
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, crystallographic characterization and homogeneous catalytic activity of novel unsymmetric porphyrins†
A. R. Antonangelo a,
K. C. M. Westrupa,
L. A. Burtb,
C. Grazia Bezzub,
T. Malewschika,
G. S. Machadoc,
F. S. Nunesa,
N. B. McKeown*b and
S. Nakagaki*a
a,
K. C. M. Westrupa,
L. A. Burtb,
C. Grazia Bezzub,
T. Malewschika,
G. S. Machadoc,
F. S. Nunesa,
N. B. McKeown*b and
S. Nakagaki*a
aLaboratório de Bioinorgânica e Catálise, Departamento de Química, Universidade Federal do Paraná, Curitiba, 81531-990, Brazil. E-mail: shirleyn@ufpr.br
bEastChem School of Chemistry, University of Edinburgh, Edinburgh, Scotland EH9 3FJ, UK. E-mail: neil.mckeown@ed.ac.uk
cCentro de Estudos do Mar, Pontal do Paraná-PR, Universidade Federal do Paraná (UFPR), Brazil
First published on 31st October 2017
Abstract
Unsymmetric porphyrins, containing both pentafluorophenyl (PFP = A) and 3,4-dimethoxyphenyl (DMP = B) substituents at the meso positions, were prepared using Lindsey's methodology. The isomeric trans-A2B2 (P1) and cis-A2B2 (P2) porphyrins together with the tris(pentafluorophenyl)porphyrin A3B (P3) were isolated using chromatography. The porphyrins were characterized by UV-VIS, 1H NMR spectroscopy, mass spectrometry, elemental analysis (C, H, N) and cyclic voltammetry (CV), and their molecular structures were confirmed by single crystal XRD. Their manganese complexes, MnP1, MnP2 and MnP3, were also synthesised and used as catalysts in cyclooctene and cyclohexane oxidation reactions under homogeneous conditions. The catalytic studies were supported by electrochemical measurements and showed that the number of electron-withdrawing substituents on the porphyrins rings influences the catalytic activity. These porphyrins may be used as precursors for the design of new materials, such as Polymers of Intrinsic Microporosity (PIMs).
Introduction
Porphyrins are macrocyclic compounds with a large extended conjugated π-system and they are often called “the pigments of life”, as they play an important role in many biological systems.1 These compounds and their metallated complexes exhibit unique properties2 and can be synthesized through various methodologies.3 The macrocycles also show great flexibility towards structural modification which makes them very interesting molecules in a number of different fields.4Metalloporphyrins, especially those containing Fe(III) and Mn(III) cations, have been extensively used as catalysts in oxidation reactions of a range of organic compounds using different oxidants. Studies have shown that the introduction of electron-withdrawing and/or bulky substituents at the ortho position of the meso-phenyl porphyrins (so called second generation porphyrins) can enhance their catalytic activity in oxidation reactions.5–7 This performance stems from the electronic effects that the substituents exert on the porphyrin ring, which enhances the electrophilicity of the active catalytic species (metal-oxo porphyrin) and also protects the ring, to some extent, against auto oxidative destruction.6,7 In this context, the free base ligand [5,10,15,20-tetrakis(pentafluorophenyl)porphyrin], [H2(TPFPP)], represents an important compound.6–8 The fluorine atoms on the phenyl ring at the four meso positions increases the stability and catalytic activity. In addition, the fluorine at para positions can be readily substituted by nucleophiles, which makes this compound a versatile platform for the design of new materials.8,9 Several materials have been recently prepared, which utilize the [H2(TPFPP)] porphyrin in a heterogeneous catalytic system.8,10–13 McKeown and co-workers used [H2(TPFPP)] porphyrin as a monomer in a polymerization reaction with 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (biscatechol) to prepare a porphyrin network PIM (so called polymer of intrinsic microporosity).12 The high surface area (up to 1000 m2 g−1) and the chemical stability of this material encourages the synthesis of new network PIMs and their application in heterogeneous oxidation catalysis.12–14
Meso substituted A2B2 trans-porphyrins bearing different functional groups are useful building blocks for the design and synthesis of new materials with a wide range of applications, such as biomimetic systems and molecular materials.15–17 The trans pattern of substitution in these kinds of porphyrins can be used to achieve new materials with well-defined structures. For example, the same polymerization reaction previously used to synthesize the porphyrin network PIM, could be employed utilizing one meso–trans A2B2 porphyrin as a preformed monomer in order to achieve new soluble ladder PIMs. Moreover, transition metal ions may also be coordinated to the porphyrin units for future investigation in catalytic membranes.14 Thus, we undertook the synthesis of a group of new unsymmetric porphyrins with either pentafluorophenyl (PFP = A) or 3,4-dimethoxyphenyl (DMP = B) at the four meso positions.
Experimental
Materials
All chemicals used in this study were purchased from Sigma-Aldrich, Merck or Fluka and were of analytical grade. Iodosylbenzene (PhIO) was synthesized according to the literature.18 The solid was carefully dried under reduced pressure and kept at 5 °C; its purity was assessed by iodometric titration.191H and 13C NMR spectra were recorded on Bruker Advance III 500 MHz spectrometers, using deuterated chloroform as solvent. Chemical shifts are reported in ppm (δ) and coupling constants (J) are given in Hz. The multiplicities of the signals in the 1H NMR spectra are abbreviated by s (singlet), d (doublet), q (quartet) and m (multiplet). Mass spectra were acquired on a Bruker Ultraflex MALDI-TOF or Thermo Electron MAT900 mass spectrometers (MALDI or EI). Electronic spectra (UV-VIS) were obtained from dichloromethane solutions on a Hewlett-Packard HP 8452A diode array spectrophotometer in the 200–800 nm range. Analysis were accomplished with a 1 cm path length cell. Fourier Transform Infrared (FTIR) spectra were measured on a Biorad 3500 GX spectrophotometer in the 400–4000 cm−1 range with a spectral resolution of 4 cm−1. Elemental analysis were obtained from a Series II 2400 Perkin Elmer Elemental Analyzer. Electron paramagnetic resonance (EPR) measurements of the powdered new manganese porphyrins were conducted on an EPR Bruker EMX MicroX spectrometer (frequency X, band 9.5 GHz) at room temperature and 77 K (liquid N2), by using perpendicular microwave polarization X-band.
Cyclic voltammetry (CV) was carried out with an IVIUM CompactStat potentiostat/galvanostat. A glassy carbon electrode was employed for the measurements at I = 0.1 mol L−1 kept constant with TBAPF6 (tetrabutylammonium hexafluorophosphate). A silver wire and a platinum wire were used as pseudo-reference and auxiliary electrodes, respectively. Ferrocene was used as an internal standard reference and potentials are reported versus the standard hydrogen electrode, SHE. Experiments were conducted with a 1.0 × 10−3 mol L−1 complex concentration in dichloromethane solutions at ambient temperature and under argon atmosphere.
Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate:triethylamine (80:20:1). The solid (pale yellow) was then triturated with hexane and filtered, yielding a colourless solid (1.29 g, 63.2% yield). Molecular formula: C17H18N2O2. 1H NMR data (500 MHz, CDCl3, 298 K) δ ppm: 7.95 (s, 2H, NH pyrrole), 6.85–6.84 (d, 1H, J = 8.1 Hz, ArH), 6.79–6.76 (m, 2H, ArH), 6.73–6.72 (m, 2H, CH pyrrole), 6.20–6.18 (q, 2H, J = 2.8 Hz, CH pyrrole), 5.98–5.96 (m, 2H, CH pyrrole); 5.46 (s, 1H, CH meso), 3.90 (s, 3H, –OCH3), 3.83 (s, 3H, –OCH3). 13C NMR data (125 MHz, CDCl3, 298 K) δ ppm: 149.1, 148.1, 134.6, 132.7, 120.3, 117.1, 111.8, 111.2, 108.5, 107.1, 56.0, 55.9, 43.6. LRMS (EI): m/z 282.1 (M+), according to the literature data.21
:ethylacetate:triethylamine (80:20:1). The solid (pale yellow) was then triturated with hexane and filtered, yielding a colourless solid (1.29 g, 63.2% yield). Molecular formula: C17H18N2O2. 1H NMR data (500 MHz, CDCl3, 298 K) δ ppm: 7.95 (s, 2H, NH pyrrole), 6.85–6.84 (d, 1H, J = 8.1 Hz, ArH), 6.79–6.76 (m, 2H, ArH), 6.73–6.72 (m, 2H, CH pyrrole), 6.20–6.18 (q, 2H, J = 2.8 Hz, CH pyrrole), 5.98–5.96 (m, 2H, CH pyrrole); 5.46 (s, 1H, CH meso), 3.90 (s, 3H, –OCH3), 3.83 (s, 3H, –OCH3). 13C NMR data (125 MHz, CDCl3, 298 K) δ ppm: 149.1, 148.1, 134.6, 132.7, 120.3, 117.1, 111.8, 111.2, 108.5, 107.1, 56.0, 55.9, 43.6. LRMS (EI): m/z 282.1 (M+), according to the literature data.21Porphyrin P1. Purple solid (0.134 g, 6.4% yield). Elemental analysis (%) calcd. for C48H28F10N4O4: C 63.03, H 3.09, N 6.12. Found: C 62.97, H 3.18, N 5.73.1H NMR data (500 MHz, CDCl3, 298 K) δ ppm = 9.04–9.03 (d, 4H, J = 4.8 Hz, Hβ-pyrrolic), 8.82–8.81 (d, 4H, J = 4.8 Hz, Hβ-pyrrolic), 7.80–7.78 (m, 4H, ArH), 7.32–7.30 (d, 2H, J = 7.9 Hz, ArH); 4.22 (s, 6H, –OCH3), 4.03 (s, 6H, –OCH3), −2.78 (s, 2H, NH). UV-VIS in CH2Cl2 at 298 K, λmax (log
ε/M−1 cm−1): 420 (5.38), 512 (4.25), 548 (3.77), 588 (3.76) and 644 (3.51). FT-IR data (cm−1, KBr pellet): 3321, 3116, 2933, 1517, 1496, 1479, 1465, 1440, 1406, 1353, 1346, 1319, 1257, 1237, 1166, 1139, 1076, 1041, 1027, 987, 923, 919, 813, 802, 779, 756, 734. MALDI-TOF-MS (m/z): 914.1 [MH]+.
Porphyrin P2. Purple solid (0.055 g, 2.6% yield). Elemental analysis (%) calcd. for C48H28F10N4O4: C 63.03, H 3.09, N 6.12, found: C 63.13, H 3.18, N 5.72.1H NMR data (500 MHz, CDCl3, 298 K) δ ppm: 9.04–9.03 (d, 2H, J = 4.8 Hz, Hβ-pyrrolic), 8.95 (s, 2H, Hβ-pyrrolic), 8.87 (s, 2H, Hβ-pyrrolic), 8.79–8.78 (d, 2H, J = 4.8 Hz, Hβ-pyrrolic), 7.80–7.76 (m, 4H, ArH), 7.31–7.29 (d, 2H, J = 8.1 Hz, ArH), 4.21 (s, 6H, –OCH3); 4.03 (s, 6H, –OCH3), −2.70 (s, 2H, NH). UV-VIS in CH2Cl2 at 298 K, λmax (log
ε/M−1 cm−1): 421 (5.39), 514 (4.24), 550 (3.74), 588 (3.78) and 642 (3.22). FT-IR data (cm−1, KBr pellet): 3321, 3105, 2931, 1515, 1498, 1479, 1463, 1440, 1404, 1350, 1321, 1257, 1238, 1166, 1139, 1120, 1081, 1041, 1027, 987, 918, 865, 802, 761, 742. MALDI-TOF-MS (m/z): 914.1 [MH]+.
Porphyrin P3. Purple solid (0.01 g, 0.5% yield). Elemental analysis (%) calcd. for C46H19F15N4O2: C 58.49, H 2.03, N 5.93, found: C 57.06, H 1.84, N 5.68.1H NMR data (500 MHz, CDCl3, 298 K) δ ppm: 9.06–9.05 (d, 2H, J = 4.8 Hz, Hβ-pyrrolic), 8.90 (m, 4H, Hβ-pyrrolic), 8.83–8.82 (d, 2H, J = 4.8 Hz, Hβ-pyrrolic), 7.79–7.77 (m, 2H, ArH); 7.32–7.30 (d, 1H, J = 8.0, ArH), 4.22 (s, 3H, –OCH3), 4.03 (s, 3H, –OCH3), −2.79 (s, 2H, NH). UV-VIS in CH2Cl2 at 298 K, λmax (log
ε/M−1 cm−1):415 (5.41), 510 (4.31), 544 (3.71), 586 (3.90) and 638 (3.30). FT-IR data (cm−1, KBr pellet): 3321, 3107, 2933, 1517, 1498, 1483, 1467, 1438, 1402, 1352, 1342, 1319, 1257, 1240, 1166, 1143, 1078, 1058, 1045, 1027, 989, 925, 918, 802, 756, 736. MALDI-TOF-MS (m/z): 944.1 [MH]+.The free base porphyrins P1, P2 and P3 were metallated by a methodology described in the literature23 using manganese(II) acetate as metal salt. The process was monitored by TLC and UV-VIS techniques. The complexes were purified by column (CH2Cl2 and MeOH) and the excess of salt was removed by washing the metalloporphyrins solids with plenty of warm water. The new metalloporphyrins, designated MnP1, MnP2 and MnP3, respectively, were characterized by UV-VIS spectroscopy (ESI: Fig. S7†) and EPR technique was used to confirm the oxidation state of manganese and also to confirm the absence of manganese(II) acetate (ESI: Fig. S8†).
