
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpeditious synthesis of the fused hexacycle of puberuline C via a radical-based cyclization/translocation/cyclization process†
Koichi
Hagiwara
,
Toshiki
Tabuchi
,
Daisuke
Urabe
and
Masayuki
Inoue
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp; Fax: +81-3-5841-0568
First published on 18th March 2016
Abstract
The fused 6/7/5/6/6/6-hexacyclic ring system of puberuline C was assembled in 18 steps from 2-(ethoxycarbonyl)cyclohexanone. After the azabicyclo[3.3.1]nonane derivative was sequentially coupled with propargyl magnesium bromide, 2-iodo cyclopentenone and allyl bromide, the pentacycle was constructed in a single step via a radical-based cyclization/translocation/cyclization process. The C11-bridgehead radical generated via C–Br homolysis participated in a 7-endo cyclization, and the 1,5-hydrogen translocation of the resultant radical was followed by transannular 6-exo cyclization to simultaneously realize the construction of the two rings and the introduction of the five contiguous stereocenters. The last 6-exo cyclization was induced by the Mukaiyama aldol reaction, and the C16–ketone was stereoselectively reduced by the action of SmI2/t-BuOH, leading for the first time to the synthesis of the entire hexacycle of puberuline C.
Plants of the genera Aconitum and Delphinium have been used for centuries in traditional oriental medicines for their anti-inflammatory, analgesic, and anti-rheumatic activities.1 Efforts to identify the pharmacologically important compounds in these plant families have resulted in the determination of over 600 structurally complex C19–diterpenoid alkaloids, of which aconitine is a representative example (Scheme 1).2 In 2009, puberuline C was isolated from a traditional Chinese medical plant, Aconitum barbatum var. puberulum,3 and was found to belong to the C19–diterpenoid alkaloid family. Its architecturally complex hexacyclic system is composed of fused 6/7/5/6/6/6-membered (A/B/C/D/E/F) rings containing one nitrogen group and six oxygen-based polar functionalities. These structural features significantly increase the challenge of chemically synthesizing puberuline C.4
 | ||
| Scheme 1 Structures of aconitine and puberuline C, and a retrosynthetic plan of the model target compound 1. | ||
Puberuline C differs structurally from the majority of C19–diterpenoid alkaloids in its C17-bond connection. Specifically, the C8–17 bond of puberuline C constitutes the six-membered F-ring, while the C7–17 bond of aconitine forms a five-membered counterpart. To date, considerable synthetic effort has been focused on the aconitine-type alkaloids,5,6 culminating in the historic total syntheses of talatisamine, chasmanine and 13-desoxydelphonine by Wiesner's group,7 neofinaconitine by Gin's group,8 and weisaconitine D and liljestrandinine by Sarpong's group.9 In sharp contrast, the molecular framework of puberuline C has not been chemically assembled.10 In this manuscript, we describe the efficient synthesis of the unique hexacyclic ring system of puberuline C by utilizing a radical-based cyclization/translocation/cyclization process and the Mukaiyama aldol reaction as the two key transformations.
To devise a novel strategy for the total synthesis of puberuline C, we designed a simplified puberuline C (1) as a model target compound (Scheme 1). Although the oxidation states at the C1, 5, 6, 7 and 18 positions of 1 are different from those of puberuline C, 1 retains its entire hexacyclic framework and nine stereocenters (C4, 8, 9, 10, 11, 13, 14, 16, 17). In our retrosynthesis, the three ring structures of 1 were disconnected at the C10–11, C13–16 and C8–17 bonds to identify 2 as a pivotal intermediate. Compound 2 was proposed as the precursor of the bridgehead radical at C11. We presumed that the bridgehead radical would enable stereoselective construction of the sterically hindered C10–11 bond because of its potent reactivity, stereochemically predestined nature, and high orthogonality to diverse polar functional groups.11 In the synthetic direction, the C11-bridgehead radical generated through C–Br homolysis of triene 2 would chemoselectively react with the C9–10 double bond of the C-ring enone, leading to the formation of the seven-membered B-ring. After radical cyclization, the remaining C16–16′ and C7–8 double bonds were to be utilized to connect the C13/16 and C8/17 atoms to transannulate the six-membered D- and F-rings of 1, respectively, thereby establishing the stereochemistry of these four positions. As the radical precursor 2 has only three stereocenters (C4, 5, 11), the route to 2 would be greatly simplified. Highly unsaturated 2 would be prepared through a series of carbon chain extensions by step-wise attachment of three achiral units (five-membered C-ring A, B and C) to the chiral 6/6-fused AE-ring 3.
