
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymorphism-dependent aggregation induced emission of a push–pull dye and its multi-stimuli responsive behavior†
Chiara
Botta
 *a,
Sara
Benedini
b,
Lucia
Carlucci
c,
Alessandra
Forni
*de,
Daniele
Marinotto
c,
Andrea
Nitti
b,
Dario
Pasini
*bf,
Stefania
Righetto
ce and
Elena
Cariati
*ce
*a,
Sara
Benedini
b,
Lucia
Carlucci
c,
Alessandra
Forni
*de,
Daniele
Marinotto
c,
Andrea
Nitti
b,
Dario
Pasini
*bf,
Stefania
Righetto
ce and
Elena
Cariati
*ce
aISMAC-CNR, Istituto per lo Studio delle Macromolecole - Consiglio Nazionale delle Ricerche, Via Corti 12, 20133 Milano, Italy. E-mail: c.botta@ismac.cnr.it
bDepartment of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it
cDepartment of Chemistry, University of Milano, via Golgi 19, 20133 MilanoItaly. E-mail: elena.cariati@unimi.it
dISTM-CNR, Istituto di Scienze e Tecnologie Molecolari – Consiglio Nazionale delle Ricerche, via Golgi 19, 20133 Milano, Italy. E-mail: alessandra.forni@istm.cnr.it
eINSTM Research Unit, University of Milano, Italy
fINSTM Research Unit, University of Pavia, Italy
First published on 14th December 2015
Abstract
A comprehensive optical investigation of 1,1-dicyano-2,2-bis(4-dimethylaminophenyl)ethylene (1) is presented. The compound crystallizes in four different forms all displaying AIE behavior. The crystalline forms A and B are yellow-orange-emissive, while C and D are green-emissive. On the basis of X-ray structural analysis, the weak intermolecular interactions account for restricted internal rotations, leading to fluorescence enhancement in the crystals; however, the difference in emission color is ascribed to the various conformations of the molecules in the four crystalline forms. In addition, the emission color of crystals of A can be tuned by heating and grinding, that of B by grinding only, while crystals of C show chronochromic behavior. An explanation for such a rich variety of luminescence behavior is formulated here through the use of steady state and time resolved photoluminescence, X-ray diffraction analysis and DFT and TDDFT calculations. The involved chromic mechanism appears to be mainly associated with surface defects induced by the external stimuli rather than an amorphization process, as frequently observed for other stimuli responsive compounds.
Introduction
Organic materials that exhibit strong emission in the aggregated state have aroused much interest in recent years because of their potential applications in optical and optoelectronic devices such as light-emitting diodes,1 optical waveguides2 and optically pumped lasers.3 D–π–A type dyes usually exhibit unique fluorescence properties due to their intramolecular charge transfer (ICT) transitions, which endow them with tuned electronic states under various conditions.4However, aggregation-caused quenching (ACQ) often takes place in the condensed phases for most D–π–A luminogens,5 limiting their real-world applications. In recent years, Tang and other groups have reported several types of organic molecules which show an unusual luminescence behavior; they are weak or non-emissive in solution, but show enhanced efficiency in the solid states.6 Such aggregation-induced-(enhanced) emission (AI(E)E) is frequently ascribed to restricted internal rotations (RIR).6,7 For applications in optoelectronic devices as well as sensors8 and memories,9 emissive materials whose emission intensities and colors can be tuned are highly desirable. However, variation of the solid state luminescence color is less affordable than in solution due to the difficulty in obtaining different stable phases in the aggregated state with varied molecular conformations. This goal can be reached either by obtaining a compound crystallizing in different polymorphs or by altering the molecular geometry and/or packing mode by external stimuli, especially heat,10 solvent vapor11 and mechanical perturbation12 (including shearing, grinding, smashing or elongation), allowing to control solid state luminescence (thermo-, vapo- and mechanochromism respectively). In some cases, the emission properties show variations in time, at room temperature and without the need for external stimuli, a behavior named chronochromism. This process was reported for AIE materials recovering their initial state from a metastable state generated by an external stimulus (self-healing effect)13,14 or for materials that undergo spontaneous transition towards a more stable crystal phase.15
In some cases the chromic variations are associated with changes in the crystal packing (different polymorphs), more frequently to a switch between crystalline/amorphous phases. More intriguing are fluorophores displaying sharp variations in the emissive properties while maintaining the same crystal structure. Few examples of this chromic behavior have been reported so far but a definitive explanation is still lacking. Possible explanations rely on the particle size and the surface structure of solids.16a In particular, different electronic processes have been hypothesized in the bulk and at the surface of microcrystals so that two emission centers, one in the middle and another in the periphery of the solids, are responsible for the whole luminescence.16,17
We have previously reported on the efficient AIE properties of a series of “push–pull” dyes possessing common structural features: a trisubstituted ethylene core, decorated with two carboxylate esters and a 4-dialkylaminophenyl branch as the third substituent.18a Studying in detail one of the AIE active compounds using ultrafast pump–probe spectroscopy combined with calculations, we have given direct evidence that the restriction of the intramolecular rotation is the key process for switching on the AIE properties.19
In this paper, we report the interesting emissive behavior of 1,1-dicyano-2,2-bis(4-dimethylaminophenyl)ethylene, 1 (see Scheme 1), displaying AIE and structure dependent chromic properties. In particular, four different crystalline forms have been isolated: the yellow-orange-emissive form A, the yellow-emissive form B and the green-emissive forms C and D, the latter containing a co-crystallized CH2Cl2 molecule. Their stimuli responsive emissive behavior is analyzed here by fluorescence spectroscopy, X-ray diffraction analysis and computational methods.
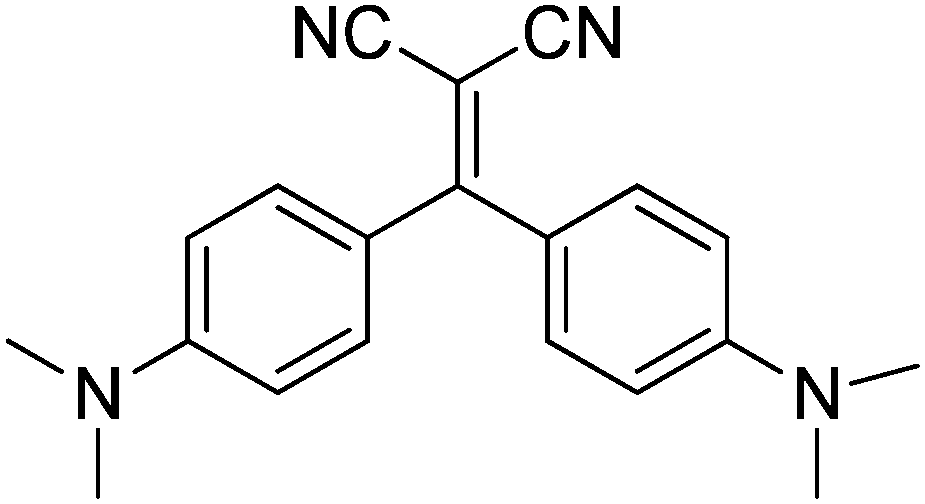 | ||
| Scheme 1 Chemical structure of compound 1. | ||
Experimental
UV-Vis absorption spectra were obtained using a Perkin Elmer Lambda 900 spectrometer and PL spectra using a SPEX 270 M monochromator equipped with a N2 cooled charge-coupled device excited using a monochromated 450 W Xe lamp. Spectra were corrected for the instrument response. Photoluminescence quantum yields (PL QYs) of solutions were obtained by using quinine sulfate or anthracene as standard. PL QYs of crystals and powders were obtained by using a home-made integrating sphere, as previously reported.20 Fluorescence microscopy images were collected using a Nikon Eclipse TE2000-U inverted confocal microscope. Excitation was performed using a 100 W Hg lamp and a 330–380 nm band-pass excitation filter or a 405 nm laser. Spectra were recorded by connecting the confocal microscope with the monochromator using an optical fiber.Lifetime measurements and Time Resolved Emission Spectra (TRES) were performed using an Edinburgh Picosecond Pulsed Diode Laser EPL-UV 375 (Edinburg Instruments Ltd) at a repetition rate of 20 MHz to 50 kHz and a pulse width of 66 ps. The dynamics of emission decay were monitored by using the FLS980s time-correlated single-photon counting.
