
High impact of the reducing agent on palladium nanomaterials: new insights from X-ray photoelectron spectroscopy and oxygen reduction reaction†
Yaovi Holade*,
Christine Canaff,
Suzie Poulin,
Têko W. Napporn*,
Karine Servat and
K. Boniface Kokoh
Université de Poitiers, UMR CNRS 7285, 4, rue Michel Brunet, B27, TSA 51106, 86073 Poitiers cedex 09, France. E-mail: yaovi.holade@univ-poitiers.fr; teko.napporn@univ-poitiers.fr; Fax: +33 549 45 39 67; Tel: +33 549 45 3580
First published on 25th January 2016
Abstract
Palladium has exceptional affinity with hydrogen and the evolution of the surface of its nanomaterials prepared from chemical methods over time is still unclear. Here, the reducing agent effect on Pd nanomaterials and their long-term chemical stability were scrutinized by X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). The subsequent impact on the catalytic properties was examined using the electrochemical oxygen reduction reaction (ORR). We have discovered that the nature of the reducing agent has noteworthy effects on the final composition of Pd nanomaterials prepared from chemical methods. The surface state of the nanomaterials prepared by using sodium borohydride as reducing agent (Pd/C–NaBH4) is radically different from those obtained from L-ascorbic acid (Pd/C–AA). In addition to pure metal, two oxides were identified: PdO and PdOx (x > 1). XRD analysis has upheld the presence of PdO only in Pd/C–NaBH4, thus underpinning the conclusion that NaBH4 has drastically changed the Pd structure. Furthermore, the reducing agent substantially affects the electrocatalytic properties. The ORR starts with enhanced kinetics (E > 1 V vs. RHE) by a 4-electron process, producing p(H2O2) < 0.5% associated with excellent durability over 5000 cycles. Both catalysts outperform all reported data for Pd electrocatalysts. The novelty of this work is combining ex/in situ XPS and XRD analyses together with ORR as a catalytic model. Overall, this work represents a clear development in our understanding of Pd affinity towards hydrogen and paves new ways for the successful synthesis of Pd-based nanomaterials free from hydrides and oxides, and having impressive catalytic activities.
1. Introduction
In 2010, L. Croft was reporting in Nature Chemistry, “Nobel prize 2010: Prestige for palladium” to emphasize and label the major role played by palladium-based catalysts in chemistry, highlighted by the Nobel prize committee.1 Indeed, palladium-based materials have been widely used in catalytic processes for several decades.1–4 For bulk materials, the impossibility to tune their activity and selectivity as well as the economic impact on the catalytic processes have oriented researches towards nanoscale materials. However, the physicochemical properties of a given material at either the bulk or nanoscale strongly depend on the nature of the different chemical species within the structure. Particularly in heterogeneous catalysis, a surface science, the performances in terms of activity, selectivity and long-term stability depend not only on parameters such as particle size and morphology but also on the surface state. To control effectively all these parameters, the last twenty years have been devoted to the development of efficient and advanced metal nanomaterials for application either in physics or in catalysis. To this end, various physical and chemical approaches have been initiated. The chemical methods for preparing metal nanoparticles use a suitable reducing agent or seed growth mediator. The common used reducing agent is the sodium borohydride (NaBH4), a strong reducing agent with fast kinetics. This approach has successfully used to prepare most of metal nanocrystals. In the case of Pd-based nanomaterials, the use of reducing agent such as NaBH4 or N2H4 that releases H2 in aqueous medium (hydrogen-rich reducer) is accompanied by hydrogen absorption. Indeed, it is well-known that Pd can absorb hydrogen in its lattice and forms PdHx hydrides.5–11 At bulk and nanoscale, this is accompanied by a change in the crystalline structure of the metal.5,7,11–14 To avoid this hydrogen absorption, L-ascorbic acid (AA) that does not releases H2 has been recommended.15–17 The peculiar affinity of Pd with hydrogen and the subsequent effect on the physical structure is well-documented.8,18–22 Indeed, Pd-based compounds have been earnestly deemed as alternative options for hydrogen storage devices. However, there is still missing information about the effect of the reducing agent on the final chemical composition and the surface properties of Pd-based nanomaterials.Furthermore, when a metal is exposed to ambient air, it intuitively undergoes a chemical oxidation process because of the oxygen in air. This leads to the formation of a very thin layer, so-called passivation layer, which protects the bulk metal from deep oxidation process. Depending of the metal, the chemical stability of the different oxide species is variable and unclear. The nature of the synthetic route, meaning the kind of reducing agent, can also has notable, if not significant effect on the final composition and properties of the obtained metal nanomaterials. Especially, in the case of Pd, the presence of hydride species PdHx could weaken the metallic bond Pd–Pd, hence affecting the electronic properties of the material and definitely subjects it to the chemical oxidation leading to various oxides. The most likely of these species is the palladium(II) oxide (PdO). Indeed, the stability of the superior oxides such as the palladium(IV) oxide (PdO2, palladium dioxide) or palladium(VI) oxide (PdO3, palladium trioxide) in their anhydrous form is unlikely, since they are unstable.23–27 Some authors, however, proposed that PdO2 particles could be stabilized by other oxide matrices such as Al2O3,24 SnO2,28 and PdO.29
Very recently, in situ X-ray diffraction (XRD) coupled with temperature desorption investigations have shown that Pd nanoparticles obtained by NaBH4 as reducing agent present larger lattice parameter.17 More importantly, the programmed thermal desorption process coupled with in situ XRD has demonstrated that the use of NaBH4 as reducing agent leads to PdHx species whereas AA gives exclusively Pd nanoparticles. This phenomenon had a noteworthy impact on the electrocatalytic properties of Pd nanomaterials towards carbon monoxide and glucose electrooxidation reactions in alkaline medium. Here, we aim to scrutinize their surface state by X-ray photoelectron spectroscopy (XPS) to determine whether the use of NaBH4 or AA induces a strong modification of the surface composition of Pd nanomaterials. Palladium-based nanomaterials are fabricated as alternatives for platinum in heterogeneous catalysis. As the latter is a surface science, the catalytic performances of nanomaterials in terms of activity and long-term stability are directly linked to the surface state. Then, to study their chemical stability at ambient temperature and pressure over time, we performed again XPS measurements on the same sample after 2 years of storage. Finally, the catalytic properties of the as-synthesized nanomaterials were examined by considering the electrochemical oxygen reduction reaction (ORR) in alkaline medium. Both XPS and ORR investigations have released new unexpected insights on the reduction agent impact on Pd nanomaterials surface state and electrocatalytic properties.
2. Experimental
2.1 Preparation of the palladium-based nanomaterials
Different kinds of Pd nanomaterials have been prepared using two different reducing agents: sodium borohydride (NaBH4, 99%) and L-ascorbic acid (AA, ≥99%) that were purchased from Sigma-Aldrich®. The preparation method is the “Bromide Anion Exchange, BAE”30,31 one, a soft and simple approach which does not required any organic molecule as surfactant or capping agent and enable the effective preparation of well-dispersed nanocrystals with a good chemical yield (>90%). Typically, 61.4 mg of potassium tetrachloropalladate(II) (K2PdCl4, 99%, purchased from Sigma-Aldrich®) were dissolved with 100 mL of ultrapure water in a reactor that is in a thermostatted bath at 25 °C. Then, 32.7 mg of potassium bromide (KBr, ≥99%, purchased from Sigma-Aldrich®) were added under vigorous stirring followed by 80 mg of Vulcan XC 72R carbon black (thermally pre-treated32) under constant ultrasonic homogenization for 45 min to target a metal loading of 20 wt%. Then, the reducing agent, NaBH4 (0.1 M, 15 mL) or AA (0.1 M, 13 mL) was added dropwise under vigorous stirring to synthesize the Pd/C–NaBH4 or Pd/C–AA nanomaterials, respectively. The mixture is kept at 40 °C for 2 h. Finally, Vulcan-supported Pd nanomaterials were filtered, rinsed several times with ultrapure water, and dried in an oven at 40 °C for 12 h. It should be noticed that 30 wt% Pt/C prepared in the same way and as detailed elsewhere32 was used as reference for ORR experiments.2.2 Physicochemical characterization of the nanomaterials
The total metal loading was determined from thermogravimetric analysis (TGA). Experiments were conducted on TA Instruments SDT Q600 apparatus by thermally heating ca. 5 mg of the sample (contained in an alumina crucible) under air flow of 100 mL min−1 from 25 to 900 °C following a 5 °C min−1 temperature rate. X-ray diffraction (XRD) measurements were performed using an ENPYREAN (PANanalytical) diffractometer in Bragg–Brentano (θ–θ) configuration with a copper tube powdered at 45 kV and 40 mA (Cukα1 = 1.54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 060 Å and Cukα2 = 1.54443 Å). The interference from the kβ component was suppressed thanks to a nickel filter, installed in a secondary optic. XRD data were collected from 15° to 140° in diffraction angle (in 2θ) in step mode, with steps of 0.05° and fixed acquisition time of 2 min per step. High-resolution transmission electron microscopy (HRTEM) observations were conducted on a TEM/STEM JEOL 2100 UHR (200 kV) microscope equipped with a LaB6 filament. The samples were prepared by dispersing nanomaterials in ethanol and then deposited onto copper grid.
060 Å and Cukα2 = 1.54443 Å). The interference from the kβ component was suppressed thanks to a nickel filter, installed in a secondary optic. XRD data were collected from 15° to 140° in diffraction angle (in 2θ) in step mode, with steps of 0.05° and fixed acquisition time of 2 min per step. High-resolution transmission electron microscopy (HRTEM) observations were conducted on a TEM/STEM JEOL 2100 UHR (200 kV) microscope equipped with a LaB6 filament. The samples were prepared by dispersing nanomaterials in ethanol and then deposited onto copper grid.
