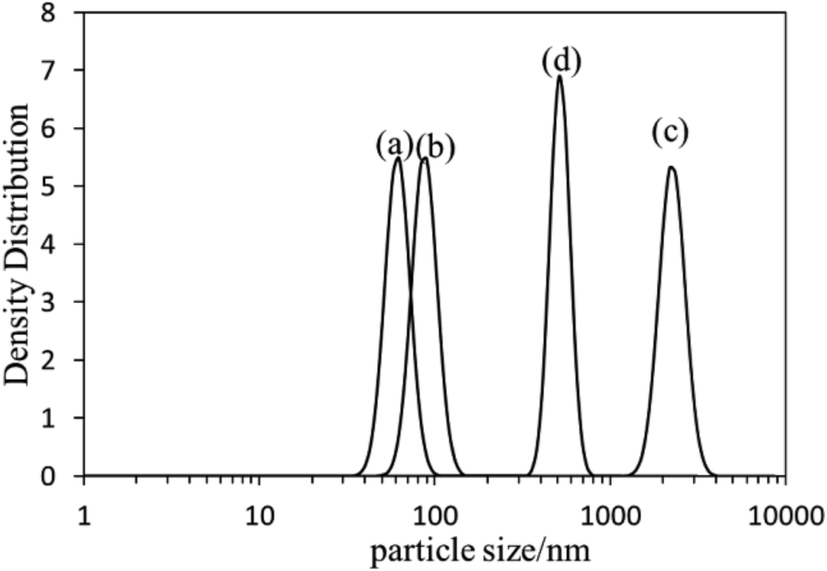 | ||
| Fig. 4 DLS profiles of poly(hexamethylene citramide) particles in (a) pH = 5.5 buffer; (b) pure water; (c) pH = 6.5 buffer; (d) pH = 8.5 buffer. | ||
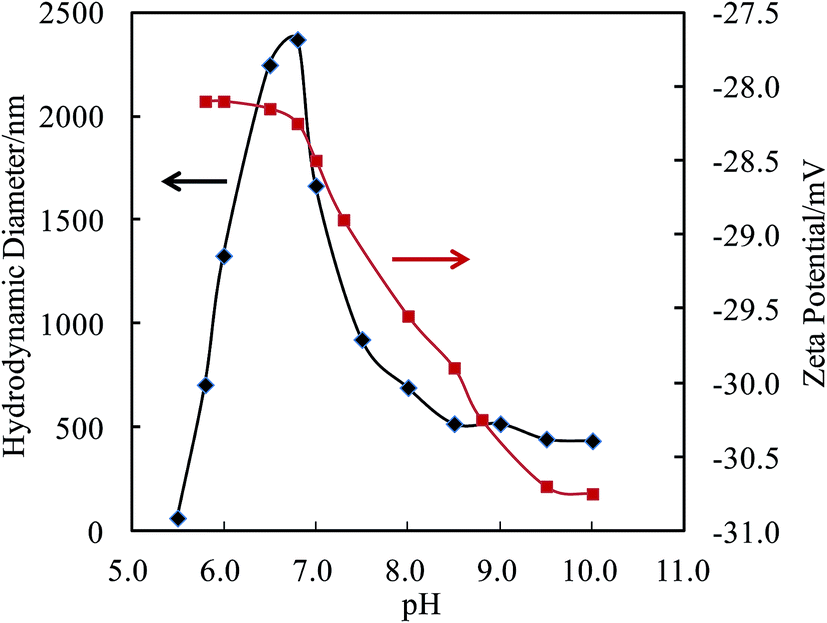 | ||
| Fig. 5 Hydrodynamic diameters and zeta-potentials of the particles in solution vs. pH values. | ||
In Fig. 5, analysis of the zeta-potentials of the particles shows that the negative charge of the particle surfaces drops from −28.1 to −30.7 mV when the solution pH is reduced from 5.7 to 10.0. No isoelectric points were observed in the pH range measured, which differs from polypeptides or proteins. A significant drop in the zeta-potential starts near a pH point of 6.8. Citric acid has pKa1 = 3.13, pKa2 = 4.76, pKa3 = 6.40, which means that in solution, citric acid is fully protonated at pH 2.0, partially deprotonated at pH 4.0–6.0, and fully deprotonated at pH 7.5.37 The high pKa3 value is attributed to the stabilization of the final proton in the acid by a COO− group of a neighboring molecule through strong intermolecular H bonds, resulting in a stable conjugate structure of R–COOH⋯−OOC–R, which opposes the removal of the final proton. Therefore, the drop in the zeta-potential occurring near the pH point of 6.8 can be related to the breakup of interchain or intrachain R–COOH⋯−OOC–R bridges. Considering the pKa of the terminal amine = 9.5, the gradual decrease of the zeta-potential between pH 6.8 and 9.5 in Fig. 5 illustrates that removal of the proton from the R–COOH⋯−OOC–R conjugate group from the citric acid units and deprotonation of terminal NH3+ occur in this wide range and overlap at ca. pH 8.0. The zeta-potential data shows three different ionization states and surface structures in acidic, intermediate and basic pH values.