Diffraction data for single crystals of A–D were collected on a Bruker Smart Apex II CCD area detector using graphite-monochromated Mo-Kα radiation. Data reduction was made using SAINT programs; absorption corrections based on multiscan were obtained using SADABS.21 The structures were solved using SHELXS-9722 and refined on F2 by full-matrix least-squares using SHELXL-97.22 All the non-hydrogen atoms were refined anisotropically, hydrogen atoms were included as ‘riding’ and not refined. Powder patterns were recorded on Philips PW1820 and Bruker AXS D8 diffractometers (Cu-Kα radiation, λ = 1.5405 Å) in the 5–40° 2θ range (0.02° and 1.5 s per step). All calculations have been performed using the Gaussian suite of programs.23 Geometry optimization of the ground state (S0) has been performed at the PBE0/6-311++G(d,p)24 level of theory, starting from the experimental X-ray geometry of polymorph A. We imposed a C2 symmetry constraint since the free optimization led to a quasi-symmetrical structure. Standard vertical Time Dependent (TD) DFT calculations were then carried out at the C2 optimized geometry using the same functional and basis set to determine the excited state properties. Up to 20 vertical excitations from S0 have been evaluated, including both singlet and triplet excited states. The lower energy singlet excited state (S1) was then submitted to TD-PBE0/6-311++G(d,p) geometry optimization to evaluate the emission from S1 to S0. Excited state optimization required to remove the C2 symmetry constraint to get a stationary point. The performance of the PBE0 functional in describing the electronic and optical properties of the present compound has been assessed together with that of other exchange–correlation functionals (CAM-B3LYP and M062X) generally suggested, besides PBE0, as the outperforming ones for optical property calculations on organic chromophores25 (see ESI†). All calculations were carried out in toluene using the conductor-like polarizable continuum model, CPCM.26 For TDDFT calculations the linear response approach has been adopted.27
Results and discussion
Molecular optical properties
Compound 1 was synthesized modifying a literature procedure (see ESI†).28 UV-vis absorption and emission spectra of 1 in different solvents are reported in Fig. 1. Positive solvatochromism is observed for both, being stronger for the emission spectra indicating that the excited state is more polar than the ground one.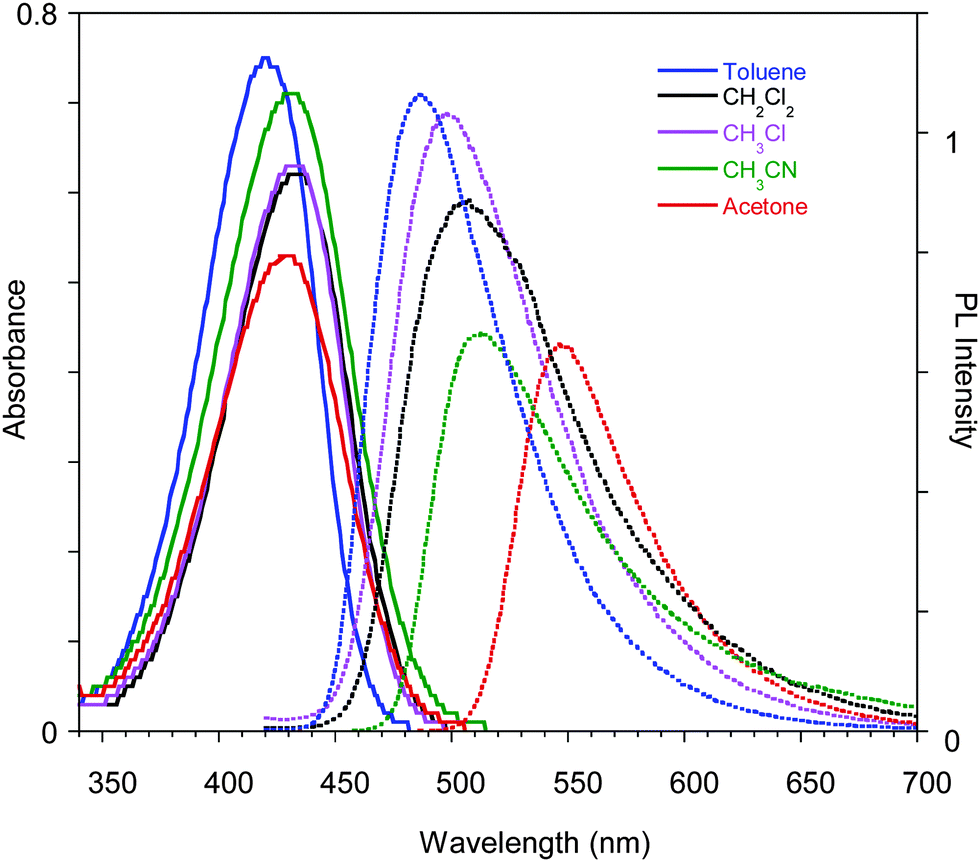 | ||
| Fig. 1 Optical absorption and emission spectra of 1 in different solvents. | ||
The compound's emission QY in good solvents is very low (<0.1%), but an intensification of the emission is observed after introducing poor solvents (Fig. 2a and Fig. S4, ESI†). This observation suggests an Aggregation Induced Emission (AIE) behavior associated with the formation of aggregates of 1 by addition of poor solvents, as reported earlier.18 The fluorescence intensity of 1 in acetone and acetone/water mixtures (retaining a concentration of 10−5 M) with increasing water (non-solvent) fraction up to 90% are reported in the inset of Fig. 2a. The emission intensity (QY of about 0.07%) of the solutions is not affected by the non-solvent addition up to 80% water fraction. At higher water concentrations the emission intensity increases (by a factor of 25 at 90% water content), the band shape sharpens and slightly (5 nm) red-shifts. These variations start in the first minutes after non-solvent addition to the acetone solution with a temporal evolution that depends on the water content, as recently reported.14 The fastest variations are observed for the highest water volume: the maximum of the emission intensity is obtained in about 40 and 20 min for water fractions of 85 and 90%, respectively. At longer times a reduction in the emission intensity is observed, probably due to the precipitation of large aggregates, while the emission retains its sharp spectral shape. The time evolution of the emission spectra for the 85% water content solution is reported as an example in Fig. 2a.
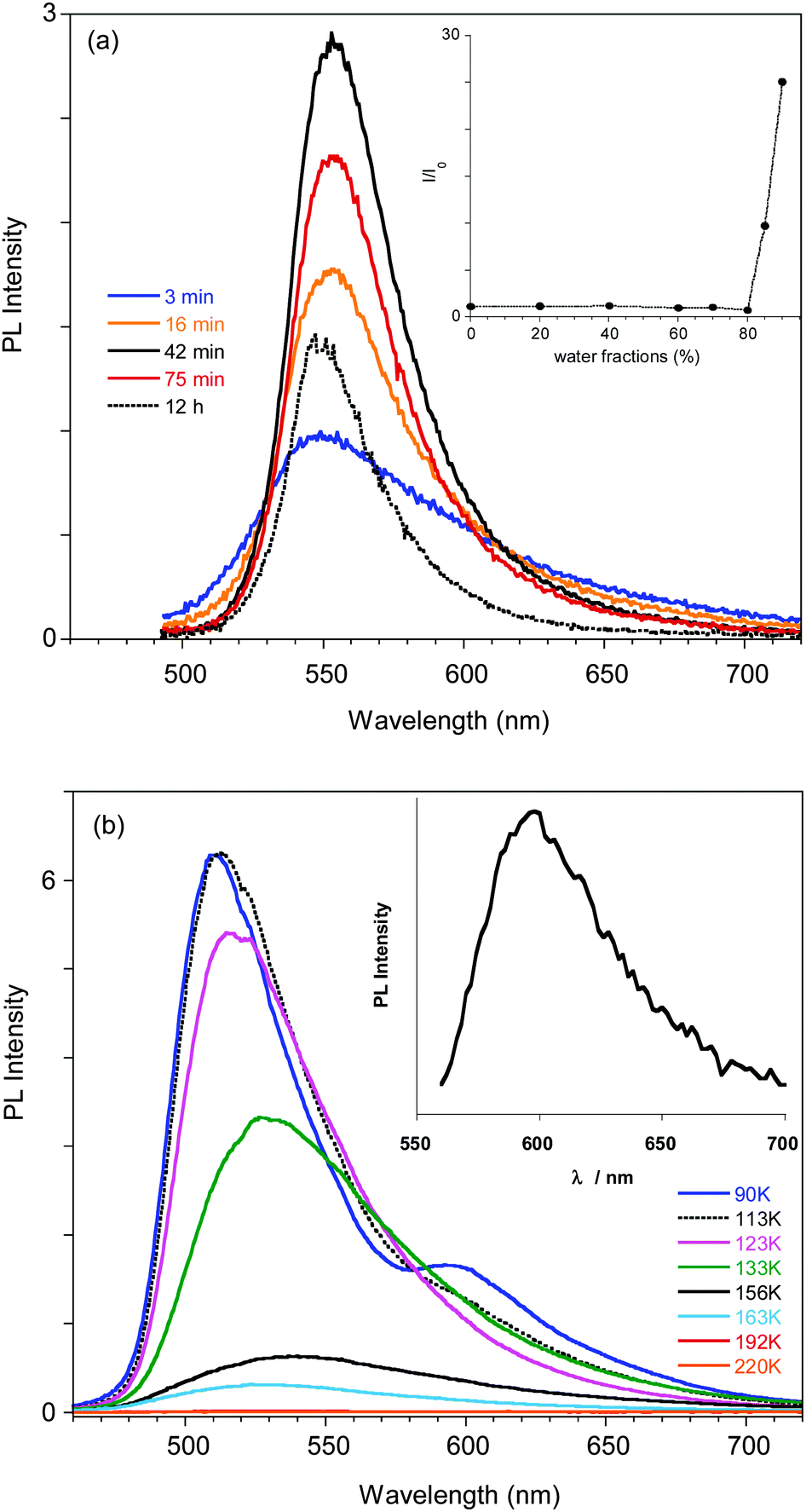 | ||
| Fig. 2 (a) Emission spectra of a solution of 1 in acetone/H2O (concentration 1 × 10−5 M) with 85% water measured at different delay times from water addition. In the inset the intensity of the emission for different water fractions (normalized with respect to the intensity at 0% water) is reported, as measured just after water addition. (b) Emission spectra of a THF solution of 1 at different temperatures. In the inset the PL emission at 77 K measured at 2.5 μs delay is reported. | ||
In order to support a Restricted Intramolecular Rotation (RIR) mechanism at the origin of the emission intensification, as is often the case for AIE fluorophores, the spectra of a diluted (3.5 × 10−5 M) good solvent (THF) solution at room temperature and at temperatures down to the solidification point of the solvent have been collected (see Fig. 2b). While at room temperature the emission is very weak, below the solidification point of the solvent (about 165 K), where the molecular motions are blocked, an increase in the emission intensity is observed, reaching a factor of about 300 at 110 K. Below 120 K the PL spectrum of the solution shows a narrowing of the profile and blue-shifts to 510 nm at 80 K. Moreover, the appearance of a new band at 595 nm is observed below 100 K.
