
Chemical modification of gold electrodes via non-covalent interactions†
Brian R.
Lydon
,
Alex
Germann
and
Jenny Y.
Yang
*
Department of Chemistry, University of California, Irvine, CA 92697-2025, USA. E-mail: j.yang@uci.edu
First published on 9th March 2016
Abstract
Chemically modifying electrode surfaces with redox active molecular complexes is an effective route to fabricating tailored functional materials. Surface modification has generally required the installation of reactive functional groups for direct covalent attachment that can present synthetic challenges. An alternative, milder method that utilizes π-interactions to physisorb the molecular complex onto a surface is described herein. Firstly, a gold electrode was modified with pyrene via covalent thiolate bonds. A pyrene-functionalized ferrocene was then physisorbed onto the pyrene-modified gold electrode. X-ray photoelectron spectroscopy, infrared spectroscopy, and cyclic voltammetry were used to demonstrate successful physisorption of the pyrene-functionalized ferrocene onto the pyrene-modified gold surface. Physisorption is attributed to pyrene–pyrene (π) interactions, as the ferrocene compound was not observed after identical treatment of a clean gold electrode surface. Additionally, cyclic voltammetry demonstrates facile electron transfer between the electrode and ferrocene through the non-covalent interactions at the interface. Since this approach of surface modification only requires functionalizing the target molecular complex with the relatively inert pyrene functionality, it broadens the range of experimentally accessible molecular precursors for chemically modified electrodes.
 Jenny Y. Yang | Jenny Yang earned her B.S. from the University of California, Berkeley working with Professor Jeffrey R. Long, and her Ph.D. in Inorganic Chemistry at the Massachusetts Institute of Technology with Professor Daniel G. Nocera. After a postdoctoral appointment at the Pacific Northwest National Laboratory (PNNL) with Dr. Daniel L. DuBois she worked as a staff scientist in the Center for Molecular Electrocatalysis at PNNL and the Joint Center for Artificial Photosynthesis at the California Institute of Technology. She began her appointment as an Assistant Professor of Chemistry at the University of California, Irvine in the summer of 2013. Her research interests include the discovery and study of inorganic electrocatalysts for the generation and utilization of chemical fuels. These studies have primarily focused on the effect of secondary coordination sphere interactions and the effect of thermochemical properties on catalytic activity. |
Introduction
Electrode surfaces modified with redox-active molecules have demonstrated significant utility in fundamental electron transfer studies and the development of tailored electroresponsive materials.1–8 Preparation of chemically modified electrodes typically requires attachment of the target molecular complex to the surface via covalent bonds. However, the reactive functional groups used to form covalent bonds to electrode surfaces (i.e. thiols to metallic surfaces, carboxylic acids and phosphonic acids to metal oxides)9 are often synthetically incompatible with molecular complexes containing open coordination sites or sensitive functional groups. The technical challenges inherent to this approach have limited the scope of molecular complexes that can be used for electrode surface modification.In contrast, physisorption of pyrene functionalized molecular complexes to carbon nanotubes or graphitic electrodes (i.e. highly oriented pyrolytic graphite) has been very successful. The π–π interaction at the interface demonstrates facile electron transfer between the electrode and molecular complexes. Additionally, pyrene is relatively inert and has already been successfully installed on a broad range of molecular complexes and sensitive biological molecules with desirable catalytic, redox, or sensing properties.10–24 However, this approach of non-covalent attachment had been limited to modifying carbon based electrodes with aromatic character.
We report herein a method of extending non-covalent attachment of molecular complexes onto non-carbon electrodes by pre-functionalizing the surface with covalently bound pyrene, as shown in Scheme 1. Again, the relatively unreactive nature of pyrene results in synthetically accessible derivatives that can be used to form covalent bonds to a variety of surfaces. A pyrene-functionalized molecular complex is then physisorbed onto the surface through π-interactions.