The effect of different NaCl salt concentrations on the DLS and zeta potentials of the polymer has been analyzed, and the related data are shown in Table 2S in ESI.† Overall, there is no significant effect on the DLS and zeta potentials, when the NaCl concentration varies from 20 mM to 170 mM.
TEM analysis of the particles formed under different pH conditions and in pure water are displayed in Fig. 6, which all reveals that the polymer forms spherical particles (Fig. 4S in ESI† shows additional TEM images). The size of the particles (1145 particles) in multiple TEM images was measured and their distribution is also displayed in Fig. 6. A unimodal distribution of the particles with an average size of 21.0 ± 6.5 nm was found from the TEM images of the nanoparticles. The hydrodynamic diameter (Dh) of the particles in pure water is 90 nm (Fig. 4b), larger than the average particle size (21 nm) obtained from TEM, indicating that the polycitramide can swell significantly. DLS measures the particles in hydrated states, rather than the dried state when measured by TEM. In addition, DLS reflects the aggregated states of the particles (particles associated with each other).
 | ||
| Fig. 6 TEM image of poly(hexamethylene citramide) nanoparticles formed under different pH conditions (top left, pH = 5.5; top right, pH = 7.0; bottom left, pH 9.0) and in pure water (bottom right), and the corresponding particle size distribution in pure water. | ||
The above pH dependent zeta-potential profiles and Dh distributions indicate that on going from pH 6.0 to 7.0, the protons in the R–COOH⋯−OOC–R bridges are gradually deprotonated, but the terminal –NH3+ species remain to be protonated. The repulsive force may surpass the H-bonding forces between the COOH groups and those between the amide groups, depending on the pH. The NH3+ and R–COO− species on the surfaces and the R–COOH groups in the cores may change their populations with the pH. Intermolecular association increases with increasing electrostatic interactions between the –NH3+ and −OOC–R groups. The driving forces for the core formation of the nanoparticles are the delicate interchain electrostatic force and H-bonding forces. Below pH 6.5, the noncovalent cross-linking through cooperative backbone amide H-bonding units as well as the H-bonding pairs of the R–COOH⋯HOOC–R from the adjacent citramide moieties dominate in the nanoparticles.25 Both acid groups in the R–COOH⋯HOOC–R pair act as proton acceptor and donor, forming a large 8-membered ring with two H-bonds. Such a kind of H-bonding is the strongest one found within neutral species, and termed a resonance-assisted hydrogen bond.38,39 Above pH 7.5, the R–COOH⋯−OOC–R conjugates are essentially all broken up, giving rise to negatively charged surface of the particles when the terminal NH3+ groups are deprotonated. At every pH point, H-bonds between the amide groups and/or between the hydroxyl groups at the tertiary carbon atoms provide critical forces for stabilizing the cores, like in folded globular proteins or in multimers of folded protein molecules.