A deeper analysis of the low temperature emissive behavior performed by time resolved spectroscopy (see ESI†) revealed a fast decay (biexponential with 3.70 ns of average lifetime) for the 510 nm peak while much slower decay (with an average lifetime of 0.93 μs) is measured at 620 nm, suggesting a phosphorescence origin of the low energy band. The emission spectrum recorded at long delay times (2.5 μs) in order to cut the fluorescence emission is shown in the inset of Fig. 2b, confirming the phosphorescence origin of the 595 nm peak observed in the steady state spectrum at temperatures below 100 K.
A phosphorescence band is known to appear in aromatic amines, particularly in N-alkylamines,29 and has been ascribed to the not strict parallelism between the lone-pair orbital on the nitrogen atom and the π-electron system of the aromatic ring, which determines nonvanishing terms in the matrix elements of spin–orbit coupling.30 The increase of the CT character of the lower-lying electronic states is expected to increase the transition moment from the first excited triplet state T1 and the ground state S0.30 In the presently investigated push–pull compound 1, the N-dimethyl groups are only slightly rotated with respect to the benzene rings to which they are bonded,31 but the large CT character of the low energy transitions (vide infra) is expected to result in non-null phosphorescence yield, detectable only at low temperature.
Quantum chemical calculations
The 421 nm absorption of 1 in toluene is well reproduced by TD-PBE0/6-311++G(d,p) calculations on the optimized PBE0/6-311++G(d,p) geometry (see Fig. 3) which provided, in the same solvent, excitation energies S0 → S1 and S0 → S2 at 415 and 408 nm, respectively, characterized by similar oscillator strengths (f = 0.71 and 0.47, respectively) and associated almost totally (99%) with HOMO−1 → LUMO and HOMO → LUMO transitions, respectively. The involvement of two transitions in the observed absorption band is confirmed by a slight skewness of the absorption profiles (see Fig. 1).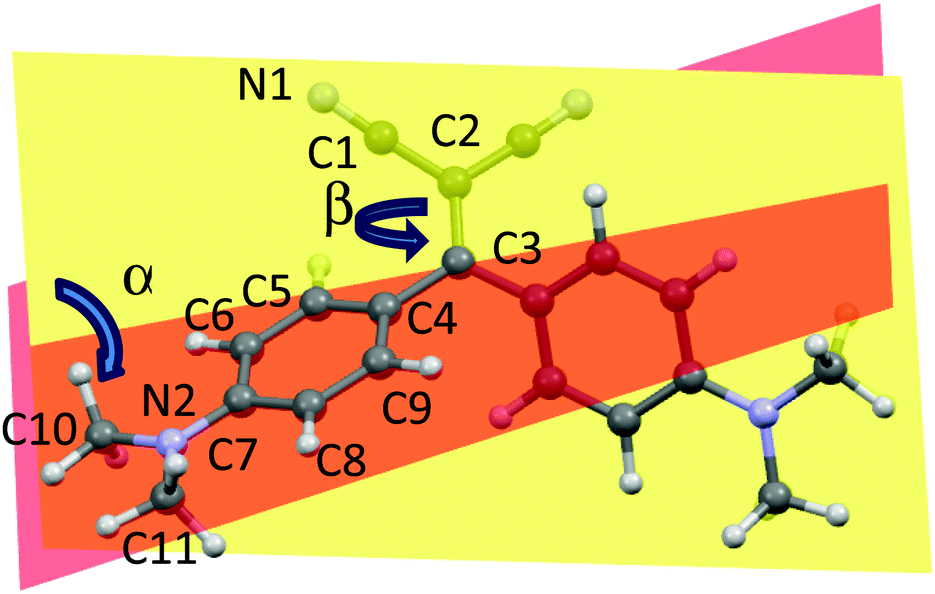 | ||
Fig. 3 Optimized PBE0/6-311++G(d,p) geometry of 1 with atom numbering scheme. α is the dihedral angle between the planes through the carbon atoms of the phenyl rings, plotted in yellow and orange, respectively; β is the C1–C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3–C4 torsion angle. C3–C4 torsion angle. | ||
Though very close in energy, the two transitions have a different nature because the two involved occupied MOs, HOMO−1 and HOMO, have quite different delocalization schemes (see Fig. S3 (ESI†) for a plot of the Frontier MOs, FMOs). While HOMO−1 is concentrated on the electron-donor dimethylamino-phenyl groups, the HOMO is delocalized on the whole molecule, though with larger contributions on the electron-donor groups. The LUMO is as well distributed on the whole molecule with larger contributions on the electron-acceptor CN groups. Such FMO delocalization scheme, which is almost invariably reproduced by other DFT functionals (see Fig. S3, ESI†), appears to be different from that usually found in other push–pull cross-conjugated architectures, which are characterized by spatially disjoint distributions of HOMOs and LUMOs.32 Both transitions involve a charge transfer from the push to pull moieties of the molecule, with an increase of the computed dipole moment from the ground (μg = 13.9 D) to S1 and S2 excited states (μe = 19.1 and 15.7 D, respectively), in agreement with the observed stronger solvatochromism of the excited state with respect to the ground state.
To investigate the origin of the proposed AIE behavior of 1, TD-PBE0/6-311++G(d,p) optimization of the S1 excited state in toluene has been performed, leading to a stationary state characterized by a remarkable conformational rearrangement of the molecule. Starting from the optimized S0 geometry, where the dimethylamino-phenyl groups are significantly tilted with respect to one another (the dihedral angle α between the least-squares planes through the phenyl carbon atoms measures 65.1°), optimization of S1 leads to a geometry where one dimethylamino-phenyl group is coplanar with the two cyano groups, forming a highly conjugated system with them, while the other one is almost perpendicular to this plane (see Fig. S5, ESI†). Emission from S1, computed at 587 nm, principally (99%) involves a transition from the LUMO, which is delocalized on the conjugated system, to the HOMO, which is distributed essentially on the perpendicular dimethylamino-phenyl group (see Fig. S5 (ESI†) for a FMO plot of the S1 optimized state). Owing to the perpendicular arrangement of the two molecular groups in the excited state and the consequent negligible FMO spatial overlap, the oscillator strength of the emission is zero. These results are in agreement with the experimental observation of the poor emissive behavior of 1 in good solvents and further support the hypothesis that the AIE behavior is connected to a RIR mechanism, which prevents the formation of the highly conjugated non-emissive excited state.
Further TDDFT calculations have been performed on 1 at the ground-state geometry optimized in toluene, by including triplet excitations in order to support the hypothesis of phosphorescence emission experimentally detected at low temperature. The T2 state, computed at 509 nm, was found to be relatively close to the S1 state (ΔES1T2 = 0.53 eV) (see Scheme 2) and share with it the same character (mainly HOMO−1 → LUMO transition, see above), where the HOMO−1 orbital has a large contribution (32%) from the lone-pair electrons of the amine nitrogen atoms. To the first order, spin–orbit coupling is forbidden between states with the same configuration,33 and this, in addition to the non-negligible ΔES1T2 gap, should further decrease the efficiency of the S1 → T2 process in the present compound. These results are in agreement with the observed low phosphorescence yield, unlike room temperature phosphorescent organic compounds34 characterized by quite smaller ΔES1T2 and different S1–T2 configurations. Finally, the T1 state, having the same character as S2 (mainly HOMO → LUMO transition), was computed at 587 nm, very close to the experimental phosphorescence band (595 nm).
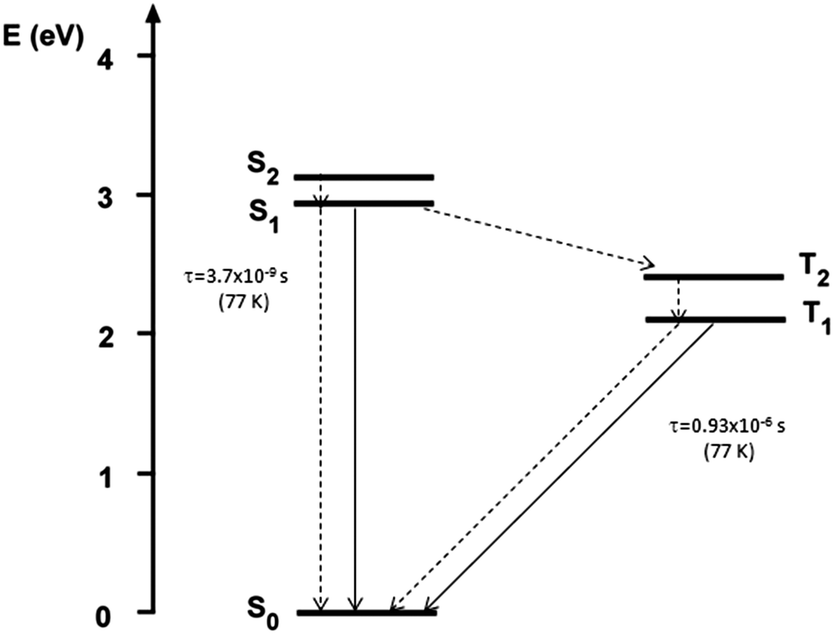 | ||
| Scheme 2 Jablonski diagram of compound 1 (τ values are measured in THF at 77 K). | ||
Crystal structures
Interestingly, four kinds of crystals of 1 were obtained by slow precipitation from a solution of dichloromethane/hexane. The crystals are characterized by different morphologies and absorption and emission colors. In particular, orange (orange emissive) crystals of prismatic shape (referred to as A), orange rod-like (yellow emissive) crystals (referred to as B), green-yellow (green emissive) needles (referred to as C) and yellow (green emissive) plate (referred to as D) were isolated and their crystal structure determined (see Table S2, ESI†). The crystal structures of two forms of 1, synthesized through alternative routes by Suzuki et al.35 and Bures et al.,36 were previously published without optical characterization. Polymorph A displays a crystal structure corresponding to that previously determined by Suzuki et al.; it belongs to the C2/c space group with half a molecule in the asymmetric unit (a.u.). Crystals of B as well belong to the C2/c space group with half a molecule in the a.u., while crystals of C belong to the non-centrosymmetric P21 space group with two molecules in the a.u. Crystals of D,36 which include dichloromethane as a co-crystallized solvent, belong to the P21/c space group with one molecule in the a.u. A distinctive feature of the molecular structures of A–D is their twisted conformation due to sterical hindrance both between the CN and the dimethylamino-phenyl substituents and between the phenyl rings. Three geometrical factors can play in a concerted way to reduce such hindrance (see Fig. 3): (i), the (N)C–CC–C(Ph) torsion angle β; (ii), the reciprocal tilting of the phenyl rings, which can be quantified through the dihedral angle α between the least-squares planes through the phenyl carbon atoms; and (iii) the central double bond, which is significantly elongated with respect to the value of 1.331(9) Å reported for (C2)–CC–(C2) unconjugated bonds37 denoting a high conjugation degree for the present compound. It is to be pointed out that, owing to the cross-conjugated architecture of 1,38 the phenyl rings, connected with each other by two single bonds, are separately conjugated to each CN group, as well as the CN groups are separately conjugated to each phenyl ring. The observed conformational differences in the four crystals (see Table 1), though small, are associated with a different conjugation degree between the molecular moieties connected through the CC double bond, as indicated by the reported bond lengths. In particular, the lower the dihedral angle between the phenyl rings, the larger the distortion around the double bond and greater the cross-conjugation. The different conformations of A–D are expected to reflect into different excitation and emission spectra. As evident from parameters in Table 1, polymorphs A and B possess similar molecular conformations quite different from those of C and D.