XPS was used to probe and characterize the surface and oxidation states of the nanomaterials. Analyses were performed in a high vacuum chamber (pressure ≤ 9 × 10−8 Pa) on a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic radiation source Al Mono (Alkα: 1486.6 eV) operating at 150 W (15 kV and 10 mA). Survey spectra were recorded with a step of 1 eV (transition energy: 160 eV). Based on the collected basic information, high-resolution XPS spectra were collected at a step of 0.1 eV (transition energy: 20 eV). XPS peaks were fitted using Gaussian–Lorentzian (GL) profile function and asymmetry determined from an etched bulk Pd metal. The Tougaard (U2T) background has been chosen. It should be noted that a systematic subtraction of the background noise preceded the normalization of the spectra and the measurement of binding energies (BE) was corrected based on the energy of C1s at 284.4 eV (Vulcan XC 72R carbon). The Fermi level was also probed. However, the XPS spectra presented herein use C1s as reference. Palladium oxide (PdO) from Alfa Aesar (anhydrous 99.9%, Ref. 11040) and palladium foil were used as references materials. Quantifications were done from the corresponding XPS peak area after correction with suitable sensitivity factors and the CASA XPS software was used for peak fitting.
2.3 In situ X-ray photoelectron spectroscopy analysis
The in situ XPS measurements were undertaken to remove the entire oxides at Pd surface before analyzing the sample and comparing it to a Pd foil. The sample was firstly introduced in a XPS chamber under a gas atmosphere composed of hydrogen and argon (H2 at 5 vol% in argon). Then, the targeted plateau temperature of 400 °C was reached by heating the sample at 5 °C min−1 temperature rate. After 1 h of operation at 400 °C, the furnace was cooled towards the room temperature. Then, a high vacuum (pressure ≤ 9 × 10−8 Pa) was applied before recording finally the XPS spectra as indicated above.2.4 Electrochemical measurements
The ORR experiments were carried out using a ring rotating disc electrode (RRDE) from “PINE Instrument Company” (AFMSRX modulated speed rotator mounted with AFDT22 electrodes) including a glassy carbon disc (0.196 cm2) and platinum ring (0.11 cm2). The tests were fulfilled in a conventional three-electrode cell using a bi-potentiostat (AUTOLAB PGSTAT302N, Netherlands). The electrolyte solution is 0.1 M potassium hydroxide (KOH, ACS reagent ≥85%, purchased from Sigma-Aldrich®). The mercury–mercury oxide (MMO) Hg|HgO|(KOH 0.1 M) from Radiometer analytical (number XR400) having E0 = 0.172 V vs. Standard Hydrogen Electrode (SHE) was used as reference electrode and is separated from the solution by a Haber–Luggin capillary tip. Here, the potentials were scaled versus the Reversible Hydrogen Electrode (RHE) using ERHE = −0.939 V vs. MMO. To prepare the catalytic ink, 180 μL of isopropanol (ACS reagent, ≥99.8%) and 20 μL of Nafion suspension (5 wt%) from Sigma-Aldrich® were ultrasonically mixed. Then, 2.4 mg of metal/C powder were added. Finally, 6 μL of the homogeneous catalytic ink were deposited onto the freshly polished glassy carbon disk, and the solvent was evaporated in a stream of ultrapure nitrogen (N2, Air Liquide, ultra-pure) at room temperature. The metal loading of the disc is 66 and 62 μgPd cm−2 for Pd/C–AA and Pd/C–NaBH4, respectively. A slab of glassy carbon (6.48 cm2 geometrical area) was used as counter electrode. During ORR experiments, the ring potential was held at 1.2 V vs. RHE and that of the disc was run at 5 mV s−1 from 1.1 to 0.35 V vs. RHE at different RRDE speeds. For cyclic voltammetry, the solution was completely deoxygenated with N2 for 30 min. For ORR polarization curves, the solution was saturated with pure oxygen (O2, Air Liquide, ultra-pure) for 30 min. The accelerated potential cycling test (APCT) under either N2 or O2 atmosphere is a fast way to evaluate the stability/durability of a catalyst.33–37 The APCT experiments were performed under O2 atmosphere by cycling the electrode potential at 100 mV s−1 within the potential range of 0.6–1 V vs. RHE for 5000 successive cycles. All the electrochemical measurements were conducted at 22 ± 2 °C.3. Results and discussion
3.1 HRTEM observations
Fig. 1a shows a typical TEM image of the pre-treated Vulcan XC 72R carbon.32 The particles size goes from 10 to 40 nm (Fig. 1b) with a mean particles size of 22 nm and quasi-spherical shape (see HRTEM in Fig. 1c). A mean value of 30 nm is currently used for the pristine Vulcan. As shown in ref. 32, this treatment enables increasing significantly the specific surface area (BET, Brunauer–Emmett–Teller) of Vulcan from 262 to 322 m2 g−1, meaning 23% gains. Corollary, a 2.2-fold increase of the electrochemical active surface area (ECSA) of Pt/C was also reported.32 Table 1 shows the concrete evidence of sulphur in Vulcan, up to 1.2 wt%. This value is comparable to that of other types of carbon black (1.3 wt%). The heat treatment has removed a fraction and the remaining sulphur might therefore be in another form impregnable by this method. Furthermore, there is an increase of nitrogen amount. This excludes a simple contamination from atmospheric air during the analysis since its amount has notably increased from 0.18 to 0.74 wt% for Ketjenblack EC-600JD carbon. The CHNS mass balance not being 100 wt%, we suppose that there are other species not detected by this method, likely oxygen since the existence of hydrogen is a tangible evidence. Therefore, the treatment (under nitrogen atmosphere) has eliminated some oxygenated groups (surface) as well as sulphur and introduced nitrogen. These findings partly explain the difference in BET and Raman spectroscopy32 analysis. Such structural modifications, however minimal, may have a significant effect on the catalytic properties, as discussed in ref. 32.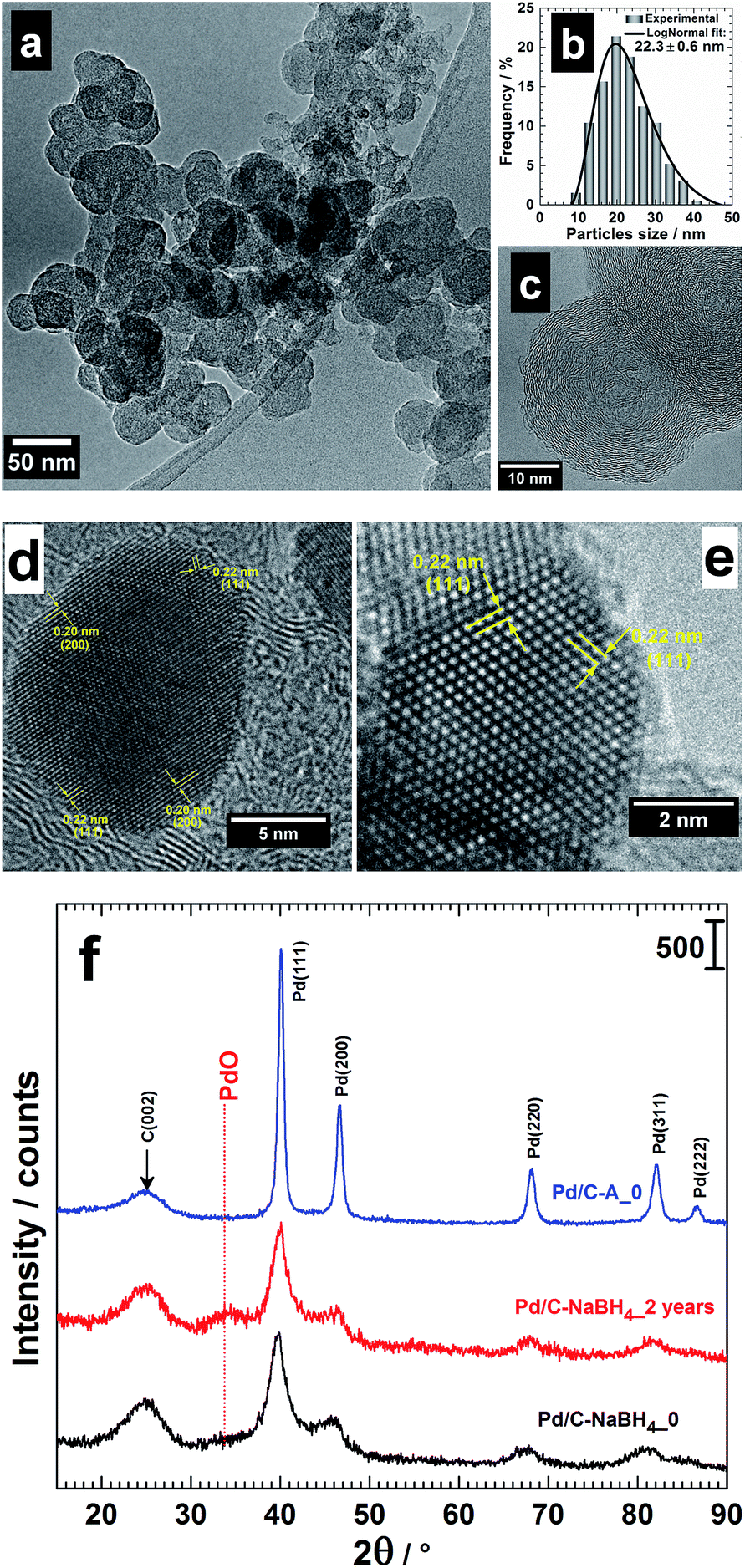 | ||