Single crystal X-ray diffraction
Single crystal X-ray diffraction data were collected on either a i19-FFD-air (fixed Chi) Pilatus M2 at the Diamond Light source Beam line I19-1 (P1) or a Rigaku Oxford Diffraction Super Nova (P2 and P3) diffractometer using synchrotron (0.6889 Å) or Cu-Kα (1.5418 Å) radiation, respectively. The crystal temperature was controlled using an Oxford Cryo systems Cryo stream 700+ low temperature device for all of the samples. All data were processed using Crysalis Pro. Crystal structure solutions were achieved using intrinsic phasing with ShelXT and refined using least squares minimization with the ShelXL refinement package interfaced with Olex2.24,25 In structures P2 and P3 N-bound hydrogens were identified from a difference Fourier map and refined with appropriate geometric restraints. C-bound H atoms in all structures were placed in calculated geometric positions and refined using the riding model. Crystallographic data for structures P1, P2 and P3 have been deposited at the Cambridge Crystallographic Data Center. CCDC: 1483009, 1536642 and 1536643. Further crystallographic data can be found in the ESI.†Catalytic studies of MnP1, MnP2 and MnP3 using (Z)-cyclooctene and cyclohexane as substrates
The catalytic activity and efficiency of the new metalloporphyrins was tested in the oxidation reactions of (Z)-cyclooctene (previously purified on alumina column) and cyclohexane with iodosylbenzene (PhIO). Furthermore, the two associated symmetric metalloporphyrins, (5,10,15,20-tetrakis-pentafluorophenyl)porphyrin manganese(III), MnTPFPP, and 5,10,15,20-tetrakis-(3,4-dimethoxyphenyl)porphyrin manganese(III), MnTDMPP, were also used as catalysts for comparison.The catalytic reactions were performed in a 1.5 mL glass flask equipped with a magnetic stirrer, in a dark chamber. The solvent mixture (acetonitrile/dichloromethane, ACN:DCM 1:1, v/v) and the substrates were purged with argon for 15 min. The catalysts (MnP1, MnP2, MnP3, MnTPFPP and MnTDMPP) and the oxidant PhIO (catalyst/PhIO at a molar ratio of 1:10) were added in the reaction flask and they were also purged with argon for 15 min. Then, the mixture of solvents (ACN:DCM, 1:1) was added to the reactions flasks, followed by addition of the substrates (Z)-cyclooctene or cyclohexane. The molar ratio (catalyst/PhIO/substrate) used was 1:10:1000 which was calculated based on the molecular mass of the metalloporphyrins (about 0.5 mg of metalloporphyrins were used for each reaction). The oxidation reactions were performed under magnetic stirring for 1 h, at room temperature, in the absence of light. After 1 h, the excess of iodosylbenzene was consumed by addition of sodium sulfite and the reaction mixture was transferred to a volumetric flask (2 mL). The resulting solution containing the reaction products was analyzed by gas chromatography using bromobenzene or 1-pentanol as internal standards. The products from the oxidation reactions were quantified with an Agilent 6850 gas chromatograph (flame ionization detector) equipped with a 30 m long DB-WAX capillary column with 0.25 mm internal diameter (J&W Scientific). Product yields were based on the quantity of PhIO added to each reaction.
Results and discussion
Porphyrin synthesis
It was anticipated that the porphyrin meso trans-A2B2(P1) would be synthesized using Lindsey's methodology,20,22 between 5-(3,4-dimethoxyphenyl)dipyrromethane and pentafluorobenzaldehyde (Scheme 1). Analysis of the crude product mixture revealed that the desired porphyrin P1 was prepared along with the cis-A2B2 isomer P2 and the tris-pentafluorophenylporphyrin A3B (P3) (Scheme 1). The formation of P2 and P3 is probably associated with scrambling of the phenyl substituents due to acidolysis and recombination of dipyrromethanes.26,27 The yield for the desired porphyrin (P1) was low (6.4%), but respectable in comparison with that of the isomeric unsymmetric porphyrin 5,15-bis(3,5-dimethoxyphenyl)-10,20-bis(pentafluorophenyl) (3.8%), prepared using the mixed aldehyde methodology.28 | ||
| Scheme 1 Schematic representation of the free-base porphyrins preparation. | ||
The 1H NMR spectroscopy was fundamental for differentiation between the trans-A2B2 (P1) and cis-A2B2 (P2) isomers (Table 1 and ESI: Fig. S3–S5†). The D2h symmetry of trans-A2B2 molecule (P1) gives specific signal patterns with two doublets for the β-pyrrole protons at 9.0 and 8.8 ppm, while the lower symmetry in the cis-A2B2 (P2) compared to the trans P1 isomer29 gives rise to two singlets at 8.9 and 8.8 ppm and two doublets at 9.0 and 8.7 ppm. In the tris-A3B molecule (P3) the β-pyrrole protons are split up into two doublets at 9.0 and 8.8 ppm and one multiplet at 8.9 ppm. These results are in agreement with the basic patterns of splitting of the β-pyrrole protons in unsymmetric porphyrins as reported for Meng and co-workers.29
| H-β-pyrrolic | ArH | –OCH3 | NH | |
|---|---|---|---|---|
| Trans-A2B2 (P1) | 9.04–9.03 (d, 4H) 8.82–8.81 (d, 4H) | 7.80–7.78 (m, 4H) 7.32–7.30 (d, 2H) | 4.22 (s, 6H) 4.03 (s, 6H) | −2,78 (s, 2H) |
| Cis-A2B2 (P2) | 9.04–9.03 (d, 2H) 8.95 (s, 2H) 8.87 (s, 2H) 8.79–8.78 (d, 2H) | 7.80–7.76 (m, 4H) 7.31–7.29 (d, 2H) | 4.21 (s, 6H) 4.03 (s, 6H) | −2,70 (s, 2H) |
| Tris-A3B (P3) | 9.06–9.05 (d, 2H) 8.9 (m, 4H) 8.83–8.82 (d, 2H) | 7.79–7.77 (m, 2H) 7.32–7.30 (d, 1H) | 4.22 (s, 3H) 4.03 (s, 3H) | −2,79 (s, 2H) |
The UV-VIS spectra of the porphyrin macrocycle are consistent with the D2h micro-symmetry according to the Four-Orbital Model developed by Gouterman (ESI: Fig. 6†).30 The spectra of the free base porphyrins shows an intense absorption near 400 nm (Soret band) and four less intense bands (Q bands) labeled as IV, III, II and I, between 450 and 700 nm. In general, electron-donating substituents lead to red shifts for both the Soret and Q bands.30–35 The electronic coupling between electron-donating groups and the porphyrin core in P1–P3 decreases the HOMO–LUMO energy gaps and causes a red shift in its spectra compared to the free base porphyrin H2TPFPP (ESI: Fig. 6†).
