
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-driven CO2 reduction on a hybrid photocatalyst consisting of a Ru(II) binuclear complex and a Ag-loaded TaON in aqueous solutions†
Akinobu
Nakada
,
Takuya
Nakashima
,
Keita
Sekizawa
,
Kazuhiko
Maeda
and
Osamu
Ishitani
*
Department of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1-NE-1 O-okayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: ishitani@chem.titech.ac.jp
First published on 23rd March 2016
Abstract
A hybrid photocatalyst consisting of a Ru(II) binuclear complex and a Ag-loaded TaON reduced CO2 by visible light even in aqueous solution. The distribution of the reduction products was strongly affected by the pH of the reaction solution. HCOOH was selectively produced in neutral conditions, whereas the formation of HCOOH competed with H2 evolution in acidic conditions. Detailed mechanistic studies revealed that the photocatalytic CO2 reduction proceeded via ‘Z-schematic’ electron transfer with step-by-step photoexcitation of TaON and the photosensitizer unit in the Ru(II) binuclear complex. The maximum turnover number for HCOOH formation was 750 based on the Ru(II) binuclear complex under visible-light irradiation, and the optimum external quantum efficiency of the HCOOH formation was 0.48% using 400 nm monochromic light with ethylenediaminetetraacetic acid disodium salt as a sacrificial reductant. Even in aqueous solution, the hybrid could also convert visible-light energy into chemical energy (ΔG0 = +83 kJ mol−1) by the reduction of CO2 to HCOOH with methanol oxidation.
Introduction
The development of photocatalytic systems for CO2 reduction is an attractive research target in the field of conversion of solar energy into chemical energy, the so-called artificial photosynthesis. Artificial photosynthetic reactions have various potential functions; one of these is to use water as both an electron source and as a solvent because water is an abundant and low-cost material. Since both CO2 and water are very stable compounds, these photocatalytic systems should have both strong reduction and oxidation power. Utilization of visible light is another important function for artificial photosynthesis because it covers ca. 50% of the solar energy, whereas the light in the UV region (λ < 400 nm) is very minor (<6%). However, there are few visible-light-driven photocatalysts for CO2 reduction which function well in water.Multinuclear Ru(II) and/or Re(I) diimine (N^N) complexes with a redox photosensitizer (PS) and a catalyst (CAT) unit, the so-called supramolecular photocatalysts, have attractive abilities as photocatalysts for CO2 reduction because of their high efficiencies and selectivities for reducing CO2 to HCOOH and CO not only in organic solution1–9 but also in aqueous solution.10,11 Since proton reduction to H2 is a more thermodynamically favorable reaction than CO2 reduction, this specific selectivity is a superior property for constructing an artificial photosynthesis system with CO2 reduction in aqueous solution. However, the photocatalytic systems constructed with only metal complexes generally require a strong reductant such as NADH model compounds2,5–9 and benzimidazoline derivatives1,3,4,11 because the excited metal complexes have relatively weak oxidizing power. To add the stronger photooxidizing power, the metal complex photocatalyst should be combined with another photocatalyst for the oxidation reaction.
Some powder semiconductor photocatalysts with much stronger oxidizing power have been reported, which can oxidize even water involving reduction of electron acceptors.12 Metal oxynitrides are typical examples; they have sufficient positive valence band potential to oxidize weak reductants and relatively small band gaps to utilize visible light.13
Based on these investigations regarding the strong and weak points of different types of photocatalysts, we have developed novel hybrid photocatalytic systems where supramolecular photocatalysts connect with metal oxynitride photocatalysts to utilize both the outstanding features of high selectivity and efficiency for CO2 photoreduction (supramolecular site) and strong photooxidizing power (semiconductor site). Visible-light irradiation to the hybrid photocatalysts consisting of a Ru(II) binuclear complex (RuRu) with [Ru(N^N)3]2+ as the PS unit and Ru(N^N)(CO)2Cl2 as the CAT unit, which was adsorbed on a tantalum(V) oxynitride (TaON) photocatalyst in pure methanol without any other reductant under a CO2 atmosphere, caused the catalytic formation of HCOOH as a reduced product of CO2 and formaldehyde as an oxidized product of methanol (MeOH).14 Using CaTaO2N instead of TaON in the hybrid achieved high selectivity of HCOOH formation (>99%) in dimethylacetamide–triethanolamine mixed solution; meanwhile, the photocatalytic reaction requires a sacrificial electron donor.15 These reactions are driven via the two-step photoinduced electron transfer mechanism, the so-called ‘Z-scheme’, as shown in Scheme 1: (1) step-by-step photoexcitation of the semiconductor and the Ru(II) PS unit occurs; (2) the valence band holes are consumed by a reductant; (3) conduction band electrons in the semiconductor transfer to the excited state of the PS unit, producing one-electron-reduced species (OERS) of PS; (4) intramolecular electron transfer from the OERS of the PS unit to the ground state of the CAT unit occurs, producing the reduced CAT unit and (5) CO2 reduction proceeds on the reduced CAT.