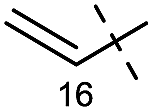
The synthesis of 1 commenced by preparing the known material 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 (Scheme 2). 2-(Ethoxycarbonyl)cyclohexanone (4) was first brominated to 5, which was then treated with ethylamine and formaldehyde to induce a double Mannich reaction, giving rise to 3 equipped with the C11–bromo group. Propargyl magnesium bromide C, which was prepared from 3-bromopropyne, magnesium turnings, and catalytic ZnBr2,13 then attacked the C5–ketone of 3 to afford the three carbon extended 6 as the major product (dr = 3.2:1). In this reaction, the tertiary amine of 3 would assist the stereoselective delivery of the organomagnesium reagent from the β-face of the molecule through chelation. Next, three Pd-catalyzed reactions from 6 completed the synthesis of the key intermediate 2. The five-membered C-ring A was attached to the terminal alkyne of 6 through a Sonogashira coupling14 [PdCl2(PPh3)2 and CuI], providing tricyclic compound 7. Upon treatment of 7 with PdCl2(PPh3)2 and n-Bu3SnH,15 the C8 position of the internal triple bond was regioselectively functionalized with the n-Bu3Sn group to form 8, presumably because the C8 position was less sterically shielded by the bulky azabicyclic AE-ring. Additionally, stereoselective hydrostannylation defined the syn-relationship of the AE- and C-rings, which later served as an important structural factor to facilitate 7-endo cyclization. The thus introduced C8–stannyl moiety of 8 was changed to the allyl group by π–allyl Stille coupling. Namely, Pd2(dba)3·CHCl3 and Ph3As16,17 effected the coupling between 8 and allyl bromide B to furnish triene 2. Therefore, only six steps were required for the conversion of monocycle 4 to tricycle 2, which possesses all the requisite carbons for the synthesis of the skeleton of 1. To prepare for the key radical cyclization, substrates 9 and 11a were derivatized from 2via chemoselective manipulations of the C8–allyl group. Dihydroxylation of the least sterically shielded olefin of triene 2 afforded triol 9 as a 1:1 C16–diastereomixture. Alternatively, dibenzyl acetal 11a was formed by oxidative glycol-cleavage with H5IO6, followed by In(OTf)3-catalyzed acetalization with benzyl alcohol.18
12 (Scheme 2). 2-(Ethoxycarbonyl)cyclohexanone (4) was first brominated to 5, which was then treated with ethylamine and formaldehyde to induce a double Mannich reaction, giving rise to 3 equipped with the C11–bromo group. Propargyl magnesium bromide C, which was prepared from 3-bromopropyne, magnesium turnings, and catalytic ZnBr2,13 then attacked the C5–ketone of 3 to afford the three carbon extended 6 as the major product (dr = 3.2:1). In this reaction, the tertiary amine of 3 would assist the stereoselective delivery of the organomagnesium reagent from the β-face of the molecule through chelation. Next, three Pd-catalyzed reactions from 6 completed the synthesis of the key intermediate 2. The five-membered C-ring A was attached to the terminal alkyne of 6 through a Sonogashira coupling14 [PdCl2(PPh3)2 and CuI], providing tricyclic compound 7. Upon treatment of 7 with PdCl2(PPh3)2 and n-Bu3SnH,15 the C8 position of the internal triple bond was regioselectively functionalized with the n-Bu3Sn group to form 8, presumably because the C8 position was less