Furthermore, it is well established that the relative intensity of the four Q bands is dependent on the substituent groups at the macrocycle periphery. According to the relative intensities, the four visible Q bands are classified as etio, rhodo, oxorhodo, and phyllo.30,36 The porphyrins P1–P3 give a phyllo spectra (with intensities IV > II > III > I), which is in agreement with meso-phenyl porphyrins spectra. The reduction in the intensities of bands III and I observed in the spectrum of the porphyrin P3 may be attributed to the restricted rotation of the phenyl groups resulting from steric interaction of the ortho-fluorine substituent and the β-pyrrole hydrogens.36
The metalation process of the free base porphyrins P1, P2 and P3 with manganese(II) acetate was monitored by recording the electronic spectra at different reaction times. The bathochromic shift observed in the Soret band is characteristic of Mn(III) porphyrins, which confirms the formation of MnP1, MnP2 and MnP337 (ESI: Fig. S7†). These new metalloporphyrins were also characterized by solid state EPR (ESI: Fig. S8†) and no signal was observed at room temperature or 77 K, in agreement with presence of Mn(III) in the porphyrin core.5,38 Indeed, the Mn(III) ions has four unpaired d electrons with S = 2, typically featuring a pronounced Jahn–Teller distortion which results in substantial spin–orbit coupling.38 Thereby, the coupling and rapid spin relaxation process results in absence of signal in mononuclear Mn(III) EPR spectra even at 77 K (under perpendicular microwave polarization).5,38
Furthermore, the EPR confirms that the excess of manganese(II) acetate, which shows a signal in g = 2 (ESI: Fig. S8†), was removed during the purification of the new metalloporphyrins (through column and washing with warm water).
Single crystal X-ray diffraction
Each one of the free base porphyrins P1, P2 and P3 was successfully characterized by single crystal XRD that allowed unambiguous confirmation of each structure. Suitable crystals were grown via slow vapour diffusion of methanol into chloroform solution. Crystal data, data collection and structure refinement can be found in the ESI.† Selected crystallographic and geometric parameters for each structure are also shown in Table 2. Crystals of P1, P2 and P3 form monoclinic unit cells belonging to the space groups P21/c, I2/a and Cc, respectively. The ORTEP39 and molecular crystal packing diagrams for each porphyrin are shown in Fig. 1.| P1 | P2 | P3 | |
|---|---|---|---|
| Empirical formula | C50H30Cl6F10N4O4 | C50H30Cl6F10N4O4 | C47H22F15N4O3 |
| Formula weight | 1153.48 | 1153.48 | 975.68 |
| Temperature/K | 100 | 200 | 120 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | I2/a | Cc |
| a/Å | 13.948(3) | 12.0821(2) | 11.5360(2) |
| b/Å | 15.6771(17) | 17.9808(2) | 26.8989(6) |
| c/Å | 11.2006(16) | 22.7546(3) | 15.0809(3) |
| β/° | 109.177(18) | 96.0320(10) | 109.961(2) |
| Volume/Å3 | 2313.2(7) | 4915.97(12) | 4398.56(16) |
| Z | 2 | 4 | 4 |
| 2Θ range for data collection/° | 2.996 to 40.268 | 6.278 to 152.246 | 6.240 to 152.16 |
| Reflections collected | 8341 | 39378 |
35755 |
| Independent reflections | 2340 [Rint = 0.0857, Rsigma = 0.0726] | 5127 [Rint = 0.0661, Rsigma = 0.0289] | 7617 [Rint = 0.0660, Rsigma = 0.0400] |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0725, wR2 = 0.1897 | R1 = 0.0659, wR2 = 0.1874 | R1 = 0.1043, wR2 = 0.2897 |
| Final R indexes [all data] | R1 = 0.1160, wR2 = 0.2565 | R1 = 0.0708, wR2 = 0.1943 | R1 = 0.1079, wR2 = 0.3001 |
| CCDC deposition number | 1483009 | 1536642 | 1536643 |
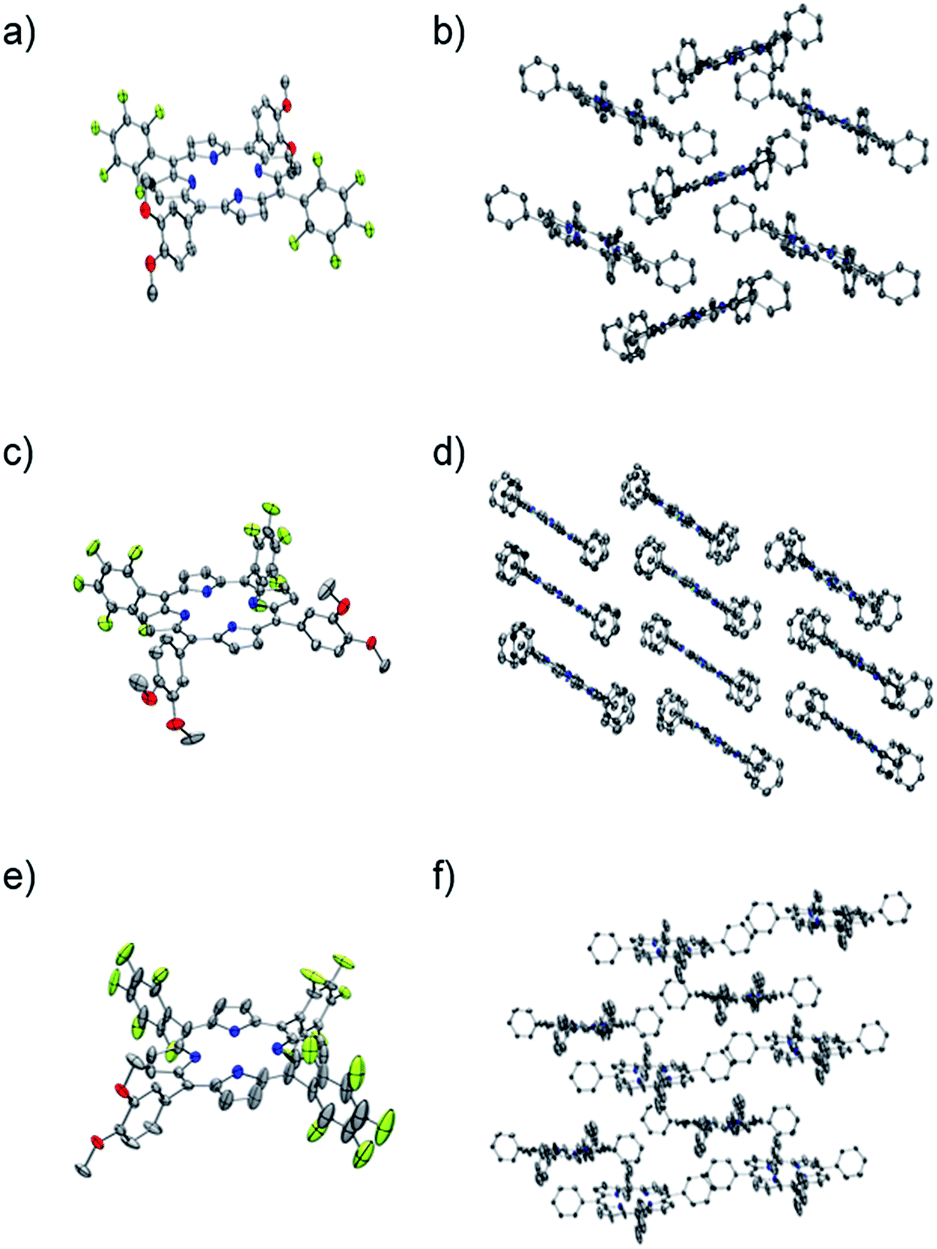 | ||
| Fig. 1 ORTEP molecular diagrams of (a) P1, (c) P2 and (e) P3 where the solvent molecules and hydrogens have been omitted for clarity. Thermal ellipsoids are also shown at 50% probability for P1 and P2 and 35% probability for P3. Crystal packing diagrams for (b) P1, (d) P2 and (f) P3 where the solvent molecules and hydrogen, oxygen and fluorine atoms have been omitted for clarity. Thermal ellipsoids are also shown at 50% probability for P1 and P2 and 35% probability for P3. | ||
The unsymmetric units of P1 and P2 contain one-half of each molecule, along with one molecule of chloroform. The porphyrin rings are both relatively flat, with the largest deviation from planarity being 0.04 Å and 0.09 Å for P1 and P2 respectively, demonstrating little difference for the cis or trans-isomer. The dihedral angle between the DMP substituents and the porphyrin ring appear to be relatively similar for both P1 and P2, varying from 66.4 to 67.9°, with the PFP groups of P1 also within this range. However, the PFP groups of P2 adopt a close to perpendicular orientation to the macrocycle, with an angle of 86.0°.