C–C(Ph) torsion angles (β, °), dihedral angles (α, °) between the least-squares planes through the phenyl rings and selected bond lengths (Å) for A–Da
| β | α | CC |
C–C(CN) | C–C(Ph) | |
|---|---|---|---|---|---|
| a See also Fig. 3 for the definition of geometrical parameters α and β. b Average over the absolute values of the four torsions around the double bonds of the two independent molecules, 10.2(4), 8.8(4) and −13.3(4), −15.1(3)°, respectively. c Average over the two non-equivalent torsions around the double bond, 15.5(1) and 14.1(1). d Average over the values observed for the two independent molecules, 75.8(1) and 67.9(1)°, respectively. e Average over: 1.370(3), 1.382(3) Å. f Average over: 1.438(3), 1.432(4), 1.433(4), 1.440(3) Å. g Average over: 1.469(3), 1.463(3), 1.457(3), 1.474(3) Å. h Average over: 1.430(4), 1.436(4) Å. i Average over: 1.460(4), 1.468(4) Å. | |||||
| C | 11.8(1)b | 71.8(1)d | 1.376(3)e | 1.436(3)f | 1.466(3)g |
| D | 14.8(1)c | 62.8(1) | 1.381(3) | 1.433(3)h | 1.464(3)i |
| B | 18.0(1) | 59.4(1) | 1.390(2) | 1.431(2) | 1.461(2) |
| A | 19.9(1) | 58.0(1) | 1.387(2) | 1.429(2) | 1.459(2) |
The twisted conformations of A–D rule out the presence of strong intermolecular π–π stacking interactions, excluding the formation of H- or J-aggregates. On the other hand, the weak intermolecular interactions found in A–D structures (a partial view of the crystal packing of A is reported in Fig. 4 as an example) are enough to fix the molecular conformations in the crystal structures activating the RIR mechanism. In both A and B structures, molecules are arranged along chains connected through weak C–H⋯N hydrogen bonds with weak π–π stacking interactions connecting the chains with each other (see Fig. 4 for crystal packing of A). In structures of C and D, on the other hand, a quite dense intermolecular interaction network, based on several weak C–H⋯N and C–H⋯π interactions, is observed (see ESI† for a detailed description of crystal packing of A–D and Fig. S6–S8 (ESI†) for a partial view of the crystal packing of B–D). The structural analysis on the four crystal forms A–D highlights, also in the molecular packing, similarities between A and B, which are both markedly different from the packing of C and D (see Fig. S9 and S10, ESI†). Such structural similarity is reflected in a similar emissive spectral range for A and B.
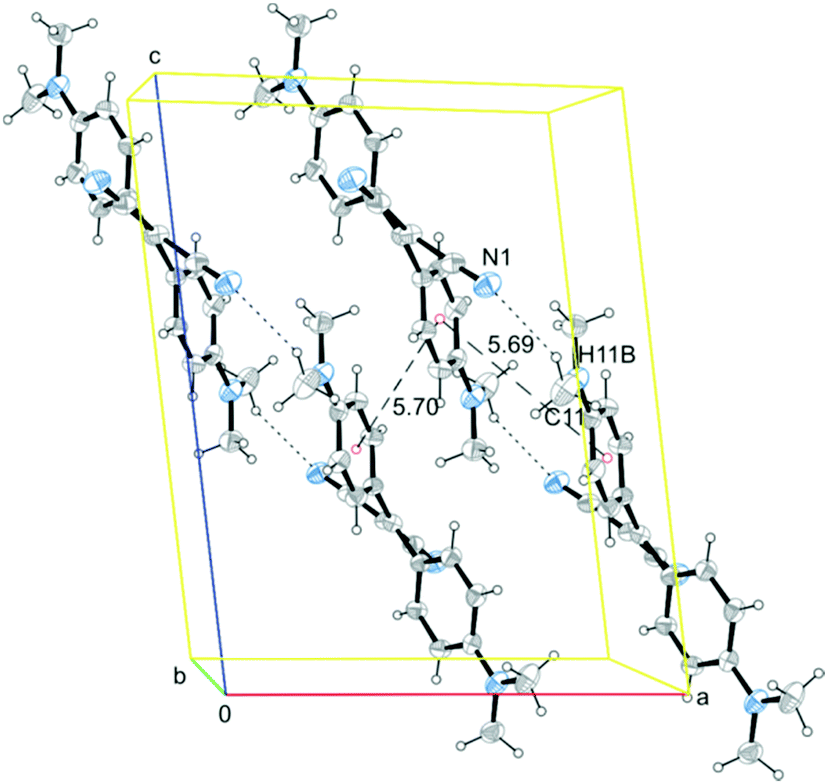 | ||
| Fig. 4 Partial view of the crystal packing of A (along the b axis direction), where selected hydrogen bonds (short dashes) and centroids-connecting lines (long dashes) are included. Ellipsoids are drawn at the 30% probability level. | ||
In order to evaluate the influence of molecular and crystal structures on the optical properties, we have performed PBE0/6-311++G(d,p) calculations on 1 by freezing the torsion angle β to selected values and relaxing all the other freedom degrees (see Table 2). Looking at the reported computed bond lengths, the degree of π-electron cross-conjugation is expected to increase with increasing β angle and decreasing α angle, in the same way as observed in the X-ray crystal structures. The results of TDDFT calculations, also reported in Table 2, are indicative of a red-shift of the computed absorption wavelengths with increasing conjugation degree, suggesting a negligible influence of the crystal packing on the optical behavior.