| Fig. 1 (a) TEM image, (b) particles size distribution (fitted using the log-normal function) and (c) HRTEM micrograph of Vulcan XC 72R carbon thermally treated at 400 °C under N2 atmosphere for 4 h. HRTEM micrographs of the as-prepared (d) Pd/C–AA and (e) Pd/C–NaBH4 and their corresponding low-resolution XRD patterns (f). The pattern of Pd/C–NaBH4_2 years is included in (c) to highlight the presence of PdO. | ||
| wt% (±0.01) | Pristine | Pre-treated |
|---|---|---|
| Carbon (C) | 86.87 | 96.69 |
| Hydrogen (H) | 0.12 | 0.02 |
| Nitrogen (N) | 0.83 | 1.17 |
| Sulphur (S) | 1.19 | 1.00 |
| Total | 89.01 | 98.88 |
The synthesized Pd nanomaterials using either AA (Pd/C–AA) or NaBH4 (Pd/C–NaBH4) as reducing agent have been firstly characterized by TEM.17 HRTEM images are shown in Fig. 1d and e. Pd/C–AA nanomaterials have an average particles size of 8 nm (Fig. 1d). The HRTEM micrograph of Pd/C–NaBH4 (an average particles size of 3 nm) is displayed in Fig. 1e with a zone axis of [101]. Overall, the nanoparticles tend to adopt a truncated octahedron shape with different degrees of truncation. The Fourier transform of these images enable obtaining the interplanar spaces d(hkl) (lattice spacing) for the corresponding crystallographic orientation of (hkl) facets. For Pd/C–AA, the lattice spacing d(hkl) = 0.20 and 0.22 nm can be assigned to the (200) and (111) planes of face-centred cubic (fcc) Pd. For Pd/C–NaBH4, only d(hkl) = 0.22 nm for (111) comes from Fig. 1e. Considering the supported nanoparticles, the formation of the facets (111) and (100) is thermodynamically more favourable since the surface energy (γ) associated with different crystallographic planes follows the order: γ(111) < γ(100) < γ(110).38 The polyhedron corresponding to the more thermodynamic stable morphology for a supported-nanoparticle with a fcc crystal symmetry is a truncated octahedron.38–40 It is composed of eight hexagonal faces and six square faces having (111) and (100) facets, respectively. Obtaining high Miller indices such as (200) instead of (100) in the case of Pd/C–AA nanomaterials suggests that the BAE synthesis method offers favourable thermodynamic conditions. It should be noticed that within the same material, different forms might coexist. Indeed, the presence of support such as Vulcan carbon does not enable controlling perfectly the final morphology of the nanocrystal. The difference in the particles size distribution and exposed crystallographic facets comes mostly from the nature of the reducing agent. It is well known that, in contrast to NaBH4, AA promotes the formation of large nanoparticles. Indeed, AA is a weak reducing agent, whereas NaBH4 has strong reduction abilities.16,17,41,42
3.2 XRD analysis of the nanomaterials
XRD analyses had been undertaken to determine whether another species (oxides) are formed when the reducing agent is changed. Then, the behavioural changes in terms of the chemical composition after 2 years of storage were investigated by XRD. The freshly prepared nanomaterials named as Pd/C–AA_0 and Pd/C–NaBH4_0 were first characterized (Fig. 1f). Both materials present a peak at approximately 25 °C, assigned to Vulcan XC 72R carbon, used as support of Pd nanoparticles.32 The others belong to Pd(111), Pd(200), Pd(220), Pd(311) and Pd(222). At this stage, all the palladium in both materials is metallic. But, a closer examination of Pd/C–NaBH4_0 diffractogram shows a very small shoulder towards 2θ = 34°. More importantly, its intensity increases notably on the sample after 2 years, Pd/C–NaBH4_2 years. The pattern of Pd/C–AA_2 years was unchanged. In other words, the amount of another palladium species rather than Pd metal has noteworthy increased only for Pd/C–NaBH4_2 years sample. It might be PdO since the stability of higher oxides such as PdO2 or PdO3 is unlikely in their anhydrous forms. To better elucidate that, high-resolution XRD of both aged samples was performed: Fig. 2b for Pd/C–AA_2 years and Fig. 2c for Pd/C–NaBH4_2 years; Fig. 2a being the XRD pattern of bulk Pd metal (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, PDF # 00-046-1043). In addition and as we suspected mostly PdO contribution on Pd/C–NaBH4_2 years sample, its XRD pattern was recorded (Fig. 2d).
m, PDF # 00-046-1043). In addition and as we suspected mostly PdO contribution on Pd/C–NaBH4_2 years sample, its XRD pattern was recorded (Fig. 2d).
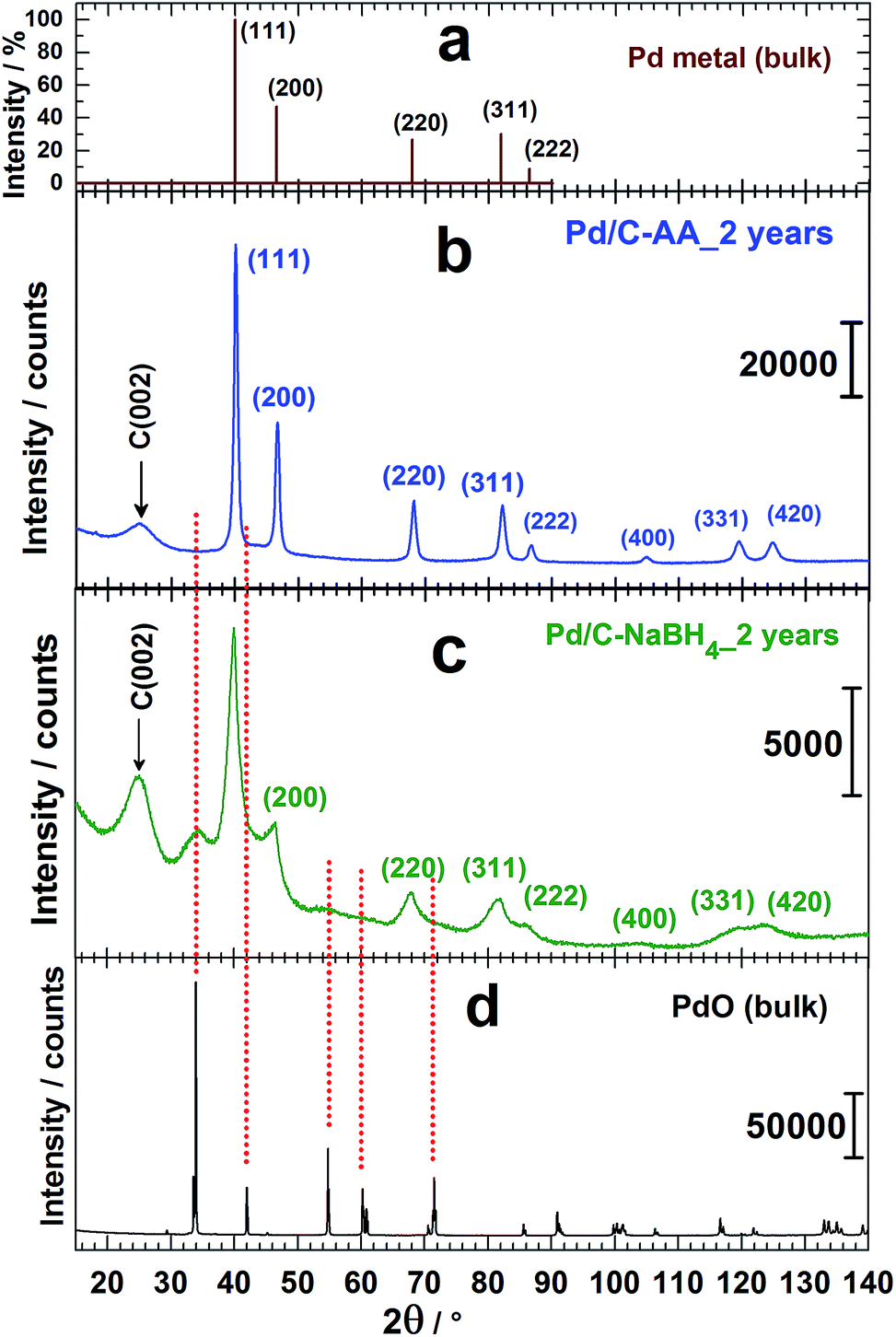 | ||
| Fig. 2 From top to down: high-resolution XRD patterns of (a) bulk Pd metal (space group: Fmm, PDF # 00-046-1043), (b) Pd/C–AA_2 years, (c) Pd/C–NaBH4_2 years and (d) PdO (anhydrous, 99.9%: Alfa Aesar, Ref. 11040). | ||
As it can be seen from the high-resolution XRD patterns, only the sample Pd/C–NaBH4_2 years presents a foreign and broad peak around 2θ = 34° corresponding to an interplanar spacing of 0.263 nm. This peak fits with PdO as it has been drawn in Fig. 2. It is assigned to the crystallographic plane (011) of PdO having a tetragonal symmetry (space group: P42/mmc, PDF # 00-043-1042). These results are in agreement with those from Kibis et al.29 using HRTEM; who also observed (002) for 0.266 nm (2θ = 33.6°). In addition, they reported the presence of PdO2 (d(101) = 0.253 nm), confirmed by XPS analysis.29 In our case, this broad peak at ∼34° could include contribution from the non-stoichiometric palladium oxidized species PdOx (x > 1).29 To validate such hypothesis, other methods like XPS need to be performed. It should be noticed that the absence of palladium oxides peaks on the XRD pattern of Pd/C–AA does not exclude totally their presence in the sample. However, it endorses completely the conclusion that the oxides amount is very insignificant in the bulk sample (detection limit of XRD).