 | ||
| Scheme 1 Hybrid powder photocatalyst of the Ru(II) binuclear complex adsorbed on Ag-modified TaON (RuRu/Ag/TaON). | ||
Along with the Z-scheme hybrid photocatalysts, another powder hybrid photocatalyst consisting of a mononuclear metal complex as the CAT and a semiconductor such as carbon nitride16–18 or nitrogen-doped Ta2O5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19,20 working as a PS have been developed for use in CO2 reduction.
19,20 working as a PS have been developed for use in CO2 reduction.
However, these hybrid photocatalysts were investigated only in organic solutions; we do not have any information on their photocatalytic activity in water. In this work, the photocatalytic activity of the hybrid photocatalyst of Ag-modified TaON and the Ru(II) binuclear complex (RuRu/Ag/TaON, Scheme 1) was investigated for the first time in aqueous solutions containing electron donors, and we observed that RuRu/Ag/TaON photocatalyzed efficient CO2 reduction with high durability. This Z-schematic hybrid photocatalyst could also drive an uphill reaction, i.e. CO2 reduction with methanol as a reductant, in a water–methanol mixed solution.
Results and discussion
A hybrid photocatalyst of Ag-modified TaON and a Ru(II) binuclear complex RuRu/Ag/TaON was synthesized according to a reported method.14 Typically, the loaded amount of silver and RuRu were 1.5 wt% and 3 μmol g−1, respectively, except for the experiment corresponding to Fig. 5. The obtained materials were characterized by diffuse reflectance spectroscopy (DRS), X-ray diffraction (XRD), emission spectroscopy and Fourier-transform infrared (FT-IR) spectroscopy, as shown in Fig. 1 and S1–S3, ESI.† The XRD patterns of TaON, Ag/TaON and RuRu/Ag/TaON confirm that the crystal structure of TaON was not changed during the attachment procedures of silver and RuRu on TaON (Fig. S1a, ESI†). The typical diffraction peak at 2θ = 38.1° is attributed to metallic silver; this peak appears in the spectra of Ag/TaON and RuRu/Ag/TaON (Fig. S1b, ESI†). Fig. 1 shows DRS spectra of the hybrids RuRu/Ag/TaON, Ag/TaON and TaON along with RuRu/Al2O3, which is a model of RuRu. A broad absorption band was observed in the cases of Ag/TaON and RuRu/Ag/TaON, which is due to surface plasmon resonance of the metallic silver particles on the surface of TaON. RuRu/Ag/TaON also exhibited an absorption attributable to the Ru(II) photosensitizer unit (Fig. 1 and S4, ESI†). A dispersion of RuRu/Ag/TaON in water showed emission with λem = 629 nm by photoexcitation at λex = 444 nm, which is attributable to phosphorescence from the triplet metal-to-ligand charge transfer (3MLCT) excited state of the Ru(II) PS unit as well as phosphorescence from RuRu dissolved in water (Fig. S2, ESI†). IR absorption bands corresponding to the CO stretching vibrations of the Ru(II) CAT unit were observed at 2061 and 1997 cm−1 in the FT-IR spectrum of RuRu/Ag/TaON (Fig. S3, ESI†). These spectroscopic results indicate that the structure of RuRu was maintained after adsorption on Ag/TaON.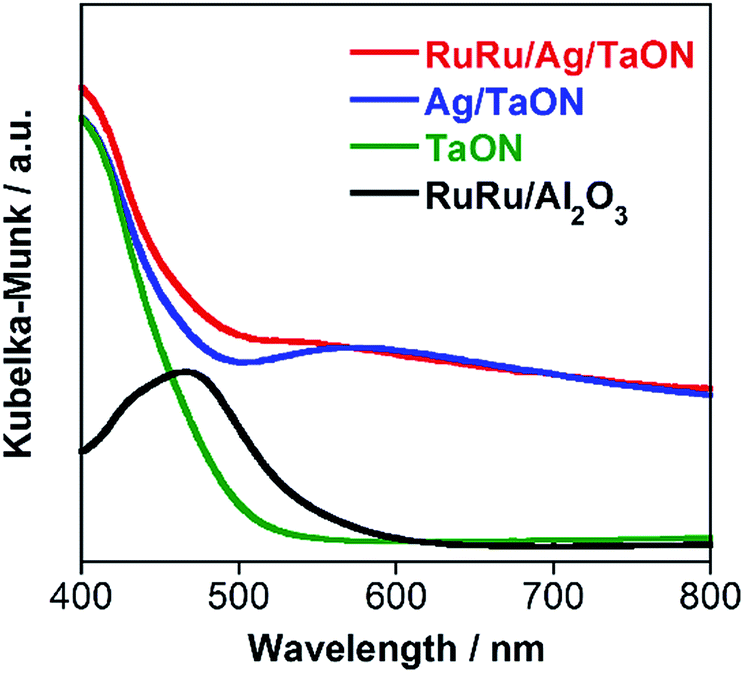 | ||
| Fig. 1 DRS of RuRu/Ag/TaON (red), Ag/TaON (blue), TaON (green) and RuRu/Al2O3 (black). | ||
As a typical run, a powder of RuRu/Ag/TaON (4 mg) was dispersed in aqueous solution (4 mL) containing ethylenediaminetetraacetic acid disodium salt (EDTA·2Na, 10 mM) and irradiated at λex > 400 nm under a CO2 atmosphere. After 24 h irradiation, formic acid, H2 and a small amount of CO were produced with turnover numbers (TON) of 750 (8.5 μmol), 1240 (14.2 μmol) and 30 (0.3 μmol), respectively (Fig. 2a). The external quantum yields (Φ) of the photocatalytic reaction were ΦHCOOH = 0.47% and ΦH2 = 0.54% using 400 nm monochromatic light. In contrast, formic acid was produced with much higher selectivity (85%) by addition of Na2CO3 (0.1 M) to the reaction solution (Fig. 2b), although TONHCOOH (620) and ΦHCOOH (0.23%) were lower than those in the absence of Na2CO3. Details of this difference are described in a later part of this paper.