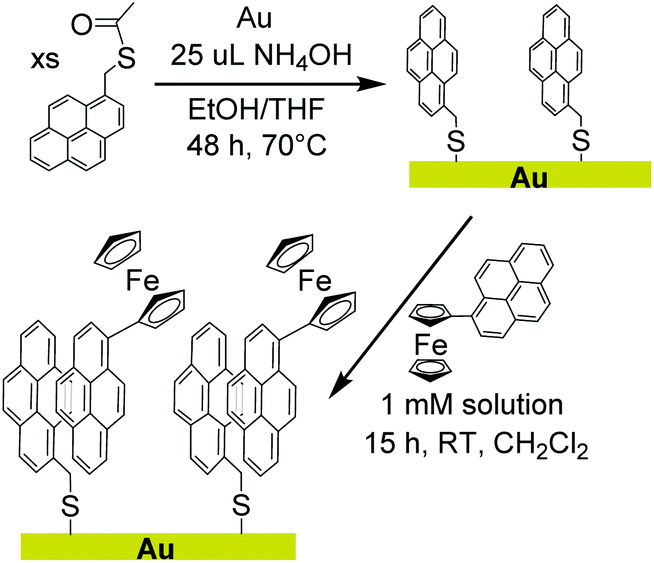 | ||
| Scheme 1 | ||
To demonstrate the feasibility, stability, and facile electron transfer of this new method of surface modification, a pyrene monolayer was established on a gold foil electrode. A pyrene-functionalized ferrocene with well-defined redox properties was used to interrogate physisorption and electron transfer at the interface. The studies confirm that covalently bound pyrene can sufficiently alter the surface properties of the electrode to allow physisorption and electron transfer to pyrene-functionalized molecular complexes.
This method represents a more benign route to functionalize electrodes with sensitive molecular complexes, effectively expanding the scope of molecular species that can be used to modify non-carbon surfaces. This approach would be generally useful for the broad range of applications for chemically modified electrodes in chemical sensing, energy conversion and storage, molecular electronics, and electrochromic materials.
Experimental
General experimental procedures
1H NMR spectra were recorded on a Bruker CRYO500 (500 MHz) spectrometer at 20 °C. All 1H chemical shifts have been internally calibrated to the monoprotio impurity of the deuterated solvent. 13C NMR spectra were recorded on a Bruker CRYO500 (126 MHz) at 20 °C.All electrochemical experiments were carried out in air in 0.1 M perchloric acid solutions. Cyclic voltammetry experiments were performed with a Pine Wavedriver 10 potentiostat using Aftermath software. All potentials are referenced to Ag/AgCl unless otherwise noted. Electrospray ionization (ESI) mass spectra were collected using an ESI LC-TOF Micromass LCT. High resolution mass spectra were obtained from a Waters (Micromass) LCT premier #1.
Fourier transform infrared (FTIR) surface spectra of the gold electrodes and compound 1 were collected on a Jasco FTIR-4700 – ATR-PRO ONE in air. FTIR of molecular compound 2 was recorded on a Thermo Scientific Nicolet iS5 spectrometer with an iD5 ATR attachment in a nitrogen filled glovebox. Samples were prepared by evaporating a dichloromethane solution of the sample onto the ATR crystal.
X-ray photoelectron spectra were acquired with a Kratos Analytical AXIS Supra spectrometer utilizing monochromatic Al Kα radiation (1486.7 eV, 250 W) under ultra-high vacuum (UHV) conditions (∼10−9 Torr). The binding energies were referred to the Au 4f7/2 signal at 84.0 eV. Survey scans were used for elemental composition of the gold surface to demonstrate purity while high resolution region scans provided information on chemical shifts and position of peaks for specific elements. Spectral data was analysed using Computer Aided Surface Analysis for X-ray Photoelectron Spectroscopy (CasaXPS). Shirley or linear backgrounds were used and Gaussian-Lorentzian lineshapes were used to model peaks. An offset Shirley background was required only for the Fe 2p XP spectrum shown in Fig. 1.