C–C(Ph) torsion angle (β, °)a
| β | α | CC |
C–C(CN) | C–C(Ph) | λ abs(f), assignments |
|---|---|---|---|---|---|
| a See Fig. 3 for geometrical parameter definitions. b Value corresponding to the optimized geometry. | |||||
| 0 | 69.2 | 1.385 | 1.422 | 1.462 | 412 (0.63), H−1 → L (99.1%); 409(0.46), H → L (99.1%) |
| 10 | 66.3 | 1.387 | 1.422 | 1.459 | 413 (0.69), H−1 → L (99.1%); 407(0.46), H → L (99.1%) |
| 13.85b | 65.1 | 1.388 | 1.421 | 1.458 | 415 (0.71), H−1 → L (99.2%); 408(0.47), H → L (99.2%) |
| 20 | 63.0 | 1.391 | 1.421 | 1.455 | 419 (0.76), H−1 → L (99.2%); 410(0.47), H → L (99.2%) |
| 30 | 59.2 | 1.399 | 1.418 | 1.451 | 431 (0.83), H−1 → L (99.3%); 424(0.46), H → L (99.3%) |
Polymorphism-dependent fluorescence properties
Crystals of A–D show an increase of more than one order of magnitude of the PL quantum efficiency with respect to the solution, according to an AIE mechanism. In addition, crystals of A and B display irreversible emissive mechanochromism, crystals of A also show reversible thermochromic behavior while crystals of C show chronochromism. Crystals of D undergo a phase transition to form B upon standing in air or, more rapidly, upon mild thermal treatment (see Scheme 3).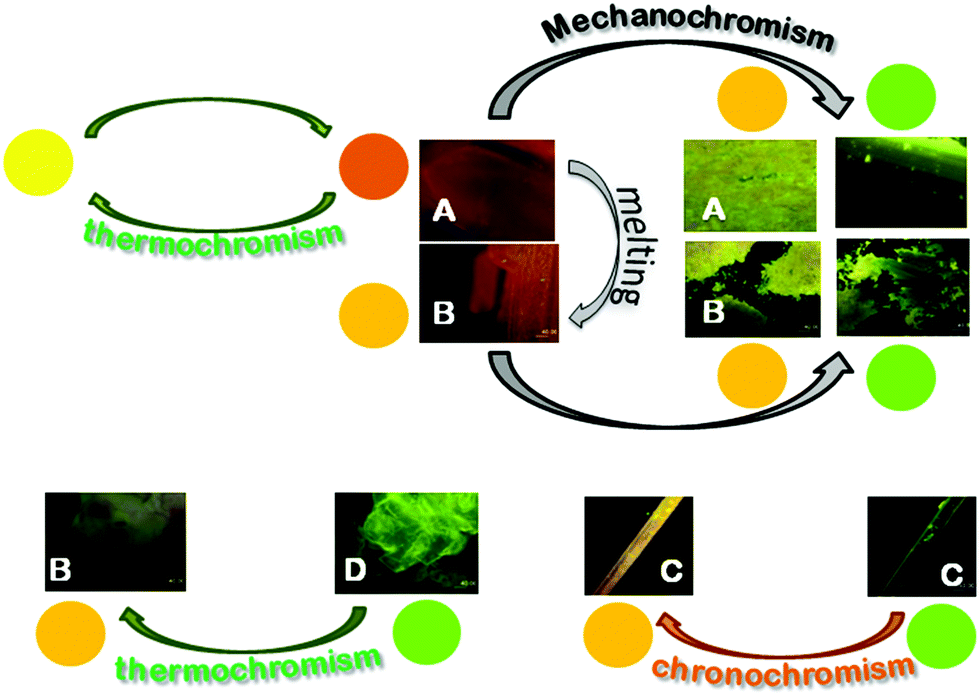 | ||
| Scheme 3 Representation of the chromic processes of crystals A–D. | ||
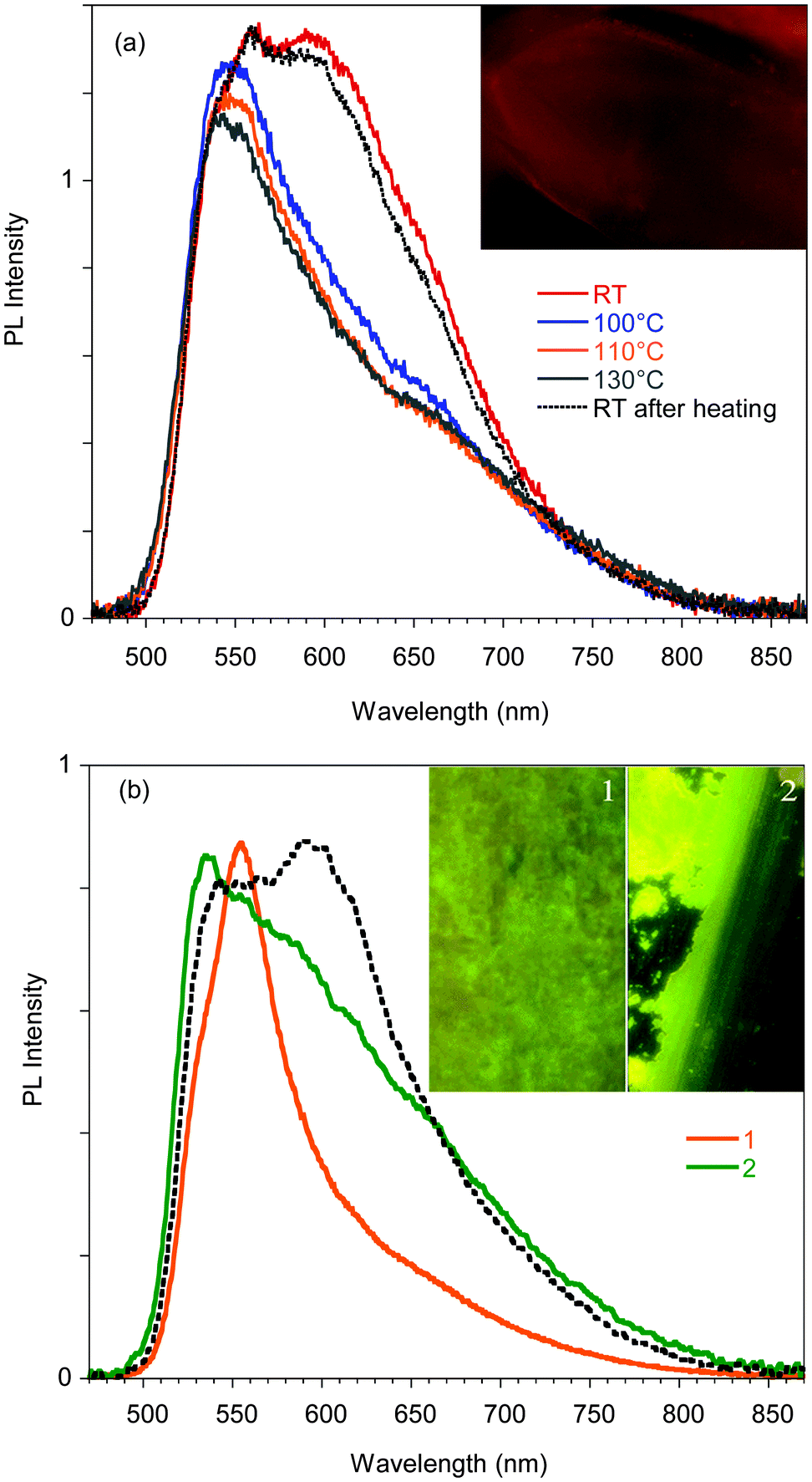 | ||
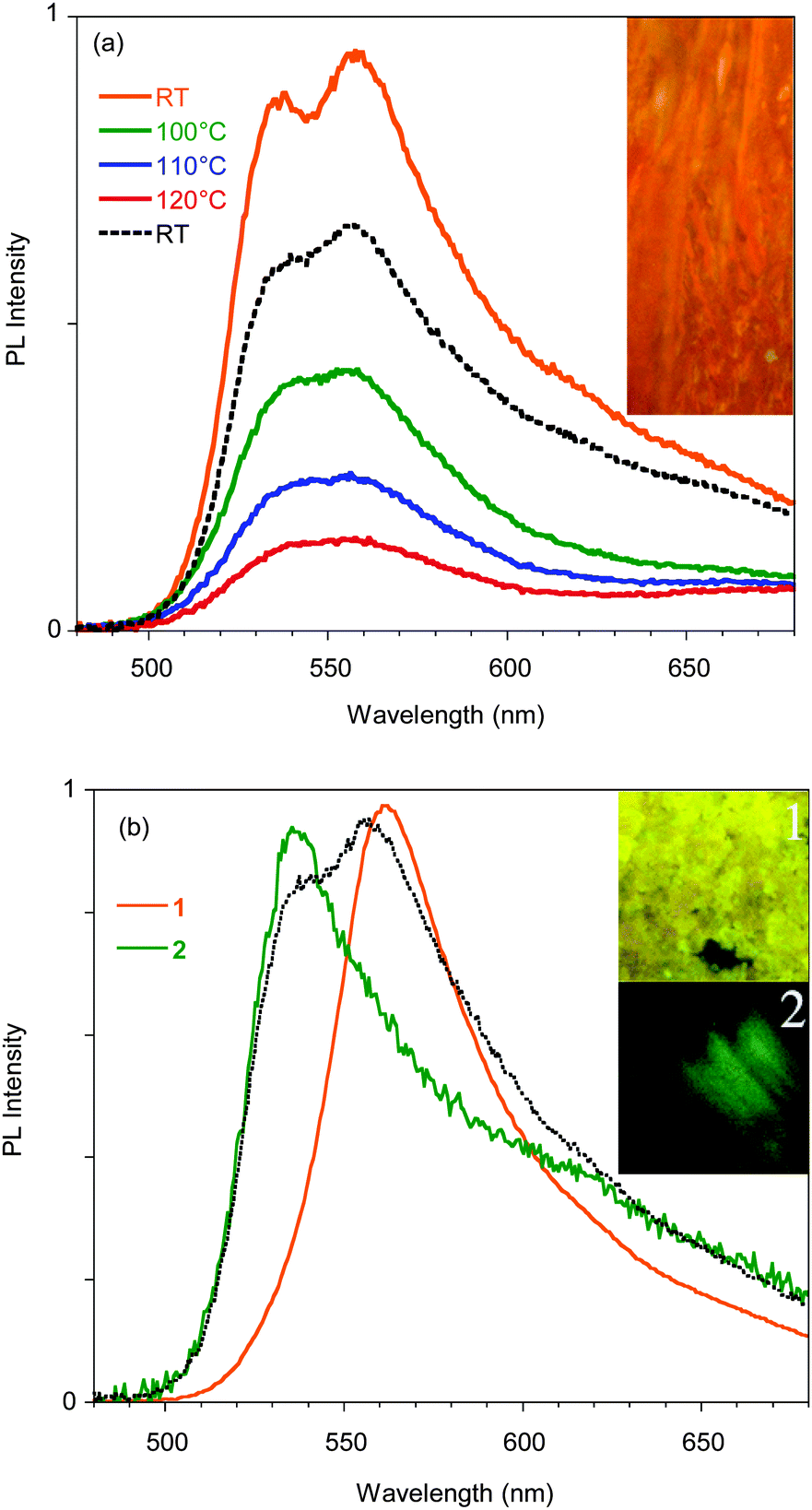
| Fig. 5 (a) PL spectra of a single crystal of A recorded during a heating–cooling cycle. Inset: Microscopy image of a 360 × 320 μm portion of the crystal. (b) Normalized PL spectra of a single crystal of A before (dotted line) and after mild (1) grinding and smashing. (2). Inset: Pictures taken using a confocal fluorescence microscope during the grinding process. Images size is 80 × 140 μm. | ||
The emission is composed of two contributions at about 595 and 550 nm. In order to investigate the possible phosphorescence origin of the 595 nm emission (as observed in the glassy solution), we have performed time resolved analysis of the crystal emission. These measurements confirmed the fluorescence origin of all the spectral components of the crystal emission (average lifetimes of about 1.50 ns, independent of the emission wavelength, see ESI†).