 | ||
| Fig. 2 Time courses of HCOOH (red), H2 (blue) and CO (green) formation by visible-light (λ > 400 nm) irradiation to RuRu/Ag/TaON (4 mg) in EDTA·2Na (10 mM) aqueous solution (4 mL) without (a) and with (b) Na2CO3 (0.1 M) under a CO2 atmosphere. | ||
The carbon source of HCOOH was confirmed by an isotope-labeling experiment. A red line in Fig. 3 shows the 1H NMR spectrum of the reaction solution after the photocatalytic reaction under the same condition as that described above, except using 13CO2 instead of ordinary CO2. A doublet attributed to H13COOH was mainly observed at 8.31 ppm (1JCH = 196 Hz), with a small singlet attributed to H12COOH. In contrast, only a singlet of H12COOH was observed for the photocatalysis under ordinary CO2 atmosphere (a blue line in Fig. 3). Based on the areas of these peaks, we calculated that 97% of HCOOH was formed by reduction of CO2 in the photocatalytic reaction. Notably, this value is comparable with the purity of the 13CO2 used (99%).
 | ||
| Fig. 3 1H NMR spectra of the aqueous reaction solutions (1 mL) containing RuRu/Ag/TaON (4 mg) and EDTA·2Na (10 mM), measured after 24 h irradiation at λex > 400 nm under 13CO2 (red) and 12CO2 (blue) atmospheres. | ||
Table 1 summarizes the results of the photocatalytic reactions using various hybrids in aqueous solution containing EDTA·2Na (10 mM). Irradiation to RuRu/Ag/Al2O3, where Al2O3 was used as an insulator instead of TaON, did not yield any reduction products (entry 2, Table 1). The oxidizing power of the excited photosensitizer unit in RuRu was evaluated by emission measurements using EDTA·2Na as a quencher (Fig. S5, ESI†). Only 7% of the emission from the 3MLCT excited state of the PS unit of RuRu on the surface of Al2O3 was quenched by 10 mM of EDTA·2Na. These results suggest that EDTA·2Na mainly supplies electrons to the Ag/TaON unit in the photocatalytic reaction using RuRu/Ag/TaON. After the photocatalytic reaction using RuRu/Ag/TaON, we could not observe N2 generation by gas chromatography. Furthermore, there were no differences in either the binding energy for the Ta4p peak or the ratio of areas for Ta4p and N1s of TaON in RuRu/Ag/TaON before and after the photocatalytic reaction by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S6, ESI†). These observations indicate that the TaON unit in RuRu/Ag/TaON did not decompose during the photocatalytic reaction, which occasionally becomes a problem in some photocatalytic systems because it consumes photo-generated holes by the decomposition of TaON itself (eqn (1)).21–25
| 2N3− + 6h+ → N2 | (1) |
| Entry | Photocatalyst | Product/μmol (TON) | ||
|---|---|---|---|---|
| HCOOH | CO | H2 | ||
| a Dispersion of a photocatalyst (4 mg) in an EDTA·2Na (10 mM) aqueous solution (4 mL) was irradiated at λex > 400 nm for 15 h. b Ru(Cat) = cis-Ru{4,4′-(CH2PO3H2)2-2,2′-bipyridine}(CO)2Cl2. c Ru(PS) = [Ru(dmb)2{4,4′-(CH2PO3H2)2-2,2′-bipyridine}](PF6)2. | ||||
| 1 | RuRu/Ag/TaON | 7.0 (600) | 0.3 (28) | 11.4 (978) |
| 2 | RuRu/Ag/Al2O3 | N.D. | N.D. | N.D. |
| 3 | Ag/TaON | N.D. | N.D. | 0.4 (—) |
| 4 | RuRu/TaON | 1.2 (103) | 0.2 (16) | 5.0 (420) |
| 5 | Ru(Cat) /Ag/TaON | <0.1 (—) | N.D. | <0.1 (—) |