sterically shielded by the bulky azabicyclic AE-ring. Additionally, stereoselective hydrostannylation defined the syn-relationship of the AE- and C-rings, which later served as an important structural factor to facilitate 7-endo cyclization. The thus introduced C8–stannyl moiety of 8 was changed to the allyl group by π–allyl Stille coupling. Namely, Pd2(dba)3·CHCl3 and Ph3As16,17 effected the coupling between 8 and allyl bromide B to furnish triene 2. Therefore, only six steps were required for the conversion of monocycle 4 to tricycle 2, which possesses all the requisite carbons for the synthesis of the skeleton of 1. To prepare for the key radical cyclization, substrates 9 and 11a were derivatized from 2via chemoselective manipulations of the C8–allyl group. Dihydroxylation of the least sterically shielded olefin of triene 2 afforded triol 9 as a 1:1 C16–diastereomixture. Alternatively, dibenzyl acetal 11a was formed by oxidative glycol-cleavage with H5IO6, followed by In(OTf)3-catalyzed acetalization with benzyl alcohol.18
 | ||
| Scheme 2 Synthesis of the three substrates for the radical reaction. Reagents and conditions: (a) Br2, Et2O, 0 °C to RT; (b) aq. HCHO, aq. EtNH2, MeOH, 0 °C to RT; (c) C, ZnBr2 (5 mol%), CH2Cl2, −78 °C, 49% (dr at C5 = 3.2:1); recrystallization 29% for 6 (3 steps); (d) A, PdCl2(PPh3)2 (5 mol%), CuI (10 mol%), i-Pr2NEt, CH3CN, 0 °C, 65%; (e) n-Bu3SnH, PdCl2(PPh3)2 (5 mol%), THF; (f) B, Pd2(dba)3·CHCl3 (4 mol%), Ph3As (16 mol%), THF, 60 °C, 59% (2 steps); (g) OsO4 (10 mol%), NMO, THF, H2O, 81% (dr = 1:1); (h) H5IO6, THF, 0 °C, 72%; (i) BnOH, In(OTf)3, CH2Cl2, 0 °C, 79%. Bn = benzyl, dba = dibenzylideneacetone, NMO = N-methylmorpholine-N-oxide, THF = tetrahydrofuran. | ||
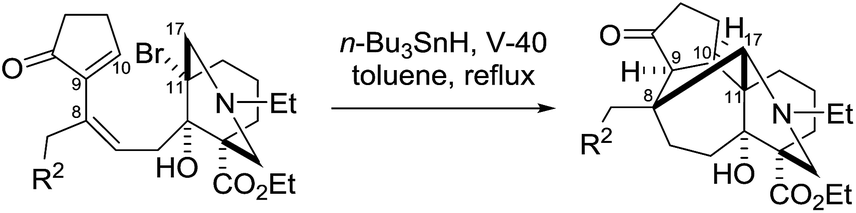
Remarkably, the single radical reaction of 2 promoted cyclization not only of the requisite seven-membered B-ring, but also of the six-membered F-ring (entry 1, Table 1). Treatment of tricycle 2 with n-Bu3SnH and V-40 in refluxing toluene provided pentacycle 12, with the formation of two C–C bonds (C10–11, C8–17) and five stereocenters (C8, 9, 10, 11, 17). The low yield (29%) of 12 from 2 was attributed to involvement of the C16–olefin in undesired radical pathways. Consequently, compounds 9 and 11a with no C16 radical acceptor were next submitted to the same reaction conditions (entries 2 and 3): the yields of the corresponding pentacyclic products 13 and 14a from 9 and 11a, respectively, almost doubled (55% for 13 and 54% for 14a).19,20 After the reaction in entry 2, 13 was obtained as C16–epimeric hemiacetals (13α:13β = 1:1.5) due to addition of the C16-secondary hydroxy group to the C14–ketone. The structure of crystalline C16-β-alcohol 13β was unambiguously determined by X-ray crystallographic analysis (Fig. 1), which revealed its unusually complicated shape. On the other hand, the newly formed ring systems of 12 and 14a were established by judicious NMR analyses and chemical derivatizations.21
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | R2 | Product | Yield |
|
a Conditions: substrate (1.0 equiv.), n-Bu3SnH (5 equiv.), V-40 (0.4 equiv.), toluene (0.02 M), reflux. n-Bu3SnH and V-40 (0.2 equiv.) in toluene were added by syringe pump over 3 h. Reactions were performed on a 0.10 mmol scale.
b
9 was used as a 1:1 C16–diastereomixture.