3.3 XPS analysis of the nanomaterials
The XPS studies were targeting two major objectives, the effect of the reducing agent on: (i) the surface chemical composition of the synthesized Pd-based nanomaterials and (ii) the catalyst aging. To elucidate that, we conducted XPS analyzes on the freshly prepared samples (Pd/C–NaBH4_0, Pd/C–AA_0) and after two years for the same batches. It is worth mentioning that, after 2 years, the syntheses were replicated (Pd/C–AA_0-new and Pd/C–NaBH4_0-new) and analyzed again by XPS to check their XPS. Obviously, the same results are obtained for the initial samples. In other words, XPS of Pd/C–AA_0-new is same as Pd/C–AA_0, and Pd/C–NaBH4_0-new same as for Pd/C–NaBH4_0”. Of course, Pd/C–AA_0-new is radically different from Pd/C–NaBH4_0-new. This confirms the excellent reliability and reproducibility of the preparation method. The high-resolution XPS spectra of the core level Pd 3d for samples Pd/C–NaBH4_0 and Pd/C–AA_0 are displayed in Fig. 3a and b, respectively. Those for the aged samples Pd/C–NaBH4_2 years and Pd/C–AA_2 years are shown in Fig. 3c and d, respectively. The extracted important data are summarized in Table 1. The observed doublets for the same band Pd 3d are related to the spin–orbit splitting, characterized by the j = l ± 1/2 quantum number (l = 2 for d). It should be pointed out that not all peaks in XPS spectra are due to simple photoelectric transitions. Secondary peaks may appear in a spectrum due to Auger transitions, plasmon structures, shake-up and shake-off energy loss processes. Indeed, the peak that appears at 347.1 eV in all spectra (Fig. 3) is attributed to Pd plasmon.23,29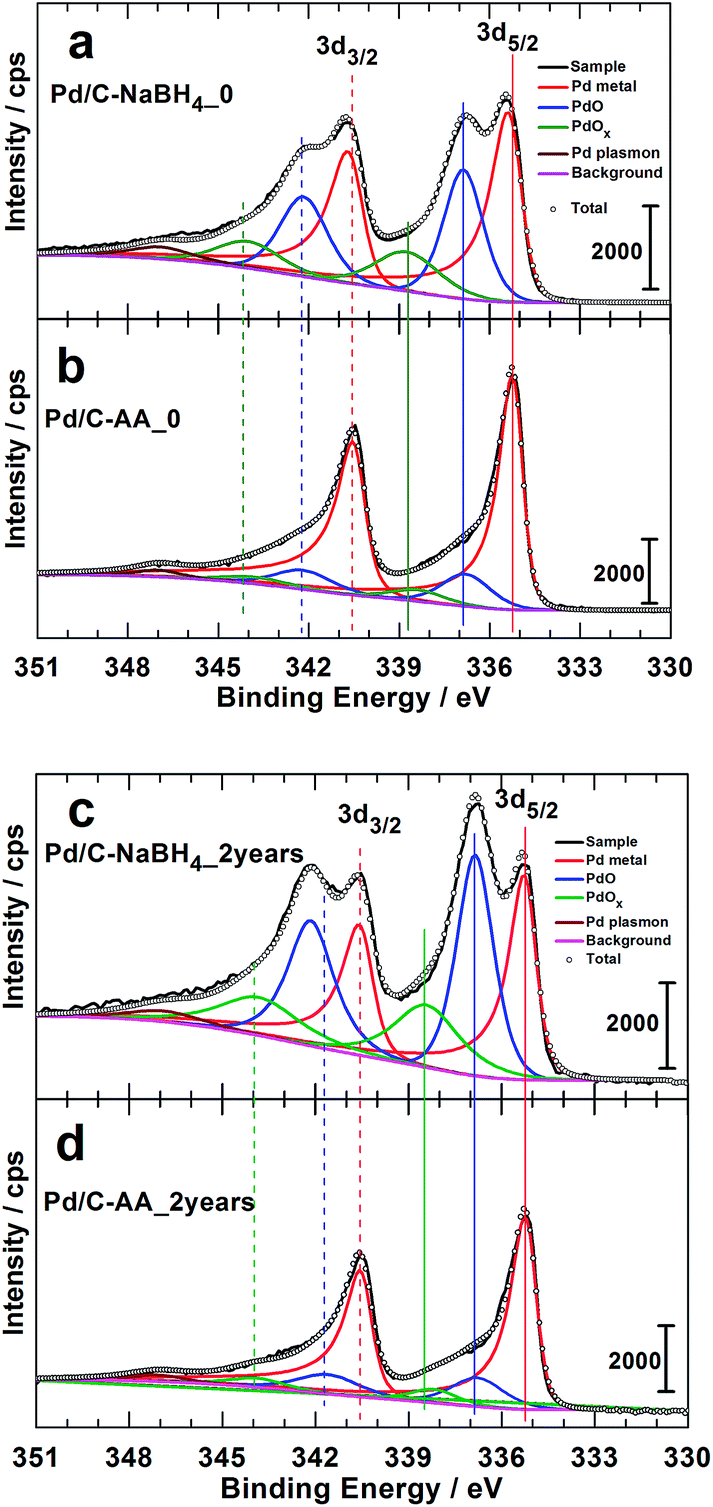 | ||
| Fig. 3 High resolution Pd 3d XPS spectra of the as-prepared materials: freshly synthesized (a) Pd/C–NaBH4_0, (b) Pd/C–AA_0, and 2 years aged (c) Pd/C–NaBH4_2 years, (d) Pd/C–AA_2 years. | ||
The doublet peak, related to Pd metal with a Pd 3d5/2 binding energy (BE) of 335.3 ± 0.1 eV and Pd 3d3/2 BE of 340.6 ± 0.1 eV is clearly evident, in agreement with the literature.23,25,29,43,44 The peak at ca. 336.8 eV is assigned to the Pd 3d5/2 of PdO species. Indeed, except the value of 336.3 eV,25 BEs of 336.5–336.7 eV (ref. 23 and 26) and 337.1–337.2 eV (ref. 26, 29 and 44–46) have been reported for Pd 3d5/2 level of PdO. According to Kibis et al.,29 PdO component shifts to the higher BE when the electron take-off angle is decreased. In addition to these two well-known and characterized XPS peaks of palladium, there is another important peak between 338.3 and 338.8 eV depending on the sample. This peak could belong to superior palladium oxides PdOx (x > 1). The BE of Pd 3d5/2 for the compound PdO2 has been reported to be in the range of 337.5–339.3 eV.25,26,28,29,47 The team of L. Victori identified PdO3 by electrochemical oxidation approach in 1 M KOH with its Pd 3d5/2 ∼ 337.6–337.7 eV.27,48 Obtaining superior oxides of Pd is utterly possible by electrochemical methods in aqueous medium;25,27,48–52 for electrode potentials higher than 1.4 V vs. RHE.53 As indicated in the Introduction section, this strongly oxidized palladium(IV) oxide PdO2 is known to be unstable in its anhydrous form. Nevertheless, it was postulated that PdO2 species are stabilized on the surface of nanocrystalline Al2O3 (ref. 24) and SnO2.28
The more plausible explanation in the absence of these matrices has been provided in 2012 by the group of Vladimir I. Boronin which supposed that PdO2 species are localized within the block boundaries of PdO.23,29 They strengthened their hypothesis by an observed decrease in the take-off angle which leads to an increase in PdO2 component intensity. In this case, palladium at Pd2+ state acts as a stabilization matrix for the Pd4+ species. This conclusion could be valid for our case where PdO species have been firstly observed by XRD on Pd/C–NaBH4_2 years sample and then confirmed by XPS analysis. We adopt the notation of PdOx (x > 1), as earlier proposed by the Vladimir I. Boronin's group.