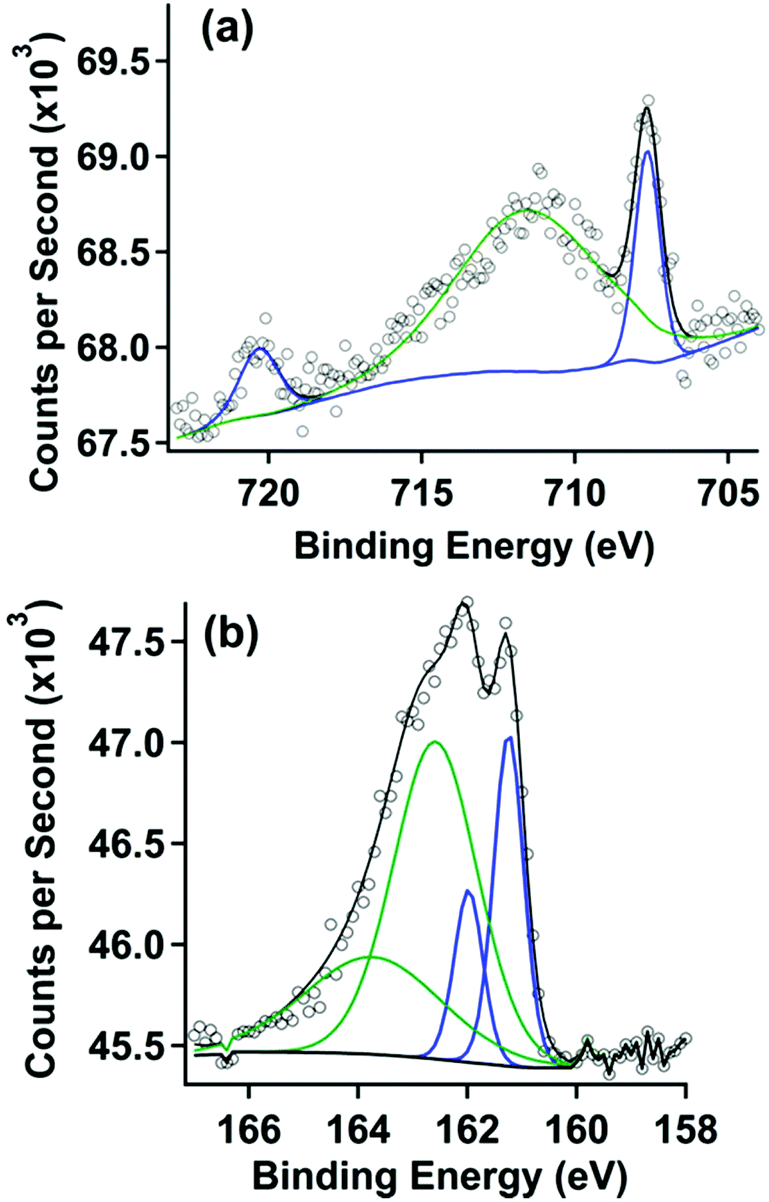 | ||
| Fig. 1 (a) Fe 2p XP spectrum and (b) S 2p XP spectrum of a gold surface after covalent attachment with pyrene followed by treatment in a 1 mM solution of 2. | ||
Synthesis and materials
All reactions and manipulations were performed under a nitrogen atmosphere using standard Schlenk techniques or a Vacuum Atmospheres glovebox unless otherwise indicated. Gold foil, 0.01 mm thickness, 99.9% pure, was purchased from Goodfellow. Solvents, with the exception of isopropanol and methanol, were dried using an activated alumina column. Water was purified by a Barnstead NANOpure ultrapure water system. All glassware was cleaned by immersion in concentrated chromic acid solution prepared using a literature procedure.25 Sonication was performed in a SPER scientific ultrasonic cleaner (42 MHz). 1-(Bromomethyl)pyrene26 was prepared using a previously published procedure.27 All other chemicals were purchased and used without further purification.Synthesis of molecular precursors
Preparation of chemically modified gold electrodes
All glassware used in the following preparations were cleaned in a concentrated chromic acid bath solution (chromerge) for at least 1 hour and then rinsed thoroughly with water before being dried in an oven.Results and discussion
Synthesis
The thioacetate functionalized pyrene (1) was synthesized for covalent attachment onto gold. Compound 1 was isolated by nucleophilic substitution of 1-bromomethylpyrene with potassium thioacetate and was characterized by 1H NMR (Fig. S1†), 13C NMR (Fig. S2†), and high resolution mass spectrometry as described in the Experimental section. Thioacetate derivatives have greater stability than their thiol counterparts because they are not prone to oxidation to form disulfides or sulfoxides, making them easier to isolate and purify.28 The thioacetate was deprotected using a strong base (NH4OH) in situ prior to forming a covalent attachment to the surface. The pyrene-functionalized ferrocene (2) was synthesized via a Pd catalysed cross-coupling reaction and was characterized by 1H NMR (Fig. S3†), 13C NMR (Fig. S4†), and high resolution mass spectrometry.Electrode surface analysis by X-ray photoelectron spectroscopy
X-ray photoelectron (XP) spectroscopy was used to characterize the clean gold electrode surface (survey scan, Fig. S5†). As expected, no peaks are observed in the S 2p (Fig. S6†) and Fe 2p (Fig. S7†) region. After covalent attachment of the thiol-functionalized pyrene, the XP spectra also showed no features in the Fe 2p (Fig. S8†) region except a minor peak consistent with Fe(III) oxide, which we attribute to trace impurities from the solvents used in sample preparation. Peaks in the S 2p (Fig. S9†) region were consistent with thiol covalently bound to gold.29 These features are also observed after treatment with pyrene-functionalized ferrocene, and are described in more detail below.The gold substrate modified by covalent attachment of pyrene was then soaked in a solution of 2 in dichloromethane and rinsed with