c Product 13 was obtained as the hemiacetal forms (13α:13β = 1:1.5). V-40 = 1,1′-azobis(cyclohexanecarbonitrile).
|
||||
| 1 | 2 |
|
12 | 29% |
| 2 | 9 |

|
13 | 55% |
| 3 | 11a |
|
14a | 54% |
 | ||
| Fig. 1 X-ray structure of C16-β-alcohol 13β. | ||
The present cascade pathway involves three essential radical reactions: 7-endo cyclization at C10 and C11, 1,5-radical translocation from C7 to C17, and 6-exo cyclization at C17 and C8. This intricate reaction course is illustrated in Scheme 3A using the transformation of 11a to 14a as an example. The bridgehead radical Int-1a is first generated from 11a by the action of the stannyl radical. To maximize the SOMO/LUMO interaction, the electron rich C11-radical of Int-1a selectively reacts with the electron deficient sp2-carbon atom at C10, thereby forming the C10–11 bond of Int-2Aa through a 7-endo cyclization. The C10-stereochemistry is controlled at this stage, while the configuration of the fixed C11-bridgehead position is retained. The vinylogous ketone moiety of Int-2Aa bestows an electron deficient character to the C7-radical, which preferentially reacts with the electron rich N-α C–H bond at the proximal C17 position via facile 1,5-hydrogen abstraction.22 Next, a transannular 6-exo cyclization of the translocated nucleophilic C17-radical of Int-3a with the electrophilic C8–9 double bond produces Int-4a. This intramolecular radical addition occurs within the fused rings to introduce the correct C8,17-stereogenic centers. Finally, the radical process is terminated by the hydrogenation of Int-4a from the convex face of the BC-ring to generate the C9-stereocenter of 14a.
 | ||
| Scheme 3 (A) Plausible reaction pathway for the radical cascade reaction. (B) Calculated energy diagram for the cascade reaction. The changes in Gibbs free energy from Int-1b were calculated at the UM06-2X/6-31G(d) level and are shown in kcal mol−1 (298 K, 1 atm, gas phase). (C) The pink dotted line represents the C10–11 bond being formed, and the cyan dotted line represents the C14O–C15H interaction in the TS-1A and TS-1B structures. | ||
The multiple radical reactions proposed above were evaluated by DFT calculations of the energy diagram at the UM06-2X/6-31G(d) level of theory (Scheme 3B).23 To facilitate the calculations, we used the structurally abbreviated radical intermediates, in which R2 and R3 were changed from CH(OBn)2 and Et to H and Me, respectively (see 11a and 11b). The calculated activation energy from Int-1b to Int-2Ab is smaller than that from the same Int-1b to the C10-epimeric Int-2Bb (ΔG‡ = +8.2 kcal mol−1 for TS-1A, +9.4 kcal mol−1 for TS-1B), supporting the observed C10-stereoselectivity of the 7-endo cyclization. The higher energy of TS-1B than TS-1A would originate from the close C14O–C15H contact in TS-1B (2.42 Å, Scheme 3C) within the sum of the van der Waals radii (2.72 Å) in comparison with that in TS-1A (2.68 Å). After formation of the stable delocalized radical Int-2Ab through the endothermic reaction (−23.2 kcal mol−1), 1,5-hydrogen abstraction (Int-2Ab → Int-3b) requires a relatively large activation energy (+19.5 kcal mol−1) and gives a less stable intermediate (+5.8 kcal mol−1). However, TS-3 is lower in energy than TS-2 by −6.6 kcal mol−1. Thus, the forward reaction from Int-3b to Int-4b is favored over the reverse reaction to Int-2Ab. Furthermore, 6-exo cyclization gives the thermodynamically preferred radical Int-4b, rather than Int-3b (−15.1 kcal mol−1). The gain in energy of the overall process from the starting radical Int-1b to the end radical Int-4b is −32.5 kcal mol−1, corroborating the high efficiency of the present cyclization/translocation/cyclization process.