Furthermore, Fig. 3 highlights two major facts. It clearly shows that the Pd/C–NaBH4 sample is more oxidized than its counterpart Pd/C–AA. In other words, the synthesis using AA as reducing agent further enables to inhibit the oxidation of palladium nanoparticles. The quantitative analysis results are reported in Table 2 and illustrated by Fig. 4. For the material Pd/C–NaBH4_0, the surface atomic composition related to palladium species is 53 surf. at% Pd metallic and 47 surf. at% Pd oxides (PdO + PdOx). After 2 years, the amount of oxidized palladium reaches 61 surf. at%. In the same time, Pd/C–AA_0 has only 20 surf. at% Pd oxides that is 23 surf. at% after 2 years. As the XRD did not highlight any oxides on Pd/C–AA sample, we postulate that the oxidized palladium amount of 23 surf. at% concerns a very thin layer at the surface. It is worth of mentioning that smaller particles tend to be oxidized easily. Consequently, it could be possible that the particles size may have a contribution on the stability of Pd/C–NaBH4 that surface is more oxidized than Pd/C–AA one. Truly speaking, we did not exclude totally this possibility. However, this intuitive easily oxidation of smaller particles is not sufficient to explain the present large disparity. As aforementioned in the Introduction section, the particular affinity of palladium in a hydrogen environment is a well-document science and the strong ability of Pd nanoparticles to absorb hydrogen in their network is well known. It has been demonstrated that palladium hydride nanostructures can be synthesized by direct exposing to hydrogen gas13 or in NaBH4 solution.54,55 Furthermore, platinum and gold nanomaterials have been prepared using both NaBH4 and AA as reducing agents. The XPS results have shown that Pt/C–NaBH4 and Au/C–AA do not present any oxide species and only a very small amount on Au/C–NaBH4 sample.56 The interesting thing is that Pt/C–NaBH4 (ref. 56) has quite smaller particles size than Pd/C–NaBH4. Therefore, we believe that the major contribution comes from the Pd affinity towards hydrogen. Then, 30 wt% Pd/C–NaBH4 sample was prepared and we also noticed that it has an important amount of oxides at its surface. It is well known that when a noble metal is exposed to ambient air, a thin protective layer covers its surface from deep oxidation. In this case, a high-resolution XRD patterns cannot detect the oxides, as it is undoubtedly the case of Pd/C–AA sample.
| Species | Pd 3d core-level: (BE deviation: ±0.1 eV) | Pd/C–AA | Pd/C–NaBH4 | |||
|---|---|---|---|---|---|---|
| Initial | After 2 years | Initial | After 2 years | |||
| Pd metal | BE (eV) | 3d3/2 | 340.6 | 340.6 | 340.7 | 340.6 |
| 3d5/2 | 335.3 | 335.3 | 335.4 | 335.3 | ||
| surf. at% from 3d5/2 | 80 | 77 | 53 | 39 | ||
| PdO | BE (eV) | 3d3/2 | 342.3 | 341.8 | 342.2 | 342.2 |
| 3d5/2 | 336.8 | 336.9 | 336.8 | 336.8 | ||
| surf. at% from 3d5/2 | 14 | 16 | 32 | 40 | ||
| PdOx (x > 1) | BE (eV) | 3d3/2 | 344.3 | 344.0 | 344.2 | 344.1 |
| 3d5/2 | 338.6 | 338.3 | 338.8 | 338.5 | ||
| surf. at% from 3d5/2 | 6 | 7 | 15 | 21 | ||
| Pd plasmon: BE (eV) | 347.1 | 347.1 | 347.1 | 347.1 | |
| Summary: Pd species (surf. at%) | Pd metallic | 80 | 77 | 53 | 39 |
| Pd oxidized | 20 | 23 | 47 | 61 | |
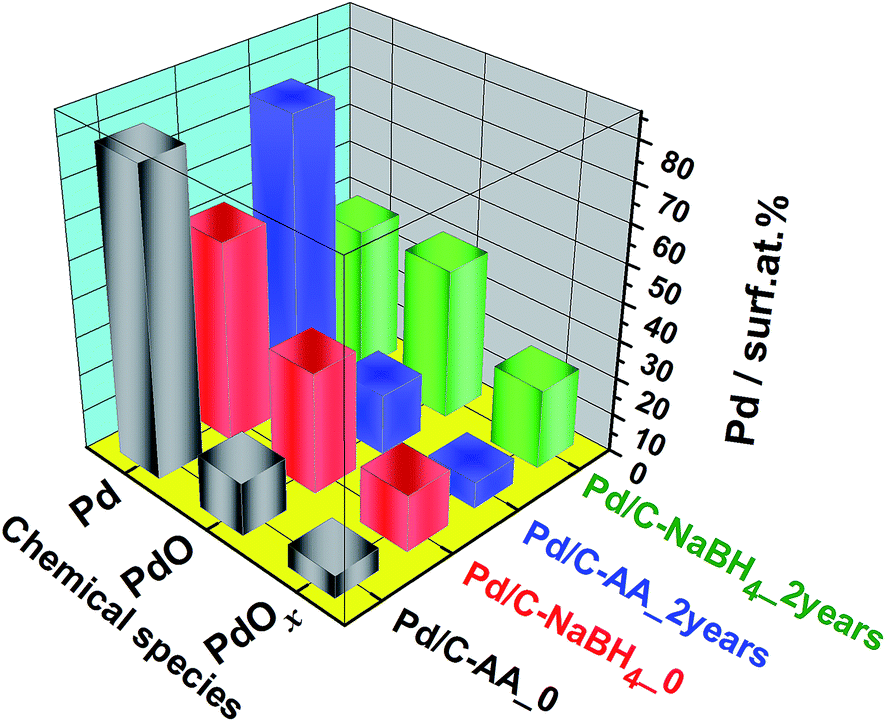 | ||
| Fig. 4 Quantitative data from the high-resolution XPS measurements on the freshly synthesized (Pd/C–AA_0, Pd/C–NaBH4_0) and 2 years aged (Pd/C–NaBH4_2 years, Pd/C–NaBH4_2 years) nanomaterials. | ||
Finally and to explain these observations, the hypothesis of the hydride PdHx, in agreement with our previous in situ XRD measurements,17 that can weaken the Pd–Pd metallic bond because of Pdδ+–Hδ− species is formulated. Otherwise, L-ascorbic acid used as reducing agent further prevents the chemical oxidation of palladium nanoparticles. This surface analysis by XPS reveals that the reducing agent plays a crucial role concerning the chemical stability of Pd nanoparticles. A reducing agent such as NaBH4 that releases hydrogen gas (H2) and protons (H+) in aqueous medium during the chemical preparation of nanoparticles changes drastically the surface state of palladium nanomaterials with a high amount of oxides that increases importantly over time.
It has been shown that heating Pd/C–NaBH4 at 150 °C enables to remove efficiently hydrogen absorbed during the synthesis.17 Here, in order to remove the entire oxides from nanoparticles surface, a heating process (400 °C in the presence of H2 gas) of the samples after 2 years and coupled with in situ XPS was performed. This thermal process is well known as an efficient method to reduced palladium oxides. The collected XPS spectra further denoted as Pd/C–NaBH4_2 years-[H2] and Pd/C–AA_2 years-[H2] are depicted in Fig. 5. The confrontation with Pd foil shows that oxides are successfully reduced into Pd metal with its BE of Pd 3d5/2 level at ca. 335.2 eV. Finally, the novelty of this work supported by data represents a clear development in our understanding of Pd–H affinity and paves new ways toward Pd metal free from hydrides and oxides.
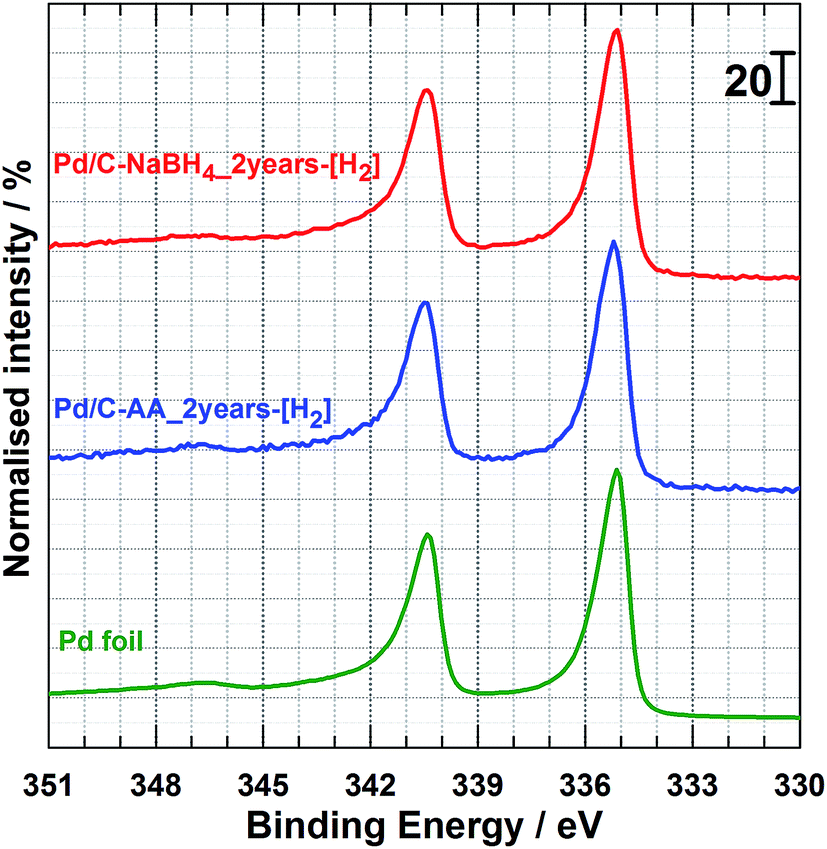 | ||
| Fig. 5 From top to down: in situ high-resolution XPS spectrum of Pd 3d core level for the as-reduced Pd/C–NaBH4_2 years and Pd/C–AA_2 years samples under H2/Ar atmosphere at 400 °C. Pd foil is given as reference material. | ||
3.4 Electrocatalytic properties towards ORR
To gain further insights on the catalytic properties on the initially synthesized Pd/C–NaBH4 and Pd/C–AA nanomaterials, we performed the electrochemical ORR in alkaline medium. The electrochemistry of Pd was firstly investigated in N2-saturated 0.1 M KOH. The common used APCT consists of the accelerated potential cycling from 0.6 to 1 V vs. RHE at 100 mV s−1 scan rate in O2-saturated supporting electrolyte.33,57 Alternatively, electrode potentials are run at larger potential window in supporting electrolyte without O2.37,58,59 The cathode ORR catalyst works continuously under O2 atmosphere during the operation of the fuel cell (>0.7 V vs. RHE)60 hence the first protocol was adopted. The cyclic voltammograms before and after the APCT measurements are reported in Fig. S1 (ESI†). The current densities were normalized by the electrochemical surface area (ECSA) of palladium, determined using the PdO/Pd redox couple, well explained elsewhere.17 The positive scan is characterized by the adsorption of hydroxyl species onto Pd surface around 0.6–0.7 V vs. RHE follows by the first PdO monolayer formation; which ends at 1.45 V vs. RHE with a faradaic charge of 424 μC cm−2.17,30,49,53 The cathodic variation of the potential shows a main PdO reduction peak in the electrode potential range of 0.7–0.6 V vs. RHE depending on the particles size, the value of the upper potential limit and the scan rate. Previously, it has been found that the specific ECSA obtained from the PdO peak reduction method is 60 m2 g−1 for Pd/C–AA and 81 m2 g−1 for Pd/C–NaBH4, that has been explained