The porphyrin macrocycles in the crystal structures of P1 and P2 are arranged in a herring bone and off-set stacked crystal packing motif, respectively (Fig. 1b and d). Analysis of the packing in the crystal of P1 reveals a variety of close intermolecular interactions between each of the macrocycles, of which C⋯H, C⋯O, H⋯N, F⋯H, F⋯C and F⋯F interactions are present. Despite the similarities in the chemical composition of P1 and P2, fewer types of interactions could be observed in the crystal structure of P2, attributed to the relative positions of the substituted phenyl substituents. The crystal structure of P2 contains C⋯F, F⋯H, F⋯C and O⋯H close interactions, with the O⋯H close contacts generated by the association between the methoxy substituents of each porphyrin (Fig. S12-ESI†).
The unsymmetric unit of P3 contains a complete porphyrin macrocycle with one molecule of methanol. The ring demonstrates a much larger deviation of planarity compared to P1 and P2, with a maximum deviation from planarity of 0.351 Å. The two PFP substituents both exhibit significant disorder that could be modelled over two positions but still demonstrate a large amount of libration as shown on the thermal ellipsoids. The disorder appears to coincide with the presence of small voids around the PFP substituents, that when combined account for 5.7% (251 Å3) of the unit cell, as calculated by PLATON VOID.40
Further analysis with PLATON SQUEEZE40 calculated a residual electron count of 47 e− that corresponds to another possible 0.6 molecules of methanol per molecule of P3. However, their scattering contribution to the overall structure refinement was not removed. The dihedral angle between the substituted phenyl groups varies from 68.4° to 86.3°, with the maximum dihedral angles observed for the two disordered PFP rings. The macrocycles within the crystal structure of P3 are arranged in a slip-stacked formation (Fig. 1f), which is arranged via a number of O⋯H, C⋯H, F⋯H, F⋯N, F⋯C and N⋯H close interactions and, as expected, a large number of the intermolecular interactions observed involve fluorine.
Cyclic voltammetry of H2P
Fig. 2 shows the cyclic voltammograms (CV) of the free base porphyrins in CH2Cl2 solutions. H2Ps showed two quasi-reversible oxidations and two quasi-reversible reductions. The oxidations exhibited E1/2 potentials that are referred to the SHE as follows: +1.671 V and +1.836 V for P1, +1.632 V and +1.866 V for P2 and +1.829 V and +2.023 V for P3. Reductions were observed at E1/2: −0.942 V and −0.536 V for P1, −0.972 V and −0.608 V for P2 and −0.853 V and −0.447 V for P3.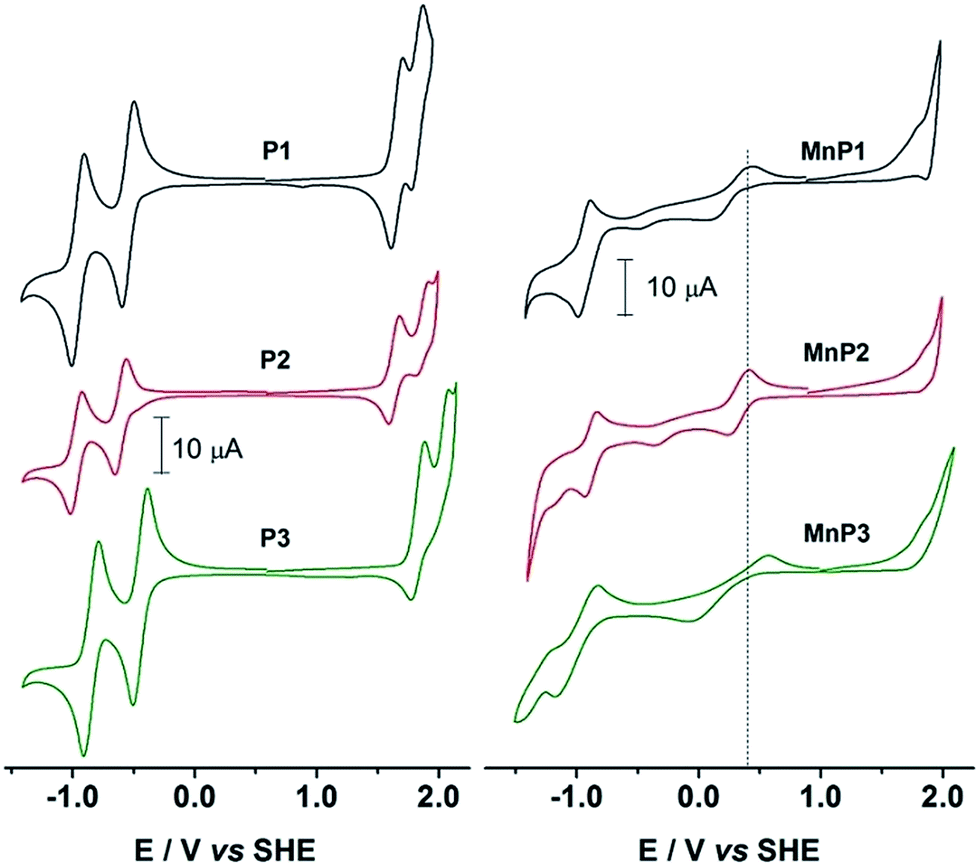 | ||
| Fig. 2 Cyclic voltammograms of H2P and MnP in dichloromethane containing 0.1 mol L−1 TBAPF6. Scan rate at 100 mV s−1. Initial anodic scan. | ||
These values clearly show a similar redox behavior for P1 and P2, which is expected since they have the same substituents, and differ only on their geometric configuration, cis or trans, thus offering the same overall outcome to the electronic density of the porphyrin. In contrast, the unsymmetric porphyrin P3 exhibited a significant resistance to oxidation, in agreement with the presence of a higher number of fluoro-atoms, which causes a strong inductive electron withdrawing effect. The mean potential difference between the two oxidations and between the two reductions for H2Ps are 0.198 V and 0.392 V, respectively. This result is in accordance with analogue cases in the literature for successive monoelectronic ring-centered processes, explained by the formation of porphyrin π-cation/anion radicals and dications/dianions as depicted in Scheme 2.41,42 Furthermore, the potential difference between the first oxidation and the first reduction, ΔE1/2(oxi–red), is a good estimate of the band gap between the frontier orbitals HOMO and LUMO. It was found the following order of decreasing ΔE1/2(oxi–red): P3 (2.276 V) > P2 (2.240 V) > P1 (2.207 V), in accordance with the higher resistance of P3 towards oxidation. The values for P3 are slightly higher than the 2.23 V observed for the meso-tetraphenylporphyrin [H2(TPP)],43 and lower than 2.34 V for 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin [H2(TPFPP)]. All electrochemical data are collected in Table 3.