Having constructed the five fused ring system with the seven stereocenters, the remaining tasks for the synthesis of the target 1 were the construction of the six-membered D-ring and the introduction of the C13,14 and 16 stereogenic centers (Scheme 4). We anticipated that an intramolecular aldol reaction between the C14–ketone and C16–aldehyde of 15a would stereoselectively form the C13–16 bond. The aldol substrate 15a was readily prepared from hemiacetal 13β through oxidative cleavage by H5IO6. However, treatment of 15a under acidic or basic conditions (e.g., aq. HCl/dioxane or DBU/benzene) resulted in either decomposition or recovery of 15a. These negative results were rationalized by DFT calculations of 15b and 16b, in which Et (R3) was replaced with Me. Although the reacting C13 and C16 atoms of 15b are in close proximity, C13–16 bond formation significantly increases the potential energy of 16b (+17.6 kcal mol−1). The bond angle (θ = 101.3°) between C14–13 and C13–16 of 16b deviates significantly from the ideal value, indicating its unusually strained character. Accordingly, the retro-aldol reaction from 16a to 15a would readily occur, even when 16a was produced.
 | ||
| Scheme 4 Attempted D-ring formation by the aldol reaction. The relative Gibbs free energy values were calculated at the UM06-2X/6-31G(d) level (298 K, 1 atm, gas phase). | ||
The above data and considerations led us to employ an irreversible Mukaiyama aldol reaction for cyclization of the strained D-ring (Scheme 5).24,25 To realize the transformation, an alternative aldol substrate 17 was designed to have the silyl enol ether as the nucleophile and the dibenzyl acetal as the activatable electrophile. The requisite TBS enol ether structure of 17 was regioselectively constructed by applying TBSOTf and Et3N to C14–ketone 14a. A number of Lewis acids (e.g., SnCl2, Sn(OTf)2, ZnBr2, BF3·OEt2, AlCl3 and TiCl4) activated the acetal moiety, yet failed to give the requisite product because the C14–oxygen atom instead of the C13 atom reacted with the C16–cation. Eventually, it was found that SnCl4 attained the requisite C13–16 bond formation. Treatment of 17 with SnCl4 in CH2Cl2 at −78 to 0 °C permitted effective construction of hexacycle 19, with installation of the C13 and 16-stereocenters. Therefore, capping the C16-hydroxy group with the benzyl group indeed inhibited the retro-aldol type reaction of adduct 19 under the reaction conditions. As illustrated by 18, the newly generated C16-stereochemistry of 19 would originate from nucleophilic attack of the silyl enol ether from the Si-face of the oxocarbenium ion, which would be fixed by the binding of SnCl4 between the oxygen and nitrogen atoms.
 | ||
| Scheme 5 Synthesis of the hexacyclic ring system of puberuline C. Reagents and conditions: (a) TBSOTf, Et3N, CH2Cl2, 0 °C, 90%; (b) SnCl4, CH2Cl2, −78 to 0 °C, 46%; (c) NaBH4, MeOH, 0 °C, 96%; (d) MeI, t-BuOK, THF, 0 °C, 93%; (e) H2, Pd/C, aq. HCl, MeOH, 95%; (f) AZADO (15 mol%), CuCl (15 mol%), 2,2′-bipyridine (15 mol%), DMAP (30 mol%), CH3CN, air, 82%; (g) NaBH4, MeOH, 0 °C, 50%; (h) SmI2, t-BuOH, HMPA, THF, 22:24 = 1:8, 75% for 24; (i) MeI, t-BuOK, THF, 0 °C, 93%. AZADO = 2-azaadamantane N-oxyl, HMPA = hexamethylphosphoric triamide, TBS = tert-butyldimethylsilyl, Tf = trifluoromethanesulfonyl. | ||
The synthesis of 1 was finalized by functional group manipulations at the C14 and C16 positions (Scheme 5). The C14–ketone of 19 was stereoselectively reduced from the convex face to provide the hydroxy group of 20. Alcohol 20 was in turn converted to methyl ether 21 using MeI and t-BuOK. Then, the C16-configuration was inverted by an oxidation/reduction sequence. After removal of the benzyl group from 21, the resultant hydroxy group of 22 was chemoselectively oxidized to the ketone within 23 in the presence of tertiary amine using the reagent combination AZADO/CuCl/2,2′-bipyridine/DMAP under air.26 However, the NaBH4 reduction of 23 only afforded its precursor 22: hydride selectively attacked from the convex face to