Phosphorescence is detected only upon lowering the temperature of the crystal and shows a structured spectrum with the main peak at 620 nm and an average lifetime of 1.82 μs (see Fig. S11, ESI†). Interestingly, when crystal A is gradually heated up to 130 °C, the 595 nm component disappears, but it is restored when the crystal is cooled back to room temperature. The reversibility of this thermochromic behavior seems reasonable to be ascribed to the presence of different surface emission centers in the crystals rather than to a phase transition. The observation that the lower energy component disappears at high temperature suggests its origin from a smoother surface region,16 which is perturbed at high temperature and recovered at room temperature. In addition, A displays an evident mechanochromic behavior. In fact, as shown in Fig. 5b, when a single crystal of A is ground with a spatula, yellow-orange (555 nm) fragments are obtained. Upon smashing, the emission color is further blue-shifted to 535 nm, a value quite close to that observed for the diluted glassy solution. The PL quantum efficiency of the single crystal is equal to 3%, increasing to 4% upon mild grinding, while it decreases below 1% upon smashing when the emission shifts to green. The original 595 nm emission cannot be restored by performing annealing treatment of the ground crystals.
The mechano-induced changes of A were also monitored by X-ray powder diffraction (XRPD) after different grinding steps (see Fig. 6). The XRPD pattern of very slightly ground crystals of A shows intense and sharp reflection peaks, in good agreement with the simulated pattern of polymorph A. After further grinding, the XRPD profile shows the same but significantly enlarged and attenuated signals attributable to the partial amorphization of the solid. At this stage, the number of surface defects is expected to be greatly increased leading to the quenching of the low energy emission component in the PL spectra.
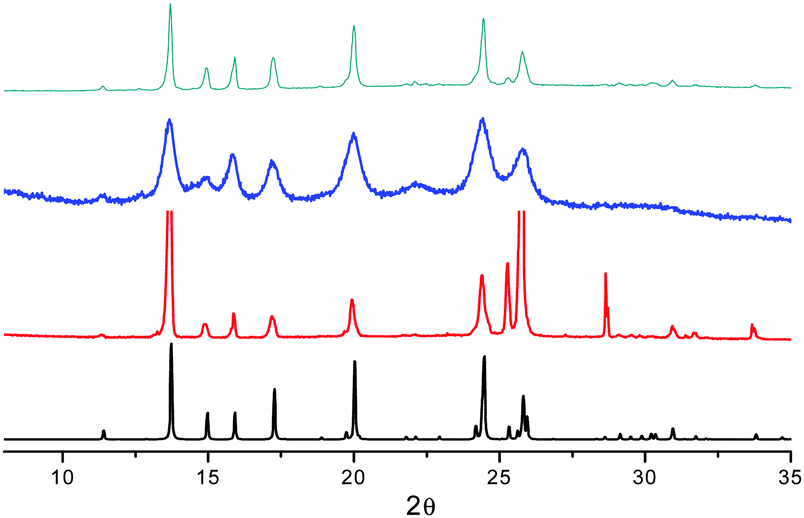 | ||
| Fig. 6 XRPD patterns monitoring the grinding process for compound A: simulated from crystal structure data (black); crystals after mild (red) and thorough (blue) grinding; powders obtained by fast precipitation (cyan). | ||
When A is obtained as powder by fast precipitation from a CH2Cl2/hexane solution of 1, as confirmed by XRPD (see Fig. 6), a narrow emission centered at 560 nm and PL quantum efficiency of 11% are observed suggesting a stronger emissive behavior in samples with more defective surfaces (see Fig. S13, ESI†). Surprisingly, in the attempt to prepare the amorphous sample by melting powders of A at 260 °C and rapidly quenching the melt with liquid nitrogen, highly crystalline powders of B were obtained (XRPD evidence) and the emission broadened and slightly red-shifted (see Fig. S12 and S13, ESI†).
 | ||
| Fig. 7 (a) PL spectra of a single crystal of B during heating and after the heating cycle (dotted line), inset: microscopy image of a 80 × 190 μm portion of the crystal. (b) Normalized PL spectra of a single crystal of B before (dotted line) and after mild grinding (1) and smashing (2). Inset: Pictures taken using a confocal fluorescence microscope during the grinding process. The size of the images of inset is 80 × 80 μm. | ||
The mechanochromic behavior of a single crystal of B was also investigated. When roughly ground with a spatula, a single and sharp emission at 560 nm is observed. Upon smashing the emission is shifted to 535 nm (see Fig. 7b). This behavior and the trend in the PLQYs are very similar to what was observed for A.
The mechanochromic emission changes of B were also monitored by XRPD after different grinding steps (see Fig. 8). The XRPD pattern of roughly ground crystals of B shows intense and sharp reflection peaks, in good agreement with the simulated pattern of polymorph B. Contrarily to polymorph A, the XRPD profile seems less affected by further grinding indicating a lower tendency of B to form an amorphous phase.
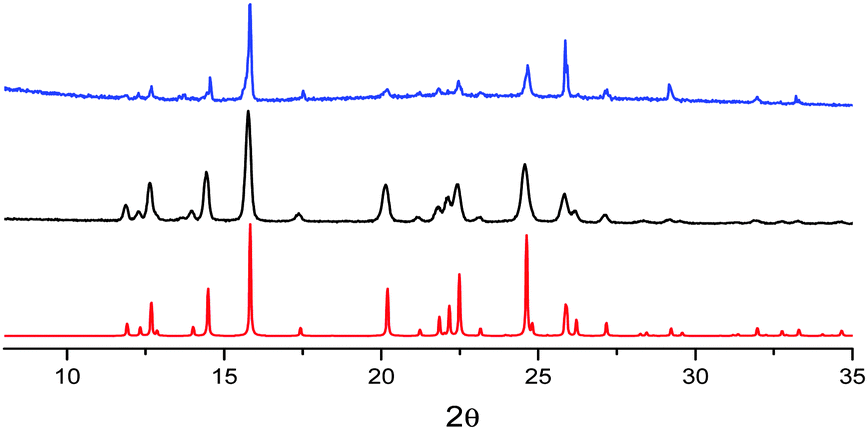 | ||
| Fig. 8 XRPD patterns monitoring the grinding process for compound B: simulated from crystal structure data (red); crystals after mild (black) and thorough (blue) grinding. | ||
The observation of an identical emissive behavior from roughly ground A and B, characterized by different crystal structures, supports the hypothesis of surface defects at the origin of the yellow emission.
Surprisingly, upon standing in air for several days, the emission color of crystals of C shifts to yellow (560 nm) as shown in Fig. 9. Upon thermal annealing of the latter sample a further red-shift of the emission is observed. This chronochromic behavior, different from previous findings in the literature,13,14 is not associated with a phase transition as indicated by single crystal X-ray diffraction analysis which gave the same unit cell parameters as the freshly prepared crystals of C. A different emissive behavior associated with the same crystal structure has been previously observed for other crystals and ascribed to different surface structures (rough or smooth morphology).16 It is reasonable that in our case the harsh ambient conditions (humid and very hot summer) have somehow modified the surface morphology leading to the formation of defects characterized by an emissive color different from that of the bulk, as previously reported.16c,d Further experiments in support of this hypothesis are planned.