acetonitrile, a solvent in which 2 is also soluble. The XP spectra for the chemically modified electrode are shown in Fig. 1. The XP spectrum of the Fe 2p region in Fig. 1a displays two peaks at 707.6 and 720.3 eV, which correlate to the Fe 2p3/2 and Fe 2p1/2 emissions, respectively. These features are consistent with spectra taken of surfaces with covalently bound ferrocene.30,31 A broad third peak is seen at 711.6 eV, attributed to the impurity acquired during the monolayer formation process described above (Fig. S8†).
The XP spectrum of the S 2p region is shown in Fig. 1b. A doublet in this spectrum was observed at binding energies of 161.2 and 162.2 eV. Modelling this doublet revealed two sets of doublets correlating to two different sulphur species. The doublet at binding energies of 162.2 eV and 163.4 eV, representing S 2p3/2 and 2p1/2 peaks respectively, and are consistent with sulphur species forming Au–S bonds.29 The lower intensity doublet at 161.2 eV appears at roughly half the concentration of the covalently-bound thiol described above. The lower intensity doublet does not correlate to peaks that would result from X-ray damage, unbound thiol, or a gold metal sulphide. Another low intensity peak at 168.6 eV (not shown) is apparent in all samples, and is attributed to a contaminant metal sulphate species that could not be removed from the surface through the cleaning methods described. However the XPS data confirms that (1) the thiol-functionalized pyrene is covalently attached to the gold surface and (2) the pyrene-functionalized ferrocene is present on the surface.
To confirm that physisorption of the pyrene-functionalized ferrocene is due to the presence of pyrene covalently attached to the gold surface, a clean gold electrode was also prepared and treated with 2 using the exact same procedure used in the prior experiment. The XP spectrum of this sample, shown in Fig. S10,† did not show any peaks that correspond to iron, but instead matched the spectrum of the clean gold surface. The absence of 2 on the surface indicates that it is washed off of the surface after treatment when there is no pyrene covalently attached to the gold. In contrast, the interaction between the pyrene at the surface and the pyrene functionalized on the ferrocene is sufficient to maintain physisorption to the surface, even after the acetonitrile wash.
Electrode surface analysis by infrared spectroscopy
ATR-FTIR measurements were used to characterize the modified gold electrodes. The infrared vibrational spectra for isolated 1 and 2 are shown in Fig. S11 and S12,† respectively. The clean gold electrode (Fig. S13†) did not show any significant vibrational stretches and was used as the background subtraction for subsequent measurements.An infrared spectrum of the gold surface treated with thiol-functionalized pyrene followed by physisorption of the pyrene functionalized ferrocene (2) (Fig. 2) displayed peaks at 2925 and 2853 cm−1, in the range of an aromatic C–H stretch as well as an sp3 C–H stretch from the bound thiol.