Supramolecular–polymer complexes by cooperative H-bonding between main chains
To investigate the potential patterns of the hydrogen bonding in the nanoparticles cores, selected segments of the polycitramides were simulated using quantum chemical calculations and the results shown in Fig. 7 have confirmed the formation of the above cooperative H-bonding interactions. In Fig. 7, highly specific H-bonding pairs are found in the hydrophilic citramide moieties from density functional theory-based calculations, namely, between the backbone amide units and between the hydroxyl groups in the chain. In the protonated states (Fig. 7a), two adjacent citramide moieties in the PHMC chains form two anti-parallel pairs of H-bonding pairs, one pair of the amide groups in donor–acceptor mode while the other pair in acceptor–donor mode. In the deprotonated states (Fig. 7b), interestingly, one pair of relatively short H-bonding appears between the –OH groups from the tertiary carbon atoms whereas the H-bonding interactions between the amide groups become weaker (corresponding to longer O⋯⋯H distances). The hydroxyl groups in the citramide moieties are in the α-positions of the carbonyl groups and relatively labile. The calculations explain transition of the cooperative H-bonds at low and high pH values, and therefore, changes of the main chain H-bonding strengths at varying pH. In Fig. 7c, the supramolecular-polymer complexes within the particles are depicted and proposed to be stabilized by cooperative H-bonding between the main chains.40,41 A triple H-bonding motif is latent in the linear polymers.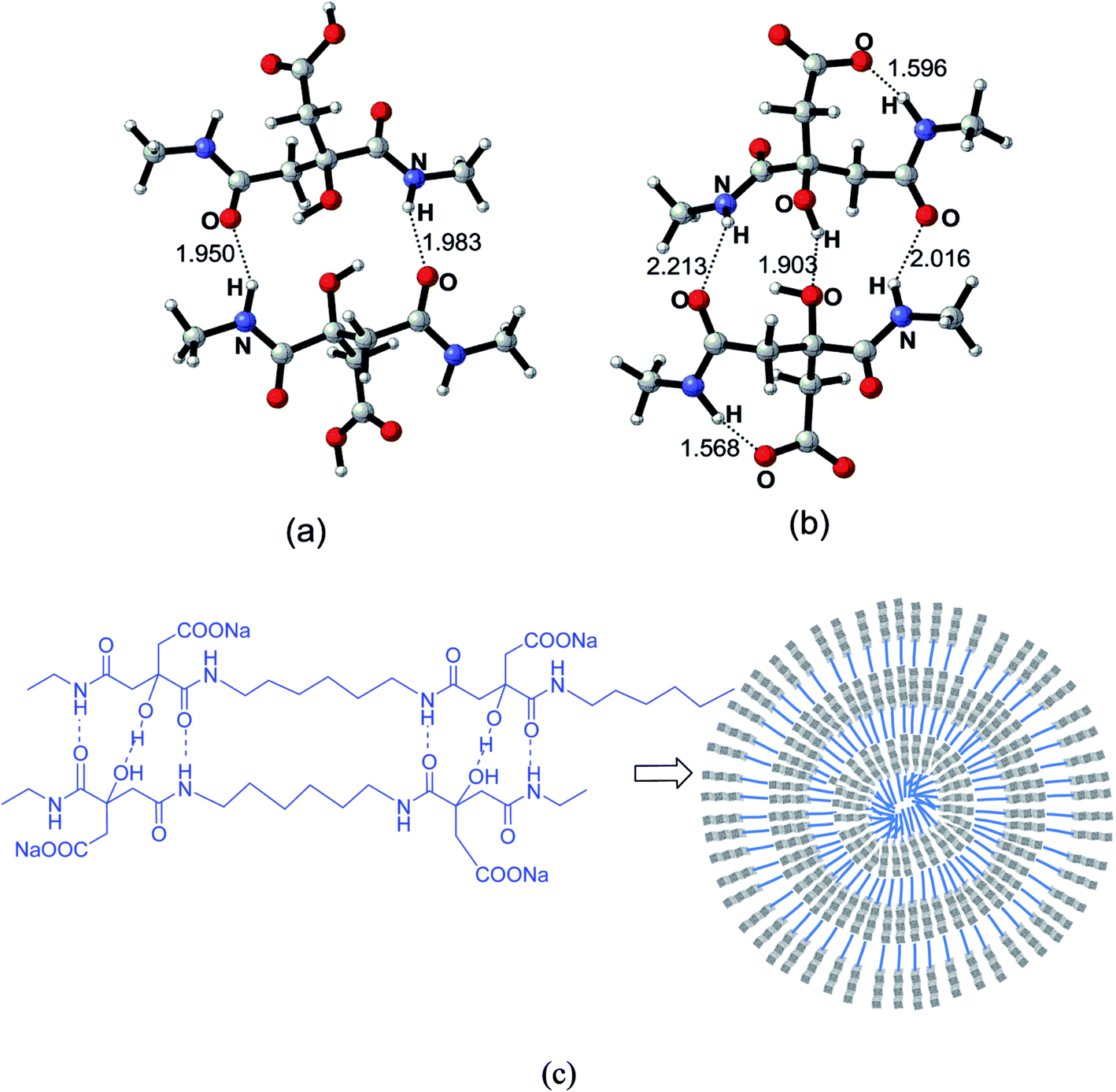 | ||
| Fig. 7 Supramolecular–polymer complex through the noncovalent cross-linking of cooperative backbone amide H-bonding units and between the hydroxyl groups in the chain. The two COOH groups from the adjacent citramide moieties in the PHMC chain are in protonated states (a) and in deprotonated states (b). The bond distances are in angstrom. (c) Illustrative scheme of a spherical particle using the supramolecular–polymer complexes. | ||