| 6 | Ru(PS) /Ag/TaON | <0.1 (—) | N.D. | 4.2 (371) |
Silver particles have been reported to act as a co-catalyst for CO2 reduction on some semiconductor photocatalysts which require irradiation of UV light.26–34 However, Ag/TaON without RuRu did not photocatalyze CO2 reduction at all (entry 3 in Table 1), indicating that the silver particles of RuRu/Ag/TaON did not work as a co-catalyst for CO2 reduction. However, loading silver to the surface of TaON dramatically enhanced the photocatalytic activity of RuRu/Ag/TaON, particularly for CO2 reduction (compare entries 1 and 4, Table 1). It has been reported that loading of Ag on the surface of a hybrid photocatalyst RuRu/CaTaO2N enhances the photoinduced electron transfer from the conduction band of CaTaO2N to the excited states of the Ru photosensitizer unit.15 A similar phenomenon should accelerate the photocatalytic ability of RuRu/Ag/TaON in the present system.
Use of the mononuclear model complex of the CAT unit (Ru(Cat)) instead of RuRu drastically lowered the photocatalytic activity of the hybrid (entry 5, Table 1). This is reasonable because the electron transfer from the conduction band of TaON (ECBM = −1.31 V)14 to Ru(Cat) (Eredp = −1.46 V vs. Ag/AgCl at pH 7)14 is an endergonic reaction. A hybrid without the catalyst unit (Ru(PS)/Ag/TaON), i.e. a mononuclear model complex of the PS unit (Ru(PS)) adsorbed on Ag/TaON, produced a catalytic amount of H2 with a very small amount of HCOOH (entry 6, Table 1). There have been some reports that [Ru(N^N)3]2+-type complexes decompose via photoinduced-ligand-substitution reactions to produce [Ru(N^N)2(X)(Y)]]n+-type complexes,35,36 and the product [Ru(N^N)2(X)(Y)]]n+ acts as a catalyst for both H2 evolution and CO2 reduction with the residual [Ru(N^N)3]2+ as the photosensitizer.10,11,37 From these control experiments and the emission quenching measurements, we can conclude that all of the units in the hybrid photocatalyst RuRu/Ag/TaON are necessary for the efficient photocatalytic reduction of CO2. RuRu/Ag/TaON worked via the Z-schematic electron-transfer mechanism from EDTA·2Na to the Ru catalyst unit with visible-light photoexcitation of both TaON and the Ru photosensitizer unit with the assistance of the Ag particles on the surface of TaON, followed by the CO2 reduction proceeding on the Ru catalyst unit, as shown in Scheme 1.
The effects of coexistent ions and the pH of the reaction solution on the photocatalytic activity were examined in detail with a series of additional salts to the reaction solution. Table 2 summarizes the results using EDTA·2Na (10 mM) as an electron donor, including the produced amounts of the reduction products, the selectivity of CO2 reduction (selCO2) and the desorption ratios of the surface-bound RuRu (ηdes). Addition of Na2CO3 (entry 2 in Table 2), K2CO3 (entry 3) and Na2HPO4 (entry 4), which changed the pH of the reaction solution to between 6.5 and 7.0, dramatically improved the selectivity of CO2 reduction. On the other hand, the change in ion strength of the reaction solution should not be a reason for this change in selectivity because the selectivity did not change in reaction solutions containing various concentrations of NaH2PO4 (34–35%, pH = 4.4, entries 5–7), where the pH was similar to that without the salts (37%, pH = 4.3, entry 1). Fig. 4a (plots of entries 1–8 and 11) exhibit clear trend that higher pH increased the selectivity of CO2 reduction unrelated to the ion strength of the solution; a more basic solution suppresses the evolution of H2, probably because of the lower proton concentration in the reaction solution.