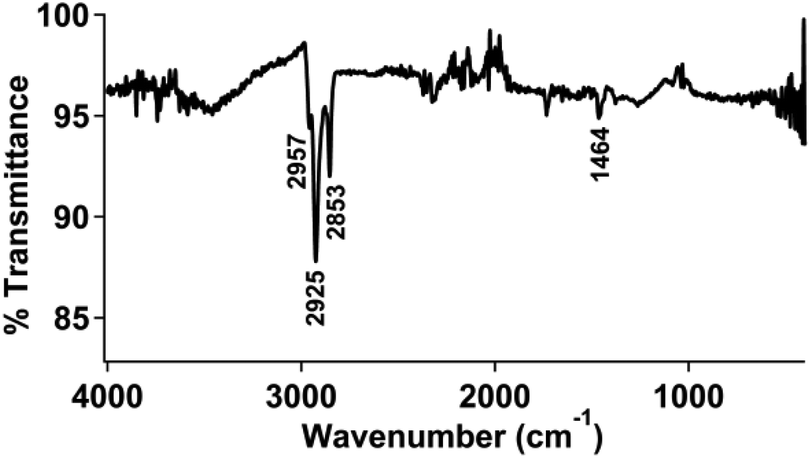 | ||
| Fig. 2 FTIR spectrum of a pyrene-modified gold electrode with 2 physisorbed to the surface. | ||
We also examined a clean gold electrode prepared and treated with 2 using the same procedure described above. The infrared spectrum of this sample, shown in Fig. S14,† did not show any significant absorption in the covalent organic region, but instead appeared very similar to the spectrum of a clean gold surface. Again, this supports that the covalently-bound pyrene facilitates physisorption of the pyrene-functionalized ferrocene.
Electrochemical characterization of modified electrodes
The electrode materials were investigated using cyclic voltammetry in 0.1 M perchloric acid. The cyclic voltammograms of clean gold electrodes, as well as electrodes modified with pyrene, did not show any features between 0.3 and 0.7 V vs. Ag/AgCl, as shown in Fig. S15 and S16,† respectively. However, upon treatment of the latter surface with pyrene-functionalized ferrocene (2), a reversible couple is observed (E1/2 = 495 mV) (Fig. 3a).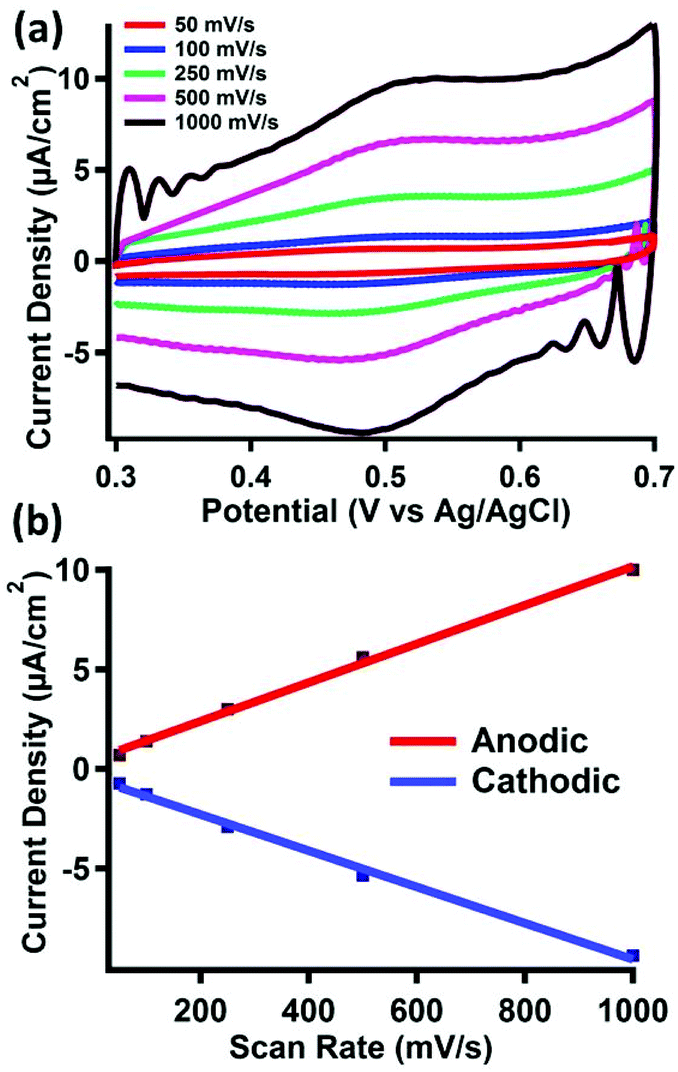 | ||
| Fig. 3 Electrochemical characterization of 2 non-covalently bound to a pyrene-modified gold surface. (a) Variable scan rate cyclic voltammogram of a chemically modified gold working electrode in 0.1 M HClO4 solution with Ag/AgCl reference and a glassy carbon auxiliary electrode. (b) Current density vs. scan rate plot from the cyclic voltammogram in (a). | ||
Cyclic voltammograms of 2 taken under homogeneous conditions in acetonitrile displays a 1 e− reversible couple, as shown in Fig. S17–S19.† Due to the insolubility of 2, we were unable to perform a similar homogeneous cyclic voltammogram in 0.1 M perchloric acid to compare to the redox event we observe after physisorption to the surface. However, the E1/2 value we observe is consistent with similar reports of surface bound ferrocene species measured in aqueous solvents.32 The scan rate also displays a linear dependence on the peak anodic or cathodic current (Fig. 3b), indicating fast electron transfer to a surface-bound species.