Further quantum chemical calculations indicate that the stable H-bonds between the amide backbones of the polycitramides are formed without involvement of water molecules. Precipitation of water molecules is energetically less favored than that between two selected segments of the polycitramides (see Fig. 5S and 6S in ESI†).
Metal chelation of the PHMC particles
Finally, from the application point of view, the metal chelating capability of the polyelectrolyte particles is evaluated since they retain metal binding and sequestration functionalities. Removal of metals from aqueous systems or soil with functionalized nanoparticles is an important technique in environmental remedy.42–44Table 1 shows the particles have very high chelating capabilities for metals such as Ca2+ and Pb2+, very close to recently reported polycitramides.32 Poly(aspartic acid) (PASP) polymers have chelating capacities of 54.0–65.0 mg g−1 for Ca2+ ion,45 while polyesters prepared from citric acid and D-sorbitol have even lower chelating capacities of 13–26 mg g−1 for Ca2+ ion in a recent study.46 Commercial chelating resins, e.g., Amberlite 200, Duolite GT-73, and Lewatit TP 207, have chelating capacities of 122–352 mg g−1 for Pb2+ ion.47 It is believed that both the COOH side group and the amide group can provide binding sites for the Ca and Pb metals, giving rise to the enhanced chelation capacities.| Ca2+ | Pb2+ | |||
|---|---|---|---|---|
| Metal ion concentration/mmol L−1 | 1.00 | 0.100 | 1.00 | 0.100 |
| Metal ion concentration after chelation/mmol L−1 | 0.60 ± 0.09 | 0.041 ± 0.007 | 0.51 ± 0.19 | 0.020 ± 0.003 |
| Metal ion chelating capability/mg g−1 | 160 ± 36 | 23.8 ± 2.7 | 1015.9 ± 391.4 | 166.7 ± 6.7 |
The effect of the initial Ca and Pb metal concentrations on the chelating capacity is also shown in Table 1. Increasing the Ca and Pb metal concentrations in the range of 0.100 to 1.00 mmol L−1 can increase the uptake values. Energy dispersive spectroscopic (EDS) analysis results of the polymers after they are chelated by Ca or Pb metals (Fig. 7S in ESI†) confirm that the polymers are chelated by Ca and Pb metals.