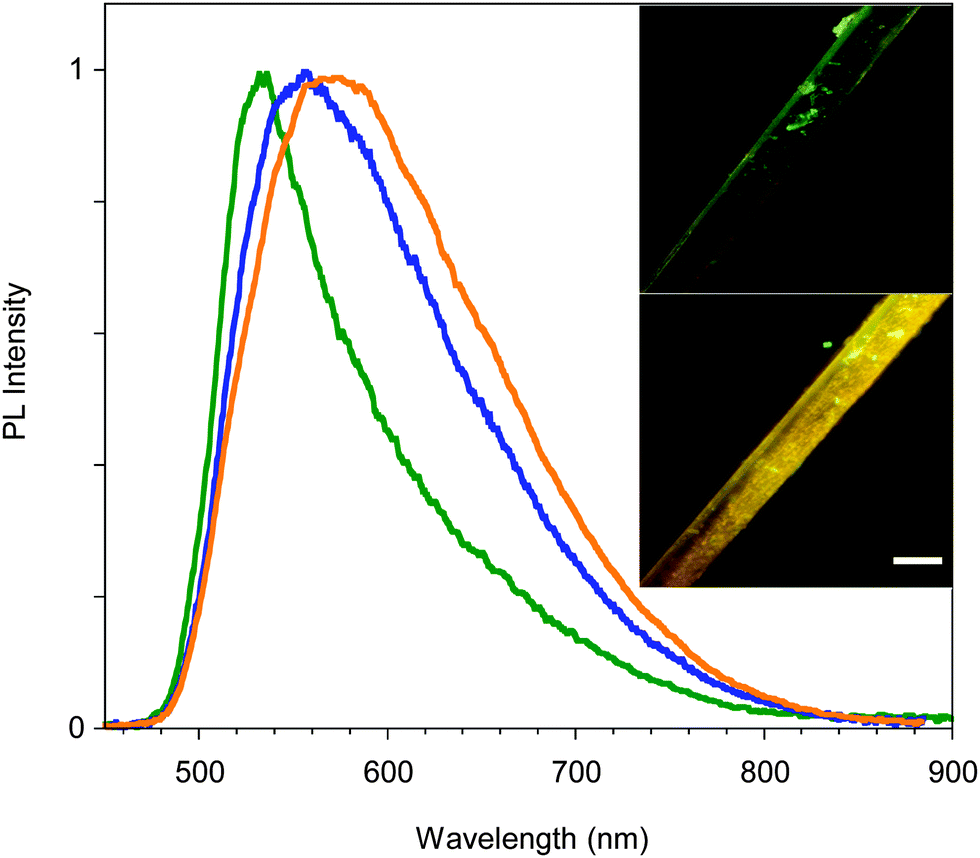 | ||
| Fig. 9 PL spectra of crystal C: as-prepared (green line), after 20 days (blue line), and the latter after thermal annealing (orange line). In the inset the picture of a portion of one crystal as-prepared (top) and after 20 days (bottom); scale bar is 40 μm long. | ||
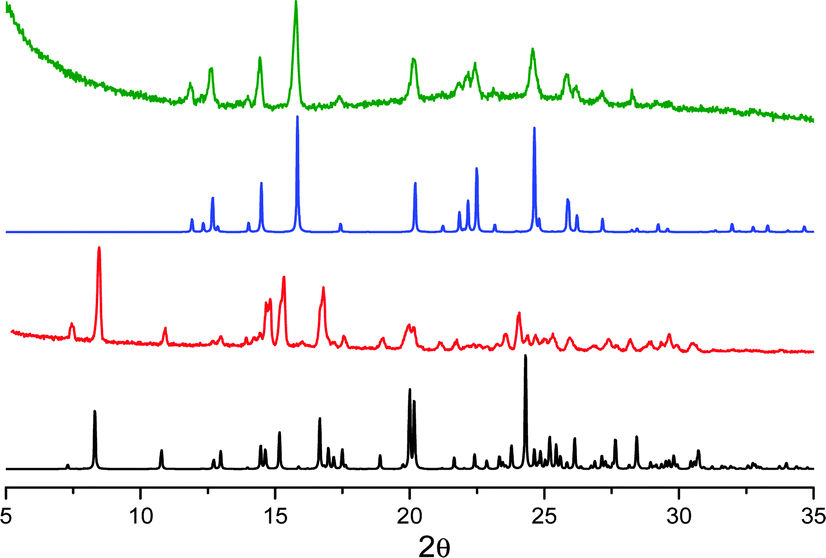 | ||
| Fig. 10 XRPD patterns monitoring the desolvation process of compound D and its transformation to B: simulated from crystal structure data for D (black); powders of D freshly prepared (red); simulated from crystal structure data for B (blue); powders of D left in the air (green). | ||
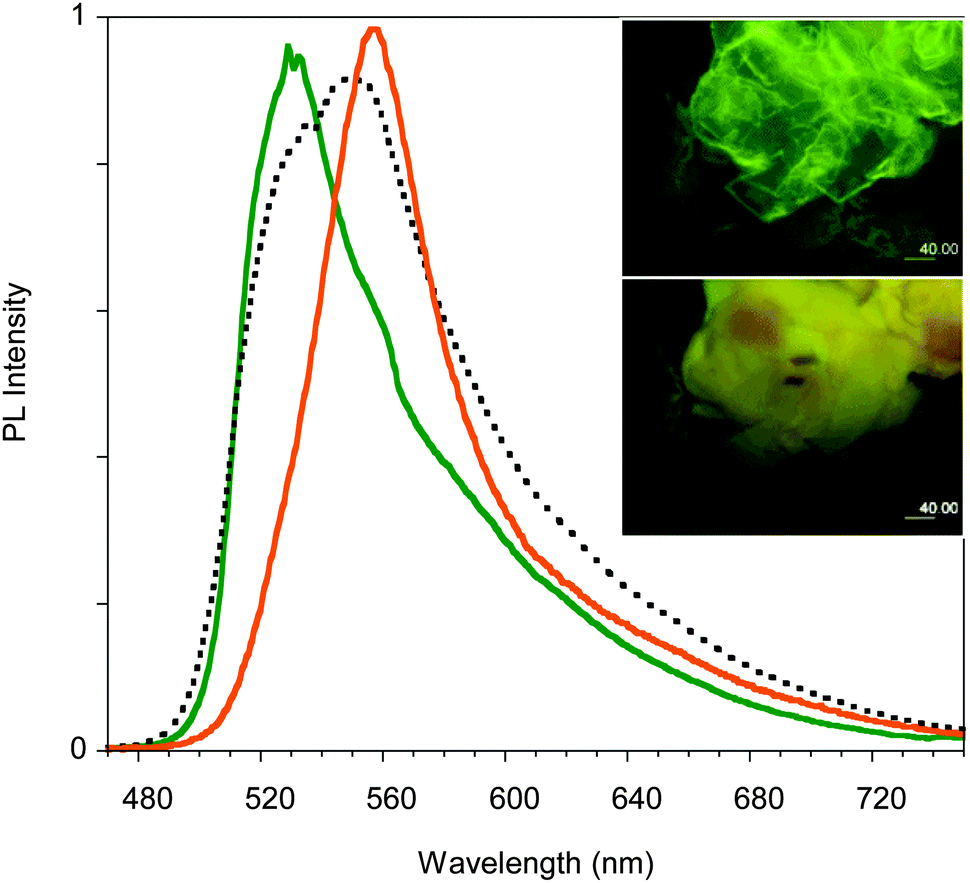 | ||
| Fig. 11 PL spectra of crystals of D before thermal treatment (green line), after treatment at 50 °C for 10 min (dotted black line) and 25 min (orange line). Inset: Images taken at the microscope before (top) and after (bottom) thermal treatment. | ||
Conclusions
Compound 1 was synthesized and its emissive behavior was investigated at both molecular and aggregate levels. Four different crystalline phases have been isolated, two of them already known while the other two reported here for the first time. In particular one of the new structures belongs to an acentric space group.All these phases display AIE (aggregation-induced emission) features and emissive behavior which can be correlated with the molecular geometry as confirmed by DFT and TDDFT calculations, indicating a red-shift of the emission with increasing conjugation degree, as imposed by the crystalline environment. In particular, emission is shifted from the green region for the less conjugated phases C and D to the yellow and yellow-orange regions for the more conjugated B and A, respectively. Analogous behavior has been previously reported for another polymorphism-dependent emissive system characterized by a twisted conformation around a double bond,14 for which the greater red-shift of the emission maxima was shown by the more conjugated conformer.
Interestingly, crystals of polymorph A, characterized by a two-component emission at room temperature, display both thermo- and mechanochromic emissive behavior. The low energy component is reversibly quenched at high temperature due to the decreased contribution from smooth surface regions. The number of defective surface centers is also increased by the grinding process, which induces crystal size reduction and amorphization. At manual smashing both phases A and B show emission properties similar to those of the glassy solution of 1. C does not display thermo- and mechanochromic behavior but chronochromic properties. D is easily transformed into B either spontaneously or by mild thermal treatment due to solvent loss. The D to B transformation represents the only emission color change associated with a phase transition. All the other emission color changes reported in the present study seem to be ascribable to a variation of the crystal surface morphology; in particular, upon grinding, surface defects are formed in which molecules with a relaxed conformation emit a different color.
Acknowledgements
The use of instrumentation purchased through the Regione Lombardia – Fondazione Cariplo joint SmartMatLab Project is gratefully acknowledged. DP acknowledges grants by MIUR (PRIN 2009-A5Y3N9), INSTM-Regione Lombardia and ENI S.p.A. for partial support of this work. C. B. thanks Regione Lombardia for funding (project “Tecnologie e materiali per l'utilizzo efficiente dell'energia solare” decreto 3667/2013). Prof. G. Terraneo is acknowledged for the use of the Bruker AXS D8 powder diffractometer at Politecnico di Milano and for his help in collecting the spectra.Notes and references
- (a) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., 2014, 126, 2151 CrossRef; (b) Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC; (c) X. Wang, Y. Zhou, T. Lei, N. Hu, E. Q. Chen and J. Pei, Chem. Mater., 2010, 22, 3735 CrossRef CAS; (d) W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed; (e) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- (a) C. Zhang, Y. S. Zhao and J. N. Yao, Phys. Chem. Chem. Phys., 2011, 13, 9060 RSC; (b) Q. Liao, H. B. Fu and J. N. Yao, Adv. Mater., 2009, 21, 4153 CrossRef CAS; (c) J. Y. Zheng, Y. L. Yan, X. P. Wang, Y. S. Zhao, J. X. Huang and J. N. Yao, J. Am. Chem. Soc., 2012, 134, 2880 CrossRef CAS PubMed; (d) Y. L. Lei, Q. Liao, H. B. Fu and J. N. Yao, J. Am. Chem. Soc., 2010, 132, 1742 CrossRef CAS PubMed.
- (a) V. Bulovi, V. G. Kozlov, V. B. Khalfin and S. R. Forrest, Science, 1998, 279, 553 CrossRef; (b) X. F. Duan, Y. Huang, R. Agarwal and C. M. Lieber, Nature, 2003, 421, 241 CrossRef CAS PubMed; (c) Y. S. Zhao, A. D. Peng, H. B. Fu, Y. Ma and J. N. Yao, Adv. Mater., 2008, 20, 1661 CrossRef CAS; (d) I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272 CrossRef CAS PubMed; (e) X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao, Y. Wu, Z. Xu, Y. Zhao, H. Fu and D. Zhang, Adv. Funct. Mater., 2012, 22, 4862 CrossRef CAS.