in the terms of particles size difference.17 Here, we focused on the Pd nanoparticles stability/durability under strong corrosive and oxidizing conditions through the APCT over 5000 cycles. The determined ECSA was normalized with respect to the initial value and displayed in Fig. 6a. It is worth mentioning that the same trend was observed by evaluating ECSA at either 50 mV s−1 or 5 mV s−1 scan rate. After the ACPT, Pd/C–NaBH4 electrode material loses ca. 30% of its initial ECSA contrariwise to Pd/C–AA that gains 4%. In other words, L-ascorbic acid enables obtaining more durable palladium electrode materials. There are several reasons that explain the present results and include the particles size (agglomeration) and the metal dissolution effects.59,61–64 Here, Pd nanoparticles being smaller in Pd/C–NaBH4 than Pd/C–AA (3 nm versus 8 nm), the as-indicated phenomenon could be more exalted on Pd/C–NaBH4. In addition, the contribution of the oxidized palladium species must not be excluded in the case of Pd/C–NaBH4 where their amount is nearly 50 surf. at%.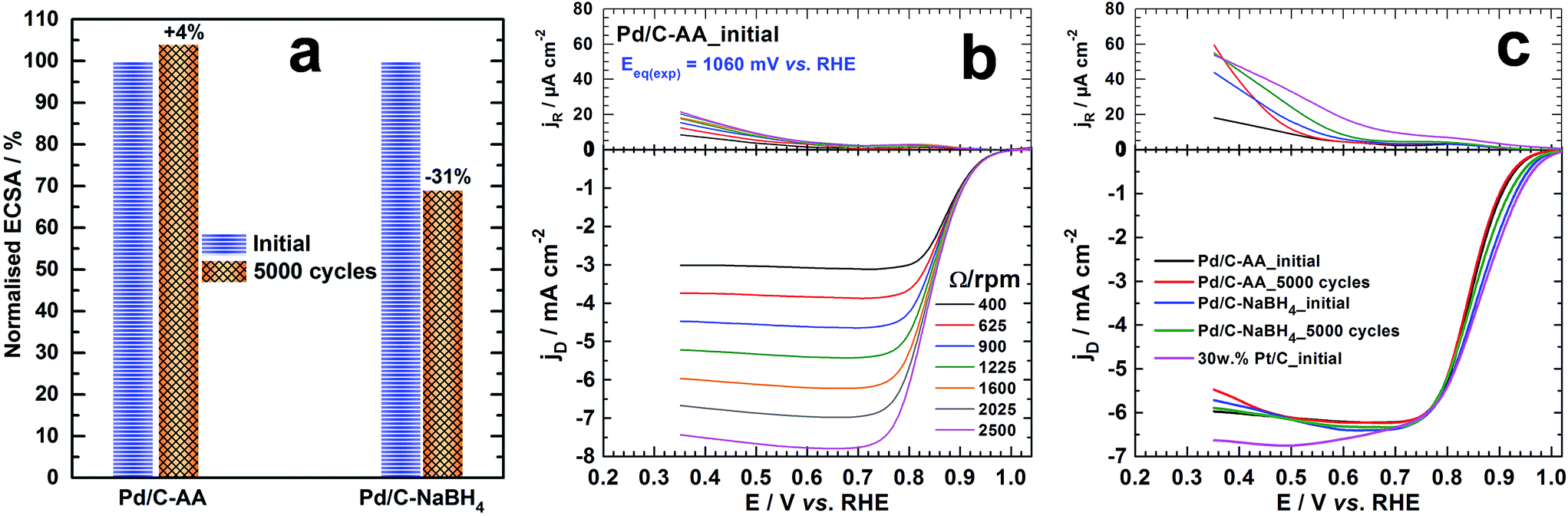 | ||
| Fig. 6 (a) The normalized ECSA behaviour before and after the 5000 cycles of accelerated potential cycling test (APCT) from 0.6 to 1 V vs. RHE at 100 mV s−1 scan rate in O2-saturated 0.1 mol L−1 KOH: note that ECSA was evaluated at 50 mV s−1 and 5 mV s−1 scan rates and had the same trend. ORR polarization curves recorded in O2–saturated 0.1 mol L−1 KOH solution at the ring (top) and disc (bottom) for 5 mV s−1 scan rate: (b) Pd/C–AA catalyst at different speeds of the RRDE; (c) different catalysts before (initial) and after APCT (5000 cycles) at RRDE speed of 1600 rpm. For panels (b) and (c), the current densities are obtained with the geometry surface area of the ring (0.11 cm2) and disc (0.196 cm2). | ||
The ORR polarization curves (disc and ring) for Pd/C–AA electrode material before (Pd/C–AA_initial) and after ACPT (Pd/C–AA_5000 cycles) are shown in Fig. 6b and S2a (ESI†). Those for Pd/C–NaBH4_initial and Pd/C–NaBH4_5000 cycles are reported in Fig. S2b and S2c (ESI†), respectively. The fundamental relationships of ORR in aqueous medium are well explained in the ESI.† As resumed in Table S1 (ESI†), the theoretical equilibrium potential for ORR (at zero current) in 0.1 M KOH is Eeq. = 1.185 V vs. RHE; corresponding to O2/HO− redox couple (see calculations in ESI†). The experimentally measured values (inserted in Fig. 6b and S2 (ESI†)) are Eeq.(exp) = 1.060 V vs. RHE (Pd/C–AA_initial), 1.047 V vs. RHE (Pd/C–AA_5000 cycles), 1.053 V vs. RHE (Pd/C–NaBH4_initial) and 1.071 V vs. RHE (Pd/C–NaBH4_5000 cycles). Hence, the overpotential is ≤0.14 V on these electrocatalysts. This insight is supported by the good behaviour where ORR starts at E > 1 V vs. RHE on all electrodes as on pure Pt/C (Fig. 6c and S2d (ESI†)), outperforming all reported Pd/C catalysts for ORR in aqueous medium.65–68 More importantly, Fig. 6c shows that the ORR polarization curve recorded at 1600 rpm on Pd/C–AA_5000 cycles is superimposed with Pd/C–AA_initial. Meanwhile, Pd/C–NaBH4_5000 cycles shows a positive shift. These results are in agreement with the ECSA measurements and support the good performance of Pd/C–AA.
The determined hydrogen peroxide (H2O2) amount is depicted in Fig. 7a (see ESI† for calculations: is in the form of HO2− in alkaline medium). The ultimate goal for ORR materials is to understate H2O2 production from incomplete ORR in the electrode potential from 1 to 0.8 V vs. RHE; thereby enhancing the faradaic yield towards H2O.60 Even after the APCT, p(H2O2) < 0.5% on the Pd/C electrode materials. In other words, robust ORR catalysts have been successfully synthesized. The Koutecky–Levich plots from the ORR polarization curves at different electrode potentials are reported in Fig. S3 (ESI†). It enables to determine directly the kinetic current density jk from the intercepts, expressed in mA per cm2 of disc and referred as the “absolute activity”. Then, and as recommended,34,60 it was normalized as: (i) the “area-specific activity” As (mA per cm2 Pd) by dividing the absolute activity by the surface-area enhancement factor and (ii) the “mass activity” Am (mA per μg of Pd) by dividing the absolute activity by the Pd loading. The surface-area enhancement factor (also referred as the electrochemical surface roughness or electrode roughness) is the electrochemical surface area of the Pd catalyst measured and divided by the planar area of the sample (cm2 Pd per planar cm2). The results at 0.85 V vs. RHE are shown in Fig. 7b. Initially, As = 0.43 mA cmPd−2 (Am = 0.086 mA μgPd−1) for Pd/C–AA and 0.13 mA cmPd−2 (Am = 0.116 mA μgPd−1) for Pd/C–NaBH4. Importantly, As and Am represent at least 2-fold and 245-fold enhancement compared to Pd/C–NaBH4 prepared from the water-in-oil method.65 Unfortunately, there are few reports on literature where full ORR kinetics are performed in alkaline medium for comparison.66,69,70 Furthermore, Pd/C–AA_5000 cycles exhibits better limiting current density, jL (Fig. S4 (ESI†)) that is ca. 40% higher than Pd/C–NaBH4_5000 cycles. The Tafel plots (Fig. 7c) show two slopes: 72–83 mV dec−1 at high potentials (low overpotential) and 123–144 mV dec−1 at low potentials (high overpotential). The determined exchange current density at high potentials is j0 = 0.4 × 10−3 mA cm−2 (Pd/C–AA_initial), 0.3 × 10−3 mA cm−2 (Pd/C–AA_5000 cycles), 0.6 × 10−3 mA cm−2 (Pd/C–NaBH4_initial) and 0.5 × 10−3 mA cm−2 (Pd/C–NaBH4_5000 cycles). At low potentials, j0 ∼ 200 × 10−3 mA cm−2 initially and ∼160 × 10−3 mA cm−2 after APCT. Given the strong susceptibility of ORR towards the particles size,71–73 a huge increase of kinetic parameters such as jk, jL and j0 was expected for Pd/C–NaBH4 (mean particles size of 3 nm) electrode material than Pd/C–AA (mean particles size of 8 nm) one. Indeed, according to this effect, a decrease of particles size from 8 to 3 nm must be accompanied by an increase of the mass activity by at least a factor of 2 in terms of Am. Surprisingly, we found quite same value of Am ∼ 0.1 mA mgPd−1 on both catalysts. We attribute this unexpected decrease of Pd/C–NaBH4 activity to its surface modification by oxidized palladium atoms as it has been regardless shown above by XPS and XRD investigations. Furthermore, a value of j0 = 11 × 10−3 mA cm−2 has been reported for Pd/C at low potential.65 Hence, the present Pd/C catalysts exhibit very high ORR kinetics by an order of magnitude. This radical enhancement of the electrocatalytic activity of Pd/C synthesized from Bromide Anion Exchange method is explained by the absence of any organic contaminants or protective ligands at their surface. These organic molecules are currently used as capping agent or surfactant to control the nanocrystal morphology and size. Unfortunately, the retained molecules decrease notably the catalytic performances of the obtained electrodes due to the inaccessibility of some catalytic sites, which are obviously blocked.