 | ||
| Scheme 2 Schematic representation of monoelectronic process in the formation of porphyrin π-cation/anion radicals and dications/dianions. | ||
| Eox1 | Eox2 | ΔEox | Ered1 | Ered2 | ΔEred | (Eox1 − Ered1)d | Ea (MnIII/MnII) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Epa.b Epc.c (Epa1 − Epc1).d HOMO–LUMO gap; n.o. = not observed; tw = this work. | |||||||||
| H2TPP | +0.93 | +1.26 | 0.33 | −1.29 | −1.62 | 0.33 | 2.22 | — | 41 |
| H2TPFPP | +1.838a | +2.123a | 0.285 | −0.543 | −0.952 | 0.409 | 2.340c | — | tw |
| H2TDMPP | +1.299 | +1.433 | 0.134 | −0.965 | −1.267 | 0.302 | 2.264 | tw | |
| P1 | +1.671 | +1.836 | 0.165 | −0.536 | −0.942 | 0.406 | 2.207 | — | tw |
| P2 | +1.632 | +1.866 | 0.234 | −0.608 | −0.972 | 0.364 | 2.240 | — | tw |
| P3 | +1.829 | +2.023 | 0.194 | −0.447 | −0.853 | 0.406 | 2.276 | — | tw |
| MnP1 | +1.8a | n.o. | — | −0.48b | −0.926 | 0.45 | 2.3 | +0.440a | tw |
| MnP2 | +1.8a | n.o. | — | −0.35b | −0.882 | 0.53 | 2.2 | +0413a | tw |
| MnP3 | +1.8a | n.o. | — | n.o. | −0.999 | — | — | +0.558a | tw |
Cyclic voltammetry of MnP
The cyclic voltammograms (CV) of manganese(III) porphyrins showed irreversible oxidations starting at +1.8 V, which were assigned to the electrogenerated cation radicals and the dications of the macrocycle ring (Fig. 2). An irreversible reduction of MnP, probably related to the irreversible oxidations, was seen in the range of −0.35 V to −0.48 V along with quasi-reversible reduction processes between −0.926 V and −0.999 V to produce the monoanion radical and dianions porphyrins, in that order. Anodic peak potentials of the quasi-reversible electron transfer centered in the manganese ion were observed at +0.440 V, +0.413 V and +0.558 V, for MnP1, MnP2 and MnP3, respectively. The overall electron transfer sequence can be summarized in the following steps: [MnIIP]2− → [MnIIP˙]− → [MnIIP] → [MnIIIP]+ → [MnIIIP˙]+ → [MnIIIP]2+.44,45Ucoski and co-workers46 reported the electrochemistry of the 5,10,15,20-tetrakis(1,3-benzodioxole)-MnIII porphyrin in dichloromethane and observed a E1/2 = −0.61 V vs. SHE for the MnIII/MnII couple. This result agrees with the donor character of the dioxole substituent, compared to the substituents reported in this work, making the oxidation of the manganese center an easier process in that case. Also for comparison, E1/2 of MnIII/MnII(TPFPP) and of MnIV/MnIII(TPFPP) were observed in acetonitrile at +0.05 and at +1.75 V vs. SHE, repectively.47
Catalytic studies of MnP1, MnP2 and MnP3 using (Z)-cyclooctene and cyclohexane as substrates
The efficiency of the porphyrins MnP1, MnP2 and MnP3 and the model compounds MnTPFPP and MnTDMPP as catalysts in homogeneous oxidation reactions was tested using the substrates (Z)-cyclooctene and cyclohexane and iodosylbenzene (PhIO) as an oxidant (Fig. 3). It has been previously shown that electron-withdrawing substituents at the ortho position of the meso-phenyl substituents in the porphyrin ring can enhance the catalytic activity of these compounds for oxidation reactions.6,7 Hence, MnTPFPP was used as a model catalyst because it is recognized as a highly efficient catalyst in homogeneous oxidation reactions for various substrates.48,49 Its enhanced performance can be attributed mainly to its greater stability under oxidative conditions, resulting from the electron-withdrawing nature of the PFP substituent, which protects, to some extent, the self-destruction of the porphyrin ring. On the other hand, MnTDMPP was used as an opposing model catalyst for comparison because it contains only the highly electron-donating DMP substituents. | ||
| Fig. 3 Structures of porphyrins MnP1, MnP2 and MnP3, and the model compounds MnTPFPP, MnTDMPP used as catalysts for (Z)-cyclooctene and cyclohexane oxidation reactions. | ||
The molecule (Z)-cyclooctene is a useful diagnostic substrate as it is easily oxidized in the presence of metalloporphyrins, with (Z)-cycloocteneoxide usually being the sole product of reaction.48–50
The catalytic activity of the new manganese(III) porphyrins MnP1, MnP2 and MnP3 using (Z)-cyclooctene as a substrate resulted in similar product yields. The new unsymmetric manganese(III) porphyrins also showed analogous catalytic activity to that observed for the completely symmetric porphyrin MnTPFPP (∼86%) (Fig. 4). This result suggests that the presence of two (MnP1 and MnP2) or one (MnP3) 3,4-dimethoxy groups in the structure of the new synthesized metalloporphyrins does not seem to interfere in their catalytic activity for the oxidation of (Z)-cyclooctene. However, the catalytic activity of the completely symmetric MnTDMPP was lower (∼73%), which can be attributed to the absence of electron-withdrawing substituent in this porphyrin.50
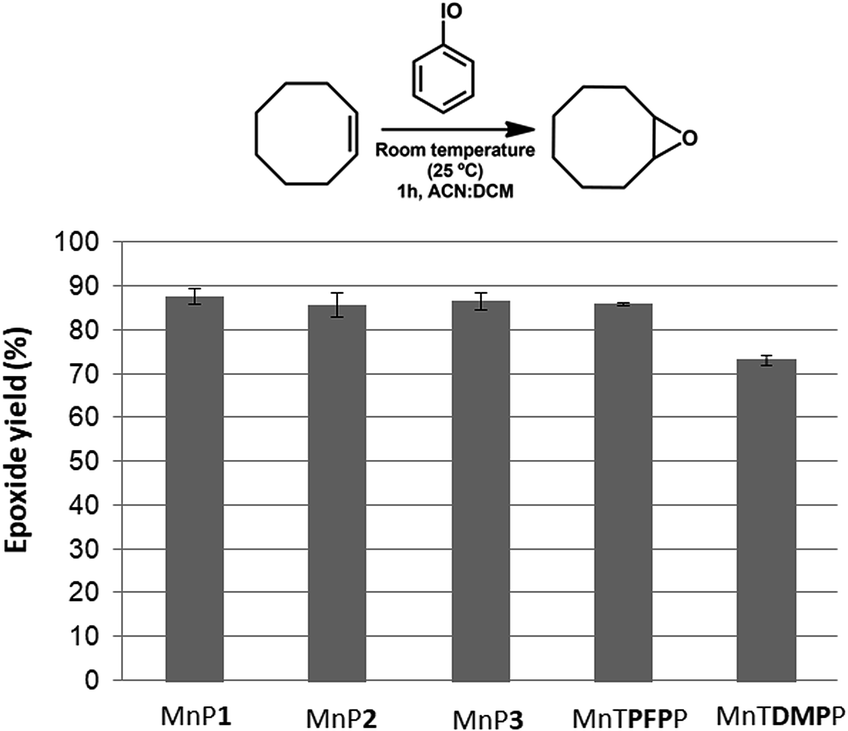 | ||