Conclusions
A novel type of supramolecular–polymer complex based on a polyelectrolyte chain is synthesized by using step-growth polymerization of hydrophilic and hydrophobic monomers. In water, the polycitramides with electrostatic forces, hydrophobic forces, hydrogen bonding structures have a unique self-organization profile. The nanoparticles show the inter-conversion of the ionizable species and changes of the core forming force strengths at varying pH. Noncovalent cross-linking of cooperative hydrogen-bonding units between the citramide moieties and between the hydroxyl groups stabilizes the self-assembled structures of the polyelectrolyte chains, leading to supramolecular–polymer complexes. In addition, changes in distributions of acidic and basic groups on the surface layers in various pH buffer solutions may tether the particles into aggregated states near pH 6.5–7.0. Similar forces for the self-assembly are known in the folding and misfolding of certain polypeptides and proteins. However, without an isoelectric point, the nanoparticles in the present study are far more stable than self-assembled polypeptide or proteins which have less flexible main chains. The principle and approach proposed in this study can be used for generating new polymer particles with bio-inspired nanostructures for metal sequestration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21674063, 21202102 and 20974063) and Natural Science Foundation of Zhejiang Province (No. LY12E03001). All calculations in this study were performed at Shenzhen Supercomputer Center.References
- J. Rodríguez-Hernández, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- T. Akagi, M. Baba and M. Akashi, Polymer, 2007, 48, 6729–6747 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- H. Shen, L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1999, 121, 2728–2740 CrossRef CAS.
- A. S. Lee, V. Butun, M. Vamvakaki, S. P. Armes, J. A. Pople and A. P. Gast, Macromolecules, 2002, 35, 8540–8551 CrossRef CAS.
- (a) S. Liu, N. C. Billingham and S. P. Armes, Angew. Chem., Int. Ed., 2001, 40, 2328–2331 CrossRef CAS; (b) Y. Cai and S. P. Armes, Macromolecules, 2004, 37, 7116–7122 CrossRef CAS.
- (a) J. V. M. Weaver, S. P. Armes and S. Liu, Macromolecules, 2003, 36, 9994–9998 CrossRef CAS; (b) S. Wan, M. Jiang and G. Zhang, Macromolecules, 2007, 40, 5552–5558 CrossRef CAS; (c) J. F. Gohy, S. K. Varshney and R. Jerome, Macromolecules, 2001, 34, 3361–3366 CrossRef CAS.
- A. Walther, A. S. Goldmann, R. S. Yelamanchili, M. Drechsler, H. Schmalz, A. Eisenberg and A. H. E. Müller, Macromolecules, 2008, 41, 3254–3260 CrossRef CAS.
- C. Fernyhough, A. J. Ryan and G. Battaglia, Soft Matter, 2009, 5, 1674–1682 RSC.
- S. Yusa, A. Sakakibara, T. Yamamoto and Y. Morishima, Macromolecules, 2002, 35, 5243–5249 CrossRef CAS.
- S. Yusa, A. Sakakibara, T. Yamamoto and Y. Morishima, Macromolecules, 2002, 35, 10182–10188 CrossRef CAS.
- P. Piyapakorn, T. Akagi, M. Hachisuka, T. Onishi, H. Matsuoka and M. Akashi, Macromolecules, 2013, 46, 6187–6194 CrossRef CAS.
- T. Akagi, P. Piyapakorn and M. Akashi, Langmuir, 2012, 28, 5249–5256 CrossRef CAS.
- F. Petit-Agnely, I. Iliopoulos and R. Zana, Langmuir, 2000, 16, 9921–9927 CrossRef CAS.
- Y. P. Bai, L. Huang, T. Huang, J. Long and Y. Zhou, Polymer, 2013, 54, 4171–4176 CrossRef CAS.
- M. Ueda, A. Hashidzume and T. Sato, Macromolecules, 2011, 44, 2970–2977 CrossRef CAS.
- E. Di Cola, N. Plucktaveesak, T. A. Waigh, R. H. Colby, J. S. Tan, W. Pyckhout-Hintzen and R. K. Heenan, Macromolecules, 2004, 37, 8457–8465 CrossRef CAS.