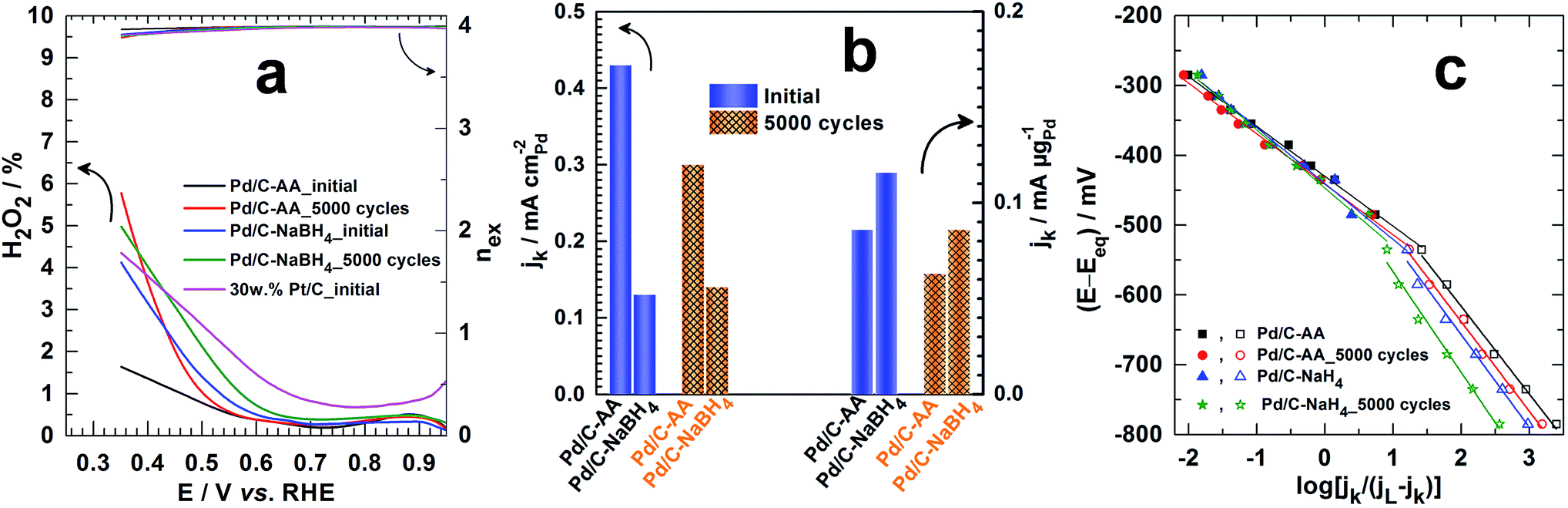 | ||
| Fig. 7 (a) The H2O2 production from incomplete ORR (left Y-axis) and the number of electrons exchanged nex (right Y-axis) determined from ORR polarization curves in O2-saturated 0.1 mol L−1 KOH at 5 mV s−1 scan rate and RRDE speed of 1600 rpm. (b) The kinetic current density jk determined from the Koutecky–Levich plots at 0.85 V vs. RHE before and after 5000 cycles of accelerated potential cycling test (APCT) from 0.6 to 1 V vs. RHE at 100 mV s−1 scan rate in O2-saturated 0.1 mol L−1 KOH: referred as the “area-specific activity” (mA per cm2 Pd) and determined by dividing the absolute activity by the surface-area enhancement factor (left Y-axis) or the “mass activity” (mA per μg of Pd) and determined by dividing the absolute activity by the Pd loading (right Y-axis). (c) Tafel plots at the different electrode materials: the limiting current density jL was determined from the “jk−1 versus potential” plots and Eeq. = 1.185 V vs. RHE in 0.1 mol L−1 KOH (see ESI†). Note: in alkaline medium, H2O2 is in the form of HO2−. | ||
As attested by the uncover data, the novelty of the present work has provided two majors advances in Materials science and catalysis and represents a deep and clear development in our understanding of palladium affinity with hydrogen. The first insight, in Materials Science, enlightens us on the fact that the reduction of palladium salt precursors by H2-based reducing agent (NaBH4, H2 or N2H4) to prepare palladium nanomaterials leads to the formation of hydrides PdHx that further promote the formation of palladium oxides. The second breakthrough, especially in the catalysis of the oxygen reduction reaction enables improving by an order of magnitude the reaction kinetics thanks to palladium nanocatalyst prepared by using L-ascorbic acid as alternative to these H2-based reducing agents. These remarkable results encapsulate definitely the strong impact of the reducing agent on the physicochemistry and catalysis of palladium-based nanomaterials prepared by chemical approaches in hydrogen gas environment.
4. Conclusions
Palladium-based nanomaterials are mostly prepared using the sodium borohydride as reducing agent (Pd/C–NaBH4), a strong and hydrogen-rich compound that can release hydrogen gas and protons during the synthesis. Given the idiosyncratic affinity of palladium with these species, the present work was aiming to elucidate by X-ray photoelectron spectroscopy (XPS) the unclear subsequent impact of NaBH4 on the final surface composition of the nanomaterials. Another reducing agent that does not involve any of these hydrogen species, L-ascorbic acid was also used to synthesize palladium nanomaterials (Pd/C–AA) as benchmark. From XPS investigations, it has been found that the majority of palladium atoms at the surface are oxidized. The presence of palladium(II) oxide PdO was upheld by XRD measurements, but only on the sample synthesized using NaBH4. In addition to PdO, the presence of superior palladium oxides PdOx (x > 1) having binding energy in the range of 338.3–338.8 eV have been observed. The reducing agent is thereby a cornerstone element during the design of chemically stable palladium nanomaterials without oxides. This unexpected disparity coming from the nature of the reducing agent has been assigned to the potency of palladium to absorb hydrogen during the synthesis. Furthermore, given the close relationship between the surface state of nanomaterials and their catalytic properties, we evaluated the potential impact of using NaBH4 to synthesize Pd catalyst. It has been firstly discovered that Pd/C–NaBH4 loses roughly 30% of its initial electrochemical active surface area (ECSA) after 5000 cycles of potential cycling under strong corrosive and degrading conditions (APCT). Meanwhile, Pd/C–AA gains 4%. Then, the catalytic performances have been considered towards the electrochemical oxygen reduction reaction (ORR), a highly surface and particles size dependent reaction. Results confirm the excellent stability of Pd/C–AA catalyst while Pd/C–NaBH4 loses activity over cycles. Otherwise, the Bromide Anion Exchange approach has drastically enhanced the ORR kinetics of Pd/C by an order of magnitude compared to similar catalyst prepared by other methods. The uncovered remarkable behaviours enhance greatly our knowledge on the Pd affinity towards hydrogen and paves new ways for successful synthesis of Pd-based nanomaterials with impressive catalytic activities while minimizing the amount of oxides. These outstanding results encapsulate definitely the palladium–hydrogen affinity in Materials Science as well as in catalysis. By providing experimental key tools at high thresholds, this work enlightens us on the physicochemistry and catalysis of palladium-based nanomaterials prepared by chemical approaches in hydrogen gas environment.Acknowledgements
Authors acknowledge financial support from the French National Research Agency (ANR) through the project “ChemBio-Energy program 2012-2015”.Notes and references
- L. Croft, Nat. Chem., 2010, 2, 1009 CrossRef CAS PubMed.
- M. Hartings, Nat. Chem., 2012, 4, 764 CrossRef CAS PubMed.
- C. Bianchini, in Interfacial Phenomena in Electrocatalysis, ed. C. G. Vayenas, Springer, New York, 2011, pp. 203–253 Search PubMed.
- A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999–12044 CrossRef CAS PubMed.
- P. Jena and C. B. Satterwaite, Electronic Structure and Properties of Hydrogen in Metals, Plenum Press, New York, 1983 Search PubMed.
- N. de Zoubov and M. Pourbaix, in Atlas d'équilibres électrochimiques: à 25 °C, ed. M. Pourbaix, Gauthier-Villars et Cie, Paris, 1963, pp. 358–363 Search PubMed.