- (a) X. Sun, Y. Liu, X. Xu, C. Yang, G. Yu, S. Chen, Z. Zhao, W. Qiu, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 10786 CrossRef CAS PubMed; (b) Y. Shirota, J. Mater. Chem., 2005, 15, 75 RSC; (c) X. Y. Shen, W. Z. Yuan, Y. Liu, Q. Zhao, P. Lu, Y. Ma, I. D. Williams, A. Qin, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 10541 CrossRef CAS; (d) X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334 CrossRef CAS; (e) F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478 CrossRef CAS PubMed; (f) T. C. Lin, G. S. He and Q. Zheng, J. Mater. Chem., 2006, 16, 2490 RSC.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed; (b) E. A. Silinsh, Organic Molecular Crystals, Springer-Verlag, Berlin, 1980 Search PubMed; (c) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- (a) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) Z. Y. Zhang, B. Xu, J. H. Su, L. P. Shen, Y. S. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654 CrossRef CAS PubMed; (c) B. Wang, Y. C. Wang, J. L. Hua, Y. H. Jiang, J. H. Huang, S. X. Qian and H. Tian, Chem. – Eur. J., 2011, 17, 2647 CrossRef CAS PubMed; (d) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- (a) X. Y. Qi, H. Li, J. W. Y. Lam, X. T. Yuan, J. Wei, B. Z. Tang and H. Zhang, Adv. Mater., 2012, 24, 4191 CrossRef CAS PubMed; (b) Z. Li, Y. Q. Dong, J. W. Y. Lam, J. X. Sun, A. J. Qin, M. Häußler, Y. P. Dong, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Adv. Funct. Mater., 2009, 19, 905 CrossRef CAS; (c) F. Mahtab, Y. Yu, J. W. Y. Lam, J. Z. Liu, B. Zhang, P. Lu, X. X. Zhang and B. Z. Tang, Adv. Funct. Mater., 2011, 21, 1733 CrossRef CAS; (d) G. Yu, S. W. Yin, Y. Q. Liu, J. S. Chen, X. J. Xu, X. B. Sun, D. G. Ma, X. W. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. H. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS PubMed; (e) J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) A. Prasanna de Silva, H. Q. Nimal Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef; (b) M. Wang, G. X. Zhang, D. Q. Zhang, D. B. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC; (c) Y. Liu, Y. H. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (d) Y. Liu, C. M. Deng, L. Tang, A. J. Qin, R. R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660 CrossRef CAS PubMed; (e) S. J. Toal, K. A. Jones, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2005, 127, 11661 CrossRef CAS PubMed; (f) C. Yu, K. H. Y. Chan, K. M. C. Wong and V. W. W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19652 CrossRef CAS PubMed; (g) M. C. L. Yeung, K. M. C. Wong, Y. K. T. Tsang and V. W. W. Yam, Chem. Commun., 2010, 46, 7709 RSC.
- (a) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789 CrossRef CAS PubMed; (b) V. I. Minkin, Chem. Rev., 2004, 104, 2751 CrossRef CAS PubMed; (c) S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346 CrossRef CAS PubMed; (d) Z. Q. Guo, W. H. Zhu, L. J. Shen and H. Tian, Angew. Chem., Int. Ed., 2007, 46, 5549 CrossRef CAS PubMed; (e) G. Y. Jiang, Y. L. Song, X. F. Guo, D. Q. Zhang and D. B. Zhu, Adv. Mater., 2008, 20, 2888 CrossRef CAS; (f) H. Li, J. X. Wang, H. Lin, L. Xu, W. Xu, R. M. Wang, Y. L. Song and D. B. Zhu, Adv. Mater., 2010, 22, 1237 CrossRef CAS PubMed; (g) Y. Q. Wen, J. X. Wang, J. P. Hu, L. Jiang, H. J. Gao, Y. L. Song and D. B. Zhu, Adv. Mater., 2006, 18, 1983 CrossRef CAS; (h) Y. L. Shang, Y. Q. Wen, S. L. Li, S. X. Du, X. B. He, L. Cai, Y. F. Li, L. M. Yang, H. J. Gao and Y. L. Song, J. Am. Chem. Soc., 2007, 129, 11674 CrossRef CAS PubMed.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685 CrossRef CAS PubMed.
- (a) X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427 RSC; (b) J. Ni, X. Zhang, Y.-H. Wu, L.-Y. Zhang and Z.-N. Chen, Chem. – Eur. J., 2011, 17, 1171 CrossRef CAS PubMed; (c) S. Jayanty and T. P. Radhakrishnan, Chem. – Eur. J., 2004, 10, 791 CrossRef CAS PubMed.
- (a) S. Yamaguchi, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2012, 22, 20065 RSC; (b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed; (c) X. Zhang, Z. Chi, X. Zhou, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. C, 2012, 116, 23629 CrossRef CAS; (d) C. Wang, S. Chen, K. Wang, S. Zhao, J. Zhang and Y. Wang, J. Phys. Chem. C, 2012, 116, 17796 CrossRef CAS; (e) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed; (f) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed; (g) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed; (h) L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918 CrossRef CAS PubMed; (i) S. Varughese, J. Mater. Chem. C, 2014, 2, 3499 RSC; (j) Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429 CrossRef CAS PubMed.
- J. Wang, J. Mei, R. Hu, J. Zhi Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed.
- P. Galer, R. C. Korošec, M. Vidmar and B. Sket, J. Am. Chem. Soc., 2014, 136, 7383 CrossRef CAS PubMed.
- (a) X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed; (b) N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409 RSC.
- (a) A. Patra, N. Hebalkar, B. Sreedhar, M. Sarkar, A. Samanta and T. P. Radhakrishnan, Small, 2006, 2, 650 CrossRef CAS PubMed; (b) Z. Lin, X. Mei, E. Yang, X. Li, H. Yao, G. Wen, C. T. Chien, T. J. Chow and Q. Ling, CrystEngComm, 2014, 16, 11018 RSC; (c) T. Han, Y. Hong, N. Xie, S. Chen, N. Zhao, E. Zhao, J. W. Y. Lam, H. H. Y. Sung, Y. Dong, B. Tong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 7314 RSC; (d) Y. Wang, I. Zhang, B. Yu, X. Fang, X. Su, Y.-M. Zhang, T. Zhang, B. Yang, M. Li and S. X.-A. Zhang, J. Mater. Chem. C, 2015, 3, 12328 RSC.
- (a) A. G. Mirochnik, E. V. Fedorenko, V. G. Kuryavyi, B. V. Bukvetskii and V. E. Karasev, J. Fluoresc., 2006, 16, 279 CrossRef CAS PubMed; (b) P. S. Hariharan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2015, 3, 8381 RSC.
- (a) E. Cariati, V. Lanzeni, E. Tordin, R. Ugo, C. Botta, A. Giacometti Schieroni, A. Sironi and D. Pasini, Phys. Chem. Chem. Phys., 2011, 13, 18005 RSC; (b) C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666 RSC; (c) F. Villafiorita-Monteleone, A. Cappelli, M. Paolino, M. Colombo, E. Cariati, A. Mura, G. Bongiovanni and C. Botta, J. Phys. Chem. C, 2015, 119, 18986 CrossRef CAS.
- T. Virgili, A. Forni, E. Cariati, D. Pasini and C. Botta, J. Phys. Chem. C, 2013, 117, 27161 CAS.
- J. Moreau, U. Giovanella, J.-P. Bombenger, W. Porzio, V. Vohra, L. Spadacini, G. Di Silvestro, L. Barba, G. Arrighetti, S. Destri, M. Pasini, M. Saba, F. Quochi, A. Mura, G. Bongiovanni, M. Fiorini, M. Uslenghi and C. Botta, ChemPhysChem, 2009, 10, 647 CrossRef CAS PubMed.
- Bruker, SMART, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01., Gaussian, Inc., Wallingford, CT, USA, 2013 Search PubMed.
- (a) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- (a) L. E. Johnson, L. R. Dalton and B. H. Robinson, Acc. Chem. Res., 2014, 47, 3258 CrossRef CAS PubMed; (b) D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708 CrossRef CAS; (b) R. Cammi and B. Mennucci, J. Chem. Phys., 1999, 110, 9877 CrossRef CAS.
- I. Shibuya, Y. Taguchi, T. Tsuchiya, A. Oishi and E. Katoh, Bull. Chem. Soc. Jpn., 1994, 67, 3048 CrossRef CAS.
- (a) V. Ermolaev, Opt. Spektrosk., 1961, 11, 492 CAS; V. Ermolaev, Opt. Spectry., 1961, 11, 266 Search PubMed; (b) S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199 CrossRef CAS.
- E. C. Lim and S. K. Chakrabarti, J. Chem. Phys., 1967, 47, 4726 CrossRef CAS.
- As determined by X-ray diffraction studies, the dihedral angles between the least-squares planes through the heavy atoms of the N-dimethyl groups and the benzene rings in the four crystal structures A–D are 4.40(9)° (A); 4.34(8)° (B); 9.1(2), 2.8(2), 8.4(2) and 7.4(2)° (C); 5.3(1) and 12.2(2)° (D).
- (a) A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397 CrossRef CAS PubMed; (b) W. C. W. Leu and C. S. Hartley, Org. Lett., 2013, 15, 3762 CrossRef CAS PubMed; (c) T. Inouchi, T. Nakashima and T. Kawai, J. Phys. Chem. A, 2014, 118, 2591 CrossRef CAS PubMed; (d) A. Felouat, A. D'Aléo, A. Charaf-Eddin, D. Jacquemin, B. Le Guennic, E. Kim, K. J. Lee, J. H. Woo, J.-C. Ribierre, J. W. Wu and F. Fages, J. Phys. Chem. A, 2015, 119, 6283 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834 CrossRef CAS.
- Y. Gong, G. Chen, Q. Peng, W. Zhang Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195 CrossRef CAS PubMed.
- T. Suzuki, K. Ono, H. Kawai and T. Tsuji, J. Chem. Soc., Perkin Trans. 2, 2001, 1798 RSC.
- F. Bureš, W. B. Schweizer, J. C. May, C. Boudon, J.-P. Gisselbrecht, M. Gross, I. Biaggio and F. Diederich, Chem. – Eur. J., 2007, 13, 5378 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal data, photoluminescence, XRPD and additional computational information. CCDC 1431281–1431284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc03352g |
| This journal is © The Royal Society of Chemistry 2016 |