- C. Malardier-Jugroot, T. G. M. van de Ven and M. A. Whitehead, J. Phys. Chem. B, 2005, 109, 7022–7032 CrossRef CAS.
- G. Garnier, M. Duskova-Smrckova, R. Vyhnalkova, T. G. M. van de Ven and J.-F. Revol, Langmuir, 2000, 16, 3757–3763 CrossRef CAS.
- C. Malardier-Jugroot, T. G. M. van de Ven, T. Cosgrove, R. M. Richardson and M. A. Whitehead, Langmuir, 2005, 21, 10179–10187 CrossRef CAS.
- J. Claracq, S. F. C. R. Santos and J. Duhamel, Langmuir, 2002, 18, 3829–3835 CrossRef CAS.
- T. D. Lazzara, T. G. M. van de Ven and M. A. Whitehead, Macromolecules, 2008, 41, 6747–6751 CrossRef CAS.
- Y. Zhu, L. Liu and J. Du, Macromolecules, 2013, 46, 194–203 CrossRef CAS.
- A. V. Dobrynin and M. Rubinstein, Macromolecules, 2000, 33, 8097–8105 CrossRef CAS.
- T. Mondal, K. Dan, J. Deb, S. S. Jana and S. Ghosh, Langmuir, 2013, 29, 6746–6753 CrossRef CAS.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS PubMed.
- K. Matsuura, K. Murasato and N. Kimizuka, J. Am. Chem. Soc., 2005, 127, 10148–10149 CrossRef CAS.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS.
- J. Dong, C. S. Atwood, V. Anderson, S. L. Siedlak, M. Smith, G. Perry and P. R. Carey, Biochemistry, 2003, 42, 2768–2773 CrossRef CAS PubMed.
- M. Jiang, W. Chen, P. Fang, S. Chen, J. Bai, Y. Huang, M. Han, P. Lu and J. Dong, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3819–3829 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, R. D. 01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS; (c) Y. Zhao and D. G. Truhlar, Chem. Phys. Lett., 2011, 502, 1–13 CrossRef CAS.
- X. Zhu, P. Lu, W. Chen and J. Dong, Polymer, 2010, 51, 3054–3063 CrossRef CAS.
- M. Bouchard, J. Zurdo, E. J. Nettleton, C. M. Dobson and C. V. Robinson, Protein Sci., 2000, 9, 1960–1967 CrossRef CAS.
- I. A. Mudunkotuwa and V. H. Grassian, J. Am. Chem. Soc., 2010, 132, 14986–14994 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- L. Evangelisti, P. Ecija, E. J. Cocinero, F. Castaño, A. Lesarri, W. Caminati and R. Meyer, J. Phys. Chem. Lett., 2012, 3, 3770–3775 CrossRef CAS PubMed.
- M. Seo, B. J. Beck, J. M. J. Paulusse, C. J. Hawker and S. Y. Kim, Macromolecules, 2008, 41, 6413–6418 CrossRef CAS.
- H. Nakade, M. Firat Ilker, B. J. Jordan, O. Uzun, N. L. LaPointe, E. B. Coughlin and V. M. Rotello, Chem. Commun., 2005, 3271–3273 RSC.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS.
- P. Liu and T. Wang, J. Hazard. Mater., 2007, 149, 75–79 CrossRef CAS.
- X. Liu, Q. Hu, Z. Fang, X. Zhang and B. Zhang, Langmuir, 2008, 25, 3–8 CrossRef.
- T. Nakato, M. Yoshitake, K. Matsubara, M. Tomida and T. Kakuchi, Macromolecules, 1998, 31, 2107–2113 CrossRef CAS.
- K. M. Doll, R. L. Shogren, J. L. Willett and G. Swift, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4259–4267 CrossRef CAS.
- Z. Reddad, C. Gerente, Y. Andres and P. Le Cloirec, Environ. Sci. Technol., 2002, 36, 2067–2073 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data of the polycitramides. See DOI: 10.1039/c7ra09205a |
| This journal is © The Royal Society of Chemistry 2017 |