| Entry | Salta | pHb | Product/μmol (TON) | selCO2c/% | η des/% | ||
|---|---|---|---|---|---|---|---|
| HCOOH | CO | H2 | |||||
| a Concentration was 0.1 M except for entries 6–8. b After purging with CO2 for 20 min. c Selectivity of CO2 reduction. d Concentration was 0.03 M. e Concentration was 0.01 M. f Concentration was 0.05 M. g Using Ag/TaON (4 mg) and Ru(bpy)2(CH3bpyCH2CH2bpyCH3)Ru(CO)2Cl2 (12 nmol). h Adsorption amount of RuRu was 1.0 μmol g−1. i Using ethylenediaminetetraacetic acid tetrasodium salt (EDTA·4Na, 10 mM) instead of EDTA·2Na. | |||||||
| 1 | None | 4.3 | 7.0 (600) | 0.3 (28) | 11.4 (978) | 37 | 17 |
| 2 | Na2CO3 | 7.0 | 4.0 (340) | N.D. | 0.7 (60) | 85 | 58 |
| 3 | K2CO3 | 7.0 | 3.7 (307) | N.D. | 0.9 (74) | 81 | 60 |
| 4 | Na2HPO4 | 6.5 | 5.6 (482) | <0.1 | 2.0 (172) | 74 | 58 |
| 5 | NaH2PO4 | 4.4 | 3.2 (257) | <0.1 | 6.1 (481) | 35 | 52 |
| 6 | NaH2PO4d | 4.4 | 3.9 (327) | <0.1 | 7.9 (658) | 34 | 37 |
| 7 | NaH2PO4e | 4.4 | 4.8 (421) | 0.2 (13) | 9.0 (791) | 34 | 32 |
| 8 | Na2HPO4f + NaH2PO4f | 6.1 | 4.8 (418) | <0.1 | 2.8 (245) | 63 | 53 |
| 9g | None | 4.3 | 0.7 (56) | N.D. | 0.5 (46) | 32 | — |
| 10h | None | 4.3 | 1.3 (350) | <0.1 | 2.9 (773) | 55 | — |
| 11i | None | 5.9 | 6.7 (589) | 0.1 (12) | 4.8 (418) | 58 | 26 |
 | ||
| Fig. 4 (a) Selectivity of CO2 reduction (selCO2) vs. pH of the reaction solution in the photocatalytic reactions. (b) Produced amount of HCOOH vs. desorption ratio of RuRu (ηdes) by the photocatalytic reactions with various concentration of NaH2PO4 (pH = 4.4). | ||
The produced amounts of HCOOH were lowered by the addition of the salts (0.1 M), regardless of the solution pH (entries 2–5). The UV-vis absorption spectra of the filtrates of the reaction solutions after the photocatalytic reactions exhibit an absorption band attributed to RuRu (Fig. S7, ESI†), indicating that RuRu partially desorbed from RuRu/Ag/Al2O3 during the photocatalytic reaction. Ru(II) diimine complexes with phosphonic acid anchor groups have been widely used as a photosensitizer in various photocatalytic systems14–19,38–41 and dye-sensitized photoelectrochemical cells.42–50 It was reported that in many cases, desorption of Ru complexes from the surface of metal oxides proceeded under visible-light irradiation in aqueous solution.51–54 The ηdess were 52–60% in the presence of the salts (0.1 M; entries 2–5), which were three times larger than those in the absence of the salts (entry 1). Higher concentration of salts in the reaction solution induced higher ηdes and lower TON (Fig. 4b), while lower concentration of salts suppressed the desorption of the metal complex and deactivation of the photocatalytic reaction (entries 1 and 6–8 in Table 2 and Fig. 4b). On the other hand, the pH of the solution and the type of added salts did not strongly affect ηdes (entries 2–5). A mixed system of Ag/TaON (4 mg) and a Ru(II) binuclear complex without the methyl phosphonate anchoring groups (12 nmol) showed much lower photocatalytic abilities (compare entry 1 and entry 9). Therefore, the addition of salts accelerated the desorption of RuRu, lowering the photocatalytic activity of RuRu/Ag/TaON. This is also supported by the following experimental data: the use of RuRu/Ag/TaON with a smaller amount of RuRu (1.0 μmol g−1) produced much smaller amounts of HCOOH and H2 (1.3 and 2.9 μmol, entry 10) compared with RuRu/Ag/TaON with 3.0 μmol g−1 of RuRu (7.0 μmol of HCOOH and 11.4 μmol of H2, entry 1). The details of the effects of the adsorbed amount of RuRu on the activity are described later. Taking into account these effects of pH and concentration of additives, higher selective HCOOH formation (58% selectivity) was obtained when ethylenediaminetetraacetic acid tetrasodium salt (EDTA·4Na, pH = 5.9; entry 11) was used instead of EDTA·2Na (pH = 4.3; entry 1) keeping high TON of 589 for HCOOH formation.