| Fig. 4 (Z)-Cyclooctene oxidation reaction using PhIO catalyzed by MnP1, MnP2, MnP3, MnTPFPP and MnTDMPP. Catalyst/PhIO/Substrate molar ratio = 1:10:1000. The product yields were calculated based on the amount of PhIO used in the reactions. Results represent reactions performed in duplicate. | ||
Cyclohexane is a less reactive substrate than (Z)-cyclooctene in oxidation reactions and when metalloporphyrins are used as catalysts, both cyclohexanol and cyclohexanone are obtained as major products.10,48–50 The novel metalloporphyrins reported here have shown alcohol selectivity in the cyclohexane oxidation (Fig. 5), which is in agreement with reported results of homogeneous catalysis using other metalloporphyrins.48–50
 | ||
| Fig. 5 Cyclohexane oxidation reaction using PhIO catalysed by MnP1, MnP2, MnP3, MnTPFPP and MnTDMPP. Catalyst/PhIO/Substrate molar ratio = 1:10:1000. The product yields for cyclohexanol (alcohol) and cyclohexanone (ketone) were calculated based on the amount of PhIO used in the reactions. Results represent reactions performed in duplicate. | ||
For the cyclohexane reactions it was possible to observe a trend in the catalytic activity of the fluorine-substituted metalloporphyrins which increased with an increasing number of pentafluorophenyl substituents present in the porphyrin: MnP1 ∼ MnP2 (∼40% yield for alcohol) < MnP3 (∼49%) < MnTPFPP (∼54%). The low catalytic activity of the MnTDMPP (∼14%) under the reaction conditions is associated with the absence of bulky or electron-withdrawing substituents at the ortho position of the meso-phenyl macrocycle, which is in agreement with results obtained previously using a similar porphyrin structure.50
The oxidation peak potentials (Epa) values of MnP1 (+0.440 V), MnP2 (+0.413 V) and MnP3 (0.558 V) (Table 3) correlates well with their increasing catalytic activity, in that order, towards the oxidation of cyclohexane (Fig. 5). The similarity of Epa for MnP1 and MnP2 agrees with the close yields (∼40%) observed for the cyclohexanol production, while MnP3 showed a 49% yield for the alcohol. The higher reduction potential of MnP3 compared to the other metalloporphyrins herein reported means that the active intermediate containing the high valent manganese51–53 IV or V Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (O)P3 is a better oxidant agent due to the increased number of pentafluorophenyl groups in the porphyrin composition, thus, in the course of the reaction, it favors the oxygen transfer to the substrate. Furthermore, the pentafluorophenyl substituents protect the porphyrin from oxidative damage, increasing the turnover numbers and helps increase the yields of the oxidation reactions.
(O)P3 is a better oxidant agent due to the increased number of pentafluorophenyl groups in the porphyrin composition, thus, in the course of the reaction, it favors the oxygen transfer to the substrate. Furthermore, the pentafluorophenyl substituents protect the porphyrin from oxidative damage, increasing the turnover numbers and helps increase the yields of the oxidation reactions.
Conclusions
In this work the synthesis, characterization and crystal structure of three unsymmetrical free base porphyrins (P1, P2 and P3) are reported. The catalytic activity of the metallated unsymmetric porphyrins MnP1, MnP2 and MnP3, together with the electrochemical studies, showed that the number of pentafluorophenyl groups on the metalloporphyrin structure (two or three) has a direct influence on their catalytic activity in homogeneous medium. The compound MnP3, which has three pentafluorophenyl in its structure, showed better catalytic results, similar to the MnTPFPP, and also higher reduction potential compared to the other metalloporphyrins herein reported. These porphyrins are interesting precursors for the design of well-defined new materials, with a focus towards the construction of PIMs that combine high surface area with recognized catalytic properties of metallated porphyrins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) – Science without Borders, Fundação Araucária and Universidade Federal do Paraná (UFPR) for financial support and the authors thank the EPSRC (grant EP/N01331X/1) for funding. Kátia C. M. Westrup thanks Fundação Araucária and Capes for the pos-doctoral scholarship (Convênio 396/2014 – Projeto 41060 – SIT 21045). The authors are also grateful to The University of Edinburgh and acknowledge beam line I19 of the Diamond Light Source (DLS), UK, for assistance in obtaining crystallographic data.References
- L. R. Milgrom, The Colours of Life. An introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, Oxford, 1997 Search PubMed.
- K. M. Smith, Porphyrins and metalloporphyrins: A New Edition Based on the Original Volume, ed. J. E. Falk, Elsevier Sci Pub Co, New York, 1975 Search PubMed.