generate the undesired β-oriented C16–alcohol. Conversely, SmI2, a one-electron reducing agent, produced the correct C16–epimer.27,28 When 23 was treated with SmI2 in THF/HMPA in the presence of t-BuOH, the α-oriented C16-alcohol 24 was obtained as the major product (22:24 = 1:8). It is noteworthy that the use of H2O or MeOH instead of t-BuOH decreased the stereoselectivity for 24 (22:24 = 1.5:1),29 indicating that the slower protonation of the C16-carbanion intermediate had a beneficial effect for generating 24. Lastly, methylation of the C16-hydroxy group of 24 provided the targeted puberuline C skeleton 1 with its ten contiguous stereocenters.21
In summary, we have accomplished the expeditious synthesis of the entire 6/7/5/6/6/6-membered ring system 1 of the C19-diterpenoid alkaloid puberuline C [18 steps from 2-(ethoxycarbonyl)cyclohexanone (4)]. The six ABCDEF rings and the ten stereogenic centers were constructed by a series of powerful transformations. First, the double Mannich reaction cyclized the AE-ring 3, with implementation of the two tetrasubstituted carbons (C4, 11). Second, carbon elongation from 3 to 2 was realized by nucleophilic addition of C and palladium-catalyzed couplings of A and B, introducing the C5-stereocenter and the C-ring structure. Third, the C11-bridgehead radical reaction of the highly unsaturated substrate 11a underwent a 7-endo cyclization/1,5-radical translocation/6-exo cyclization process to form the B- and F-rings, establishing the stereochemistries of the two quaternary (C8, 11) and three tertiary carbons (C9, 10, 17) in a single step. Fourth, the Mukaiyama aldol reaction, followed by hydride and one-electron reductions, constructed the D-ring and the C13,14,16-stereocenters. Importantly, detailed DFT-calculations fully validated the reaction course and the stereoselective outcome of the salient radical cascade reaction. Further synthetic studies of puberuline C based on the newly developed strategy are underway in our laboratory.
Acknowledgements
This research was financially supported by the Funding Program a Grant-in-Aid for Scientific Research (A) to M.I. (JSPS) and (C) (JSPS) and on Innovative Areas (MEXT) to D.U. A fellowship from JSPS to K.H. is gratefully acknowledged. We thank Dr Kenji Yoza (Bruker AXS) for X-ray crystallographic analysis.Notes and references
- F.-P. Wang and Q.-H. Chen, The C19-Diterpene Alkaloids, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Elsevier Science, New York, 2010, vol. 69, p. 1 Search PubMed.
- K. Wiesner, M. Götz, D. L. Simmons, L. R. Fowler, F. W. Bachelor, R. F. C. Brown and G. Büchi, Tetrahedron Lett., 1959, 1, 15 CrossRef.
- (a) L.-M. Sun, H.-L. Huang, W.-H. Li, Z.-D. Nan, G.-X. Zhao and C.-S. Yuan, Helv. Chim. Acta, 2009, 92, 1126 CrossRef . Khairitdinova reported the isolation of the same compound from Aconitum septentrionale K., and designated it as septontrionine. In this paper, we used puberuline C as the compound name; (b) E. D. Khairitdinova, E. M. Tsyrlina, L. V. Spirikhin, A. A. Balandina, S. K. Latypov and M. S. Yunusov, Russ. J. Org. Chem., 2008, 44, 536 CrossRef.
- For selected reviews on total synthesis of terpenoids, see: (a) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44 CrossRef; (b) R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2007, 72, 4293 CrossRef PubMed; (c) T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396 CrossRef PubMed; (d) D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207 CrossRef PubMed.
- For representative synthetic studies on aconitine-type alkaloids, see: (a) J. L. van der Baan, J. W. F. K. Barnick, G. van Beek and F. Bickelhaupt, Tetrahedron, 1992, 48, 2773 CrossRef; (b) G. A. Kraus and S. Kesavan, Tetrahedron Lett., 2005, 46, 1111 CrossRef; (c) D. F. Taber, J.-L. Liang, B. Chen and L. Cai, J. Org. Chem., 2005, 70, 8739 CrossRef PubMed; (d) R. M. Conrad and J. Du Bois, Org. Lett., 2007, 9, 5465 CrossRef PubMed; (e) H. Cheng, F.-H. Zeng, D. Ma, M.-L. Jiang, L. Xu and F.-P. Wang, Org. Lett., 2014, 16, 2299 CrossRef PubMed.