Fig. 5 shows the external quantum efficiencies for photocatalytic HCOOH production (ΦHCOOH) using RuRu/Ag/TaON with various loading amounts of RuRu. The ΦHCOOH increased with increasing loading amount of RuRu from 1.0 to 3.0 μmol g−1 and then reached plateau with the maximum values of ΦHCOOH = 0.48% at 8.3 μmol g−1. This is probably why the separation of the electron–hole pairs in TaON was accelerated because of the electron transfer from the conduction band to RuRu. The loading amount of 3.0 μmol g−1 might be sufficient to produce this effect. Notably, ΦHCOOH is the highest value obtained for photocatalytic CO2 reduction using semiconductor–photosensitizer–catalyst triad systems to date.
 | ||
| Fig. 5 Relationship between ΦHCOOH and loading amount of RuRu in the photocatalytic reaction using RuRu/Ag/TaON (30 mg) and EDTA·2Na (10 mM) in aqueous solution (10 mL) with 400 nm monochromatic light irradiation under a CO2 atmosphere. | ||
We have already reported that RuRu/Ag/TaON can use methanol as a reductant for CO2 reduction in pure methanol.14 This is important because CO2 reduction with methanol oxidation producing HCOOH as a reduced product of CO2 and HCHO as an oxidized product of methanol (eqn (2)) is an endergonic reaction (ΔG0 = +83 kJ mol−1); in other words, the visible-light energy is converted into chemical energy via the photocatalytic CO2 reduction reaction. As the next step, in this study, we investigated whether the same endergonic CO2 conversion reaction can proceed even in aqueous solution. Fig. 6 shows a time course of the TONs of both reduction products (HCOOH and H2) and an oxidation product (formaldehyde) in a photocatalytic reaction using RuRu/Ag/TaON in a H2O–MeOH mixed solution (4:1 v/v) without any other reductants. HCOOH and H2 were produced continuously and TONHCOOH reached 17 at 3 h of irradiation. Formaldehyde was also formed, whose produced amount corresponded to the total of HCOOH and H2 (Fig. 6 inset). This indicates that the overall reaction of the CO2 reduction can be represented in eqn (2).
| CO2 + CH3OH → HCOOH + HCHO | (2) |
 | ||
| Fig. 6 Time courses of HCOOH (red), H2 (blue) and HCHO (green) formation along with the sum of HCOOH and H2 (black broken line) in the photocatalytic reaction: RuRu/Ag/TaON (4 mg) in a H2O–MeOH (4:1 v/v) mixed solution (4 mL) was irradiated by visible light (λ > 400 nm) under a CO2 atmosphere. Inset shows enlarged time courses until 4 h irradiation. | ||
However, further irradiation induced less production of formaldehyde than the sum of HCOOH and H2 (Fig. 6). We employed a 13CO2 labelling experiment to clarify the carbon sources of HCOOH. Fig. 7a shows the 1H NMR spectrum of the filtered reaction solution after irradiation for 48 h; a doublet signal with 1JCH = 204 Hz and a singlet at 8.21 ppm are attributed to the methine protons of H13COOH and H12COOH, respectively. From this spectrum, we estimated that the main carbon source of HCOOH was CO2 (86%), although there were other carbon sources (14%). To gather information on the other carbon sources, a similar photocatalytic reaction was conducted using 2-propanol (i-PrOH) instead of methanol. This photocatalytic system also yielded HCOOH with TONHCOOH = 58 after 15 h of irradiation but did not give any HCHO. Fig. 7b shows the result of a 13CO2 labelling experiment using i-PrOH as the reductant; the 1H NMR spectrum of the filtered reaction solution after 48 h irradiation exhibits that the HCOOH was completely produced from CO2. Therefore, when methanol was used as the reductant, partial HCOOH produced in the photocatalytic reduction was probably generated by further oxidation of HCHO, which was produced by oxidation of the methanol. This is also supported by the following result: the photocatalytic oxidation of MeOH using TaON as a photocatalyst and AgNO3 as a sacrificial oxidant in aqueous solution containing MeOH yielded not only HCHO as a main product but also HCOOH as a minor one (Fig. S8, ESI†). This minor formation process of HCOOH should contribute to determining the product distribution after a certain amount of HCHO was generated in the reaction solution. As described above, the ‘mismatch’ between the amount of HCHO and the total amount of HCOOH and H2 was initially observed after a 6 h irradiation, and a longer irradiation increased this mismatch.