- B. Kappesser, U. Stuhr, H. Wipf, J. Weißmüller, C. Klos and H. Gleiter, J. Alloys Compd., 1995, 231, 337–342 CrossRef CAS.
- G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 2014, 13, 802–806 CrossRef CAS PubMed.
- D. G. Narehood, S. Kishore, H. Goto, J. H. Adair, J. A. Nelson, H. R. Gutiérrez and P. C. Eklund, Int. J. Hydrogen Energy, 2009, 34, 952–960 CrossRef CAS.
- M. Suleiman, J. Faupel, C. Borchers, H. U. Krebs, R. Kirchheim and A. Pundt, J. Alloys Compd., 2005, 404–406, 523–528 CrossRef CAS.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. Diaz Fernandez, G. Nalin, D. Switlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS PubMed.
- A. Baldi, T. C. Narayan, A. L. Koh and J. A. Dionne, Nat. Mater., 2014, 13, 1143–1148 CrossRef CAS PubMed.
- R. Bardhan, L. O. Hedges, C. L. Pint, A. Javey, S. Whitelam and J. J. Urban, Nat. Mater., 2013, 12, 905–912 CrossRef CAS PubMed.
- N. de Zoubov and M. Pourbaix, in Atlas d'équilibres électrochimiques: à 25 C, ed. M. Pourbaix, Gauthier-Villars et Cie, Paris, 1963, pp. 358–363 Search PubMed.
- J. Turkevich and G. Kim, Science, 1970, 169, 873–879 CAS.
- B. Lim, M. Jiang, J. Tao, P. H. C. Camargo, Y. Zhu and Y. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS.
- Y. Holade, T. W. Napporn, C. Morais, K. Servat and K. B. Kokoh, ChemElectroChem, 2015, 2, 592–599 CrossRef CAS.
- J. A. Eastman, L. J. Thompson and B. J. Kestel, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 84–92 CrossRef CAS.
- C. M. Ghimbeu, C. Zlotea, R. Gadiou, F. Cuevas, E. Leroy, M. Latroche and C. Vix-Guterl, J. Mater. Chem., 2011, 21, 17765–17775 RSC.
- L. Schlapbach and J. P. Burger, J. Phys., Lett., 1982, 43, 273–276 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- M. Suleiman, N. M. Jisrawi, O. Dankert, M. T. Reetz, C. Bähtz, R. Kirchheim and A. Pundt, J. Alloys Compd., 2003, 356–357, 644–648 CrossRef CAS.
- L. S. Kibis, A. I. Titkov, A. I. Stadnichenko, S. V. Koscheev and A. I. Boronin, Appl. Surf. Sci., 2009, 255, 9248–9254 CrossRef CAS.
- K. Otto, L. P. Haack and J. E. deVries, Appl. Catal., B, 1992, 1, 1–12 CrossRef CAS.
- K. S. Kim, A. F. Gossmann and N. Winograd, Anal. Chem., 1974, 46, 197–200 CrossRef CAS.
- M. R. Mucalo and C. R. Bullen, J. Mater. Sci. Lett., 2001, 20, 1853–1856 CrossRef CAS.
- J. M. Tura, P. Regull, L. Victori and M. D. de Castellar, Surf. Interface Anal., 1988, 11, 447–449 CrossRef CAS.
- J. Kappler, N. Bârsan, U. Weimar, A. Dièguez, J. L. Alay, A. Romano-Rodriguez, J. R. Morante and W. Göpel, Fresenius. J. Anal. Chem., 1998, 361, 110–114 CrossRef CAS.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, J. Phys. Chem. C, 2012, 116, 19342–19348 CAS.
- Y. Holade, C. Morais, S. Arrii-Clacens, K. Servat, T. W. Napporn and K. B. Kokoh, Electrocatalysis, 2013, 4, 167–178 CrossRef CAS.
- Y. Holade, K. Servat, T. W. Napporn and K. B. Kokoh, Electrochim. Acta, 2015, 162, 205–214 CrossRef CAS.
- Y. Holade, C. Morais, K. Servat, T. W. Napporn and K. B. Kokoh, Phys. Chem. Chem. Phys., 2014, 16, 25609–25620 RSC.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- Y. Li, F. Quan, L. Chen, W. Zhang, H. Yu and C. Chen, RSC Adv., 2014, 4, 1895–1899 RSC.
- A. Marcu, G. Toth and R. J. Behm, Fuel Cells, 2014, 14, 378–385 CrossRef CAS.
- M. Lahmani, C. Bréchignac and P. Houdy, Les nanosciences: 2. Nanomatériaux et nanochimie, Berlin, Paris, France, 2012 Search PubMed.
- A. Wieckowski, E. R. Savinova and C. G. Vayenas, Catalysis and Electrocatalysis at Nanoparticle Surfaces, Marcel Dekker, Inc., New York, USA, 2003 Search PubMed.
- C. R. Henry, Prog. Surf. Sci., 2005, 80, 92–116 CrossRef CAS.
- L. Wang, C. Hu, Y. Nemoto, Y. Tateyama and Y. Yamauchi, Cryst. Growth Des., 2010, 10, 3454–3460 CAS.
- B. Lim, H. Kobayashi, P. C. Camargo, L. Allard, J. Liu and Y. Xia, Nano Res., 2010, 3, 180–188 CrossRef CAS.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 387–394 CrossRef CAS.
- T. H. Fleisch and G. J. Mains, J. Phys. Chem., 1986, 90, 5317–5320 CrossRef CAS.
- T. H. Fleisch, G. W. Zajac, J. O. Schreiner and G. J. Mains, Appl. Surf. Sci., 1986, 26, 488–497 CrossRef CAS.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 395–401 CrossRef CAS.
- G. A. Shafeev, J. M. Themlin, L. Bellard, W. Marine and A. Cros, J. Vac. Sci. Technol., A, 1996, 14, 319–326 CAS.
- V. Chausse, P. Regull and L. Victori, J. Electroanal. Chem. Interfacial Electrochem., 1987, 238, 115–128 CrossRef CAS.
- M. Grden, M. Lukaszewski, G. Jerkiewicz and A. Czerwinski, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef CAS.
- A. J. Zhang, M. Gaur and V. I. Birss, J. Electroanal. Chem., 1995, 389, 149–159 CrossRef.
- V. I. Birss, V. H. Beck, A. J. Zhang and P. Vanýsek, J. Electroanal. Chem., 1997, 429, 175–184 CrossRef CAS.
- A. E. Bolzan, J. Electroanal. Chem., 1995, 380, 127–138 CrossRef.
- A. Czerwiński, J. Electroanal. Chem., 1994, 379, 487–493 CrossRef.
- D. W. Murphy, S. M. Zahurak, B. Vyas, M. Thomas, M. E. Badding and W. C. Fang, Chem. Mater., 1993, 5, 767–769 CrossRef CAS.
- T.-H. Phan and R. E. Schaak, Chem. Commun., 2009, 3026–3028 RSC.
- Y. Holade, K. Servat, J. Rousseau, C. Canaff, S. Poulin, T. W. Napporn and K. B. Kokoh, J. Electrochem. Soc., 2015, 162, H929–H937 CrossRef CAS.
- J. Liu, X. Sun, P. Song, Y. Zhang, W. Xing and W. Xu, Adv. Mater., 2013, 25, 6879–6883 CrossRef CAS PubMed.
- Z.-Z. Jiang, Z.-B. Wang, Y.-Y. Chu, D.-M. Gu and G.-P. Yin, Energy Environ. Sci., 2011, 4, 728–735 CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- C. Martins, P. Fernández, H. Troiani, M. Martins, A. Arenillas and G. Camara, Electrocatalysis, 2014, 5, 204–212 CrossRef CAS.
- J. C. Meier, I. Katsounaros, C. Galeano, H. J. Bongard, A. A. Topalov, A. Kostka, A. Karschin, F. Schuth and K. J. J. Mayrhofer, Energy Environ. Sci., 2012, 5, 9319–9330 CAS.
- L. Tang, X. Li, R. C. Cammarata, C. Friesen and K. Sieradzki, J. Am. Chem. Soc., 2010, 132, 11722–11726 CrossRef CAS PubMed.
- F. Hiraoka, K. Matsuzawa and S. Mitsushima, Electrocatalysis, 2013, 4, 10–16 CrossRef CAS.
- D. Diabaté, T. W. Napporn, K. Servat, A. Habrioux, S. Arrii-Clacens, A. Trokourey and K. B. Kokoh, J. Electrochem. Soc., 2013, 160, H302–H308 CrossRef.
- V. Mazumder, M. Chi, K. L. More and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 9368–9372 CrossRef CAS PubMed.
- J. H. Shim, J. Kim, C. Lee and Y. Lee, Chem. Mater., 2011, 23, 4694–4700 CrossRef CAS.
- S. J. Hwang, S. J. Yoo, J. Shin, Y.-H. Cho, J. H. Jang, E. Cho, Y.-E. Sung, S. W. Nam, T.-H. Lim, S.-C. Lee and S.-K. Kim, Sci. Rep., 2013, 3, 1309 Search PubMed.
- M. Yun, M. S. Ahmed and S. Jeon, J. Power Sources, 2015, 293, 380–387 CrossRef CAS.
- M. C. Oliveira, R. Rego, L. S. Fernandes and P. B. Tavares, J. Power Sources, 2011, 196, 6092–6098 CrossRef CAS.
- A. Anastasopoulos, J. C. Davies, L. Hannah, B. E. Hayden, C. E. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973–1982 CrossRef CAS PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORR calculations and additional plots. See DOI: 10.1039/c5ra24829a |
| This journal is © The Royal Society of Chemistry 2016 |