- For reviews on synthetic studies of the related diterpenoid alkaloids, see: (a) K. J. Goodall, D. Barker and M. A. Brimble, Synlett, 2005, 1809 Search PubMed; (b) E. C. Cherney and P. S. Baran, Isr. J. Chem., 2011, 51, 391 CrossRef PubMed; (c) A. M. Hamlin, J. K. Kisunzu and R. Sarpong, Org. Biomol. Chem., 2014, 12, 1846 RSC; (d) X.-Y. Liu and Y. Qin, Asian J. Org. Chem., 2015, 4, 1010 CrossRef; (e) G. Zhu, R. Liu and B. Liu, Synthesis, 2015, 47, 2691 CrossRef.
- Total synthesis of aconitine-type alkaloids from Wiesner's group. Talatisamine: (a) K. Wiesner, T. Y. R. Tsai, K. Huber, S. E. Bolton and R. Vlahov, J. Am. Chem. Soc., 1974, 96, 4990 CrossRef; (b) K. Wiesner, Pure Appl. Chem., 1975, 41, 93 CrossRef. Chasmanine: (c) S.-F. Lee, G. M. Sathe, W. W. Sy, P.-T. Ho and K. Wiesner, Can. J. Chem., 1976, 54, 1039 CrossRef; (d) T. Y. R. Tsai, C. S. J. Tsai, W. W. Sy, M. N. Shanbhag, W. C. Liu, S. F. Lee and K. Wiesner, Heterocycles, 1977, 7, 217 CrossRef; (e) K. Wiesner, T. Y. R. Tsai and K. P. Nambiar, Can. J. Chem., 1978, 56, 1451 CrossRef. 13–Desoxydelphonine: (f) K. Wiesner, Pure Appl. Chem., 1979, 51, 689 CrossRef.
- Y. Shi, J. T. Wilmot, L. U. Nordstrøm, D. S. Tan and D. Y. Gin, J. Am. Chem. Soc., 2013, 135, 14313 CrossRef PubMed.
- C. J. Marth, G. M. Gallego, J. C. Lee, T. P. Lebold, S. Kulyk, K. G. M. Kou, J. Qin, R. Lilien and R. Sarpong, Nature, 2015, 528, 493 CrossRef PubMed.
- For conversion of the naturally occurring aconitine-type alkaloids to the puberuline frameworks, see: (a) T. Amiya and T. Shima, Bull. Chem. Soc. Jpn., 1967, 40, 1957 CrossRef; (b) O. E. Edwards, Can. J. Chem., 1981, 59, 3039 CrossRef; (c) P. Tang, L. Wang, Q.-F. Chen, Q.-H. Chen, X.-X. Jian and F.-P. Wang, Tetrahedron, 2012, 68, 5031 CrossRef.
- α-Alkoxy bridgehead radicals were utilized for the intra- and intermolecular reactions in our laboratory: (a) D. Urabe, H. Yamaguchi and M. Inoue, Org. Lett., 2011, 13, 4778 CrossRef PubMed; (b) D. Urabe, M. Nagatomo, K. Hagiwara, K. Masuda and M. Inoue, Chem. Sci., 2013, 4, 1615 RSC; (c) K. Murai, S. Katoh, D. Urabe and M. Inoue, Chem. Sci., 2013, 4, 2364 RSC; (d) D. Kamimura, D. Urabe, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 5122 CrossRef CAS PubMed; (e) M. Nagatomo, M. Koshimizu, K. Masuda, T. Tabuchi, D. Urabe and M. Inoue, J. Am. Chem. Soc., 2014, 136, 5916 CrossRef CAS PubMed; (f) T. Asaba, Y. Katoh, D. Urabe and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 14457 CrossRef CAS PubMed; (g) M. Nagatomo, K. Hagiwara, K. Masuda, M. Koshimizu, T. Kawamata, Y. Matsui, D. Urabe and M. Inoue, Chem.–Eur. J., 2016, 22, 222 CrossRef CAS PubMed.