 | ||
| Fig. 7
1H NMR spectra of reaction solutions (2 mL) containing RuRu/Ag/TaON (8 mg) in (a) H2O–MeOH (4:1 v/v) and (b) H2O–iPrOH (4:1 v/v) after a 48 h irradiation with visible light (λ > 400 nm) under 13CO2 (red) and 12CO2 (blue) atmospheres. | ||
Experiments
General procedures
UV-vis absorption spectra were measured with a JASCO V-565 spectrophotometer. X-ray diffraction was measured with a Rigaku MiniFlex 600. FT-IR spectra were measured at 1 cm−1 resolution with a JASCO FT/IR-610 spectrophotometer. Emission spectra were measured at 298 ± 0.1 K with a JASCO FP-6500 spectrofluorometer. Emission lifetimes were measured with a Horiba FluoroCube 1000U-S time-correlated single-photon-counting system (the excitation source was a nano-LED 440L, and the instrument response was less than 1 ns).Materials
RuRu/Ag/TaON was synthesized according to a literature procedure.14 Briefly, an AgNO3 (137 μM) aqueous solution (10 mL) was added dropwise to a dispersion (100 mg) of TaON in water (10 mL), followed by stirring for 2 h. Then the suspension was evaporated and the residue was heated at 473 K for 1 h under a H2 atmosphere to obtain 1.5 wt% Ag-modified TaON (Ag/TaON). Then, a moderate amount of Ag/TaON was soaked in an acetonitrile solution of the Ru(II) binuclear complex (RuRu) for 3 h to obtain RuRu/Ag/TaON. The adsorption amount was estimated by the UV-vis absorbance changes of the solution before and after soaking (Fig. S4, ESI† shows an example of a RuRu adsorbed sample with a loading amount of 3 μmol g−1).Ag/Al2O3 and Ag/TiO2 were prepared by the same impregnation–hydrogenation method followed by adsorption of RuRu as RuRu/Ag/TaON for Al2O3 (AEROXIDE Alu C, AEROSIL) and TiO2 (AEROXIDE TiO2 P25, AEROSIL), respectively.
Tap water was purified using a Millipore Elix Essential 3 UV system and used on the same day. Methanol was used after distillation. Absolute 2-propanol was purchased from Kanto Chemical Co., Inc. and used without purification. Other materials were reagent-grade quality and were used without further purification.
Photocatalytic reactions
A suspension of photocatalyst (4 mg) in a reaction solution (4 mL) was prepared in an 8 mL test tube (i.d. = 8 mm) and purged with CO2. The suspensions were irradiated by stirring using a photocatalytic reactor (Koike Precision Instruments) at λ > 400 nm with a high-pressure Hg lamp combined with a NaNO2 aqueous solution filter. The temperatures of the solutions were controlled at 298 ± 2 K using an EYELA constant temperature system (CTP-1000) during irradiation. The quantum yield for HCOOH and H2 formation was evaluated in a reaction cell containing RuRu/Ag/TaON (30 mg) in a reaction solution (10 mL), which was irradiated with 400 nm monochromatic light using a 300 W Xe-lamp (Asahi Spectrum MAX-303) with a band pass filter (fwhm = 10 nm). The gaseous reaction products, i.e. CO and H2, were analyzed by a GC-TCD (GL Science GC 323). HCOOH in the liquid phase was analyzed by a capillary electrophoresis system (Otsuka Electronics Co. Capi-3300I). HCHO was quantitated by a colorimetric analysis following a reported procedure.14We evaluated the photocatalytic activity of the hybrids by using turnover number (TON, eqn (3)), selectivity (eqn (4)) and external quantum efficiency (Φ, eqn (5)).
 | (3) |
 | (4) |
 | (5) |
13CO2 labelling experiments
13CO2 labelling experiments in EDTA·2Na (10 mM) aqueous solution were performed using a dispersion of RuRu/Ag/TaON (4 mg) in aqueous solution (1 mL) containing EDTA·2Na (10 mM) in a reaction cell. The cell was degassed using the freeze–pump–thaw method, and then 13CO2 (99%, 703 mmHg) was introduced into it. For the photocatalytic system in H2O–MeOH mixed solution, a suspension of RuRu/Ag/TaON (8 mg) in a H2O–MeOH (2 mL, 4:1 v/v) mixed solution in an 8 mL test tube was purged with 13CO2 (99%) for 20 min. The suspensions were irradiated using a photocatalytic reactor (Koike Precision Instruments) at λ > 400 nm with a high-pressure Hg lump combined with a NaNO2 aqueous solution filter. After photolysis, the reaction solution was analyzed by 1H NMR by using a JEOL ECA400II (400 MHz) system with a No-D technique following filtration.