Surface coverage of 2 was estimated by integration of the faradaic current of the anodic wave. Surface coverage was calculated between Γ = 5–28 pmol cm−2 across all samples. The same method was used to analyze the stability of 2 bound to the surface after repeated oxidation/reduction cycles (Fig. S20†). Experiments with up to 100 redox cycles did not show evidence for loss of 2 from the surface, as the Faradaic current for the cathodic and anodic events remains relatively constant. The results of this experiment, summarized in Table S1,† suggest good stability of the π–π interactions under redox conditions in acidic media.
A clean gold electrode treated with 2 under the same conditions did not show any redox active species in the same range (Fig. S21†), indicating no physisorption of the ferrocene to clean gold surfaces. This is consistent with the conclusions from the XPS and infrared spectroscopy measurements.
Conclusions
The surface characterization studies demonstrate that modification of a gold electrode by covalent attachment of pyrene permits physisorption of a pyrene-functionalized ferrocene. Furthermore, the pyrene-functionalized ferrocene is stable on the surface even after washing with solvents in which the molecular species is soluble. The electrochemical studies demonstrate that electron transfer through this interface is facile, demonstrating ideal reversible behaviour for the ferrocene redox couple.This approach towards chemically modifying a surface requires covalent attachment of pyrene to the target surface, which is generally facile because of the inert nature of pyrene and ease of installing a wide variety of functional groups. More significantly, this method permits modification of surfaces with pyrene-functionalized molecules, which are synthetically more accessible than many of the reactive functional groups traditionally used to form covalent bonds at the surface. Non-covalent attachment of molecular complexes functionalized with pyrene has demonstrated great success in modifying carbon-based electrodes; the method described herein extends this utility to alternative electrode surfaces.
Acknowledgements
The authors would like to thank Dr. Qiyin Lin, Dr. Ich Tran, and Zhongyue Luan for assistance and guidance with the XPS experiments, and Prof. Manny Soriaga and Dr. Alnald Javier for helpful discussions and advice on preparing the Au electrodes. This material is based upon work supported by the Hellman Faculty Fellows Fund and the School of Physical Sciences at UC Irvine. XPS work was performed at the UC Irvine Materials Research Institute (IMRI) using instrumentation funded in part by the National Science Foundation Major Research Instrumentation Program under grant no. CHE-1338173.Notes and references
- R. W. Murray, Acc. Chem. Res., 1980, 13, 135–141 CrossRef CAS.
- R. W. Murray, A. G. Ewing and R. A. Durst, Anal. Chem., 1987, 59, 379A–390A CAS.
- B. Ulgut and H. D. Abruña, Chem. Rev., 2008, 108, 2721–2736 CrossRef CAS PubMed.
- E. Bakker and Y. Qin, Anal. Chem., 2006, 78, 3965–3984 CrossRef CAS PubMed.
- C. E. D. Chidsey, Science, 1991, 251, 919–922 CrossRef CAS PubMed.
- C. Amatore, E. Maisonhaute, B. Schöllhorn and J. Wadhawan, ChemPhysChem, 2007, 8, 1321–1329 CrossRef CAS PubMed.