- G. A. Kraus and J. Shi, J. Org. Chem., 1991, 56, 4147 CrossRef CAS.
- H. P. Acharya, K. Miyoshi and Y. Kobayashi, Org. Lett., 2007, 9, 3535 CrossRef CAS PubMed.
- For a review, see: J. A. Marsden and M. M. Haley, Cross-Coupling Reactions to sp Carbon Atoms, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, vol. 1, p. 317 Search PubMed.
- J.-F. Betzer, F. Delaloge, B. Muller, A. Pancrazi and J. Prunet, J. Org. Chem., 1997, 62, 7768 CrossRef CAS.
- For a review, see: V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1 CrossRef CAS.
- V. Farina and B. Krishnan, J. Am. Chem. Soc., 1991, 113, 9585 CrossRef CAS.
- B. M. Smith and A. E. Graham, Tetrahedron Lett., 2006, 47, 9317 CrossRef CAS.
- For recent reviews on radical cascade reactions, see: (a) A. Baralle, A. Baroudi, M. Daniel, L. Fensterbank, J.-P. Goddard, E. Lacôte, M.-H. Larraufie, G. Maestri, M. Malacria and C. Ollivier, Radical Cascade Reactions, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons Ltd, Chichester, 2012, vol. 2, p. 729 Search PubMed; (b) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed; (c) J.-R. Chen, X.-Y. Yu and W.-J. Xiao, Synthesis, 2015, 47, 604 CrossRef CAS.
- For recent reviews on cascade reactions in natural product synthesis, see: (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed; (b) H. Pellissier, Chem. Rev., 2013, 113, 442 CrossRef CAS PubMed.
- For the structural determinations, see ESI.†.
- For selected reports on the radical translocation/cyclization process, see: (a) D. C. Lathbury, P. J. Parsons and I. Pinto, J. Chem. Soc., Chem. Commun., 1988, 81 RSC; (b) D. P. Curran, D. Kim, H. T. Liu and W. Shen, J. Am. Chem. Soc., 1988, 110, 5900 CrossRef CAS; (c) V. Snieckus, J. C. Cuevas, C. P. Sloan, H. Liu and D. P. Curran, J. Am. Chem. Soc., 1990, 112, 896 CrossRef CAS; (d) J. Robertson, M. A. Peplow and J. Pillai, Tetrahedron Lett., 1996, 37, 5825 CrossRef CAS; (e) F. Beaufils, F. Dénès, B. Becattini, P. Renaud and K. Schenk, Adv. Synth. Catal., 2005, 347, 1587 CrossRef CAS; (f) M. Fujitani, M. Tsuchiya, K. Okano, K. Takasu, M. Ihara and H. Tokuyama, Synlett, 2010, 822 CAS; (g) J. K. Vellucci and C. M. Beaudry, Org. Lett., 2015, 17, 4558 CrossRef CAS PubMed . For reviews, see: ; (h) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94 RSC; (i) F. Dénès, F. Beaufils and P. Renaud, Synlett, 2008, 2389 CrossRef.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- T. Mukaiyama, T. Izawa and K. Saigo, Chem. Lett., 1974, 323 CrossRef CAS.
- For a review on Mukaiyama aldol reaction in natural product synthesis, see: S. B. J. Kan, K. K.-H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097 CrossRef CAS PubMed.
- Y. Sasano, S. Nagasawa, M. Yamazaki, M. Shibuya, J. Park and Y. Iwabuchi, Angew. Chem., Int. Ed., 2014, 53, 3236 CrossRef CAS PubMed.
- P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693 CrossRef CAS.
- For a book, see: Organic Synthesis using Samarium Diiodide A Practical Guide, ed. D. J. Procter, R. A. Flowers II and T. Skrydstrup, RSC publishing, Cambridge, 2010 Search PubMed.
- For the effects of proton source on stereoselective reduction of ketones, see: (a) S. K. Upadhyay and S. Hoz, J. Org. Chem., 2011, 76, 1355 CrossRef CAS PubMed; (b) P. R. Chopade, E. Prasad and R. A. Flowers ll, J. Am. Chem. Soc., 2004, 126, 44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1452192. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00671j |
| This journal is © The Royal Society of Chemistry 2016 |