Conclusions
A hybrid of a supramolecular photocatalyst with both Ru(II) photosensitizer and catalyst units, and Ag-loaded TaON photocatalyzed CO2 reduction, even in aqueous solution; step-by-step photoexcitation of the Ru(II) photosensitizer unit and TaON could induce both strong reducing and oxidizing power in the hybrid photocatalyst, and relatively efficient CO2 reduction giving HCOOH proceeded with high durability in aqueous solution containing EDTA·2Na as an electron donor. This Z-scheme-type hybrid photocatalyst could also induce reduction of CO2 with methanol as the reductant giving HCOOH and HCHO even in aqueous solution, where the visible-light energy was converted into chemical energy (ΔG0 = +83 kJ mol−1).Acknowledgements
This work was supported by CREST/JST and a Grant-in-Aid for Scientific Research on Innovative Areas “Artificial photosynthesis (AnApple)” (No. 24107005) from the Japan Society for the Promotion of Science (JSPS). K. M. acknowledges the Noguchi Institute for financial support. The authors also thank AEROSIL for supplying TiO2 and Al2O3 materials.References
- Y. Tamaki, K. Koike and O. Ishitani, Chem. Sci., 2015, 6, 7213–7221 RSC.
- E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003–3012 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109–114 CrossRef CAS.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454–461 CAS.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800–1807 CrossRef CAS PubMed.
- A. Nakada, K. Koike, K. Maeda and O. Ishitani, Green Chem., 2016, 18, 139–143 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, Phys. Chem. Chem. Phys., 2013, 15, 10537–10548 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092–13097 CAS.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698–1699 RSC.
- M. Hara, G. Hitoki, T. Takata, J. N. Kondo, H. Kobayashi and K. Domen, Catal. Today, 2003, 78, 555–560 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, Energy Environ. Sci., 2011, 4, 4138–4147 CAS.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS PubMed.
- K. Maeda, R. Abe and K. Domen, J. Phys. Chem. C, 2011, 115, 3057–3064 CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 CAS.
- K. Teramura, H. Tatsumi, Z. Wang, S. Hosokawa and T. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 431–437 CrossRef CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2015, 163, 241–247 CrossRef CAS.
- T. Takayama, K. Tanabe, K. Saito, A. Iwase and A. Kudo, Phys. Chem. Chem. Phys., 2014, 16, 24417–24422 RSC.
- T. Takayama, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2015, 88, 538–543 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- N. Yamamoto, T. Yoshida, S. Yagi, Z. Like, T. Mizutani, S. Ogawa, H. Nameki and H. Yoshida, e-J. Surf. Sci. Nanotechnol., 2014, 12, 263–268 CrossRef CAS.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto, Y. Yamamoto, S. Yagi and H. Yoshida, J. Mater. Chem. A, 2015, 3, 16810–16816 CAS.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto and S. Yagi, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 359, 64–68 CrossRef CAS.
- P. E. Hoggard and G. B. Porter, J. Am. Chem. Soc., 1978, 100, 1457–1463 CrossRef CAS.
- J. Van Houten and R. J. Watts, Inorg. Chem., 1978, 17, 3381–3385 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157–173 CrossRef CAS.
- K. Maeda, M. Eguchi, S.-H. A. Lee, W. J. Youngblood, H. Hata and T. E. Mallouk, J. Phys. Chem. C, 2009, 113, 7962–7969 CAS.
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed.
- K. Maeda, G. Sahara, M. Eguchi and O. Ishitani, ACS Catal., 2015, 5, 1700–1707 CrossRef CAS.
- Y. Ueda, H. Takeda, T. Yui, K. Koike, Y. Goto, S. Inagaki and O. Ishitani, ChemSusChem, 2015, 8, 439–442 CrossRef CAS PubMed.
- B. H. Farnum, A. Nakada, O. Ishitani and T. J. Meyer, J. Phys. Chem. C, 2015, 119, 25180–25187 CAS.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- B. D. Sherman, D. L. Ashford, A. M. Lapides, M. V. Sheridan, K.-R. Wee and T. J. Meyer, J. Phys. Chem. Lett., 2015, 6, 3213–3217 CrossRef CAS.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- M. V. Sheridan, B. D. Sherman, Z. Fang, K.-R. Wee, M. K. Coggins and T. J. Meyer, ACS Catal., 2015, 5, 4404–4409 CrossRef CAS.
- L. Alibabaei, B. D. Sherman, M. R. Norris, M. K. Brennaman and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5899–5902 CrossRef CAS PubMed.
- F. Li, K. Fan, L. Wang, Q. Daniel, L. Duan and L. Sun, ACS Catal., 2015, 5, 3786–3790 CrossRef CAS.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347–2350 CrossRef CAS.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462–1469 CAS.
- E. Bae, W. Choi, J. Park, H. S. Shin, S. B. Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093–14101 CrossRef CAS.
- K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837–14847 CAS.
- K. Hanson, M. D. Losego, B. Kalanyan, G. N. Parsons and T. J. Meyer, Nano Lett., 2013, 13, 4802–4809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00586a |
| This journal is © The Royal Society of Chemistry 2016 |