- H. D. Sikes, J. F. Smalley, S. P. Dudek, A. R. Cook, M. D. Newton, C. E. D. Chidsey and S. W. Feldberg, Science, 2001, 291, 1519–1523 CrossRef CAS PubMed.
- A. D. Richard and A. B. Elmo, in Fundamentals and Applications of Chemical Sensors, American Chemical Society, 1986, ch. 14, vol. 309, pp. 245–255 Search PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- L. Yang, H. Ozawa, M. Koumoto, K. Yoshikawa, M. Matsunaga and M.-A. Haga, Chem. Lett., 2015, 44, 160–162 CrossRef CAS.
- A. Navaee and A. Salimi, J. Mater. Chem. A, 2015, 3, 7623–7630 CAS.
- N. Kong, J. J. Gooding and J. Liu, J. Solid State Electrochem., 2014, 18, 3379–3386 CrossRef CAS.
- B. Reuillard, A. Le Goff and S. Cosnier, Chem. Commun., 2014, 50, 11731–11734 RSC.
- C. Walgama and S. Krishnan, J. Electrochem. Soc., 2014, 161, H47–H52 CrossRef CAS.
- N. Lalaoui, K. Elouarzaki, A. L. Goff, M. Holzinger and S. Cosnier, Chem. Commun., 2013, 49, 9281–9283 RSC.
- A. Le Goff, B. Reuillard and S. Cosnier, Langmuir, 2013, 29, 8736–8742 CrossRef CAS PubMed.
- E. J. Parra, F. X. Rius and P. Blondeau, Analyst, 2013, 138, 2698–2703 RSC.
- S.-N. Ding, D. Shan, S. Cosnier and A. Le Goff, Chem. – Eur. J., 2012, 18, 11564–11568 CrossRef CAS PubMed.
- A. Le Goff, K. Gorgy, M. Holzinger, R. Haddad, M. Zimmerman and S. Cosnier, Chem. – Eur. J., 2011, 17, 10216–10221 CrossRef CAS PubMed.
- J. Bartelmess, B. Ballesteros, G. de la Torre, D. Kiessling, S. Campidelli, M. Prato, T. Torres and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 16202–16211 CrossRef CAS PubMed.
- S. D. Mhaske, M. Ray and S. Mazumdar, Inorg. Chim. Acta, 2010, 363, 2804–2811 CrossRef CAS.
- J. Liu, O. Bibari, P. Mailley, J. Dijon, E. Rouviere, F. Sauter-Starace, P. Caillat, F. Vinet and G. Marchand, New J. Chem., 2009, 33, 1017–1024 RSC.
- W. Birch, A. Carré and K. L. Mittal, in Developments in Surface Contamination and Cleaning, ed. R. K. L. Mittal, William Andrew Publishing, Norwich, NY, 2008, pp. 693–723 Search PubMed.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- J. L. Bartels, P. Lu, A. Walker, K. Maurer and K. D. Moeller, Chem. Commun., 2009, 5573–5575 RSC.
- J. M. Tour, L. Jones, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. Atre, J. Am. Chem. Soc., 1995, 117, 9529–9534 CrossRef CAS.
- P. Angelova, H. Vieker, N.-E. Weber, D. Matei, O. Reimer, I. Meier, S. Kurasch, J. Biskupek, D. Lorbach, K. Wunderlich, L. Chen, A. Terfort, M. Klapper, K. Müllen, U. Kaiser, A. Gölzhäuser and A. Turchanin, ACS Nano, 2013, 7, 6489–6497 CrossRef CAS PubMed.
- J. Trasobares, F. Vaurette, M. François, H. Romijn, J.-L. Codron, D. Vuillaume, D. Théron and N. Clément, Beilstein J. Nanotechnol., 2014, 5, 1918–1925 CrossRef CAS PubMed.
- L. P. Méndez De Leo, E. de la Llave, D. Scherlis and F. J. Williams, J. Chem. Phys., 2013, 138, 114707 CrossRef PubMed.
- C. E. D. Chidsey, C. R. Bertozzi, T. M. Putvinski and A. M. Mujsce, J. Am. Chem. Soc., 1990, 112, 4301–4306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for 1, 2, and the gold substrates as well as electrochemical data for 2 and the gold substrates. See DOI: 10.1039/c6qi00010j |
| This journal is © the Partner Organisations 2016 |