- S. Shanmugathasan, C. Edwards and R. W. Boyle, Tetrahedron, 2000, 56, 1025–1046 CrossRef CAS.
- K. M. Kadish, K. M. Smith and R. Guilard. The porphyrin handbook vol. 6, Applications: Past, Present and Future, Academic Press, San Diego, 2000 Search PubMed.
- V. S. Da Silva, S. Nakagaki, G. M. Ucoski, Y. M. Idemori and G. DeFreitas-Silva, RSC Adv., 2015, 5, 106589–106598 RSC.
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS.
- J. I. T. Costa, A. T. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2011, 15, 1116–1133 CrossRef CAS.
- P. Battioni, O. Brigaud, H. Desvaux, D. Mansuy and T. G. Traylor, Tetrahedron Lett., 1991, 32, 2893–2896 CrossRef CAS.
- F. B. Zanardi, I. A. Barbosa, P. C. S. Filho, L. D. Zanatta, D. L. Silva, A. O. Serra and Y. Iamamoto, Microporous Mesoporous Mater., 2016, 219, 161–171 CrossRef CAS.
- K. A. D. F. Castro, F. H. C. Lima, M. M. Q. Simões, M. G. P. M. S. Neves, F. A. A. Paz, R. F. Mendes, S. Nakagaki and J. A. S. Cavaleiro, Inorg. Chim. Acta, 2017, 455, 575–583 CrossRef CAS.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782–2783 RSC.
- A. R. Antonangelo, C. Grazia Bezzu, S. S. Mughal, T. Malewschik, N. B. McKeown and S. Nakagaki, Catal. Commun., 2017, 99, 100–104 CrossRef CAS.
- N. B. McKeown, ISRN Mater. Sci., 2012, 1–16 Search PubMed.
- N. U. Day, C. C. Wamser and M. G. Walter, Polym. Int., 2015, 64, 833–857 CrossRef CAS.
- Y. Matsuo, J. Hatano and T. Nakagawa, J. Phys. Org. Chem., 2014, 27, 87–93 CrossRef CAS.
- H. L. Anderson, Chem. Commun., 1999, 2323–2330 RSC.
- H. Saltzmann and J. G. Sharefkin, Org. Synth., 1963, 43, 60–61 CrossRef.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- C.-H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427–11440 CrossRef CAS.
- K. Singh, S. Behal and M. S. Hundal, Tetrahedron, 2005, 61, 6614–6622 CrossRef CAS.
- B. J. Littler, Y. Ciringh and J. S. Lindsey, J. Org. Chem., 1999, 64, 2864–2872 CrossRef CAS PubMed.
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, J. Inorg. Nucl. Chem., 1970, 32, 2443–2445 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. R. Geier, B. J. Littler and J. S. Lindsey, J. Chem. Soc., Perkin Trans. 2, 2001, 701–711 RSC.
- F. R. Kooriyaden, S. Sujatha, B. Varghese and C. Arunkumar, J. Fluorine Chem., 2015, 170, 10–16 CrossRef CAS.
- K. Tashiro, K. Konishi and T. Aida, J. Am. Chem. Soc., 2000, 122, 7921–7926 CrossRef CAS.
- G. G. Meng, B. R. James and K. A. Skov, Can. J. Chem., 1994, 72, 1894–1909 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- J. Dalton, L. R. Milgrom and S. M. Pemberton, J. Chem. Soc., Perkin Trans. 2, 1980, 370–372 RSC.
- C. J. P. Monteiro, M. M. Pereira, M. G. H. Vicente and L. G. Arnaut, Tetrahedron, 2012, 68, 8783–8788 CrossRef CAS.
- V. N. Nemykin and R. G. Hadt, J. Phys. Chem. A, 2010, 114, 12062–12066 CrossRef CAS PubMed.
- C. Chen and S. J. Hsieh, J. Chin. Chem. Soc., 1997, 44, 23–31 CrossRef CAS.
- J. R. Weinkauf, S. W. Cooper, A. Schweiger and C. C. Wamser, J. Phys. Chem. A, 2003, 107, 3486–3496 CrossRef CAS.
- T. P. Wijesekera and D. Dolphin, Synthetic aspects of porphyrin and metalloporphyrin chemistry, R. A. Sheldon, New York, 1994, ch. 7 Search PubMed.
- L. J. Boucher, J. Am. Chem. Soc., 1970, 92, 2725–2730 CrossRef CAS PubMed.
- M. T. Caudle, C. K. Mobley, L. M. Bafaro, R. LoBrutto, G. T. Yee and T. L. Groy, Inorg. Chem., 2004, 43, 506–514 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. Paliteiro and A. Sobral, Electrochim. Acta, 2005, 50, 2445–2451 CrossRef CAS.
- J. Y. Inisan, R. Saillard, A. Guilard, Y. Tabard and L. Mest, New J. Chem., 1998, 22, 823–830 RSC.
- K. M. Kadish, Z. Ou, R. Zhan, T. Khoury, E. Wenbo and M. J. Crossley, J. Porphyrins Phthalocyanines, 2010, 14, 866–876 CrossRef CAS.
- L. J. Boucher and H. K. Garber, Inorg. Chem., 1970, 9, 2644–2649 CrossRef CAS.
- R. D. Arasasingham and T. C. Bruice, Inorg. Chem., 1990, 29, 1422–1427 CrossRef CAS.
- G. Ukoski, S. Nakagaki and F. S. Nunes, J. Porphyrins Phthalocyanines, 2014, 18, 1–8 CrossRef.
- G. R. Friedermann, M. Halma, K. A. D. F. Castro, F. L. Benedito, F. G. Doro, S. M. Drechsel, A. S. Mangrich, M. D. Assis and S. Nakagaki, Appl. Catal., A, 2006, 308, 172–181 CrossRef CAS.
- K. A. D. F. Castro, M. M. Q. Simões, M. G. P. M. S. Neves, J. A. S. Cavaleiro, F. Wypych and S. Nakagaki, Catal. Sci. Technol., 2014, 4, 129–141 CAS.
- K. A. D. F. Castro, M. M. Q. Simões, M. G. P. M. S. Neves, J. A. S. Cavaleiro, R. R. Ribeiro, F. Wypych and S. Nakagaki, Appl. Catal., A, 2015, 503, 9–19 CrossRef.
- G. K. B. Ferreira, K. A. D. F. Castro, G. S. Machado, R. R. Ribeiro, K. J. Ciuffi, G. P. Ricci, J. A. Marques and S. Nakagaki, J. Mol. Catal. A: Chem., 2013, 378, 263–272 CrossRef CAS.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS.
- W. Nam, I. Kim, M. H. Lim, H. J. Choi, J. S. Lee and G. H. Jang, Chem.–Eur. J., 2002, 8, 2067–2071 CrossRef CAS.
- J. T. Groves, W. T. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6315–6371 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1483009, 1536642 and 1536643. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08734a |
| This journal is © The Royal Society of Chemistry 2017 |